Convergent synthesis of a hexasaccharide corresponding to the cell wall O-antigen of Escherichia coli O41†
Tamashree
Ghosh
,
Abhishek
Santra
and
Anup Kumar
Misra
*
Bose Institute, Division of Molecular Medicine, P-1/12, C.I.T. Scheme VII-M, Kolkata-700054, India. E-mail: akmisra69@gmail.com; Fax: +91-33-2355 3886; Tel: +91-33-2569 3240
First published on 20th November 2013
Abstract
A convergent synthetic strategy has been developed for the synthesis of a hexasaccharide corresponding to the O-antigen of E. coli O41 using stereoselective [3 + 3] block glycosylation strategy. The target compound was synthesized assembling a series of suitably protected monosaccharide intermediates. Thioglycoside derivatives have been used as glycosyl donors in most of the glycosylation reactions. All intermediate steps are high yielding and the glycosylation steps are highly stereoselective. A number of recently developed methodologies have been used in the synthesis.
Introduction
Escherichia coli is a facultative anaerobic gram-negative rod that belongs to the family Enterobacteriaceae.1 Although most of the E. coli strains found in the human colonic flora are usually non-pathogenic, certain strains acquire virulence and are responsible for a number of infections in humans.2 In general three types of E. coli infections are found in humans, which include (i) diarrhoea; (ii) sepsis/meningitis and (iii) urinary tract infections.3 Diarrhoeal infections are a serious concern in the developed countries with inadequate sanitation.4E. coli strains associated with diarrhoeal infections are broadly classified in five subgroups based on their mechanism of actions,5 which include enteroinvasive E. coli (EIEC), enterotoxigenic E. coli (ETEC), enteropathogenic E. coli (EPEC), enterohemorrhagic E. coli (EHEC) and enteroaggregative E. coli (EAEC). EHEC strains are also termed as Shiga-toxin producing E. coli (STEC) and verotoxin producing E. coli (VTEC), which act through the production of Shiga-like toxin during the initial stage of infection to the host.6 Among various subgroups, STEC or VTEC are responsible for several diarrhoeal outbreaks as well as haemorrhagic colitis and haemorrhagic uremic syndromes.7,8 The most well-documented E. coli strain associated STEC is E. coli O157, which has been the cause of several diarrhoeal outbreaks in the developing countries and Japan.9 The oligosaccharide repeating units of the cell wall O-antigen of the pathogenic bacterial strains play a pivotal role in the bacterial infection due to its presence in the outer layer of the cell wall.10E. coli O41 strain belongs to the STEC or VTEC category and has been associated with diarrhoeal infections and colitis.11 Zhu et al. established the structure of the hexasaccharide repeating unit of the O-antigen of E. coli O41, which is composed of two α-linked L-frucose, two β-linked D-glucosamine, α-linked D-galactose and β-linked D-glucuronic acid moieties.12 In the current scenario, the emergence of multidrug resistant bacterial strains demands for the development of novel effective antibacterial agents. In the past, glycoconjugate derivatives related to the cell wall oligosaccharides (O-antigens) have been evaluated in the development of antimicrobial agents.13–15 For the detailed biological studies of the cell wall oligosaccharides and glycoconjugate derivatives, considerable amounts of oligosaccharides are required, which cannot be accessible from the natural source and hence chemical synthesis of the oligosaccharide repeating units becomes pertinent. In this report, a convergent synthetic strategy is presented for the synthesis of a hexasaccharide corresponding to the O-antigen of E. coli O41 as its p-methoxyphenyl glycoside (Fig. 1).Structure of the hexasaccharide repeating unit of the O-antigen of Escherichia coli O41:
| →3)-[β-D-GlcpA-(1→4)]-α-L-Fucp-(1→4)-β-D-GlcpNAc-(1→3)-α-L-Fucp-(1→3)-β-D-GlcpNAc-(1→3)-α-D-Galp-(1→ |
