Macroinitiator triggered polymerization for versatile immunoassay†
Lingling
Xu
,
Liang
Yuan
and
Songqin
Liu
*
State Key Laboratory of Bioelectronics, School of Chemistry and Chemical Engineering, Southeast University, Jiangning District, Nanjing, 211189, P.R. China. E-mail: liusq@seu.edu.cn; Fax: +86-25-5209061; Tel: +86-25-52090613
First published on 6th November 2013
Abstract
A novel polymerization-based signal amplification strategy by using a dual-functional macroinitiator for ultrasensitive detection of protein was proposed. The macroinitiator composed of streptravidins, initiators, and poly(acrylic acid-co-acrylamide) was endowed with the ability of both molecular recognition and high initiation efficiency. After the immunoreaction and streptravidin–biotin specific identification, the macroinitiators were immobilized on the surface of the substrate and then triggered the generation of an activator by the electron transfer for atom transfer radical polymerization (AGET ATRP) of 2-hydroxyethyl methacrylate (HEMA). The obtained polymers altered the surface reflectivity and opacity at the location of the macroinitiator, which could be easily distinguished by the naked eye within 10 min of polymerization. The numerous hydroxyl groups in the growing polymer chains also led the surface hydrophily change after only 7 min of polymerization. The detection limit of human immunoglobulin G antigen (IgG) by contact angle measurement was 0.13 ng mL−1. The excellent performance of IgG clinical serum sample assay was further examined, showing great potential to detect other biological sample by this sensitive and easy strategy.
Introduction
The sensitive detection of trace amounts of target proteins in complex biological matrices plays an important roles in many fields, including biomedical research,1–3 clinical diagnosis,4–6 food quality control,7–9 and environmental analysis.10–12 Great efforts have been made towards exploring novel assay strategies and signal amplification methods to enhance the signal readout. Successful signal amplification strategies included applying novel detection probes, incorporating nanomaterials for increased loading of tags,13–17 making use of enzyme-assisted amplification processes,18,19 DNA-related amplification techniques,20,21 and polymerization-based amplification strategy.22–24 Among these, polymerization-based amplification strategy has recently emerged as a promising signal amplification technique in molecular diagnostic.25–30 This strategy coupled a small initiator molecule to the detection probe prior to the ligand–target binding. After the affinity interaction of DNA/protein–target, the initiators were immobilized on the substrate. The polymerization led to connect the small monomers in ahead-to-tail fashion and form a long polymer chain that contained hundreds to millions of repeating units at the location where the initiator molecule was attached. The resulting polymer materials changed the optical and electrochemical properties at this location, which make the signal easily distinguishable from the background. For example, a surface plasmon resonance method for detection of cell membrane binding proteins using functional gold nanoparticles in combination with in situ atom transfer radical polymerization (ATRP) was proposed.31 The biotin–gold nanoparticle was functionalized with ATRP initiator that triggered AGET ATRP and produced local aggregation of poly(hydroxyl-ethyl methacrylate) brush, contributing to remarkable surface plasmon resonance signal enhancement and quantitative measurement of bacterial cholera toxin at attomol level. We recently reported a strategy for ultrasensitive detection of protein based on the integration of tyramide signal amplification and polymerization-assisted signal amplification.32 The ATRP of glycidyl methacrylate was triggered by the initiator-coupled protein immobilized on the electrode surface through sandwiched immunoreactions. Growth of long chain polymeric materials provided numerous epoxy groups for subsequent coupling of horseradish peroxidase, which in turn significantly increased the loading of quantum dots-labeled tyramide in the presence of hydrogen peroxide. The increased loading of horseradish peroxidase due to the formation of polymer further significantly increased the accumulation of quantum dots, yielding 10-fold increase in detection signal in comparison with that by the unamplified method.It was reported that the total amount of grafted polymeric materials could be further increased by forming either longer polymer chains or more polymer chains.33,34 The number of polymer chains would increase by using macroinitiator. Therefore, the design and synthesis of a macromolecule possessing both molecular recognition and high initiation efficiency was highly desirable, and would benefit to the target-triggered polymerization-assisted signal amplification method. He et al.33 reported the use of polylysine as carrier to bring multiple polymerization reaction initiators to surface-affixed thiolated 15-mer peptide nucleic acid/DNA duplexes for signal amplification. Compared to the previously reported value of single-initiator-tagged DNA detection, the detection limit using macroinitiator had been improved for approximately 60 times. Bowman et al.35 designed a macrophotoinitiator by covalent linking of avidin and photoinitiator to poly(acrylic acid-co-acrylamide) for DNA detection. After a 10 min dose of light, a macroscopically observable polymer grew from the spots where biotinylated oligonucleotides were located, resulting in an observable color change from gold to blue even by naked eyes.
