Low dimensional cyano-bridged heterobimetallic M–FeIII (M = NiII, CuII) complexes constructed from Mer-[FeIII(qcq)(CN)3]− building blocks: syntheses, structures and magnetic properties†
Hongbo
Zhou
a,
Jiahao
Yan
a,
Xiaoping
Shen
*a,
Hu
Zhou
b and
Aihua
Yuan
b
aSchool of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: xiaopingshen@163.com; Fax: +86-511-8879180; Tel: +86-511-88791800
bSchool of Material Science and Engineering, Jiangsu University of Science and Technology, Zhenjiang 212003, China
First published on 29th October 2013
Abstract
Four cyano-bridged heterobimetallic trinuclear complexes, {[Ni(en)2][Fe(qcq)(CN)3]2}·2H2O (1) [en = 1,2-ethylenediamine; qcq− = 8-(2-quinoline-2-carboxamido)quinoline anion], {[Ni(teta)][Fe(qcq)(CN)3]2}·2DMF·2H2O (2) (teta = 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane, DMF = N,N-dimethylformamide), {[CuL1][Fe(qcq)(CN)3]2}·0.61H2O (3) (L1 = 3,10-dibutyl-1,3,5,8,10,12-hexaazacyclotetradecane) and {[CuL2][Fe(qcq)(CN)3]2}·4.75H2O (4) (L2 = 3,10-dipropyl-1,3,5,8,10,12-hexaazacyclotetradecane) have been synthesized and characterized both structurally and magnetically. The structural analyses reveal that 1–4 are all trinuclear centrosymmetric clusters, and the intercluster π–π interactions and hydrogen bonds extend 1–4 into high dimensional supermolecular networks. Magnetic investigation indicates that 1 and 2 exhibit intracluster ferromagnetic couplings accompanied by significant magnetic anisotropy. In contrast, 3 and 4 show unusual intracluster antiferromagnetic couplings, which could even be decoupled by a strong applied field.
Introduction
Single molecule magnets (SMM)1 and single chain magnets (SCM),2 which are called low-dimensional molecular magnets,3 have been attracting much attention from chemists and physicists because of their potential applications in molecular devices, high-density information storage, quantum computing and so on.4 To date, a great number of magnetic complexes have been investigated for the purpose of clarifying the magnetic mechanism so as to design ideal magnetic materials. Among various magnetic systems studied, coordination complexes especially the cyano-bridged magnetic assemblies are of special importance because the cyanide groups (CN−) can mediate effectively the magnetic exchanges between the spin carriers and moreover, the linear coordination mode of cyanide groups makes the structure of target systems more predictable, facilitating the structure designs and the analyses of magneto–structural correlation.5 Cyanometalates building blocks such as the hexacyanometalates [M(CN)6]q− (M = Fe, Cr, Mn, or V) have provided a large family of three-dimensional complexes known as Prussian blue analogues, which exhibit extremely high critical temperature (Tc) even up to room temperature.6 Recently, another synthetic strategy based on the modified hexacyanometalates of [(L)M(CN)p]q− (where L = polydentate N/O-ligands; M = Cr, Fe, etc.) have been investigated by introducing various polydentate ligands of L into the cyanometalates.7 Different from hexacyanometalates that are used for three-dimensional molecular magnets, the modified hexacyanometalates are designed for low dimensional systems that behave possibly as SMMs and SCMs, in which the selection of polydentate ligands L (also called capping ligands) are very important: (i) the tailored organic ligands make the self-assembly reaction more controllable, limit the oligomerization or polymerization effect and promote the formation of the low-dimensional structures. (ii) The molecular geometries of blocking group L define the molecular orbitals and affect greatly the magnetic coupling mechanism and anisotropy. (iii) The intermolecular magnetic interactions, which are also critical to low dimensional magnetism, can be readily tuned by varying the capping ligands. Especially, tricyanidoferrate of [(L)Fe(CN)3]3− has aroused intense interest because of its flexible facial (fac-) and meridian (mer-) coordination modes depending on the capping ligand L.8 The facially coordinate ligands such as hydrotris(pyrazol-1-yl)borate (Tp), hydrotris(3,5-dimethylpyrazol-1-yl)borate (Tp*), tetra(pyrazol-1-yl)borate (pzTp), and 1,3,5-triaminocyclohexane (tach) have been well investigated,9 and some of the related complexes such as [{(pzTp)Fe(CN)3}2{Ni(bipy)2}]·2H2O10 and [{(Tp*Bn)Fe(CN)3}2{Ni(DMF)4}]·2DMF9g exhibit interesting SMM behaviors. However, relatively less attentions have been paid to mer-tricyanidoferrate (Scheme 1a) although several complexes derived from mer-[Fe(L)(CN)3]− have been reported.11–14 In this work, we focus our interest on a recently reported mer-tricyanidoferrate building block, [FeIII(qcq)(CN)3]−[qcq− = 8-(2-quinoline-2-carboxamido)quinoline anion],15 from which four new cyano-bridged heterobimetallic trinuclear complexes, {[Ni(en)2][Fe(qcq)(CN)3]2}·2H2O (1) [en = 1,2-ethylenediamine; qcq− = 8-(2-quinoline-2-carboxamido)quinoline anion], {[Ni(teta)][Fe(qcq)(CN)3]2}·2DMF·2H2O (2) (teta = 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane, DMF = N,N-dimethylformamide), {[CuL1][Fe(qcq)(CN)3]2}·0.61H2O (3) (L1 = 3,10-dibutyl-1,3,5,8,10,12-hexaazacyclotetradecane) and {[CuL2][Fe(qcq)(CN)3]2}·4.75H2O (4) (L2 = 3,10-dipropyl-1,3,5,8,10,12-hexaazacyclotetradecane) have been synthesized by a chemical self-assembly method (teta, L1 and L2 are shown in Scheme 1b). Herein, we report the syntheses, crystal structures and magnetic properties of the four complexes.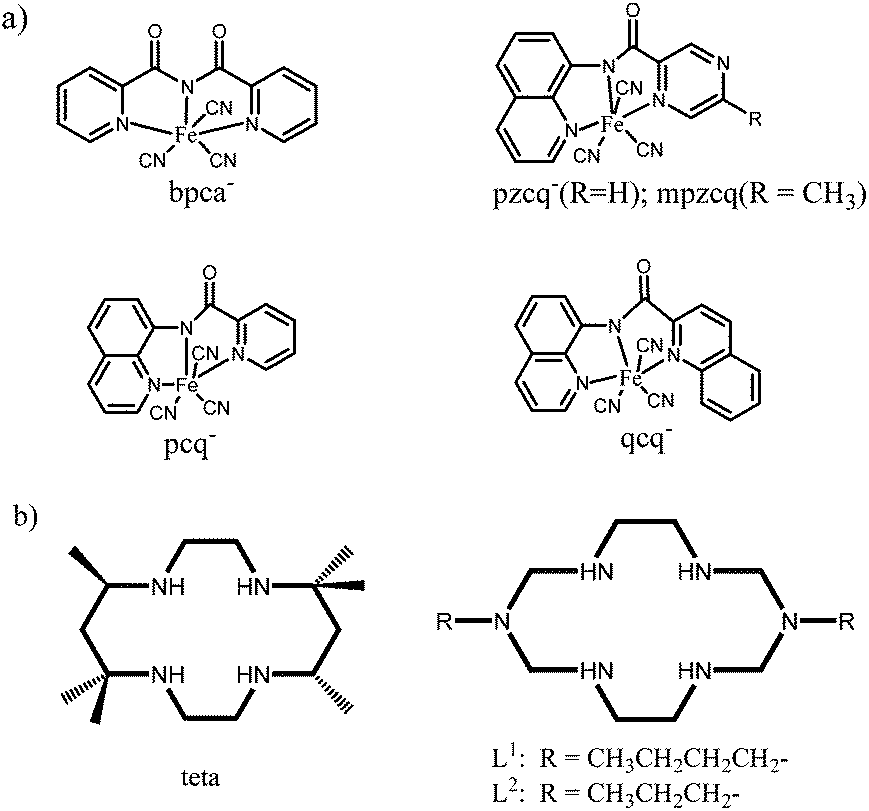 | ||
| Scheme 1 (a) Typical mer-tricyanidoferrate building blocks; (b) macrocyclic ligands of teta, L1 and L2. | ||
Experimental
Physical measurements
Elemental analyses for C, H and N were performed at a Perkin-Elmer 240C analyzer. Ni, Cu and Fe analyses were made on a Jarrell-Ash 1100 + 2000 inductively coupled plasma quantometer (ICP). IR spectra were recorded on a Nicolet FT-170SX spectrometer with KBr pellets in the 4000–400 cm−1 region. All magnetic measurements on microcrystalline samples were conducted on a Quantum Design MPMP-XL7 superconducting quantum interference device (SQUID) magnetometer. Corrections of measured susceptibilities were carried out considering both the sample holder as the background and the diamagnetism of the constituent atoms according to Pascal's tables.16Caution! Cyanides are highly toxic and perchlorate salts of metal complexes are potentially explosive. So handing them carefully with small quantities is highly suggested for the safety consideration.
Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) solution (10 mL) of [Ni(teta)](ClO4)2 (0.05 mmol) through a H-shaped tube at room temperature for about two weeks. The resulting crystals were collected, washed with H2O and CH3OH, and dried in air. Anal. found: C, 56.86; H, 5.81; N, 17. 99; Fe, 8.11; Ni, 4.23%. Calcd for C66H78Fe2NiN18O6: C, 57.04; H, 5.66; N, 18.14; Fe, 8.04; Ni, 4.22%. IR: νmax/cm−1 3435(s), 2119(m), 1633(s), 1504(w), 1461(m), 1436(s), 1388(m), 1342(m), 1214(w), 1155(m), 1097(s), 725(m).
:4) solution (10 mL) of [Ni(teta)](ClO4)2 (0.05 mmol) through a H-shaped tube at room temperature for about two weeks. The resulting crystals were collected, washed with H2O and CH3OH, and dried in air. Anal. found: C, 56.86; H, 5.81; N, 17. 99; Fe, 8.11; Ni, 4.23%. Calcd for C66H78Fe2NiN18O6: C, 57.04; H, 5.66; N, 18.14; Fe, 8.04; Ni, 4.22%. IR: νmax/cm−1 3435(s), 2119(m), 1633(s), 1504(w), 1461(m), 1436(s), 1388(m), 1342(m), 1214(w), 1155(m), 1097(s), 725(m).
X-Ray structure determination
Diffraction data were collected on a Bruker SMART APEX CCD area detector diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) with the φ and ω scan mode. Diffraction data analysis and reduction were performed with SMART, SAINT and XPREP.19 Absorption corrections were performed with SADABS.20 Structures were solved using direct method and refined by full-matrix least-squares techniques based on F2 using SHELXL-97.21 All non-hydrogen atoms of 1–4 were refined with anisotropic thermal parameters. The H atoms of all chelated ligands and DMF molecules were calculated at idealized positions, but the water–H atoms in 1 and 2 are located from difference maps with all the H atoms refined in a riding mode. The water molecules in 1 and 2 were assigned with partial occupancy factor. For 3 and 4, the water oxygen atoms (water–H atoms are not found) are not present with unit occupancy, and it is allowed a anisotropic free variable refinement with partial occupancy. The pendant groups of the macrocyclic ligands in 3 and 4 split into two different positions and a few distance or angle restraints (see refine special details in CIF file) were used in the refinement. The crystallographic data and experimental details for structural analyses are summarized in Table 1.| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Formula | C48H44Fe2NiN16O4 | C66H78Fe2NiN18O6 | C60H63.22CuFe2N18O2.61 | C58H67.5CuFe2N18O6.75 |
| F W | 1079.40 | 1389.87 | 1253.50 | 1300.01 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c |
P |
P |
| a/Å | 9.3769(16) | 25.8501(15) | 10.283(2) | 10.350(2) |
| b/Å | 10.441(2) | 16.246(2) | 12.391(3) | 12.023(3) |
| c/Å | 14.992(3) | 19.5463(13) | 13.032(3) | 12.921(3) |
| α/° | 90.311(2) | 90.00 | 107.05(3) | 101.583(3) |
| β/° | 106.740(3) | 121.075(4) | 94.67(3) | 94.969(3) |
| γ/° | 104.011(2) | 90.00 | 94.81(3) | 94.440(3) |
| V/Å3 | 1359.3(4) | 7030.8(11) | 1572.2(5) | 1561.8(6) |
| Z | 1 | 4 | 1 | 1 |
| ρ calcd/g cm−3 | 1.319 | 1.313 | 1.323 | 1.372 |
| F(000) | 556 | 2912 | 649.8 | 667 |
| θ/° | 3.09–25.91 | 3.48–26.03 | 3.15–25.00 | 1.62–24.99 |
| Index ranges | −10 ≤ h ≤ 11 | −31 ≤ h ≤ 31 | −12 ≤ h ≤ 11 | −11 ≤ h ≤ −12 |
| −12 ≤ k ≤ 12 | −19 ≤ k ≤ 20 | −14 ≤ k ≤ 14 | −13 ≤ k ≤ 14 | |
| −18 ≤ l ≤ 16 | −24 ≤ l ≤ 17 | −15 ≤ l ≤ 14 | −15 ≤ l ≤ 15 | |
| Total/unique data | 12576/5140 |
16527/6759 |
13145/5466 |
11219/5437 |
| Observed data [I > 2σ(I)] | 3169 | 5101 | 4611 | 3955 |
| R int | 0.0519 | 0.0385 | 0.0412 | 0.0355 |
| Data/restraints/parameters | 3169/0/340 | 5101/0/435 | 4611/16/388 | 3955/29/390 |
| GOF on F2 | 1.054 | 1.033 | 1.016 | 1.069 |
| R 1[I > 2σ(I)] | 0.0607 | 0.0509 | 0.0750 | 0.0480 |
| wR 2 (all data) | 0.1297 | 0.1257 | 0.1601 | 0.1356 |
Results and discussion
Synthesis and characterization
In this paper, [Fe(qcq)(CN)3]− was used as a building block to prepare the low-dimensional complexes. In fact, the solubility of A[Fe(qcq)(CN)3] (A = counter cations) is highly dependent on the counter cation selected because different solvent polarity favors different type of cations. For instance, PPh4[Fe(qcq)(CN)3] can be well dissolved in methanol but insoluble in water, while K[Fe(qcq)(CN)3] is just on the contrary. In this study, PPh4[Fe(qcq)(CN)3] is used instead of K[Fe(qcq)(CN)3] because the latter is insoluble in methanol. To confirm this point, we also used K[Fe(qcq)(CN)3] as the building block and unfortunately, no suitable crystals were obtained, indicating the subtle reaction mechanism and the important role of PPh4+ cation for the growth of the crystals. The reactions of PPh4[Fe(qcq)(CN)3] with [Ni(en)2](ClO4)2 or [Ni(teta)](ClO4)2 both gave block black crystals, which are stable in room temperature and show insolubility in water and methanol. By replacing the nickel complexes with copper precursors, deep red crystals were formed, which also exhibit high stabilities. With the same solubility consideration, DMF was used for the syntheses of 2, 3 and 4 because the precursors of [Ni(teta)](ClO4)2, [CuL1](ClO4)2 and [CuL2](ClO4)2 are more soluble in DMF than in water. IR spectroscopy for 1–4 all displayed absorption peaks in 2100–2150 cm−1, indicating the presence of cyanide groups in these complexes.22 The similarity of the IR spectra of these complexes is obviously due to the comparable structural moieties presented in the molecular units.Description of the structures
The key bond distances and angles for 1–4 are listed in Table 2. The molecular structural diagrams of 1–4 are shown in Fig. 1, and the intermolecular packing diagrams including the intermolecular short contacts are shown in Fig. S1 and S2 in ESI.†| 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|