 | ||
| Fig. 1 Structure of the synthesized hexasaccharide as its p-methoxyphenyl glycoside and its synthetic intermediates. | ||
Results and discussion
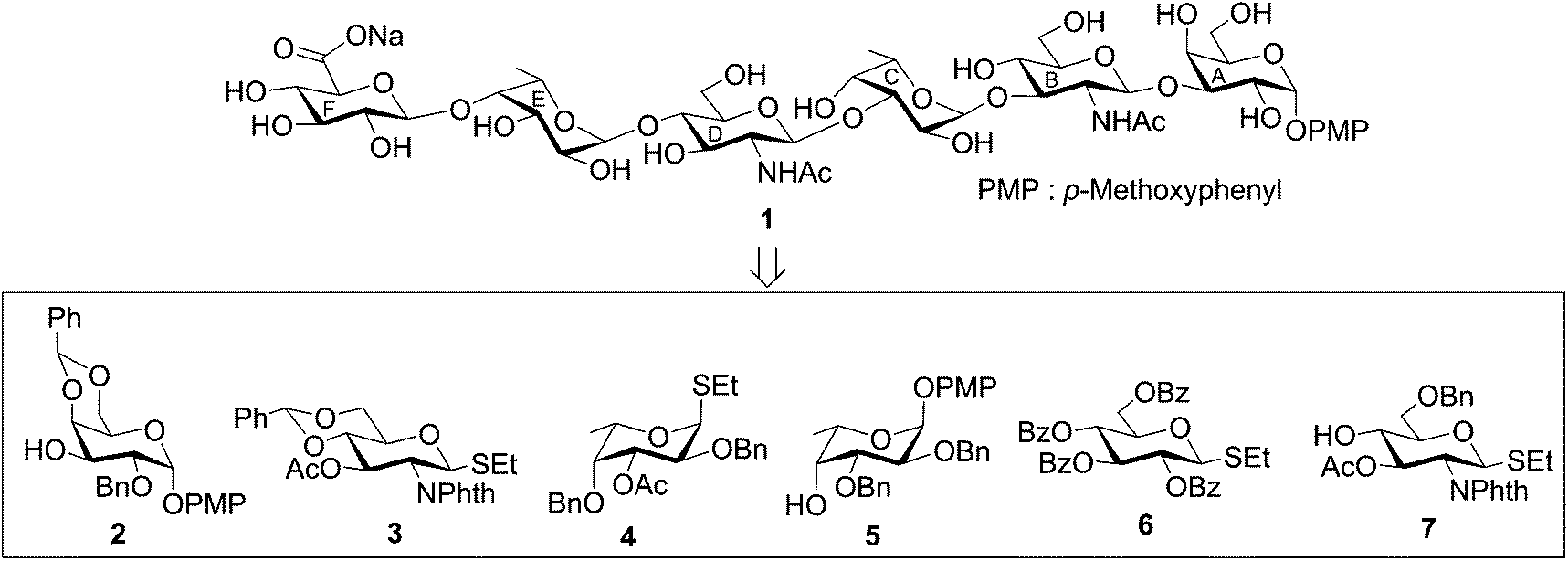
The target hexasaccharide 1 was synthesized as its p-methoxyphenyl (PMP) glycoside leaving the possibility for further glycoconjugate preparation by the oxidative deprotection of PMP group from compound 1. The synthetic strategy has been designed with the minimum number of steps required for achieving the target molecule. Thus a [3 + 3] block glycosylation strategy has been adopted involving stereoselective coupling of a trisaccharide glycosyl acceptor (11) with a trisaccharide thioglycoside donor (14). The retrosynthetic analysis of compound 1 led to a number of suitably functionalized monosaccharide intermediates 2,163,174,185,16619 and 7,20 which were prepared from the commercially available reducing sugars following reported reaction methodologies (Fig. 1). The notable features of the synthetic strategy include (a) [3 + 3] block glycosylation; (b) formation of a number of stereoselective 1,2-cis-glycosyl linkages; (c) generalized reaction conditions for the glycosylations using thioglycoside derivatives as glycosyl donors; (d) TEMPO mediated selective oxidation of primary hydroxyl group to carboxyl group at a late-stage of the synthesis, etc.p-Methoxyphenyl 4,6-O-benzylidene-2-O-benzyl-α-D-galactopyranoside (2)16 was allowed to couple stereoselectively with ethyl 3-O-acetyl-4,6-O-benzylidene-2-deoxy-2-N-phthalimido-1-thio-β-D-glucopyranoside (3)17 in the presence of a combination of N-iodosuccinimide (NIS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf)21,22 in dichloromethane to furnish disaccharide derivative (8) in 77% yield. The formation of compound 8 was confirmed from its spectral analysis [signals at δ 5.83 (d, J = 8.5 Hz, H-1B), 5.55, 5.52 (2 s, 2 PhCH), 5.11 (d, J = 3.5 Hz, H-1A) in the 1H NMR and δ 101.7 (PhCH), 100.5 (PhCH), 100.0 (C-1B), 96.8 (C-1A) in the 13C NMR spectra]. De-O-acetylation of compound 8 using sodium methoxide23 resulted in the formation of disaccharide acceptor 9 in 94% yield. Stereoselective 1,2-cis-glycosylation of compound 9 with ethyl 3-O-acetyl-2,4-di-O-benzyl-1-thio-β-L-fucopyranoside (4)18 in the presence of a combination of NIS–TMSOTf21,22 in the mixed solvent dichloromethane and diethyl ether furnished trisaccharide derivative 10 in 74% yield together with a minor quantity (∼8%) of its other isomer, which was separated by column chromatography. Stereochemistry at the glycosyl linkages in compound 10 was confirmed from the NMR spectral analysis [δ 5.67 (d, J = 8.5 Hz, H-1B), 5.57, 5.52 (2 s, 2 PhCH), 5.08 (d, J = 3.5 Hz, H-1A), 4.86 (d, J = 3.5 Hz, H-1C) in the 1H NMR spectra and δ 101.7 (PhCH), 100.3 (PhCH), 100.2 (C-1B), 98.6 (C-1C), 96.9 (C-1A) in the 13C NMR spectra]. Formation of a new 1,2-cis-glycosyl linkage in compound 10 was unambiguously confirmed using gated 1H coupled 13C NMR spectral analysis. Appearance of JC1–H1 coupling constants 169.0 Hz, 170.0 Hz and 156.0 Hz supported the presence of two axial (1,2-cis) and one equatorial (1,2-trans) glycosyl bonds in the molecule.24 Compound 10 was treated with sodium methoxide23 to give trisaccharide acceptor 11 in 96% yield, which has been used in the block glycosylation step for the preparation of hexasaccharide derivative (Scheme 1).
 | ||
Scheme 1 Reagents: (a) N-iodosuccinimide (NIS), TMSOTf, MS-4Å, CH2Cl2, −15 °C, 30 min, 77%; (b) 0.05 M CH3ONa, CH3OH, room temperature, 2 h, 94% for compound 9 and 96% for compound 11; (c) NIS, TMSOTf, MS-4Å, CH2Cl2–Et2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3; v/v), −10 °C, 30 min, 74%. :3; v/v), −10 °C, 30 min, 74%. | ||
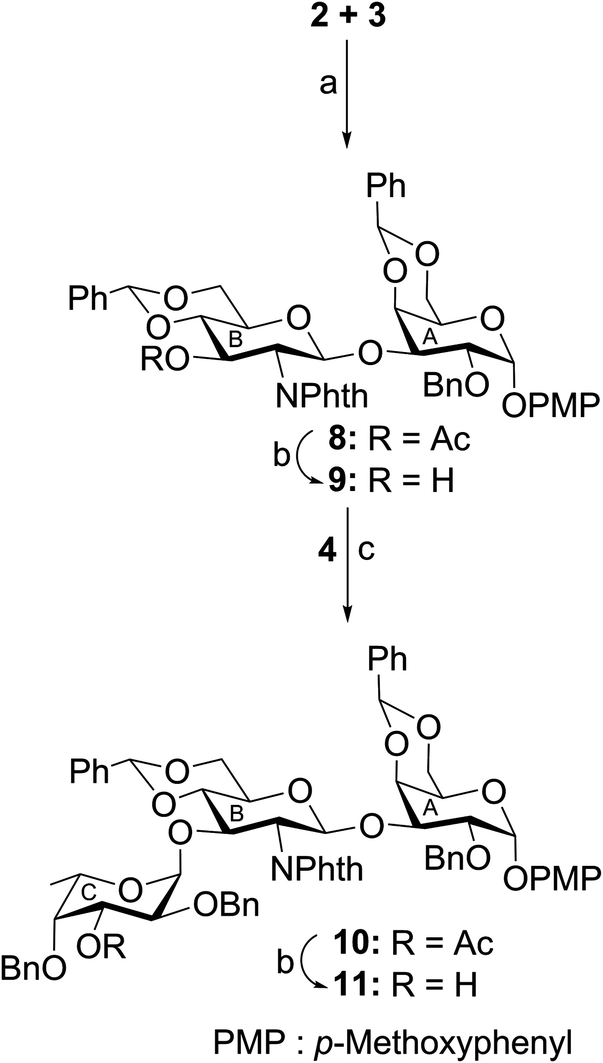