The present work was motivated by robust promising applications of macroinitiators and polymerization-based signal amplification for biosensing. The macroinitiator was prepared by coating the P(AA-AM) with two kinds of functional molecules, streptavidin and ATRP initiators N-hydroxysuccinmidyl bromoisobutyrate, which were in favor of subsequent biorecognition and numerous aggregation of monomers. The macroinitiators used in AGET ATRP was also demonstrated to be applicable for the signal amplification. To confirm the signal amplification system, sandwich immune reaction was carried out, and subsequently the above-mentioned macroinitiator was immobilized onto the substrate via biotin–streptavidin specific identification, followed by the AGET ATRP to grown PHEMA, which was visible by naked eye (Scheme 1). Qualitative analysis of IgG with visualization or contact angle measurement was achieved within 10 min.
 | ||
| Scheme 1 Conceptual depiction of visualization detection of proteins based on the sandwich immunoreaction and macroinitiator-triggered polymerization. | ||
Experimental section
Materials
Au substrates (50 Å chrome followed by 1000 Å gold on float glass) were purchased from Evaporated Metal Films (Ithaca, NY). Human immunoglobulin G antigen (IgG, Ag, 1 mg mL−1), rabbit anti-human IgG antibody (anti-IgG, Ab1, 1 mg mL−1), and biotin labeled rabbit anti-human IgG antibody (BT-anti-IgG, Ab2, 1 mg mL−1) were obtained from Boster Biological Technology Co. Ltd (Wuhan, China). Streptavidin (SA) was purchased from Promega biological techno Co., Ltd (Beijing, China). Bovine serum albumin (BSA) was obtained from SunShine BIO (Nanjing, China). 2-Hydroxyethyl methacrylate (HEMA, 99%), N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), mercaptoundecanoic acid (MUA), 2-bromoisobutyryl bromide (BriBuBr, 98%), and poly(acrylic acid-co-acrylamide) partial sodium salt (P(AA-AM), Mw: 520 kDa, acrylamide: ∼80 wt%) were purchased from Sigma-Aldrich (Shanghai, China). Copper(II) bromide (CuBr2, 98%), triethylamine (TEA), 2,2′-bipyridyl (bpy), ascorbic acid (AA) and N,N-dimethylformamide (DMF), were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). 2-Hydroxyethyl methacrylate (HEMA, 99%) was purified in-house by passing through a column with activated Al2O3 to remove the inhibitors before polymerization. All other chemicals were of analytical grade and were used as received. 0.1 M phosphate buffer (PB) was prepared by mixing 0.1 M NaH2PO4 and Na2HPO4. Twice-distilled water was used throughout the study.Instruments
Gas chromatography/attenuated total reflection infrared (GC/ATR-IR) measurement was performed on a NEXUS670 spectrometer (Thermo Nicolet Corporation, America). Scanning electron microscope (SEM) image was characterized by a SEM instrument (JEM-2100, JEOL, Japan) with an acceleration voltage of 1 kV. UV/vis absorption spectrum was measured by using a UV-2450 UV-visible spectrophotometer (Shimadzu, Japan). The electrochemical impedance spectroscopy (EIS) detection was conducted with a Vers STAT 3 (Princeton Applied Research, UK). The time-resolved surface plasmon resonance spectrometer (TR-SPR) (DyneChem HiTech Ltd. China), a single channel and prism coupling-based instrument, was used to investigate the binding performance of a target protein and immunoreactions of antigen protein onto the polymer films-modified SPR chip. 1H NMR spectra were performed by 1H NMR spectroscopy on Avance 400 MHz spectrometer (Bruker, Switzerland). Contact angle was measured by OCA15plus contact angle meter (Dataphysics Inc., Germany). Attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIAS) were carried out on a Tensor 27 (Bruker Inc., Germany) equipped with a liquid-nitrogen-cooled MCT detector.Synthesis of N-hydroxysuccinmidyl bromoisobutyrate (NHS-Br) initiator
The NHS-Br initiator was prepared according to our previous method.29,30,32 In brief, a solution of 2-bromoisobutyryl bromide (10.86 mmol) in anhydrous diethyl ether (50 mL) was cooled in an ice bath. A solution of N-hydroxysuccinimide (10.86 mmol) and TEA (16.31 mmol) in 1,4-dioxane (50 mL) was then added dropwise. After that, the reaction mixture was stirred at room temperature for 3 h, followed by filtration to remove any precipitates. The filtrate was washed with saturated NaHCO3 solution and twice-distilled water, and then dried over MgSO4. Most of the solvent was removed on a rotary evaporator. The residue was stored at 4 °C to obtain a white solid product. 1H NMR (CDCl3, 400 MHz): d = 2.87 (s, 4H), 2.08 ppm (s, 6H); 13C NMR (CDCl3, 400 MHz): d = 25.5, 30.6, 51.1, 167.5, 168.6 ppm. The as-prepared NHS-Br active ester was used in the following experiments without further purification.Synthesis of macroinitiator
The synthesis of streptavidin/initiator-functionalized P(AA-AM), also known as macroinitiator, was outlined in Scheme 2. In the first step, 0.8 mg P(AA-AM) was completely dissolved in 700 μL twice-distilled water at room temperature for 30 min, then 32 mg NHS-Br initiator dissolved in 1 mL DMF was dropped into the reaction mixture and incubated on a shaker overnight at room temperature, which allowed complete coupling and hydrolysis of excess NHS esters. After a predetermined time, the resulted mixture was ultrafiltrated at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g-force for approximately 15 min at 15 °C. It was then washed by deionized for 3 times under the same condition to remove the nonconjugated initiators, the P(AA-AM)–NHS–Br were thus obtained. In the second step, the obtained P(AA-AM)–NHS–Br was dispersed in a mixed solutions of 440 μL EDC (1 mg mL−1, in PB, pH 7.2) and 260 μL NHS (1 mg mL−1, in PB, pH 7.2) at room temperature for 30 min in order to activate the carboxyl groups of P(AA-AM), followed by addition of 150 μL of streptavidin (1 mg mL−1, in PB, pH 7.2). After reaction at room temperature under continuous shaking for 2 h, the resulted mixture was ultrafiltrated and washed at the same condition above. Thus, dual-functional macrophotoinitiators with initiation and biological recognition were obtained (80 μL) and stored at 4 °C for the subsequent polymerization.