| C1–Fe1 | 1.962(4) | C20–Fe1 | 1.968(3) | C1–Fe1 | 1.959(5) | C1–Fe1 | 1.958(4) |
| C2–Fe1 | 1.957(5) | C21–Fe1 | 1.922(3) | C2–Fe1 | 1.959(6) | C2–Fe1 | 1.955(4) |
| C3–Fe1 | 1.971(5) | C22–Fe1 | 1.950(3) | C3–Fe1 | 1.952(6) | C3–Fe1 | 1.957(4) |
| Fe1–N5 | 1.922(3) | Fe1–N2 | 1.947(2) | Fe1–N5 | 1.888(2) | Fe1–N5 | 1.890(3) |
| Fe1–N6 | 2.010(3) | Fe1–N3 | 1.964(2) | Fe1–N6 | 1.977(2) | Fe1–N6 | 1.971(3) |
| Fe1–N4 | 2.018(3) | Fe1–N1 | 2.048(3) | Fe1–N4 | 2.009(2) | Fe1–N4 | 2.001(3) |
| N1–Ni1 | 2.137(3) | N6–Ni1 | 2.112(3) | Cu1–N8 | 2.001(4) | Cu1–N8 | 2.006(3) |
| N7–Ni1 | 2.085(3) | N7–Ni1 | 2.111(2) | Cu1–N7 | 2.005(2) | Cu1–N7 | 2.014(3) |
| N8–Ni1 | 2.126(4) | N8–Ni1 | 2.073(2) | Cu1–N1 | 2.568(2) | Cu1–N1 | 2.559 (2) |
| C1–N1–Ni1 | 161.5(3) | C22–N6–Ni1 | 165.2(2) | C1–N1–Cu1 | 142.2(2) | C1–N1–Cu1 | 143.1(2) |
| N1–C1–Fe1 | 175.8(4) | N4–C20–Fe1 | 175.2(3) | N1–C1–Fe1 | 173.6 (4) | N1–C1–Fe1 | 173.7(3) |
| N2–C2–Fe1 | 178.4(4) | N5–C21–Fe1 | 179.0(3) | N2–C2–Fe1 | 177.9(4) | N2–C2–Fe1 | 178.1(3) |
| N3–C3–Fe1 | 177.8(4) | N6–C22–Fe1 | 175.7(2) | N3–C3–Fe1 | 178.1 (4) | N3–C3–Fe1 | 177.0(3) |
 | ||
| Fig. 1 Molecular structures with selected atom-labeling schemes for 1 (a), 2 (b), 3 (c) and 4 (d). (The solvents molecules are omitted for clarity). | ||
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond angles of 1 and 2 are 161.5(3) and 165.2(2)°, respectively, exhibiting obvious deviation from linearity. For the moieties of [Fe(qcq)(CN)3]−, the coordination environment of Fe center can be described as distorted octahedron, consisting of three C atoms from cyanide groups and three N atoms from qcq−. The Fe–C bond lengths are nearly close to each other (1.922(3)–1.971(5) Å). However, Fe–N bond distances [1.922(3)–2.048(3) Å] deviate from each other with the smallest value found for the Fe–N (amide) bonds [1.922(3) Å (Fe1–N5) for 1; 1.947(2) Å (Fe1–N2) for 2]. The relatively shorter Fe–N (amide) bond distances than those for Fe–N (aromatic rings) might be attributed to the strong σ-donor effect of the deprotonated amide.15 Different from the bent Ni–NC bond angles, the Fe–CN bond angles for 1 and 2 are nearly linear (175.2(3)–179.0(3)°). It is worth noting that the two planes of quinoline rings (C4–C12 and C14–C22) from qcq− in 1 are obviously noncoplanar with dihedral angle being as high as 27.6(2)°, which is significantly larger than that [3.2(5)°] found in 2, possibly owing to the different coordinating modes of [Fe(qcq)(CN)3]− in 1 and 2. For 1, the bridging cyanide (C1N1) is nearly coplanar with the quinoline rings leaving two terminal cyanides (C2N2, C2N2) in trans-mode, while that (C22N6) in 2 is basically perpendicular to the quinoline rings keeping the two terminal cyanides (C21N5, C20N4) in cis-mode. The former coordination fashion might produce larger steric effects than the latter one, and thus the quinoline rings planes in 1 are highly distorted.
C bond angles of 1 and 2 are 161.5(3) and 165.2(2)°, respectively, exhibiting obvious deviation from linearity. For the moieties of [Fe(qcq)(CN)3]−, the coordination environment of Fe center can be described as distorted octahedron, consisting of three C atoms from cyanide groups and three N atoms from qcq−. The Fe–C bond lengths are nearly close to each other (1.922(3)–1.971(5) Å). However, Fe–N bond distances [1.922(3)–2.048(3) Å] deviate from each other with the smallest value found for the Fe–N (amide) bonds [1.922(3) Å (Fe1–N5) for 1; 1.947(2) Å (Fe1–N2) for 2]. The relatively shorter Fe–N (amide) bond distances than those for Fe–N (aromatic rings) might be attributed to the strong σ-donor effect of the deprotonated amide.15 Different from the bent Ni–NC bond angles, the Fe–CN bond angles for 1 and 2 are nearly linear (175.2(3)–179.0(3)°). It is worth noting that the two planes of quinoline rings (C4–C12 and C14–C22) from qcq− in 1 are obviously noncoplanar with dihedral angle being as high as 27.6(2)°, which is significantly larger than that [3.2(5)°] found in 2, possibly owing to the different coordinating modes of [Fe(qcq)(CN)3]− in 1 and 2. For 1, the bridging cyanide (C1N1) is nearly coplanar with the quinoline rings leaving two terminal cyanides (C2N2, C2N2) in trans-mode, while that (C22N6) in 2 is basically perpendicular to the quinoline rings keeping the two terminal cyanides (C21N5, C20N4) in cis-mode. The former coordination fashion might produce larger steric effects than the latter one, and thus the quinoline rings planes in 1 are highly distorted.
Since 1 and 2 crystallize in different space groups (P-1 for 1, C2c for 2), different intermolecular packing structures are formed. As shown in Fig. S1a,† the trinuclear units of 1 interact with each other via π⋯π stacking (centroid to centroid distance: 4.2 Å) of adjacent quinoline rings along c axis, forming 1-D supramolecular chains, which then arrange in parallel with significant hydrogen bonds interactions (mediated via water–H and terminal cyanide nitrogen atoms with the maximum D–H⋯A distance shorter than 2.1 Å). The molecular units of 2 also interact with each other via π⋯π stacking of adjacent quinoline rings (centroid to centroid distance: 3.6–3.9 Å), but different from 1, a 2-D grid-like supramolecular network is formed in ab plane, and the molecules of DMF are situated in the square holes (Fig. S1b†). Moreover, hydrogen bonds interactions mediated via solvent molecules are also observed in 2 though they seem to be rather weak (minimum D–H⋯A distance larger than 3.0 Å).