In another experiment, stereoselective glycosylation of p-methoxyphenyl 2,3-di-O-benzyl-α-L-fucopyranoside (5)16 and ethyl 2,3,4,6-tetra-O-benzoyl-1-thio-β-D-glucopyranoside (6)19 in the presence of NIS–TMSOTf21,22 furnished disaccharide derivative 12 in 73% yield. Formation of compound 12 was confirmed from its spectral analysis [signals at δ 5.17 (d, J = 3.5 Hz, H-1E), 5.09 (d, J = 8.0 Hz, H-1F) in the 1H NMR and δ 101.7 (C-1F), 97.5 (C-1E) in the 13C NMR spectra]. Oxidative removal of the p-methoxyphenyl group from compound 12 using ammonium cerium(IV) nitrate (CAN)25 followed by treatment of the hemiacetal derivative with trichloroacetonitrile in the presence of DBU26 led to the formation of disaccharide trichloroacetimidate derivative 13 in 70% yield. Stereoselective 1,2-cis-glycosylation of the trichloroacetimidate derivative 13 with ethyl 3-O-acetyl-6-O-benzyl-2-deoxy-2-N-phthalimido-1-thio-β-D-glucopyranoside (7)20 following a newly developed reaction condition using nitrosyl tetrafluoroborate27 as glycosyl activator in the mixed solvent dichloromethane–diethyl ether (1:2) furnished trisaccharide thioglycoside derivative 14 in 70% yield together with a minor quantity (∼6%) of its other isomer, which was separated using column chromatography. The stereochemistry of the glycosyl linkages in compound 14 was established using NMR spectral analysis [signals at δ 5.43 (d, J = 10.5 Hz, H-1D), 4.96 (d, J = 8.0 Hz, H-1F), 4.73 (d, J = 3.0 Hz, H-1E) in the 1H NMR and δ 101.9 (C-1F) (JC1–H1 = 156.0 Hz), 100.5 (C-1E) (JC1–H1 = 169.0 Hz), 80.6 (C-1D) (JC1–H1 = 157.0 Hz) in the 13C NMR spectra] (Scheme 2). The presence of a newly formed 1,2-cis-glycosyl linkage in compound 14 was unambiguously confirmed from the JC1–H1 coupling constants in the gated 1H coupled 13C NMR spectral analysis.24
 | ||
| Scheme 2 Reagents: (a) NIS, TMSOTf, MS-4Å, CH2Cl2, −20 °C, 30 min, 73%; (b) CAN, CH3CN–H2O (9:1), 0–5 °C, 3 h; (c) CCl3CN, DBU, CH2Cl2, −5 °C, 1 h, 70%; (d) NOBF4, CH2Cl2–Et2O (1:2; v/v), −10 °C, 1 h, 70%. | ||
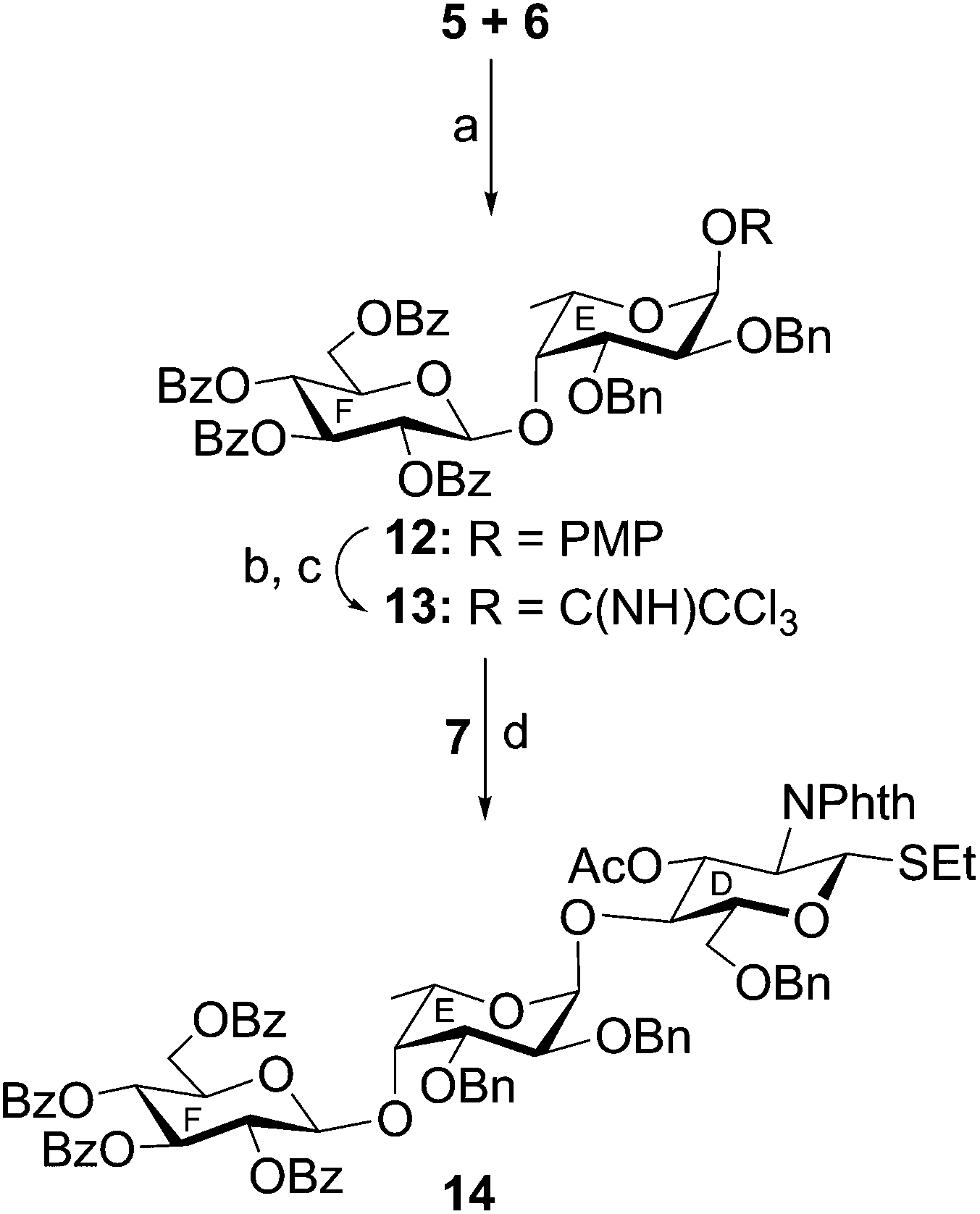
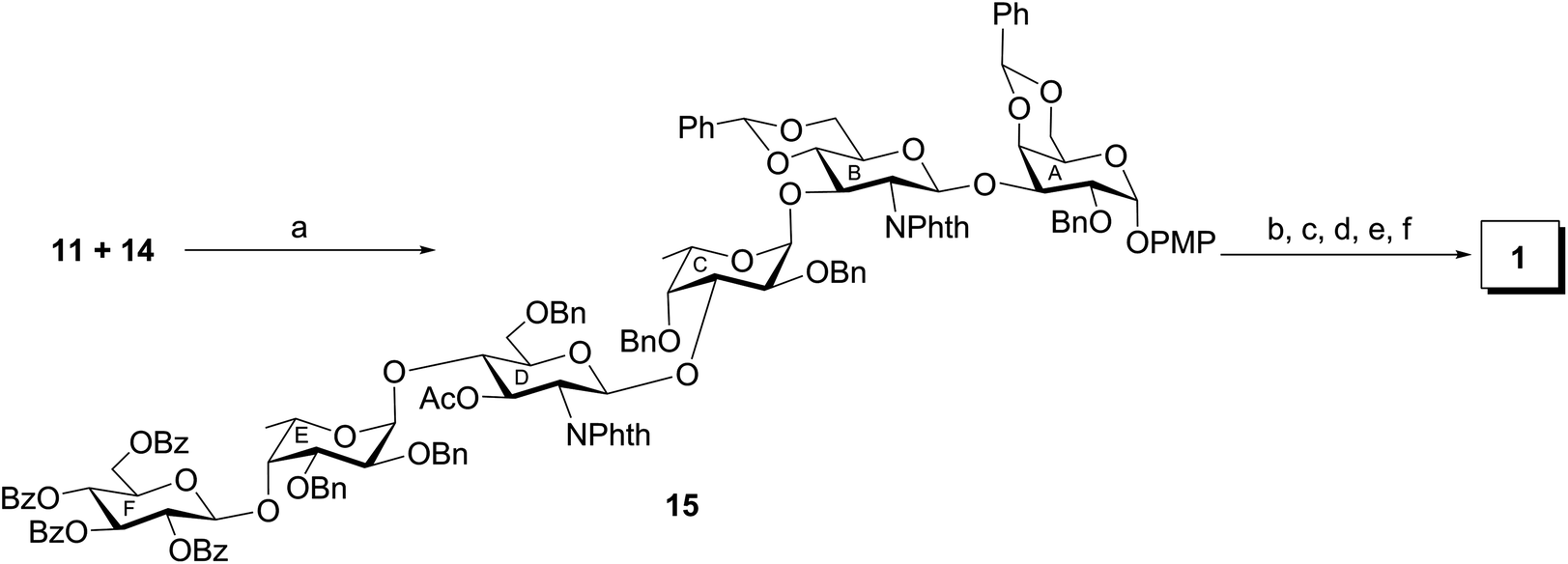
Finally, a [3 + 3] block glycosylation strategy has been adopted for the synthesis of hexasaccharide derivative (15) by the stereoselective glycosylation of compound 11 and compound 14. For this purpose, compound 11 was reacted with thioglycoside derivative 14 in the presence of NIS–TMSOTf21,22 in dichloromethane to furnish hexasaccharide derivative 15 in 66% yield. Formation of compound 15 was confirmed from its spectral analysis [signals at δ 5.48 (d, J = 8.5 Hz, H-1B), 5.46 (d, J = 8.5 Hz, H-1D), 5.05 (d, J = 3.0 Hz, H-1A), 4.97 (d, J = 8.0 Hz, H-1F), 4.71 (d, J = 3.0 Hz, H-1C), 4.51 (d, J = 3.0 Hz, 1H, H-1E) in the 1H NMR and δ 101.8 (C-1F), 100.4 (C-1B), 100.3 (C-1C), 99.2 (C-1E), 96.8 (C-1A), 95.0 (C-1D) in the 13C NMR spectra]. In order to achieve compound 1 containing D-glucuronic acid moiety, compound 15 was subjected to a series of synthetic transformations involving (a) conversion of N-phthaloyl group to acetamido group by the removal of phthaloyl group28 using hydrazine monohydrate followed by N- and O-acetylation using acetic anhydride and pyridine and de-O-acylation using sodium methoxide; (b) TEMPO mediated selective oxidation of the primary hydroxyl group to the carboxylic group;29 and (c) catalytic transfer hydrogenation30 of the oxidized hexasaccharide derivative using triethylsilane and 10% Pd–C to furnish compound 1, which was passed through a column of Dowex 50W X (Na+) and then through a Sephadex LH-20 gel column to give pure compound 1 as its sodium salt and p-methoxyphenyl glycoside in 53% overall yield. Formation of compound 1 was unambiguously confirmed from its spectral analysis [signals at δ 5.34 (br s, H-1A), 4.99, 4.95 (2 d, J = 8.5 Hz each, H-1B, H-1D), 4.62 (d, J = 8.0 Hz, H-1F), 4.35, 4.34 (2 d, J = 3.0 Hz each, H-1C, H-1E) in the 1H NMR and δ 103.1 (3C, C-1C, C-1E, C-1F), 100.6 (C-1B), 100.1 (C-1D), 98.5 (C-1A) in the 13C NMR spectra] (Scheme 3).