000g-force for approximately 15 min at 15 °C. It was then washed by deionized for 3 times under the same condition to remove the nonconjugated initiators, the P(AA-AM)–NHS–Br were thus obtained. In the second step, the obtained P(AA-AM)–NHS–Br was dispersed in a mixed solutions of 440 μL EDC (1 mg mL−1, in PB, pH 7.2) and 260 μL NHS (1 mg mL−1, in PB, pH 7.2) at room temperature for 30 min in order to activate the carboxyl groups of P(AA-AM), followed by addition of 150 μL of streptavidin (1 mg mL−1, in PB, pH 7.2). After reaction at room temperature under continuous shaking for 2 h, the resulted mixture was ultrafiltrated and washed at the same condition above. Thus, dual-functional macrophotoinitiators with initiation and biological recognition were obtained (80 μL) and stored at 4 °C for the subsequent polymerization.
 | ||
| Scheme 2 Schematic representation of the preparation of the macroinitiator. | ||
Sandwiched immunoassay for preparation of the macroinitiator-coupled anti-IgG-modified Au substrates
:3 v/v, H2SO4–H2O2) (Caution!) for 1 h to active the surface and remove inorganic and organic contaminants on the surface. For the immobilization of anti-IgG (Ab1), a self-assembled monolayer with carboxyl functional group was firstly formed on the surface of Au substrate by incubating the substrate in a saturated MUA–ethanol solution overnight. After being removed from solution and thoroughly rinsed with ethanol and H2O, the MUA-coated Au substrate was dried under a stream of N2, and then dipped in 0.4 mM EDC/0.1 mM NHS in 0.1 M, pH 7.2 PB for 30 min at 4 °C. The substrate was washed thoroughly with 0.1 M pH 7.2 PB followed by dropping 5 μL of Ab1 solution (1 mg mL−1, in 0.1 M pH 7.2 PB, denoted Ab1) onto the surface of MUA-modified Au substrate and incubating at room temperature for 1 h. Finally, the Ab1-modified Au substrate was soaked in 1% BSA solution for another 30 min to block excess active groups and nonspecific binding sites on the substrate. The substrate was then rinsed thoroughly with buffer (0.1 M, pH 7.2, PB) and stored at 4 °C for following use.
Surface-initiated AGET ATRP polymerization
In a typical surface-initiated AGET ATRP reaction, a mixture of CuBr2 (7.0 mg) and bpy (9.38 mg) was added to a glass container containing 1.6 mL of HEMA solution (1:1, v/v, HEMA–H2O). The macroinitiator-immobilized substrate was then immersed into the reaction mixture, followed by addition of 50 μL ascorbic acid (AA, 96 mg mL−1) to trigger the polymerization reaction immediately after the container was sealed. The polymerization was performed at room temperature for various periods of time, as specified in the text. The polymerization was stopped by removing the substrate from the reaction solution and rinsing with methanol, followed by washing with copious water to remove nonspecifically adsorbed monomers. Thus the Au substrate with PHEMA spots located in the site of anchored initiator was obtained.
Additionally, small molecule initiator NHS-Br was employed to directly trigger the AGET ATRP reaction with the purpose of demonstrating the amplification effect of macroinitiator. In this control experiment, the NHS-Br initiator was coupled with Ab2 at room temperature overnight according to the previous literature,30 followed by same sandwiched immunoreaction and AGET ATRP process.