C bonds are highly bent with the bond angles of 142.2(2)° for 3 and 143.1(2)° for 4, respectively. For the macrocyclic ligands L, the pendant groups in 3 (C27, C28, C29, C30) and 4 (C27, C28, C29) are highly disordered and split into two different positions, which should be originated from the average of the crystallographic sites. For the [Fe(qcq)(CN)3]− unit in 3 and 4, all the Fe–C bond lengths are highly consistent [1.952(6)–1.959(6) Å for 3, 1.955(4)–1.958(4) Å for 4], while the Fe–N bond distances [1.888(2)–2.009(2) Å for 3 and 1.890(3)–2.001(3) Å for 4] deviate from each other, which is comparable to that found in 1 and 2. For the bond angles of Fe–CN in 3 and 4, the terminal ones range from 177.0(3)° to 178.1(4)°, while bridging ones are more bent with the value of 173.6 (4)°–173.7(3). In addition, noncoplanar quinoline rings of qcq− in 3 and 4 are also found with the dihedral angles of 24.7(5)° and 23.9(2)°, respectively, which is comparable to the case of 1.
For packing structures (Fig. S2†), 3 and 4 feature the similar fashions. The clusters interact with each other via π⋯π stacking of adjacent quinoline rings [centroid to centroid distance: 3.8–4.4 Å (3) and 3.7–4.0 Å (4) ], affording 2-D supramolecular layered networks. Each layer is well separated with average separations of 8.3 Å for 3 and 7.0 Å for 4, respectively. Hydrogen bonds interactions should also widely exist in 3 and 4 although it is not possible to analysis in detail due to the disorder of solvents molecules.
Magnetic properties
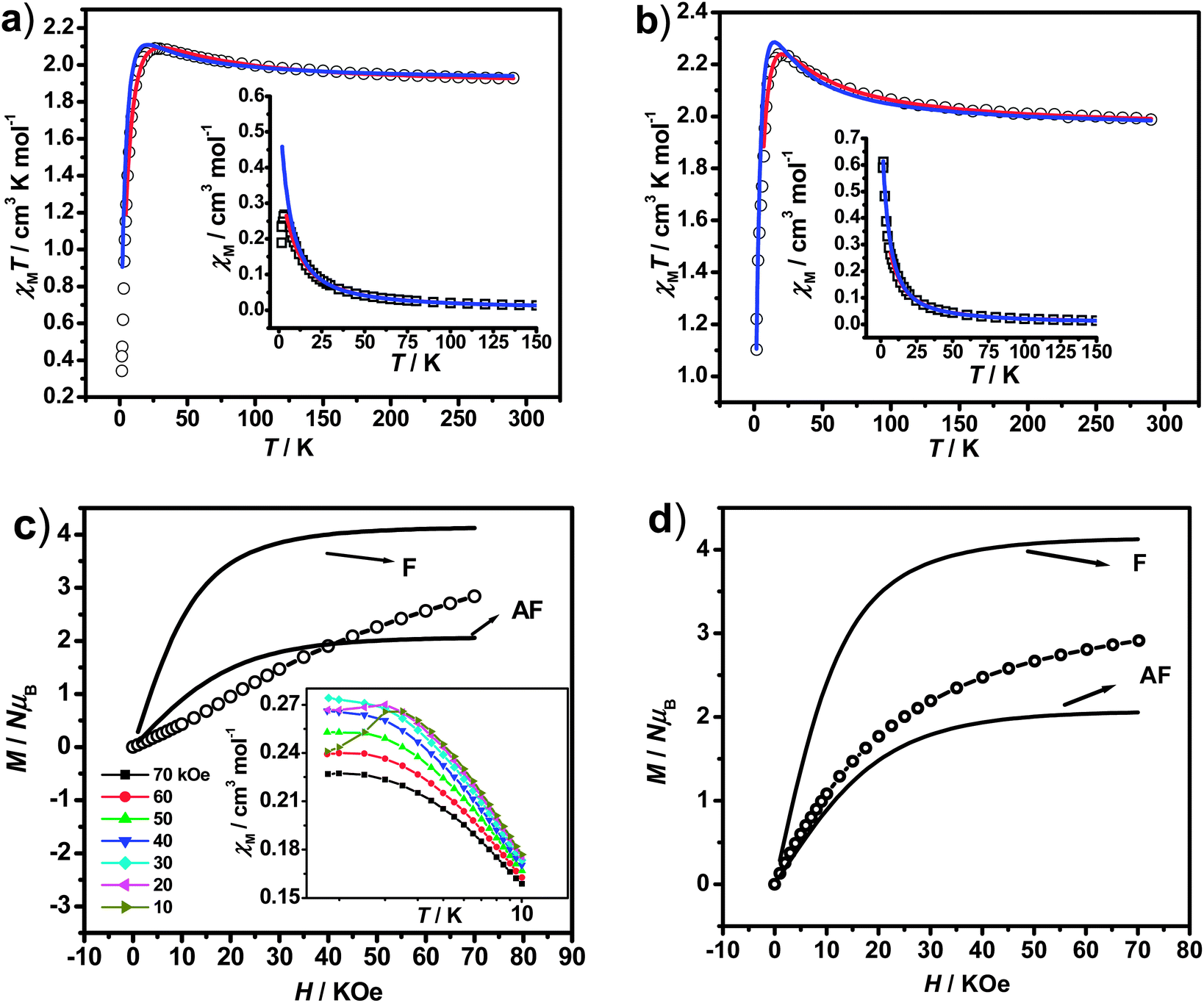 | ||
| Fig. 2 Temperature dependence of χMT and χM (inset) for 1 (a) and 2 (b) measured at 2 kOe (the red and blue solid lines represent the best fits to the models described in the text); field dependence of the magnetization for 1 (c) and 2 (d) at 1.8 K (the curves marked with F, AF represent the theoretical Brillouin curves of ferro- and antiferromagnetic states of NiFe2 unit; the inset shows the corresponding field-cooled magnetization for 1 in different applied fields). | ||
To evaluate quantitatively the coupling constants for 1 and 2, the linear trinuclear model based on Kambe's method32 considering both the intra- and intermolecular contributions has been used to fit the χMT data. Based on the Hamiltonian Ĥ = −2JSNi(SFe1 + SFe2) (J represents the intramolecular coupling constant of Ni⋯Fe interactions), the magnetic formulas deduced can be written as:
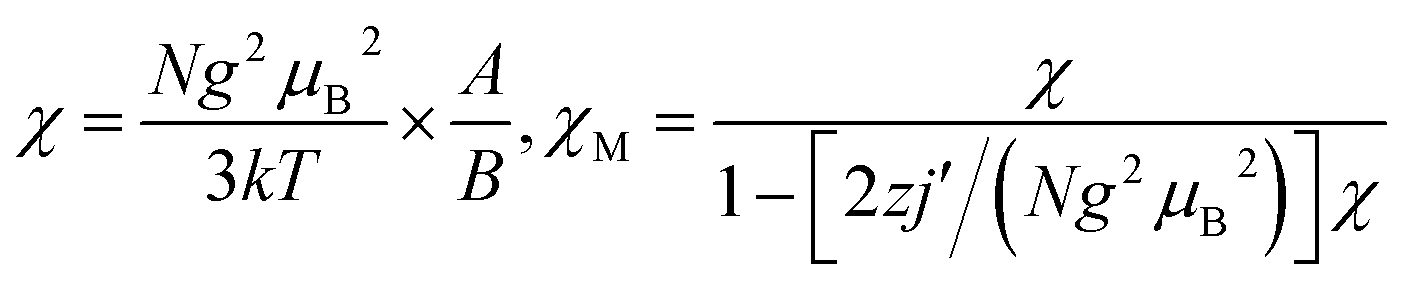
| A = 6 + 6e−2x + 30e2x; B = 3 + e−4x + 3e−2x + 5e2x |
C angles often contribute to stronger magnetic exchange coupling. In fact, the larger NiII–NC angles in FeIII(CrIII)–CN–NiII complexes can indeed decrease the overlap of magnetic orbital and lead to stronger ferromagnetic interaction. Nevertheless, there are still many complexes showing inconsistency with the NiII–NC angle-dependant rule, suggesting some other structural factors such as metal–metal separations and/or torsion angles are also relevant to the exchange coupling strength.