 | ||
| Scheme 3 Reagents: (a) NIS, TMSOTf, MS-4Å, CH2Cl2, −10 °C, 45 min, 66%; (b) NH2NH2·H2O, EtOH, 80 °C, 10 h; (c) acetic anhydride, pyridine, room temperature, 3 h; (d) 0.1 M CH3ONa, CH3OH, room temperature, 2 h; (e) (i) TEMPO, NaBr, TBAB, NaOCl, CH2Cl2, H2O, NaHCO3, 0–5 °C, 3 h; (ii) tert-butanol, 2-methyl-but-2-ene, NaClO2, NaH2PO4, room temperature, 3 h; (f) Et3SiH, 10% Pd–C, CH3OH–CHCl3 (5:1; v/v), room temperature, 10 h, overall 53%. | ||
Conclusions
In summary, a hexasaccharide (1) corresponding to the O-antigen of the cell wall polysaccharide of E. coli O41 has been synthesized as its p-methoxyphenyl glycoside and sodium salt in excellent yield using [3 + 3] block glycosylation approach. During the synthesis of the target molecule, a number of novel reaction conditions have been applied for the functional group manipulations. In most of the glycosylation reactions, thioglycoside derivatives have been used as selective glycosyl donors. The stereochemistries of the glycosyl linkages were confirmed by the NMR spectral analysis. All synthetic intermediates were solid and fully characterized using spectral analysis. A late stage TEMPO mediated selective oxidation of the primary hydroxyl group in a biphasic reaction condition efficiently furnished D-glucuronic acid moiety in the hexasaccharide derivative. All intermediate glycosylation steps were high yielding with excellent stereo control.Experimental
General methods
All reactions were monitored by thin layer chromatography over silica gel-coated TLC plates. The spots on TLC were visualized by warming ceric sulfate [2% Ce(SO4)2 in 2 N H2SO4]-sprayed plates on a hot plate. Silica gel 230–400 mesh was used for column chromatography. 1H and 13C NMR, DEPT 135, 2D COSY and 2D HSQC NMR spectra were recorded on a Bruker Avance DRX 500 MHz spectrometer using CDCl3 and D2O as solvents and TMS as internal reference unless stated otherwise. Chemical shift values are expressed in δ ppm. MALDI-MS were recorded on a Bruker Daltronics mass spectrometer. Elementary analysis was carried out on Carlo Erba analyzer. Optical rotations were measured at 25 °C on a Jasco P-2000 polarimeter. Commercially available grades of organic solvents of adequate purity are used in all reactions.p-Methoxyphenyl (3-O-acetyl-4,6-O-benzylidene-2-deoxy-2-N-phthalimido-β-D-glucopyranosyl)-(1→3)-2-O-benzyl-4,6-O-benzylidene-α-D-galactopyranoside (8)
To a solution of compound 2 (1.5 g, 3.23 mmol) and compound 3 (1.7 g, 3.51 mmol) in anhydrous CH2Cl2 (20 mL) was added MS-4Å (2 g) and the reaction mixture was allowed to stir at room temperature for 30 min under argon. The reaction mixture was cooled to −15 °C. To the cooled reaction mixture were added N-iodosuccinimide (NIS; 830 mg, 3.69 mmol) and TMSOTf (15 μL) and it was stirred at the same temperature for 30 min. The reaction mixture was diluted with CH2Cl2 (100 mL) and filtered through a Celite® bed and washed with CH2Cl2. The combined organic layer was successively washed with 5% Na2S2O3, satd. NaHCO3 and water, dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane–EtOAc (5:1) as eluent to give pure compound 8 (2.2 g, 77%). White solid; m.p. 108–110 °C [EtOH]; [α]25D +32.4 (c 1.2, CHCH3); IR (KBr): 3651, 2924, 1777, 1742, 1721, 1507, 1387, 1223, 1102, 1040, 995, 796 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.75–6.94 (m, 19H, Ar–H), 6.85, 6.74 (2 d, J = 9.0 Hz each, 4H, Ar–H), 5.95 (t, J = 9.5 Hz each, 1H, H-3B), 5.83 (d, J = 8.5 Hz, 1H, H-1B), 5.55, 5.52 (2 s, 2H, 2 PhCH), 5.11 (d, J = 3.5 Hz, 1H, H-1A), 4.47 (t, J = 8.5 Hz each, 1H, H-2B), 4.45–4.43 (m, 1H, H-5B), 4.34 (d, J = 3.0 Hz, 1H, H-4A), 4.29 (dd, J = 10.5, 3.0 Hz, 1H, H-3A), 4.24 (d, J = 12.5 Hz, 1H, PhCH2), 4.14 (d, J = 12.0 Hz, 1H, H-6aA), 3.99 (d, J = 12.0 Hz, 1H, H-6bA), 3.95 (d, J = 12.5 Hz, 1H, PhCH2), 3.90–3.80 (m, 4H, H-2A, H-4B, H-6abB), 3.74 (s, 3H, OCH3), 3.73–3.72 (m, 1H, H-5A), 1.92 (s, 3H, COCH3); 13C NMR (125 MHz, CDCl3): δ 169.9 (COCH3), 167.4 (Phth), 154.8–114.4 (Ar–C), 101.7 (PhCH), 100.5 (PhCH), 100.0 (C-1B), 96.8 (C-1A), 79.2 (C-2A), 76.5 (C-3A), 76.3 (C-4B), 74.9 (C-4A), 73.1 (PhCH2), 69.8 (C-3B), 69.1 (C-6A), 68.7 (C-6B), 66.0 (C-5B), 63.1 (C-5A), 55.6 (C-2B), 55.5 (OCH3), 20.6 (COCH3); ESI-MS: 908.3 [M + Na]+; anal. calcd for C50H47NO14 (885.30): C, 67.79; H, 5.35; found: C, 67.60; H, 5.50.
p-Methoxyphenyl (4,6-O-benzylidene-2-deoxy-2-N-phthalimido-β-D-glucopyranosyl)-(1→3)-2-O-benzyl-4,6-O-benzylidene-α-D-galactopyranoside (9)
A solution of compound 8 (2 g, 2.26 mmol) in 0.05 M CH3ONa in CH3OH (30 mL) was allowed to stir at room temperature for 2 h. The reaction mixture was neutralized with Dowex 50W-X8 (H+) resin, filtered and concentrated. The crude product was passed through a small pad of SiO2 using hexane–EtOAc (1:1) as eluent to give pure compound 9 (1.8 g, 94%). White solid; m.p. 122–124 °C [EtOH]; [α]25D +25 (c 1.2, CHCH3); IR (KBr): 3476, 2924, 2865, 1775, 1724, 1507, 1486, 1390, 1210, 1175, 1095, 1044, 993, 798 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.75–6.94 (m, 19H, Ar–H), 6.84, 6.72 (2 d, J = 9.0 Hz each, 4H, Ar–H), 5.66 (d, J = 8.5 Hz, 1H, H-1B), 5.58, 5.52 (2 s, 2H, 2 PhCH), 5.09 (d, J = 3.0 Hz, 1H, H-1A), 4.73 (t, J = 10.0 Hz each, 1H, H-3B), 4.42–4.38 (m, 2H, H-2B, H-5B), 4.33 (d, J = 2.5 Hz, 1H, H-4A), 4.29 (dd, J = 10.5, 3.0 Hz, 1H, H-3A), 4.24 (d, J = 12.5 Hz, 1H, PhCH2), 4.11 (d, J = 12.5 Hz, 1H, H-6aA), 3.99 (d, J = 12.5 Hz, 1H, H-6bA), 3.93 (d, J = 12.5 Hz, 1H, PhCH2), 3.90–3.80 (m, 2H, H-2A, H-6aB), 3.74 (s, 3H, OCH3), 3.73–3.70 (m, 2H, H-5A, H-6bB), 3.66 (t, J = 10.0 Hz each, 1H, H-4B); 13C NMR (125 MHz, CDCl3): δ 167.5 (Phth), 155.6–114.1 (Ar–C), 101.9 (PhCH), 100.3 (PhCH), 100.2 (C-1B), 96.8 (C-1A), 82.2 (C-4B), 76.5 (C-4A), 75.8 (C-3A), 75.2 (C-2A), 73.2 (PhCH2), 69.0 (C-6A), 68.7 (C-6B), 68.4 (C-5B), 66.0 (C-3B), 63.2 (C-5A), 56.7 (C-2B), 55.5 (OCH3); ESI-MS: 866.2 [M + Na]+; anal. calcd for C48H45NO13 (843.29): C, 68.32; H, 5.37; found: C, 68.12; H, 5.55.