Results and discussion
Sandwich immunoassay using macroinitiator and AGET ATRP
The high-molecular-weight copolymer P(AA-AM) with carboxyl groups and part of amino groups could covalently reacted with streptavidin and water-soluble ATRP initiators, respectively (Scheme 2). The binding of NHS-Br to P(AA-AM) was analyzed by 1H NMR (Fig. S1†). Peaks at 1.55, 1.66, 2.11 and 2.24 ppm were belonged to the protons peak of methylenes on P(AA-AM) backbone. A new peak at 1.84 ppm on curve A was attributable to the protons peak of the methyl groups in modified bromoisobutyryl groups, confirming that the initiator group had been successfully linked to P(AA-AM). After further conjugated with streptavidin, the P(AA-AM)–streptavidin displayed a specific absorption peak of protein at approximate 275 nm in the UV-vis spectra (Fig. S2†).After polymerization, a new broad peak between 3000 and 3500 cm−1 (hydroxyl stretching) accompanied with an absorption increase of carbonyl peak near 1700 cm−1, as well as peaks at 1450, 2850 and 2920 cm−1 corresponding to stretch vibration of methyl and methylene groups were clear observed in reflectance FT-IR spectra, confirmed the formation of PHEMA film (Fig. S3†). The formation of PHEMA film on the surface of Au substrate was also demonstrated by attenuated total reflection surface-enhanced infrared absorption spectroscopy as shown in Fig. 1A. The Ab1/Au/silicon-prism was incubated with 0 ng mL−1 (curve a) and 10 ng mL−1 IgG (curve b) for 30 min, followed by immersed in Ab2 solution and coupled with macroinitiator for another 30 min. In order to deduct the basal signal and obtain the ATR-SEIAS spectra of generated PHEMA directly, the spectrum of macroinitiator/Ab2/Ag/Ab1/Au/silicon-prism in PB was collected and set as the background. After that, the substrate was further performed to carry out an AGET ATRP of HEMA. For comparison, the Ab2/IgG/Ab1/Au substrate was reacted with the only SA-labeled P(AA-AM) without NHS-Br coupling, followed by afore mentioned background scanning procedure and the ATRP reaction (curve c). After polymerization, a strong IR absorption of polymer at ∼1700 cm−1 was clearly observed due to the enhanced adsorption intensity of carbonyl in polymer after subtraction of the background. No discernible polymer growth on the substrate was observed for Ab1/Au/silicon-prism in the absence of IgG and the Au/Ab1/IgG/Ab2/P(AA-AM) without NHS-Br coupling (curve a and c). All these results suggested that the generated IR signal was not resulted from the physical adsorption of monomer HEMA, but mainly due to the formed PHEMA via the sandwich immunoreaction and macroinitiator-triggered polymerization.
 | ||
| Fig. 1 (A) The ATR-SEIAS spectrum of Ab1/Au–Si prism incubated with 0 ng mL−1 (curve a) and 10 ng mL−1 IgG (curve b) for 30 min, followed by being immersed in Ab2 solution, coupled with macroinitiator and polymerization for 15 min. The ATR-SEIAS spectra of macroinitiator/Ab2/Ag/Ab1/Au/silicon-prism collected in PB was set as the background. Curve (c) was the Ab2/IgG/Ab1/Au substrate reacted with the only SA-labeled P(AA-AM) without NHS-Br coupling, followed by the same background scanning process and the ATRP reaction like curve a. (B) and (C) respectively showed the contact angles and SPR resonance angles of different Au chips including (a) bare Au, (b) MUA/Au, (c) Ab1/MUA/Au, (d) BSA/Ab1/MUA/Au, (e) Ag/Ab1/MUA/Au, (f) Ab2/Ag/Ab1/MUA/Au, (g) macroinitiator/Ab2/Ag/Ab1/MUA/Au, and (h) PHEMA/macroinitiator/Ab2/Ag/Ab1/MUA/Au, at IgG concentration of 100 μg mL−1, polymerization time of 7 min. The error bars were calculated based on three replicates. | ||
Additionally, the growing polymer chain provided numerous hydroxyl groups, which led to the change of the hydrophily of substrate, which could be monitored by contact angle measurements. Fig. 1B showed the cleaned Au substrate displayed a contact angle of 74.2°. After modified with MUA monolayer, the extended carboxylic group in MUA led to the increase of the hydrophilic property of the Au substrate, which made a remarkable decrease of the contact angle to 30.2°. The BSA and Ab1 modified Au substrates displayed slightly decrease of the contact angle to 28.5° and 25.2°, respectively. After sandwiched immunoreactions, the contact angles increased slightly, e.g., 27.3° for Ag/Ab1/MUA/Au and 25.2° for Ab2/Ag/Ab1/MUA/Au. The decrease of the contact angle for these protein modified substrate was reasonable due to the increasing surface hydrophily by the immobilized protein comparing to MUA layer. After further coupling with macroinitiator, the contact angle increased to 30.1° due to the better hydrophobic properties of the macromolecules than proteins. The small change of contact angle not only confirmed the successful immunoreactions, but also somewhat showed that the surface was partially covered by long-chain macromolecules instead of former antibody sites. Once undergoing the polymerization, an increase of the contact angle to 52.9° was observed due to the formation of much hydrophobic and rough surface (supported by SEM image in Fig. 2B) on the macromolecules coupled site. What's more, to confirm the contact angle increase wasn't caused by sandwich dissociation during polymerization, the Ab2/IgG/Ab1/Au substrate was reacted with the only SA-labeled P(AA-AM) without NHS-Br coupling and after polymerization the contact angle shows no obvious decrease compared with before polymerization (Table S1†).