| Complexes | Ni–N–C (degree) | g | J exp/cm−1 | Ref. |
|---|---|---|---|---|
| a Abbreviations used for the ligands: pzTp = tetra(pyrazol-1-yl)borate; L = 1,5,8,12-tetraazadodecane; bbp = bis(2-benzimidazolyl)pyridine dianion; L* = 3,10-bis(2-phenylethyl)-1,3,5,8,10,12-hexaazacyclotetradecane; bpb = 1,2-bis(pyridine-2-carboxamido)benzenate; dmphen = 2,9-dimethyl-1,10-phenanthroline; OTf: O3SCF3; Tp = hydrotris(pyrazol-1-yl)borate; TpmMe = tris(3,5-dimethyl-1-pyrazoyl)-methane; dbphen = 5,6-dibromo-1,10-phenanthroline; dabhctd = 1,8-di(amethylbenzyl)-1,3,6,8,10,13-hexaazacyclotetradecane. | ||||
| [(pzTp)Fe(CN)3]4[NiL]4[OTf]410DMF·Et2O | 177.4–179.1 | 2.20 | 6.6 | 9c |
| {[Ni(ntb)(MeOH)]2[Fe(bbp)(CN)3][ClO4]2}·2MeOH | 176.5 | 2.22 | 8.61 | 22 |
| [Ni(L*)][Fe(bpb)(CN)2]2·4H2O | 173.4 | 7.7 | 26 | |
| [(pzTp)Fe(CN)2(μ-CN)Ni(dmphen)2]2(ClO4)2·2CH3OH | 170.3–171.6 | 2.39 | 6.2–8.4 | 9h |
| [(pzTp)2Fe2(CN)6Ni(bipy)2]·2H2O | 169.8 | 2.31 | 4.9 | 10 |
| [(Tp)3(TpmMe)2Fe3Ni2(CN)9]ClO4·15H2O | 168.4 | 2.27 | 4.84 | 7e |
| {[Ni(teta)][Fe(qcq)(CN)3]2}·2DMF·2H2O | 165.2 | 2.11 | 4.6 | This work |
| {[Ni(en)2][Fe(qcq)(CN)3]2}·2H2O | 161.5 | 2.09 | 4.5 | This work |
| [(dbphen)2Fe2(CN)8Ni(dabhctd)]·2H2O | 154.8 | 2.07 | 0.48 | 9f |
| [(Tp)2Fe(CN)6Ni(en)2] | 153.8 | 2.25 | 1.2 | 25 |
| [(pzTp)2Fe2(CN)6Ni(L)]·1/2CH3OH | 149.2–150.7 | 2.5 | 0.9 | 10 |
To further study the low temperature magnetic behaviors, in particular for 1 which exhibits detectable magnetic ordering at very low temperature, field-dependent magnetization measurement (0–70 kOe at 1.8 K) was then performed. As shown in Fig. 2(c), the magnetization of 1 first increases linearly with the increase of external field, and then increases more rapidly until reaching about 2.84 NμB at 70 kOe, which is far from the saturation value of 4 NμB (calculated from MS = g(2SFe + SNi) with g = 2) anticipated from ferromagnetic behaviors. For comparison, the theoretical Brillouin curves of ferro- and antiferromagnetic states of units of NiIIFe2III were also plotted (Fig. 2(c)), which imply the presence of significant magnetic anisotropy in the system.35 The magnetic behaviors of 1 revealed by M–H measurement are in accordance with the χMT result, and both of them indicate the presence of antiferromagnetic phase at very low temperature, which are overcome by the external field (critical field, Hc = 20 kOe) and lead to the sigmoid shape of the magnetization. This M–H curve for 1 suggests the metamagnetic-like behavior,11,35b,36 which can be further evidenced by the measurement of susceptibility under different applied fields in the range of 1.8–10 K deduced by field-cooling magnetization (FCM) (Inset of Fig. 2(c)). Under low applied fields, the susceptibility curves show peaks at about 3.5 K, but the peaks become less prominent and invisible when the applied fields increase higher than 20 kOe, confirming the presence of metamagnetic transitions induced by the external fields. Associated with magnetic behaviors in high temperature regions, the antiferromagnetic ordering in 1 at very low temperature should derived from the intermolecular interactions mediated via π⋯π stacking or hydrogen bonds, as revealed by the structural analysis. However, no hysteresis loop can be detected for 1 in the temperature region explored, excluding the possibility of any metamagnet behaviors. Different from 1, the magnetization of 2 (Fig. 2(d)) increases steadily with the increase of external field without sigmoid shape until reaching about 2.91 NμB at 70 kOe, which is also lower than the ferromagnetic saturated value (4 NμB). The magnetization curve of 2 locates between the F and AF state of theoretical Brillouin curves, indicating the combined contributions from ferromagnetic exchanges and the single ion axial magnetic anisotropy.
 | ||
| Fig. 3 Temperature dependence of χMT and χM (inset) for 3 (a) and 4 (b) measured at 2 kOe. (The red solid line represents the best fits to the model described in the text). | ||
To evaluate the couplings constants, linear trinuclear model9f based on the Hamiltonian Ĥ = −2JSCu(SFe1 + SFe2) (J represents the intramolecular coupling constant of Cu⋯Fe interaction) has been used for fitting the data and the magnetic formulas can be expressed as (zj′ represents the intermolecular interaction):
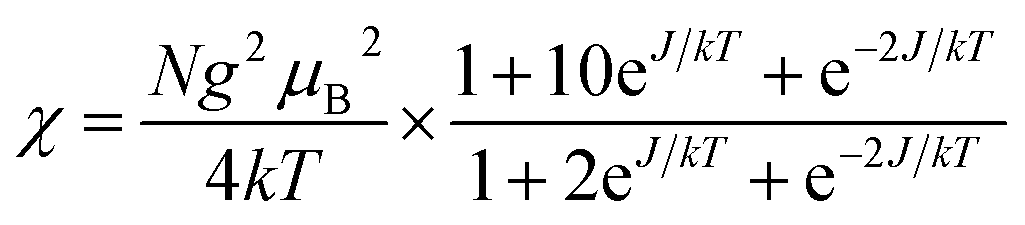
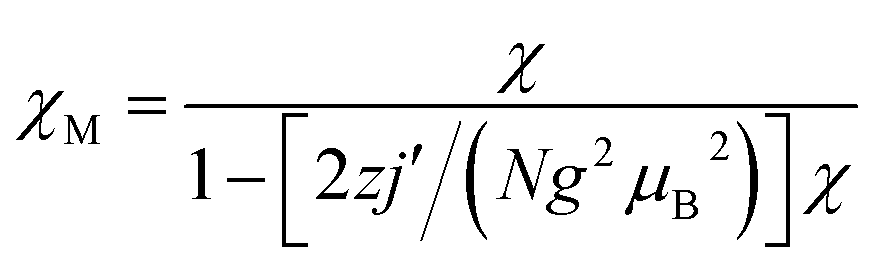
The best fits of the data of 3 and 4 in the whole temperature region gave: J = −0.40 cm−1, g = 2.18, zj′ = −0.05 cm−1, R = 2.2 × 10−4 and J = −2.56 cm−1, g = 2.21, zj′ = −0.1 cm−1, R = 1.6 × 10−4, respectively. The J value of 3 is comparable to that of the analogous CuFe2 complexes,29 but that for 4 seems to be slightly overestimated because of the pretty long Cu–Naxial bond length [2.559(2) Å]. Considering that cyanide nitrogen atoms in such systems usually occupy the axial positions of the elongated octahedral Cu surroundings, weak magnetic coupling should be expected. The fitting results further confirm the weak antiferromagnetic interactions presented in 3 and 4. It is worth noting that most FeIII–CN–CuII complexes display intramolecular ferromagnetic interactions,9f,37,38 and antiferromagnetic examples are very rare.29 The intramolecular ferromagnetic interactions in FeIII–CN–CuII systems can be easily attributed to the strict orthogonality between the magnetic orbitals of low spin FeIII ions (t2g5) and the CuII ions (eg3). However, to our knowledge, there are no systematically magneto–structural correlations investigations for antiferromagnetic magnetic exchanges of FeIII–CN–CuII systems. So the antiferromagnetic couplings mechanism for FeIII–CN–CuII systems is interesting and deserves further investigation.