p-Methoxyphenyl (3-O-acetyl-2,4-di-O-benzyl-α-L-fucopyranosyl)-(1→3)-(4,6-O-benzylidene-2-deoxy-2-N-phthalimido-β-D-glucopyranosyl)-(1→3)-2-O-benzyl-4,6-O-benzylidene-α-D-galactopyranoside (10)
To a solution of compound 9 (1.5 g, 1.78 mmol) and compound 4 (830 mg, 1.93 mmol) in anhydrous CH2Cl2–Et2O (15 mL; 1:3, v/v) was added MS-4Å (2 g) and the reaction mixture was allowed to stir at room temperature for 30 min under argon. The reaction mixture was cooled to −10 °C. To the cooled reaction mixture were added NIS (450 mg, 2.0 mmol) and TMSOTf (5 μL) and it was stirred at the same temperature for 30 min. The reaction mixture was diluted with CH2Cl2 (50 mL) and filtered through a Celite® bed and washed with CH2Cl2. The combined organic layer was successively washed with 5% Na2S2O3, satd. NaHCO3 and water, dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane–EtOAc (5:1) as eluent to give pure compound 10 (1.6 g, 74%). White solid; m.p. 118–120 °C [EtOH]; [α]25D −18 (c 1.2, CHCH3); IR (KBr): 3477, 2933, 1777, 1742, 1718, 1507, 1387, 1391, 1244, 1099, 1044, 995, 797 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.59–6.94 (m, 29H, Ar–H), 6.84, 6.71 (2 d, J = 9.0 Hz each, 4H, Ar–H), 5.67 (d, J = 8.5 Hz, 1H, H-1B), 5.57, 5.52 (2 s, 2H, 2 PhCH), 5.08 (d, J = 3.5 Hz, 1H, H-1A), 4.96 (dd, J = 10.5, 3.5 Hz, 1H, H-3C), 4.86 (d, J = 3.5 Hz, 1H, H-1C), 4.76 (t, J = 9.5 Hz each, 1H, H-3B), 4.54 (t, J = 8.5 Hz each, 1H, H-2B), 4.45–4.40 (m, 3H, H-5B, PhCH2), 4.34 (d, J = 3.5 Hz, 1H, H-4A), 4.28 (dd, J = 10.0, 3.5 Hz, 1H, H-3A), 4.24 (d, J = 12.0 Hz, 1H, PhCH2), 4.16–4.10 (m, 3H, H-6aA, PhCH2), 3.98–3.91 (m, 3H, H-6bA, H-6aB, PhCH2), 3.88–3.82 (m, 2H, H-2A, H-5C), 3.79–3.66 (m, 2H, H-4B, H-4C), 3.73 (s, 3H, OCH3), 3.69 (dd, J = 10.5, 3.5 Hz, 1H, H-2C), 3.59–3.57 (m, 1H, H-5A), 1.63 (s, 3H, COCH3), 0.75 (d, J = 6.5 Hz, 3H, CCH3); 13C NMR (125 MHz, CDCl3): δ 169.9 (COCH3), 167.6 (Phth), 154.8–114.4 (Ar–C), 101.7 (PhCH), 100.3 (PhCH), 100.2 (C-1B), 98.6 (C-1C), 96.9 (C-1A), 81.6 (C-4B), 78.6 (C-5A), 76.5 (C-4A), 75.9 (C-3A), 75.5 (PhCH2), 75.0 (C-3B), 74.9 (C-5C), 73.4 (C-3C), 73.2 (PhCH2), 73.1 (C-2C), 72.5 (PhCH2), 69.1 (C-6A), 68.8 (C-6B), 66.6 (C-5B), 66.2 (C-2A), 63.2 (C-4C), 56.0 (C-2B), 55.5 (OCH3), 20.7 (COCH3), 15.9 (CCH3); MALDI-MS: 1234.3 [M + Na]+; anal. calcd for C70H69NO18 (1211.45): C, 69.35; H, 5.74; found: C, 69.18; H, 5.95.
p-Methoxyphenyl (2,4-di-O-benzyl-α-L-fucopyranosyl)-(1→3)-(4,6-O-benzylidene-2-deoxy-2-N-phthalimido-β-D-glucopyranosyl)-(1→3)-2-O-benzyl-4,6-O-benzylidene-α-D-galactopyranoside (11)
A solution of compound 10 (1.5 g, 1.24 mmol) in 0.05 M CH3ONa in CH3OH (25 mL) was allowed to stir at room temperature for 2 h. The reaction mixture was neutralized with Dowex 50W-X8 (H+) resin, filtered and concentrated. The crude product was passed through a small pad of SiO2 using hexane–EtOAc (1:1) as eluent to give pure compound 11 (1.4 g, 96%). White solid; m.p. 80–81 °C [EtOH]; [α]25D −12.6 (c 1.2, CHCH3); IR (KBr): 3477, 2931, 2869, 1777, 1716, 1507, 1454, 1391, 1243, 1215, 1101, 1045, 995, 797, 697 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.54–6.94 (m, 29H, Ar–H), 6.84, 6.71 (2 d, J = 9.0 Hz each, 4H, Ar–H), 5.69 (d, J = 8.5 Hz, 1H, H-1B), 5.55, 5.51 (2 s, 2H, 2 PhCH), 5.09 (d, J = 3.5 Hz, 1H, H-1A), 4.91 (d, J = 3.5 Hz, 1H, H-1C), 4.74 (t, J = 9.5 Hz each, 1H, H-3B), 4.61 (d, J = 12.0 Hz, 1H, PhCH2), 4.53–4.48 (m, 2H, H-2B, PhCH2), 4.44–4.41 (m, 1H, H-5B), 4.34 (d, J = 3.0 Hz, 1H, H-4A), 4.30–4.22 (m, 3H, H-3A, PhCH2), 4.14–4.80 (m, 2H, H-5C, PhCH2), 3.99–3.87 (m, 3H, H-4C, H-6aA, PhCH2), 3.86–3.80 (m, 3H, H-2A, H-3C, H-6bA), 3.79–3.73 (m, 3H, H-4B, H-6abB), 3.74 (s, 3H, OCH3), 3.45–3.42 (m, 2H, H-2C, H-5A), 0.88 (d, J = 6.5 Hz, 3H, CCH3); 13C NMR (125 MHz, CDCl3): δ 167.7 (Phth), 154.8–114.4 (Ar–C), 101.5 (PhCH), 100.4 (PhCH), 100.2 (C-1B), 97.7 (C-1C), 96.8 (C-1A), 81.6 (C-4B), 79.8 (C-5A), 76.5 (C-2C), 76.4 (C-4A), 76.0 (C-3A), 75.4 (PhCH2), 75.0 (C-3B), 74.5 (C-3C), 73.2 (PhCH2), 72.2 (PhCH2), 70.2 (C-5C), 69.1 (C-6A), 68.8 (C-6B), 67.0 (C-5B), 66.1 (C-2A), 63.1 (C-4C), 56.1 (C-2B), 55.5 (OCH3), 16.4 (CCH3); MALDI-MS: 1192.4 [M + Na]+; anal. calcd for C68H67NO17 (1169.44): C, 69.79; H, 5.77; found: C, 69.62; H, 5.95.