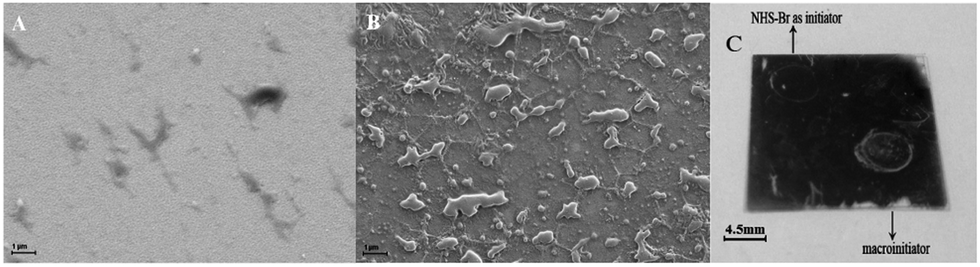 | ||
| Fig. 2 The SEM images of (A) NHS-Br-coupled Ab2*/IgG/Ab1/Au and (B) macroinitiator/Ab2/IgG/Ab1/Au substrate after polymerization, with IgG concentration of 0.1 μg mL−1 and polymerization time of 0.5 h. (C) A photograph of polymer growing on a substrate after polymerization with different initiators NHS-Br and macroinitiator. | ||
The sandwiched immunoreaction and the polymerization initiated by the macroinitiator were further confirmed by electrochemical impedance spectroscopy (EIS) (Fig. S4†) and surface plasmon resonance (SPR) measurements. The time-resolved surface plasmon resonance spectrometer was usually employed to ascertain the multiple coupling process involving protein, macroinitiator, and polymer in real time. Fig. 1C showed the SPR signal changed as a function of scanning angle on SPR chips with different modification stages. The resonance angles of the MUA/Au chip and Ab1/MUA/Au chip were 64.62° and 65.25°, respectively, which were slightly higher than that of the bare Au chip (64.34°). After the sandwiched immunoreactions, the resonance angle of the resulting Ab2/Ag/Ab1/MUA/Au chip increased to 66.09°. With respect to the following macroinitiator and polymerization of HEMA atop Ab2/MUA/Au chip process, the chips displayed higher resonance angles of 66.22° and 67.70°, respectively. Control experiments were performed by incubating of the Ab1-modified substrate with 50 μg mL−1 thrombin, 50 μg mL−1 breast cancer antigen (CA15-3), and 50 μg mL−1 α-fetoprotein (AFP), followed by same procedures including the second immunoreaction with Ab2, the coupling macroinitiator through streptavidin–biotin interaction and macroinitiator-triggered AGET ATRP. When the target changed to thrombin, AFP, and CA15-3, the resonance angle of 65.19°, 65.48° and 65.47° was very close to 65.25° for Ab1/MUA/Au chip (Fig. S5†). This confirmed that the macroinitiator could be loaded to the Au substrate through highly specific sandwich immunoreactions while not originated from physical absorption or cross-reaction.
All of the above confirmed that the immobilized macroinitiators were from a highly specific immunoassay, and the macroinitiators initiated AGET ATRP on the chip surface with high initiation efficiency. More quantitative measurements were carried out in the following contact angle and colorimetric analysis.
Detection of IgG
The current strategy using P(AM-AA) as carrier increased the loading of the NHS-Br initiator so as to in turn increase the polymerization efficiency and enhance the signal readout. Fig. 2 showed the SEM images and the photographic picture of modified Au substrate after polymerization for 0.5 h at IgG concentration of 0.1 μg mL−1. Prior to polymerization, the SEM images for both Au slice modified with immunocomplex labeled with NHS-Br directly and the BT-label immunocomplex further coupled with SA-linked macroinitiator displayed clean and smooth surface (Fig. S6A and B†). In contrast, immediately examination of the surface after 0.5 h of polymerization revealed numerous polymer islands on the surface (Fig. 2A and B), which indicated successful AGET ATRP. Moreover, a large increase of polymer density could be observed clearly from the Au slice surface that employed macroinitiator to trigger polymerization reaction, while the polymer film was sparser when NHS-Br was used as initiator. This was also revealed by the pictures as shown in Fig. 2C, where the exuberant PHEMA growth after polymerization enabled the direct visualization of the immunocomplex-macroinitiator immobilized spot, yet the NHS-Br labeled immunocomplex spot emerged little polymer on the edge. Thus, the macroinitiator signal amplification was remarkable, and the improved polymerization efficiency was attributed to the attached macroinitiator containing streptavidin for molecular recognition and NHS-Br initiator for initiating the polymerization.In the polymerization-based sensing strategy, the signal intensities both observed by naked eyes and detected by contact angle measurement, were dependent on the length and density of the polymer chains, i.e., the number of the hydroxyl groups grafted per biomolecular binding event. Due to the living polymerization reaction, AGET ATRP offered the advantage to tune the length of polymer chains by the variation of reaction time. In other words, a longer polymerization time would result in a larger amount of hydroxyl groups per initiation site on the substrate. The formed thick polymer films not only altered the surface reflectivity, but also changed the surface opacity, thus rendered the spots directly visible to the naked eye.36Fig. 3 showed the visual detection of 0.1 μg mL−1 IgG at various polymerization time. Once beyond 7 min, the clear spots could be observed on the substrate (Inset in Fig. 3). The spot coated by PHEMA looked not very uniform. This was possibly due to the irregular self-assembled MUA layer on the Au substrate. Moreover, the optimal polymerization time for visual detection was ascertained by the means of measurement of the contact angle of each spot. The values of contact angle were decreased along with the growing of reaction time, which was nearly linear at the early stage of the polymerization. The plot, however, started to level off after 10 min. This slow-down tendency in the decrease of the contact angle at later stage might be due to several reasons, including a corresponding slow-down of polymer growth from radical termination, and reduced accessibility of hydroxyls on the side chain of PHEMA due to lower AGET ATRP monomer amount and/or the steric hindrance.26 To avoid tedious procedure, the optimum polymerization time for model protein IgG detection by naked eyes was chosen to be 10 min.