Field-dependent magnetization measurement (0–70 kOe at 1.8 K) was also performed for 3 and 4 (Fig. 4) to verify the magnetic behaviors. As the external field increases, the magnetizations for 3 continuously increase until reaching 2.74 NμB at 70 kOe, which are larger than the theoretical values (1 NμB) expected for the antiferromagnetic coupled CuFe2 unit (calculated from MS = g(2SFe–SCu) with g = 2). Suggesting the paramagnetic behaviors resulting from the decoupling effect of the weak antiparallely aligned spins by magnetic fields.39 The decoupling effect can be readily reproduced by simulation of the M–H curves by Magpack (Fig. 4), where the weak antiferromagnetic coupled CuFe2 unit can indeed show as large magnetization as uncoupled ones at high applied field. By comparing the experimental magnetizations with the simulated ones, the J value for 3 corresponds approximately to −0.5 cm−1, which is in accord with the result provided by χMT–T fittings. For 4, the magnetization increases linearly until reaching 2.27 NμB at 70 kOe, which is consistent with the theoretical simulated M–H curves corresponding to J = −2.0 cm−1, indicating that the antiferromagnetic couplings are relatively stronger than 3, but could also be decoupled by external field.
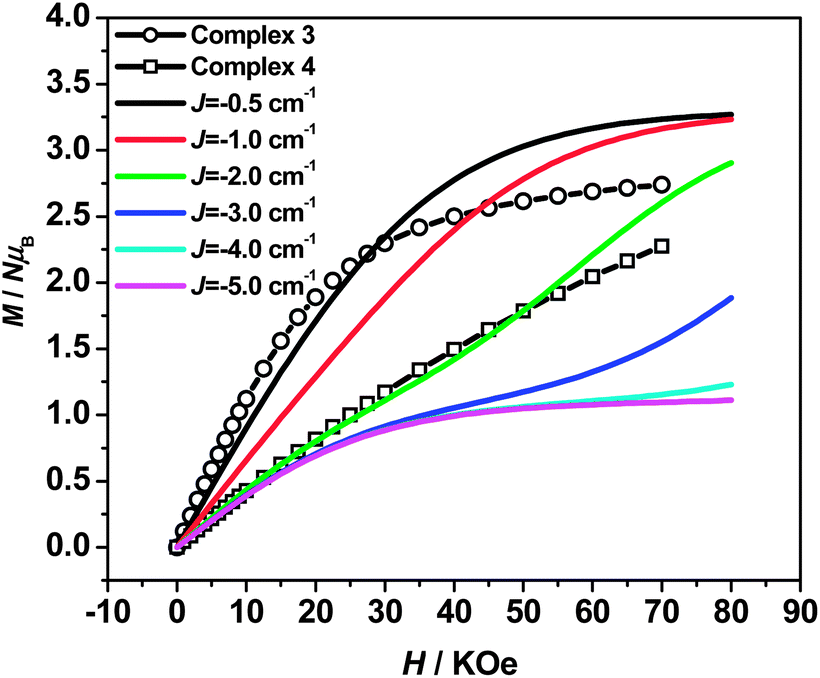 | ||
| Fig. 4 Field-dependent magnetization for 3 and 4 (0–70 kOe at 1.8 K). The solid lines represent the simulated M–H curves for CuFe2 unit assuming JFe–Cu = 0.5–5.0 cm−1 at 1.8 K. | ||
Conclusions
In this work, the mer-tricyanidoferrate building block, [FeIII(qcq)(CN)3]− has been used to successfully synthesize four new low dimensional cyano-bridged heterobimetallic complexes, which are shown as linear trinuclear NiFe2 (1, 2) and CuFe2 (3, 4) clusters. To our knowledge, these complexes are the first examples of Fe–CN–Ni(Cu) magnetic assembling using the latest designed mer-tricyanidoferrate building block, [FeIII(qcq)(CN)3]−. The magnetic investigations reveal that intramolecular ferromagnetic FeIII–CN–NiII interactions are presented in 1 and 2, while the unusual intramolecular antiferromagnetic FeIII–CN–CuII interactions were observed for 3 and 4. The magnetic behaviors of 1 and 2 were well analyzed by considering the single ion anisotropy (D) of NiII ions. Interestingly, the unusual antiferromagnetic FeIII–CN–CuII interactions were observed for 3 and 4, and the weak magnetic couplings could even be decoupled by strong applied field. The fully understanding of this unusual magnetic properties and clarifying the coupling mechanism need further investigation, related work is undergoing.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (no. 51072071, 51272094 and 1601310042), Doctoral Innovation Program Foundation of China (no. 1721310119) and the Startup Foundation for Advanced Talents of Jiangsu University (no. 11JDG106).Notes and references
- (a) C. P. Berlinguette, D. Vaughn, C. Canada-Vilalta, J. R. Galan-Mascaros and K. R. Dunbar, Angew. Chem., Int. Ed., 2003, 42, 1523 CrossRef CAS PubMed; (b) Y. Song, P. Zhang, X. M. Ren, X. F. Shen, Y. Z. Li and X. Z. You, J. Am. Chem. Soc., 2005, 127, 3708 CrossRef CAS PubMed; (c) D. E. Freedman, D. M. Jenkins, A. T. Iavarone and J. R. Long, J. Am. Chem. Soc., 2008, 130, 2884 CrossRef CAS PubMed; (d) J. J. Le Roy, M. Jeletic, S. I. Gorelsky, I. Korobkov, L. Ungur, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 3502 CrossRef CAS PubMed.
- (a) R. Clerac, H. Miyasaka, M. Yamashita and C. Coulon, J. Am. Chem. Soc., 2002, 124, 12837 CrossRef CAS PubMed; (b) H. Miyasaka, A. Saitoh, M. Yamashita and R. Clerac, Dalton Trans., 2008, 2422 RSC; (c) T. Liu, Y. J. Zhang, S. Kanegawa and O. Sato, J. Am. Chem. Soc., 2010, 132, 8250 CrossRef CAS PubMed; (d) X. Feng, J. Liu, T. D. Harris, S. Hill and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7521 CrossRef CAS PubMed; (e) V. Tangoulis, M. Lalia-Kantouri, M. Gdaniec, C. Papadopoulos, V. Miletic and A. Czapik, Inorg. Chem., 2013, 52, 6559 CrossRef CAS PubMed.