p-Methoxyphenyl (2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-2,3-di-O-benzyl-α-L-fucopyranoside (12)
To a solution of compound 5 (1.2 g, 2.66 mmol) and compound 6 (1.8 g, 2.81 mmol) in anhydrous CH2Cl2 (25 mL) was added MS-4Å (3 g) and the reaction mixture was allowed to stir at room temperature for 30 min under argon. The reaction mixture was cooled to −20 °C. To the cooled reaction mixture were added NIS (660 mg, 2.93 mmol) and TMSOTf (10 μL) and it was stirred at the same temperature for 30 min. The reaction mixture was diluted with CH2Cl2 (100 mL) and filtered through a Celite® bed and washed with CH2Cl2. The combined organic layer was successively washed with 5% Na2S2O3, satd. NaHCO3 and water, dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane–EtOAc (5:1) as eluent to give pure compound 12 (2 g, 73%). White solid; m.p. 60–62 °C [EtOH]; [α]25D −22.4 (c 1.2, CHCH3); IR (KBr): 3445, 2918, 1736, 1507, 1452, 1263, 1072, 1068, 1026, 709 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.98–7.19 (m, 30H, Ar–H), 6.87, 6.74 (2 d, J = 9.0 Hz each, 4H, Ar–H), 5.78 (t, J = 9.5 Hz each, 1H, H-3F), 5.66 (t, J = 10.0 Hz each, 1H, H-4F), 5.60 (t, J = 9.0 Hz each, 1H, H-2F), 5.17 (d, J = 3.5 Hz, 1H, H-1E), 5.09 (d, J = 8.0 Hz, 1H, H-1F), 4.86–4.78 (m, 3H, PhCH2), 4.65 (d, J = 12.0 Hz, 1H, PhCH2), 4.46 (dd, J = 12.0, 3.0 Hz, 1H, H-6aF), 4.25 (dd, J = 12.0, 4.0 Hz, 1H, H-6bF), 4.14 (dd, J = 10.0, 3.5 Hz, 1H, H-2E), 4.06 (dd, J = 10.0, 3.0 Hz, 1H, H-3E), 4.01 (br s, 1H, H-4E), 4.0–3.96 (m, 1H, H-5E), 3.73 (s, 3H, OCH3), 3.72–3.70 (m, 1H, H-5F), 0.97 (d, J = 6.5 Hz, 3H, CCH3); 13C NMR (125 MHz, CDCl3): δ 165.9, 165.8, 164.9, 164.8 (4 COPh), 154.8–114.4 (Ar–C), 101.7 (C-1F), 97.5 (C-1E), 78.9 (C-4E), 77.8 (C-3E), 75.7 (C-2E), 73.7 (PhCH2), 73.4 (C-3F), 73.1 (PhCH2), 72.7 (C-2F), 72.3 (C-5F), 69.6 (C-4F), 66.6 (C-5E), 62.9 (C-6F), 55.5 (OCH3), 16.3 (CCH3); MALDI-MS: 1051.3 [M + Na]+; anal. calcd for C61H56O15 (1028.36): C, 71.19; H, 5.48; found: C, 71.00; H, 5.69.
(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-2,3-di-O-benzyl-α-L-fucopyranosyl trichloroacetimidate (13)
To a solution of compound 12 (1.8 g, 1.75 mmol) in CH3CN–H2O (25 mL; 9:1; v/v) was added ammonium cerium(IV) nitrate (CAN; 2 g; 3.65 mmol) and the reaction mixture was allowed to stir at 0–5 °C for 3 h. The reaction mixture was diluted with CH2Cl2 (100 mL) and the organic layer was successively washed with satd. NaHCO3, water, dried (Na2SO4) and concentrated to give the crude product, which was passed through a short pad of SiO2 using hexane–EtOAc (2:1) as eluent to give the hemiacetal derivative. To a solution of the hemiacetal derivative in anhydrous CH2Cl2 (20 mL) were added CCl3CN (1.2 mL, 11.97 mmol) and DBU (50 μL) and the reaction mixture was allowed to stir at −5 °C for 1 h. The solvents were removed under reduced pressure and the crude product was passed through a short pad of SiO2 using hexane–EtOAc (9:1) as eluent to give pure compound 13 (1.3 g, 70%), which was used immediately without further characterization.
Ethyl (2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3-di-O-benzyl-α-L-fucopyranosyl)-(1→4)-3-O-acetyl-6-O-benzyl-2-deoxy-2-N-phthalimido-1-thio-β-D-glucopyranoside (14)
To a solution of compound 7 (500 mg, 1.03 mmol) and compound 13 (1.2 g, 1.12 mmol) in anhydrous CH2Cl2–Et2O (10 mL; 1:2, v/v) was added NOBF4 (140 mg, 1.2 mmol) and the reaction mixture was allowed to stir at −10 °C for 1 h under argon. The reaction mixture was diluted with CH2Cl2 (50 mL) and filtered through a Celite® bed and washed with CH2Cl2. The combined organic layer was successively washed with satd. NaHCO3 and water, dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane–EtOAc (3:1) as eluent to give pure compound 14 (1 g, 70%). White solid; m.p. 88–90 °C [EtOH]; [α]25D −28.6 (c 1.2, CHCH3); IR (KBr): 3447, 2929, 1722, 1718, 1452, 1386, 1265, 1094, 710 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.94–7.21 (m, 39H, Ar–H), 5.80 (t, J = 9.5 Hz each, 1H, H-3F), 5.64 (t, J = 10.0 Hz each, 1H, H-4F), 5.62 (t, J = 9.5 Hz each, 1H, H-2F), 5.54 (t, J = 9.5 Hz each, 1H, H-3D), 5.43 (d, J = 10.5 Hz, 1H, H-1D), 4.96 (d, J = 8.0 Hz, 1H, H-1F), 4.82 (d, J = 12.0 Hz, 1H, PhCH2), 4.76 (d, J = 12.0 Hz, 1H, PhCH2), 4.73 (d, J = 3.0 Hz, 1H, H-1E), 4.60 (d, J = 12.0 Hz, 1H, PhCH2), 4.54 (d, J = 12.0 Hz, 1H, PhCH2), 4.47 (dd, J = 12.0, 2.5 Hz, 1H, H-6aF), 4.33–4.29 (m, 2H, H-6bF, PhCH2), 4.24 (d, J = 12.0 Hz, 1H, PhCH2), 4.15 (t, J = 10.0 Hz each, 1H, H-2D), 4.01–3.99 (m, 1H, H-6aD), 3.91 (br s, 1H, H-4E), 3.89 (dd, J = 10.0, 3.5 Hz, 1H, H-2E), 3.85–3.81 (m, 1H, H-5F), 3.78–3.68 (m, 3H, H-3E, H-5D, H-5E), 3.59–3.52 (m, 2H, H-4D, H-6bD), 2.74–2.56 (m, 2H, SCH2CH3), 1.42 (s, 3H, COCH3), 1.19 (t, J = 7.5 Hz each, 3H, SCH2CH3), 0.89 (d, J = 6.5 Hz, 3H, CCH3); 13C NMR (125 MHz, CDCl3): δ 170.9 (COCH3), 167.9, 167.8 (Phth), 166.0, 165.9, 165.1, 164.7 (4 COPh), 138.7–123.5 (Ar–C), 101.9 (C-1F), 100.5 (C-1E), 80.6 (C-1D), 79.1 (C-4E), 78.4 (C-3E), 78.2 (C-4D), 77.1 (C-2E), 75.9 (C-5D), 74.2 (PhCH2), 73.9 (C-3D), 73.1 (C-3F), 73.0 (PhCH2), 72.3 (2C, C-2F, C-5F), 71.9 (PhCH2), 69.9 (C-6D), 69.7 (C-4F), 67.1 (C-5E), 63.3 (C-6F), 54.1 (C-2D), 24.3 (SCH2CH3), 20.4 (COCH3), 15.9 (CCH3), 15.0 (SCH2CH3); MALDI-MS: 1412.4 [M + Na]+; anal. calcd for C79H75NO20S (1389.46): C, 68.24; H, 5.44; found: C, 68.05; H, 5.64.