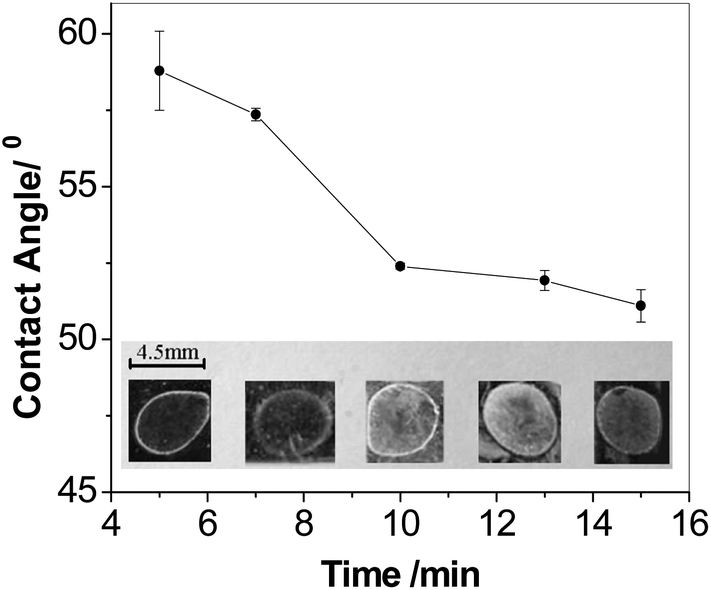 | ||
| Fig. 3 A plot of contact angles as a function of polymerization time. The error bars were calculated based on three replicates. Inset: a photograph of polymer-modified substrates with polymerization time of 5, 7, 10, 13 and 15 min (from left to right) with IgG concentration of 0.1 μg mL−1. | ||
Fig. 4A and B showed the qualitative detection of IgG by visual inspection. Under the optimum polymerization time, the concentrations of target IgG solutions varied from 100 μg mL−1 to 1 ng mL−1 were measured. It could be seen that the result of 10 ng mL−1 IgG was readily discernible to the naked eyes, while no pronounced difference could be found from neither the control spot where the IgG concentration was 0 ng mL−1 nor the spot with an IgG concentration of 1 ng mL−1. Therefore, a 10 min reaction was sufficient to be distinguished from the background (0 ng mL−1) when the concentration of analyte was higher than 10 ng mL−1. Moreover, longer reaction time like 15 min or more could easily endow the visual qualitative determination of the target proteins with lower detection limit even at nanogram level (data not shown).
 | ||
| Fig. 4 (A) Schematic diagram of test surface, spot 1 to 9 stand for IgG concentration of 100, 50, 10, 5, 1, 0.1, 0.01, 0.001 and 0 μg mL−1. (B) A photograph of a sandwich immunoreaction incubated with IgG at different concentrations corresponding to (A) after polymerization for 10 min. (C) Plot of contact angle of the test surface polymer versus IgG concentrations. Inset: linear plot for IgG determination. ATRP reaction time = 7 min. | ||
Based on the macroinitiator-triggered polymerization, less reaction time and lower detection limit could be realized by the contact angle measurement. After polymerization for 7 min and thorough washing with twice-distilled water, the substrate surface was dried under N2, followed by the contact angle measurement. The contact angle values of substrates were found to be proportional to the concentration of IgG in the incubation solutions (Fig. 4C). The contact angle decreased with the IgG concentration from 0 μg mL−1 to 100 μg mL−1. The linear equation in the concentration range of 1 ng mL−1 to 100 μg mL−1 was θCA = 56.58 − 2.05log(c/μg mL−1) with a correlation coefficient R2 of 0.991, in which θCA was the contact angle value and c was the IgG concentration in the incubation solution. The detection limit of 0.13 ng mL−1, at a signal-to-noise ratio of 3, which was comparable to what had been reported for traditional immunochemical methods and the polymerization based amplification method in recent years.26,30 According to the low detection limit and available short polymerization time (as low as 7 min), the proposed method was demonstrated to be rapid and reliable for the IgG concentration detection.