- K. R. Dunbar, Inorg. Chem., 2012, 51, 12055 CrossRef CAS PubMed.
- (a) W. Wernsdorfer, N. Allaga-Alcalde, D. N. Hendrickson and G. Christou, Nature, 2002, 416, 406 CrossRef PubMed; (b) H. L. Sun, Z. M. Wang and S. Gao, Coord. Chem. Rev., 2010, 254, 1081 CrossRef CAS; (c) F. Troiani and M. Affronte, Chem. Soc. Rev., 2011, 40, 3119 RSC; (d) S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336 RSC; (e) J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464 RSC.
- (a) M. J. Gunter, K. J. Berry and K. S. Murray, J. Am. Chem. Soc., 1984, 106, 4227 CrossRef CAS; (b) K. V. Langenberg, S. R. Batten, K. J. Berry, D. C. R. Hockless, B. Moubaraki and K. S. Murray, Inorg. Chem., 1997, 36, 5006 CrossRef; (c) R. J. Parkera, L. Spiccia, B. Moubaraki, K. S. Murray, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2000, 300, 922 CrossRef; (d) K. V. Langenberg, D. C. R. Hockless, B. Moubaraki and K. S. Murray, Synth. Met., 2001, 122, 573 CrossRef; (e) R. J. Parker, L. Spiccia, B. Moubaraki, K. S. Murray, D. C. Hockless, A. D. Rae and A. C. Willis, Inorg. Chem., 2002, 41, 2489 CrossRef CAS PubMed; (f) D. E. Freedman, D. M. Jenkins and J. R. Long, Chem. Commun., 2009, 4829 RSC; (g) P. V. Bernhardt, M. Martinez and C. Rodriguez, Inorg. Chem., 2009, 48, 4787 CrossRef CAS PubMed; (h) D. Visinescu, L. M. Toma, J. Cano, O. Fabelo, C. Ruiz-Perez, A. Labrador, F. Lloret and M. Julve, Dalton Trans., 2010, 39, 5028 RSC; (i) X. Zhu, J. W. Zhao, B. L. Li, Y. Song, Y. M. Zhang and Y. Zhang, Inorg. Chem., 2010, 49, 1266 CrossRef CAS PubMed; (j) S. Chorazy, K. Nakabayashi, K. Imoto, J. Mlynarski, B. Sieklucka and S. Ohkoshi, J. Am. Chem. Soc., 2012, 134, 16151 CrossRef CAS PubMed; (k) D. P. Dong, Y. J. Zhang, H. Zheng, P. F. Zhuang, L. Zhao, Y. Xu, J. Hu, T. Liu and C. Y. Duan, Dalton Trans., 2013, 42, 7693 RSC.
- (a) S. Ferlay, T. Mallah, R. Ouahes, P. Veillet and M. Verdaguer, Nature, 1995, 378, 701 CrossRef CAS; (b) O. Hatlevik, W. E. Buschmann, J. Zhang, J. L. Manson and J. S. Miller, Adv. Mater., 1999, 11, 914 CrossRef CAS; (c) S. M. Holmes and G. S. Girolami, J. Am. Chem. Soc., 1999, 121, 5593 CrossRef CAS.
- (a) R. Lescouezec, J. Vaissermann, F. Lloret, M. Julve and M. Verdaguer, Inorg. Chem., 2002, 41, 5943 CrossRef PubMed; (b) J. Kim, S. Han, K. I. Pokhodnya, J. M. Migliori and J. S. Miller, Inorg. Chem., 2005, 44, 6983 CrossRef CAS PubMed; (c) Z. H. Ni, H. Z. Kou, L. F. Zhang, C. Ge, A. L. Cui, R. J. Wang, Y. Li and O. Sato, Angew. Chem., Int. Ed., 2005, 44, 7742 CrossRef CAS PubMed; (d) D. Li, S. Parkin, R. Clerac and S. M. Holmes, Inorg. Chem., 2006, 45, 7569 CrossRef CAS PubMed; (e) Z. G. Gu, Q. F. Yang, W. Liu, Y. Song, Y. Z. Li, J. L. Zuo and X. Z. You, Inorg. Chem., 2006, 45, 8895 CrossRef CAS PubMed; (f) M. Shatruk, A. Dragulescu-Andrasi, K. E. Chambers, S. A. Stoian, E. L. Bominaar, C. Achim and K. R. Dunbar, J. Am. Chem. Soc., 2007, 129, 6104 CrossRef CAS PubMed; (g) F. Pan, Z. M. Wang and S. Gao, Inorg. Chem., 2007, 46, 10221 CrossRef CAS PubMed; (h) C. G. Freiherr von Richthofen, A. Stammler, H. Bogge, M. W. DeGroot, J. R. Long and T. Glaser, Inorg. Chem., 2009, 48, 10165 CrossRef PubMed; (i) I. Boldog, F. J. Munoz-Lara, A. B. Gaspar, M. C. Munoz, M. Seredyuk and J. A. Real, Inorg. Chem., 2009, 48, 3710 CrossRef CAS PubMed; (j) T. Liu, Y. J. Zhang, S. Kanegawa and O. Sato, Angew. Chem., Int. Ed., 2010, 49, 8645 CrossRef CAS PubMed; (k) S. Nastase, C. Maxim, M. Andruh, J. Cano, C. Ruiz-Perez, J. Faus, F. Lloret and M. Julve, Dalton Trans., 2011, 40, 4898 RSC; (l) I. Y. Yoo, D. W. Ryu, J. H. Yoon, A. R. Sohn, K. S. Lim, B. K. Cho, E. K. Koh and C. S. Hong, Dalton Trans., 2012, 41, 1776 RSC; (m) M. X. Yao, Q. Zheng, X. M. Cai, Y. Z. Li, Y. Song and J. L. Zuo, Inorg. Chem., 2012, 51, 2140 CrossRef CAS PubMed.
- (a) R. Gheorghe, M. Kalisz, R. Clerac, C. Mathoniere, P. Herson, Y. Li, M. Seuleiman, R. Lescouezec, F. Lloret and M. Julve, Inorg. Chem., 2010, 49, 11045 CrossRef CAS PubMed; (b) Y. H. Peng, Y. F. Meng, L. Hu, Q. Li, X. Y. Z. Li, J. L. Zuo and X. Z. You, Inorg. Chem., 2010, 49, 1905 CrossRef CAS PubMed; (c) H. Y. Kwak, D. W. Ryu, J. W. Lee, J. H. Yoon, H. C. Kim, E. K. Koh, J. Krinsky and C. S. Hong, Inorg. Chem., 2010, 49, 4632 CrossRef CAS PubMed; (d) M. Nihei, Y. Sekine, N. Suganami, K. Nakazawa, A. Nakao, H. Nakao, Y. Murakami and H. Oshio, J. Am. Chem. Soc., 2011, 133, 3592 CrossRef CAS PubMed; (e) T. Liu, D. P. Dong, S. Kanegawa, S. Kang, O. Sato, Y. Shiota, K. Yoshizawa, S. Hayami, S. Wu, C. He and C. Y. Duan, Angew. Chem., Int. Ed., 2012, 51, 4367 CrossRef CAS PubMed.