p-Methoxyphenyl (2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→4)-(2,3-di-O-benzyl-α-L-fucopyranosyl)-(1→4)-(3-O-acetyl-6-O-benzyl-2-deoxy-2-N-phthalimido-β-D-glucopyranosyl)-(1→3)-(2,4-di-O-benzyl-α-L-fucopyranosyl)-(1→3)-(4,6-O-benzylidene-2-deoxy-2-N-phthalimido-β-D-glucopyranosyl)-(1→3)-2-O-benzyl-4,6-O-benzylidene-α-D-galactopyranoside (15)
To a solution of compound 11 (720 mg, 0.61 mmol) and compound 14 (900 mg, 0.65 mmol) in anhydrous CH2Cl2 (15 mL) was added MS-4Å (2 g) and the reaction mixture was allowed to stir at room temperature for 30 min under argon and cooled to −10 °C. To the cooled reaction mixture were added NIS (160 mg, 0.71 mmol) and TMSOTf (5 μL) and it was stirred at the same temperature for 45 min. The reaction mixture was diluted with CH2Cl2 (100 mL) and filtered through a Celite® bed and washed with CH2Cl2. The combined organic layer was successively washed with 5% Na2S2O3, satd. NaHCO3 and water, dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane–EtOAc (5:1) as eluent to give pure compound 15 (1 g, 66%). White solid; m.p. 95–97 °C [EtOH]; [α]25D +12 (c 1.2, CHCH3); IR (KBr): 3477, 2932, 2869, 1775, 1714, 1507, 1454, 1392, 1243, 1213, 1100, 1045, 996 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.94–6.52 (m, 72H, Ar–H), 5.78 (t, J = 9.5 Hz each, 1H, H-3F), 5.63 (t, J = 10.0 Hz each, 1H, H-4F), 5.61 (t, J = 9.5 Hz each, 1H, H-2F), 5.54 (s, 1H, PhCH), 5.48 (d, J = 8.5 Hz, 1H, H-1B), 5.46 (d, J = 8.5 Hz, 1H, H-1D), 5.42 (t, J = 9.0 Hz each, 1H, H-3D), 5.30 (s, 1H, PhCH), 5.05 (d, J = 3.0 Hz, 1H, H-1A), 4.97 (d, J = 8.0 Hz, 1H, H-1F), 4.84 (d, J = 12.0 Hz, 1H, PhCH2), 4.78–4.72 (m, 3H, H-3B, PhCH2), 4.71 (d, J = 3.0 Hz, 1H, H-1C), 4.68–4.62 (m, 2H, H-5B, PhCH2), 4.60–4.52 (m, 2H, PhCH2), 4.51 (d, J = 3.0 Hz, 1H, H-1E), 4.48–4.44 (m, 1H, H-6aF), 4.37 (t, J = 8.5 Hz each, 1H, H-2D), 4.34 (d, J = 3.5 Hz, 1H, H-4A), 4.30–4.20 (m, 5H, H-5E, H-6aA, H-6aF, PhCH2), 4.18–4.12 (m, 2H, H-6aD, PhCH2), 4.08–3.97 (m, 4H, H-2B, H-6aB, H-6bD, PhCH2), 3.94–3.79 (m, 8H, H-2A, H-3C, H-4C, H-4E, H-5C, H-5F, PhCH2), 3.78–3.70 (m, 2H, H-3A, H-5D), 3.73 (s, 3H, OCH3), 3.69–3.62 (m, 4H, H-3E, H-4B, H-6bA, H-6bB), 3.52–3.45 (m, 2H, H-2C, H-2E), 3.22–3.20 (m, 1H, H-5A), 3.14 (t, J = 9.5 Hz each, 1H, H-4D), 1.55 (s, 3H, COCH3), 0.88–0.84 (m, 6H, 2 CCH3); 13C NMR (125 MHz, CDCl3): δ 170.1 (COCH3), 167.7 (2C) (Phth), 167.4 (2C) (Phth), 165.8, 165.7, 165.0, 164.6 (4 COPh), 154.8–114.4 (Ar–C), 101.9 (PhCH), 101.8 (C-1F), 100.5 (PhCH), 100.4 (C-1B), 100.3 (C-1C), 99.2 (C-1E), 96.8 (C-1A), 95.0 (C-1D), 80.4 (C-4D), 78.8 (C-5A), 78.3 (C-2C), 78.2 (C-3E), 77.1 (2C, C-2E, C-4A), 76.6 (C-5D), 75.9 (C-4E), 75.8 (2C, C-3B, C-4B), 75.1 (C-3A), 74.8 (PhCH2), 74.2 (PhCH2), 74.0 (PhCH2), 73.6 (C-3C), 73.2 (2C, C-3D, PhCH2), 73.1 (C-3F), 72.5 (PhCH2), 72.3 (2C, C-2F, C-5F), 72.0 (PhCH2), 71.7 (C-5C), 69.7 (2C, C-4F, C-6A), 69.1 (C-6D), 68.6 (C-6B), 67.1 (C-2A), 66.6 (C-5B), 66.2 (C-5E), 63.2 (C-4C), 63.1 (C-6F), 56.2 (C-2D), 55.5 (OCH3), 55.3 (C-2B), 20.4 (COCH3), 15.9, 15.7 (2 CCH3); MALDI-MS: 2519.8 [M + Na]+; anal. calcd for C145H136N2O37 (2496.88): C, 69.70; H, 5.49; found: C, 69.54; H, 5.68.