To evaluate the analytical reliability and the practical application potential of the proposed method, the concentrations of IgG in three clinical serum samples, which were collected from the Second Hospital of Nanjing (Jiangsu, China), were examined and the standard addition recovery tests were carried out together. For comparison, the serum samples were simultaneously detected by enzyme linked immunosorbent assay (ELASA) in the Second Hospital of Nanjing. Every serum sample was be analyzed without any pretreatment except appropriate dilution (according the detection result that the hospital offered) to the detection range with PB (0.1 M, pH 7.2). The assay results by contact angle measurement are shown in Table 1. The recoveries for the spiked samples ranged from 93% to 115% and the RSD ranged from 15% to 25%. The relative deviation obtained by comparing the results from our method described above to ELASA provided by the hospital ranged from 2.6% to 12.7%, which indicated the acceptable accuracy and precision of this signal amplification strategy in the practical bioassay.
| Sample | Added (mg mL−1) | Found (mg mL−1) | Recovery (%) | Clinical detection value (mg mL−1) | Relative deviation (%) |
|---|---|---|---|---|---|
| 1 | 0 | 15.9 ± 3.9 | 114 | 14.1 | 12.7 |
| 18.3 | 36.5 ± 6.1 | ||||
| 2 | 0 | 20.1 ± 3.1 | 93 | 18.3 | 9.8 |
| 18.3 | 37.2 ± 2.9 | ||||
| 3 | 0 | 22.9 ± 3.9 | 101 | 22.2 | 2.6 |
| 18.3 | 41.4 ± 3.4 |
Conclusions
In this work, a novel strategy for rapid and sensitive immunoassay of IgG by integrating macroinitiator and polymerization-based signal amplification was successfully developed. The poly(acrylic acid-co-acrylamide) provided numerous amino groups on the side chains for loading of NHS-Br initiators, which gave rise to an enhanced initiation efficiency even at low concentration of antigen after immobilization of macroinitiator via specific immunity. The increased loading of initiators significantly enhanced the accumulation of PHEMA at the location where the macroinitiator was attached, resulting in the changing of the surface reflectivity, opacity and the hydrophily. Utilizing the macroinitiator led to higher initiator efficiency and polymerization efficiency thus made it possible to grow large amount of polymer in short time. Obvious distinction could be observed from the background by naked eyes within 10 min polymerization and a detection limit of 0.13 ng mL−1 by contact angle measurement was achieved with 7 min polymerization. Through extending the reaction time, this method could obtain even lower detection limit. Considering the acceptable accuracy and precision of assay results for clinical human serum samples, the proposed method was easier to perform and less time-consuming. The presented strategy taking advantage of the high initiation efficiency of macroinitiator and polymerization-based signal amplification offered great promising applications in providing a sensitive, specific and potent method for wide biological detection.Acknowledgements
The project is supported by the National Basic Research Program of China (no. 2010CB732400), the Key Program (21035002) from the National Natural Science Foundation of China and the National Natural Science Foundation of China (Grant no. 20875013, 21175021).Notes and references
- K. Mitsakakis and E. Gizeli, Biosens. Bioelectron., 2011, 26, 4579–4584 CrossRef CAS PubMed.
- K. Vangala, M. Yanney, C. T. Hsiao, W. W. Wu, R. F. Shen, S. G. Zou, A. Sygula and D. M. Zhang, Anal. Chem., 2010, 82, 10164–10171 CrossRef CAS PubMed.
- K. Yang and C. Y. Zhang, Anal. Chem., 2010, 82, 9500–9505 CrossRef CAS PubMed.
- K. Knox, D. Carrigan, G. Simmons, F. Teque, Y. C. Zhou, J. Hackett, X. X. Qiu, K. C. Luk, G. Schochetman, A. Knox, A. M. Kogelnik and J. A. Levy, Science, 2011, 333, 94–97 CrossRef CAS PubMed.
- H. Dacres, J. Wang, V. Leitch, I. Horne, A. R. Anderson and S. C. Trowell, Biosens. Bioelectron., 2011, 29, 119–124 CrossRef CAS PubMed.
- Y. H. Chen, M. R. Snyder, Y. Zhu, L. J. Tostrud, L. M. Benson, J. A. Katzmann and H. R. Bergen, Clin. Chem., 2011, 57, 1161–1168 CAS.
- M. J. Whitcombe, I. Chianella, L. Larcombe, S. A. Piletsky, J. Noble, R. Porter and A. Horgan, Chem. Soc. Rev., 2011, 40, 1547–1571 RSC.