- (a) R. Lescouezec, L. M. Toma, J. Vaissermann, M. Verdaguer, F. S. Delgado, C. Ruiz-Perez, F. Lloret and M. Julve, Coord. Chem. Rev., 2005, 249, 2691 CrossRef CAS; (b) D. F. Li, S. Parkin, G. B. Wang, G. T. Yee, A. V. Prosvirin and S. M. Holmes, Inorg. Chem., 2005, 44, 4903 CrossRef CAS PubMed; (c) D. Li, S. Parkin, G. Wang, G. T. Yee, R. Clerac, W. Wernsdorfer and S. M. Holmes, J. Am. Chem. Soc., 2006, 128, 4214 CrossRef CAS PubMed; (d) L. Jiang, H. J. Choi, X. L. Feng, T. B. Lu and J. R. Long, Inorg. Chem., 2007, 46, 2181 CrossRef CAS PubMed; (e) J. Z. Gu, L. Jiang, M. Y. Tan and T. B. Lu, J. Mol. Struct., 2008, 890, 24 CrossRef CAS; (f) L. C. Kang, X. Chen, C. F. Wang, X. H. Zhou, J. L. Zuo and X. Z. You, Inorg. Chim. Acta, 2009, 362, 5195 CrossRef CAS; (g) Y. Z. Zhang, U. P. Malik, N. P. Rath, R. Clerac and S. M. Holmes, Inorg. Chem., 2011, 50, 10537 CrossRef CAS PubMed; (h) E. Pardo, M. Verdaguer, P. Herson, H. Rousseliere, J. Cano, M. Julve, F. Lloret and R. Lescouezec, Inorg. Chem., 2011, 50, 6250 CrossRef CAS PubMed.
- D. Li, R. Clérac, S. Parkin, G. Wang, G. T. Yee and S. M. Holmes, Inorg. Chem., 2006, 45, 5251 CrossRef CAS PubMed.
- T. Senapati, C. Pichon, R. Ababei, C. Mathoniere and R. Clerac, Inorg. Chem., 2012, 51, 3796 CrossRef CAS PubMed.
- Z. H. Ni, H. Z. Kou, L. F. Zhang, W. W. Ni, Y. B. Jiang, A. L. Cui, J. Ribas and O. Sato, Inorg. Chem., 2005, 44, 9631 CrossRef CAS PubMed.
- J. I. Kim, H. S. Yoo, E. K. Koh, H. C. Kim and C. S. Hong, Inorg. Chem., 2007, 46, 8481 CrossRef CAS PubMed.
- J. I. Kim, H. S. Yoo, E. K. Koh and C. S. Hong, Inorg. Chem., 2007, 46, 10461 CrossRef CAS PubMed.
- J. L. Kim, H. Y. Kwak, J. H. Yoon, D. W. Ryu, I. Y. Yoo, N. Yang, B. K. Cho, J. G. Park, H. Lee and C. S. Hong, Inorg. Chem., 2009, 48, 2956 CrossRef CAS PubMed.
- O. Kahn, Molecular Magnetism, VCH, New York, 1993 Search PubMed.
- N. F. Curtis, J. Chem. Soc., 1964, 2644 RSC.
- M. P. Suh and S. G. Kang, Inorg. Chem., 1988, 27, 2544 CrossRef CAS.
- Bruker, SMART. SAINT and XPREP: Area Detector Control and Data Integration and Reduction Software, Bruker Analytical X-ray Instruments Inc.; Madison, Wisconsin, USA, 1995 Search PubMed.
- G. M. Sheldrick, SADABS: Program for Empirical Absorption correction of Area Detector Data, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXL-97: Program for the Refinement of Crystal Structure, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. Panja, P. Guionneau, I. R. Jeon, S. M. Holmes, R. Clerac and C. Mathoniere, Inorg. Chem., 2012, 51, 12350 CrossRef CAS PubMed.
- H. Xiang, S. J. Wang, L. Jiang, X. L. Feng and T. B. Lu, Eur. J. Inorg. Chem., 2009, 2074 CrossRef CAS.
- C. F. Wang, Z. G. Gu, X. M. Lu, J. L. Zuo and X. Z. You, Inorg. Chem., 2008, 47, 7957 CrossRef CAS PubMed.
- S. Wang, J. L. Zuo, H. C. Zhou, Y. Song and X. Z. You, Inorg. Chim. Acta, 2005, 358, 2101 CrossRef CAS.
- Z. H. Ni, H. Z. Kou, Y. H. Zhao, L. Zheng, R. J. Wang, A. L. Cui and O. Sato, Inorg. Chem., 2005, 44, 2050 CrossRef CAS PubMed.
- S. Wang, J. L. Zuo, H. C. Zhou, Y. Song, S. Gao and X. Z. You, Eur. J. Inorg. Chem., 2004, 3681 CAS.
- R. Appelt and H. Vahrenkamp, Z. Anorg. Allg. Chem., 2003, 629, 133 CrossRef CAS.
- B. Zhang, Z. H. Ni, A. L. Cui and H. Z. Kou, New J. Chem., 2006, 30, 1327 RSC.
- K. J. Cho, D. W. Ryu, H. Y. Kwak, J. W. Lee, W. R. Lee, K. S. Lim, E. K. Koh, Y. W. Kwon and C. S. Hong, Chem. Commun., 2012, 48, 7404 RSC.
- (a) L. Toma, R. Lescouezec, J. Vaissermann, F. S. Delgado, C. Ruiz-Perez, R. Carrasco, J. Cano, F. Lloret and M. Julve, Chem.–Eur. J., 2004, 10, 6130 CrossRef CAS PubMed; (b) X. P. Shen, Q. Zhang, H. B. Zhou, H. Zhou and A. H. Yuan, New J. Chem., 2012, 36, 1180 RSC.
- K. Kambe, J. Phys. Soc. Jpn., 1950, 5, 48 CrossRef CAS.
- J. J. Borrts-Almenar, J. M. Clemente-Juan, E. Coronado and B. S. Tsukerblat, J. Comput. Chem., 2001, 22, 985 CrossRef.
- G. Rogez, J. N. Rebilly, A. L. Barra, L. Sorace, G. Blondin, N. Kirchner, M. Duran, J. van Slageren, S. Parsons, L. Ricard, A. Marvilliers and T. Mallah, Angew. Chem., Int. Ed., 2005, 44, 1876 CrossRef CAS PubMed.
- (a) H. Miyasaka, H. Ieda, N. Matsumoto, K. Sugiura and M. Yama-shita, Inorg. Chem., 2003, 42, 3509 CrossRef CAS PubMed; (b) H. B. Zhou, J. Wang, H. S. Wang, Y. L. Xu, X. J. Song, Y. Song and X. Z. You, Inorg. Chem., 2011, 50, 6868 CrossRef CAS PubMed.
- H. R. Wen, C. F. Wang, J. L. Zuo, Y. Song, X. R. Zeng and X. Z. You, Inorg. Chem., 2006, 45, 582 CrossRef CAS PubMed.
- M. Atanasov, C. Busche, P. Comba, F. El Hallak, B. Martin, G. Rajaraman, J. van Slageren and H. Wadepohl, Inorg. Chem., 2008, 47, 8112 CrossRef CAS PubMed.
- L. M. Toma, R. Lescouezec, J. Pasan, C. Ruiz-Perez, J. Vaissermann, J. Cano, R. Carrasco, W. Wernsdorfer, F. Lloret and M. Julve, J. Am. Chem. Soc., 2006, 128, 4842 CrossRef CAS PubMed.
- Q. Zhang, H. B. Zhou, X. P. Shen, H. Zhou and Y. Q. Yang, New J. Chem., 2013, 37, 941 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The intermolecular short contacts and packing diagrams of 1–4 in Fig. S1 and S2. CCDC 948658–948661. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra45218b |
| This journal is © The Royal Society of Chemistry 2014 |