p-Methoxyphenyl (sodium β-D-glucopyranosyluronate)-(1→4)-(α-L-fucopyranosyl)-(1→4)-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-(1→3)-(α-L-fucopyranosyl)-(1→3)-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-(1→3)-α-D-galactopyranoside (1)
To a solution of compound 15 (800 mg, 0.32 mmol) in C2H5OH (15 mL) was added hydrazine monohydrate (0.3 mL) and the reaction was allowed to stir at 80 °C for 10 h. The solvents were removed under reduced pressure and the crude product was dissolved in acetic anhydride and pyridine (4 mL, 1:1 v/v) and kept at room temperature for 3 h. Solvents were removed under reduced pressure and the acetylated product was dissolved in 0.1 M CH3ONa in CH3OH (15 mL) and the reaction mixture was allowed to stir at room temperature for 2 h. The reaction mixture was neutralized using Dowex 50W-X8 (H+) resin, filtered and concentrated under reduced pressure. To a solution of the hexasaccharide pentahydroxy derivative in CH2Cl2 (25 mL) and H2O (5 mL) were successively added 1 M aq. NaBr (3 mL), 1 M aq. TBAB (5 mL), TEMPO (150 mg, 0.96 mmol), satd. NaHCO3 (20 mL) and 4% aq. NaOCl (15 mL) and the reaction mixture was allowed to stir at 5 °C for 3 h and neutralized with 1 N HCl. To the reaction mixture were added tert-butanol (20 mL), 2-methyl-but-2-ene (20 mL; 2 M solution in THF), aq. NaClO2 (2 g per 10 mL) and aq. NaH2PO4 (2 g per 10 mL) in succession and the reaction mixture was allowed to stir at room temperature for 3 h. The reaction mixture was diluted with satd. aq. NaH2PO4 and extracted with CH2Cl2 (100 mL). The organic layer was washed with water, dried (Na2SO4) and concentrated to dryness to give the crude product, which was successively passed through a short pad of SiO2 and a short column of Dowex 50W-X8 (Na+) to give the sodium salt of glucuronic acid containing hexasaccharide derivative. To a solution of the oxidized product and 10% Pd–C (150 mg) in CH3OH (10 mL) was added Et3SiH (0.7 mL, 4.38 mmol) dropwise over 30 min and the reaction mixture was allowed to stir at room temperature for 10 h. The reaction mixture was filtered through a Celite® bed, washed with CH3OH–H2O (2:1, v/v) and concentrated under reduced pressure to give compound 1, which was passed through a column of Sephadex LH-20 gel using CH3OH–H2O (3:1) as eluent to give pure compound 1 as p-methoxyphenyl glycoside and sodium salt (200 mg, 53%). Glass; [α]25D +10 (c 1.0, CH3OH); IR (KBr): 3478, 2933, 2867, 1777, 1714, 1507, 1457, 1392, 1243, 1213, 1100, 1045, 996 cm−1; 1H NMR (500 MHz, D2O): δ 7.02–6.86 (2 d, J = 9.0 Hz each, 4H, Ar–H), 5.34 (br s, 1H, H-1A), 4.99, 4.95 (2 d, J = 8.5 Hz each, 2H, H-1B, H-1D), 4.62 (d, J = 8.0 Hz, 1H, H-1F), 4.35, 4.34 (2 d, J = 3.0 Hz each, 2H, H-1C, H-1E), 4.20–4.02 (m, 2H, H-5C, H-5E), 4.0–3.86 (m, 5H, H-2B, H-2C, H-3F, H-4A, H-4E), 3.85–3.75 (m, 10H, H-2D, H-2E, H-3C, H-3D, H-4B, H-5D, H-6abA, H-6abD), 3.69 (s, 3H, OCH3), 3.68–3.50 (m, 7H, H-3A, H-3E, H-4D, H-4F, H-5A, H-6abB), 3.46–3.25 (m, 6H, H-2A, H-2F, H-3B, H-4C, H-5B, H-5F), 2.05, 1.98 (2 s, 6H, 2 COCH3), 1.18, 1.12 (2 d, J = 6.0 Hz each, 6H, 2 CCH3); 13C NMR (125 MHz, D2O): δ 175.9, 175.7 (2 COCH3), 173.0 (COONa), 154.6–115.0 (Ar–C), 103.1 (3C, C-1C, C-1E, C-1F), 100.6 (C-1B), 100.1 (C-1D), 98.5 (C-1A), 80.6 (2C, C-3D, C-4B), 75.9 (4C, C-3A, C-4C, C-5B, C-5F), 75.5 (2C, C-3B, C-4A), 74.7 (C-3E), 73.5 (2C, C-4D, C-5A), 72.1 (C-3F), 71.2 (C-3C), 69.4 (2C, C-4F, C-5D), 69.0 (2C, C-2F, C-4E), 68.7 (2C, C-2C, C-5C), 68.3 (C-2A), 67.8 (C-5E), 66.9 (C-2E), 60.9 (C-6A), 60.5 (2C, C-6B, C-6D), 55.7 (OCH3), 55.0 (C-2B), 54.9 (C-2D), 23.7, 22.4 (2 COCH3), 15.1 (2C, 2 CCH3); MALDI-MS: 1183.3 [M + 1]+; anal. calcd for C47H71N2NaO31 (1182.39): C, 47.72; H, 6.05; found: C, 47.51; H, 6.28.
Acknowledgements
T. G. and A. S. thank CSIR, New Delhi for providing Junior and Senior Research Fellowship, respectively. This work was supported by DST, New Delhi [Project no. SR/S1/OC-83/2010].Notes and references
- S. M. Horne and K. D. Young, Arch. Microbiol., 1995, 163, 357 CrossRef CAS.
- P. B. Eckburg, E. M. Bik, C. N. Bernstein, E. Purdom, L. Dethlefsen, M. Sargent, S. R. Gill, K. E. Nelson and D. A. Relman, Science, 2005, 308, 1635 CrossRef PubMed.
- J. P. Nataro and J. B. Kaper, Clin. Microbiol. Rev., 1998, 11, 142 CAS.
- A. C. Cheng, J. R. McDonald and N. M. Thielman, J. Clin. Gastroenterol., 2005, 39, 757 CrossRef.
- R. Stenutz, A. Weintraub and G. Widmalm, FEMS Microbiol. Rev., 2006, 30, 382 CrossRef CAS PubMed.
- S. C. Kehl, J. Clin. Microbiol., 2002, 40, 2711 CrossRef.
- T. G. Boyce, D. L. Swerdlow and P. M. Griffin, N. Engl. J. Med., 1995, 333, 364 CrossRef CAS PubMed.
- A. Ezawa, F. Gocho, M. Saitoh, T. Tamura, K. Kawata, T. Takahashi and N. Kikuchi, J. Vet. Med. Sci., 2004, 66, 779 CrossRef.
- R. L. Vogt and L. Dippold, Public Health Rep., 2005, 120, 174 Search PubMed.
- A. Weintraub, Carbohydr. Res., 2003, 338, 2539 CrossRef CAS PubMed.
- J. M. Hunt, Clin. Lab. Med., 2010, 30, 21 CrossRef PubMed.
- H. Zhu, A. V. Perepelov, S. N. Senchenkova, A. S. Shashkov, L. Wang and Y. A. Knirel, Carbohydr. Res., 2012, 349, 86 CrossRef CAS PubMed.
- K. J. Doores, D. P. Gamblin and B. G. Davis, Chem.–Eur. J., 2006, 12, 656 CrossRef CAS PubMed.
- B. Kuberan and R. J. Linhardt, Curr. Org. Chem., 2000, 4, 653 CrossRef CAS.
- R. Roy, Drug Discovery Today: Technol., 2004, 1, 327 CrossRef CAS PubMed.
- T. Tanikawa, M. Fridman, W. Zhu, B. Faulk, I. C. Joseph, D. Kahne, B. K. Wagner and P. A. Clemons, J. Am. Chem. Soc., 2009, 131, 5075 CrossRef CAS PubMed.
- J. O. Kihlberg, D. A. Leigh and D. R. Bundle, J. Org. Chem., 1990, 55, 2860 CrossRef CAS.
- A. Mukherjee, M. M. Palcic and O. Hindsgaul, Carbohydr. Res., 2000, 326, 1 CrossRef CAS.
- F. Dasgupta and P. J. Garegg, Acta Chem. Scand., 1989, 43, 471 CrossRef CAS PubMed.
- D.-Q. Sun, R. Busson and P. Herdewijn, Eur. J. Org. Chem., 2006, 5158 CrossRef CAS.
- G. H. Veeneman, S. H. van Leeuwen and J. H. van Boom, Tetrahedron Lett., 1990, 31, 1331 CrossRef CAS.
- P. Konradsson, U. E. Udodong and B. Fraser-Reid, Tetrahedron Lett., 1990, 31, 4313 CrossRef CAS.
- G. Zemplén, Ber. Dtsch. Chem. Ges., 1926, 59, 1254 CrossRef.
- K. Bock and C. J. Pedersen, J. Chem. Soc., Perkin Trans. 2, 1974, 293 RSC.
- D. B. Werz and P. H. Seeberger, Angew. Chem., Int. Ed., 2005, 44, 6315 CrossRef CAS PubMed.
- R. R. Schmidt and K.-H. Jung, in Preparative Carbohydrate Chemistry, ed. S. Hanessian, Marcel Dekker Inc., New York, 1997, pp. 283–312 Search PubMed.
- A. Sau, A. Santra and A. K. Misra, Synlett, 2012, 2341 CAS.
- H.-H. Lee, D. A. Schwartz, J. F. Harris, J. P. Carver and J. J. Krepinsky, Can. J. Chem., 1986, 64, 1912 CrossRef CAS.
- L. Huang, N. Teumelsan and X. Huang, Chem.–Eur. J., 2006, 12, 5246 CrossRef CAS PubMed.
- A. Santra, T. Ghosh and A. K. Misra, Beilstein J. Org. Chem., 2013, 9, 74 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1D and 2D NMR spectra of compounds 1 and 8–15. See DOI: 10.1039/c3ra45493b |
| This journal is © The Royal Society of Chemistry 2014 |