- S. K. Vashist, D. Zheng, K. Al-Rubeaan, J. H. T. Luong and F. S. Sheu, Biotechnol. Adv., 2011, 29, 169–188 CrossRef CAS PubMed.
- R. Zeleny and H. Schimmel, Food Chem., 2010, 123, 1343–1351 CrossRef CAS PubMed.
- J. S. Lu, W. Wei, L. H. Yin, Y. P. Pu and S. Q. Liu, Analyst, 2013, 138, 1483–1489 RSC.
- W. Boonjob, M. Miro, M. A. Segundo and V. Cerda, Anal. Chem., 2011, 83, 5237–5244 CrossRef CAS PubMed.
- X. X. Zhu, A. M. Kriegel, C. A. Boustany and D. A. Blake, Anal. Chem., 2011, 83, 3717–3724 CrossRef CAS PubMed.
- L. Y. Chen, C. L. Chen, R. N. Li, Y. Li and S. Q. Liu, Chem. Commun., 2009, 2670–2672 RSC.
- K. C. Han, E. G. Yang and D. R. Ahn, Chem. Commun., 2012, 48, 5895–5897 RSC.
- G. S. Lai, F. Yan, J. Wu, C. Leng and H. X. Ju, Anal. Chem., 2011, 83, 2726–2732 CrossRef CAS PubMed.
- G. F. Jie and J. X. Yuan, Anal. Chem., 2012, 84, 2811–2817 CrossRef CAS PubMed.
- H. Zhong, Q. L. Zhang and S. S. Zhang, Chem.–Eur. J., 2011, 17, 8388–8394 CrossRef CAS PubMed.
- G. Liu, Y. Wan, V. Gau, J. Zhang, L. H. Wang, S. P. Song and C. H. Fan, J. Am. Chem. Soc., 2009, 130, 6920–6925 Search PubMed.
- F. L. Gao, J. P. Lei and H. X. Ju, RSC Adv., 2013, 3, 13163–13168 RSC.
- M. M. Ali and Y. F. Li, Angew. Chem., Int. Ed., 2009, 48, 3512–3515 CrossRef CAS PubMed.
- J. Zhou, M. D. Xu, D. P. Tang, Z. Q. Gao, J. Tang and G. N. Chen, Chem. Commun., 2012, 48, 12207–12209 RSC.
- A. Srivastava, V. Mishra, P. Singh and R. Kumar, J. Appl. Polym. Sci., 2012, 126, 395–407 CrossRef CAS.
- A. K. Bunha, J. Mangadlao, M. J. Felipe, K. Pangilinan and R. Advincula, Macromol. Rapid Commun., 2012, 33, 1214–1219 CrossRef CAS PubMed.
- Z. L. Yao and K. C. Tam, Polymer, 2012, 53, 3446–3453 CrossRef CAS PubMed.
- J. K. Lee, B. W. Heimer and H. D. Sikes, Biomacromolecules, 2012, 13, 1136–1143 CrossRef CAS PubMed.
- Y. F. Wu, S. Q. Liu and L. He, Anal. Chem., 2009, 81, 7015–7021 CrossRef CAS PubMed.
- Y. F. Wu, H. Y. Shi, L. Yuan and S. Q. Liu, Chem. Commun., 2010, 46, 7763–7765 RSC.
- Y. F. Wu, W. Wei and S. Q. Liu, Acc. Chem. Res., 2012, 45, 1441–1450 CrossRef CAS PubMed.
- L. Yuan, X. Hua, Y. F. Wu, X. H. Pan and S. Q. Liu, Anal. Chem., 2011, 83, 6800–6809 CrossRef CAS PubMed.
- L. Yuan, Y. F. Wu, H. Y. Shi and S. Q. Liu, Chem.–Eur. J., 2011, 17, 976–983 CrossRef CAS PubMed.
- Y. Liu and Q. Cheng, Anal. Chem., 2012, 84, 3179–3186 CrossRef CAS PubMed.
- L. Yuan, L. L. Xu and S. Q. Liu, Anal. Chem., 2012, 84, 10737–10744 CrossRef CAS PubMed.
- H. Qian and L. He, Sens. Actuators, B, 2010, 150, 594–600 CrossRef CAS PubMed.
- R. R. Hansen, L. M. Johnson and C. N. Bowman, Anal. Biochem., 2009, 386, 285–287 CrossRef CAS PubMed.
- H. D. Sikes, R. R. Hansen, L. M. Johnson, R. Jenison, J. W. Birks, K. L. Rowlen and C. N. Bowman, Nat. Mater., 2008, 7, 52–56 CrossRef CAS PubMed.
- X. H. Lou, M. S. Lewis, C. B. Gorman and L. He, Anal. Chem., 2005, 77, 4698–4705 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of macroinitiator, ATR-IR spectra, EIS, SPR spectra and SEM image. See DOI: 10.1039/c3ra45504a |
| This journal is © The Royal Society of Chemistry 2014 |