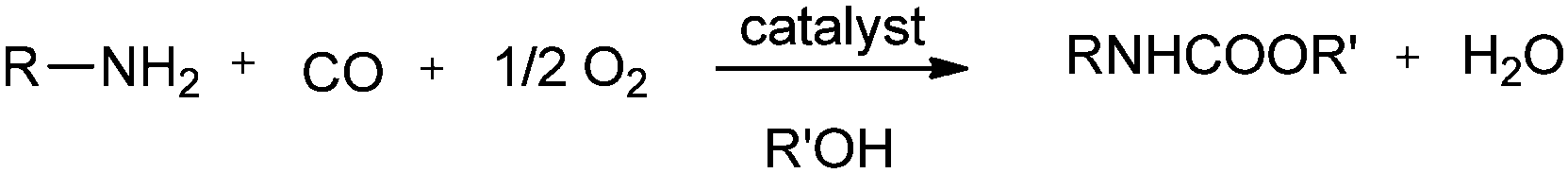
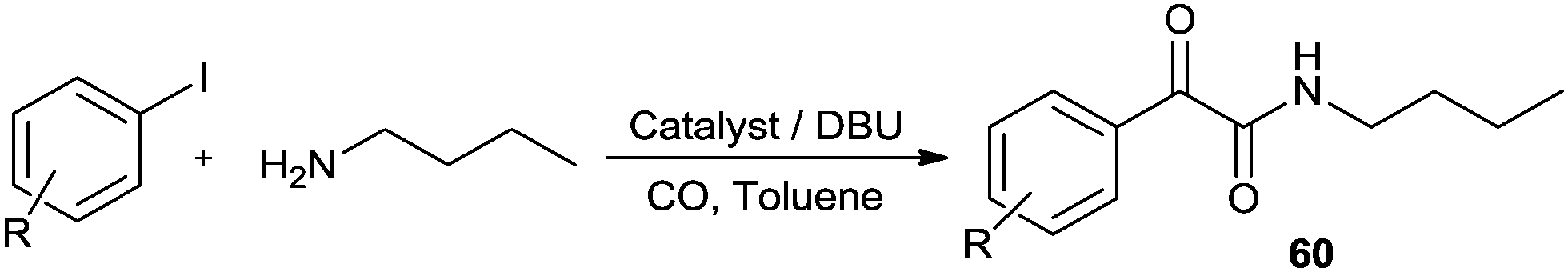
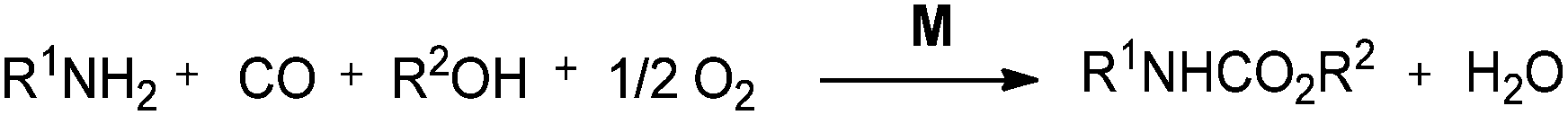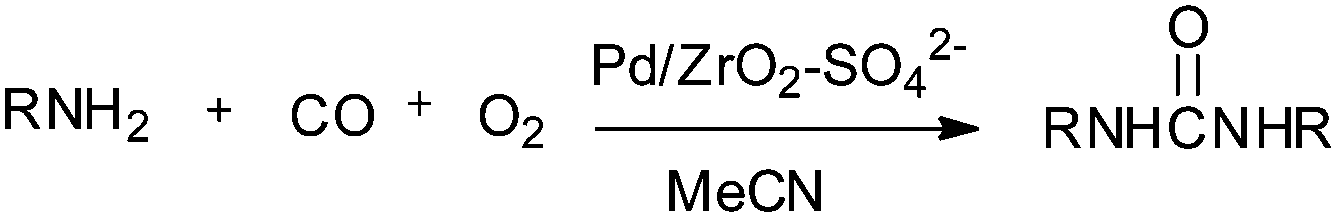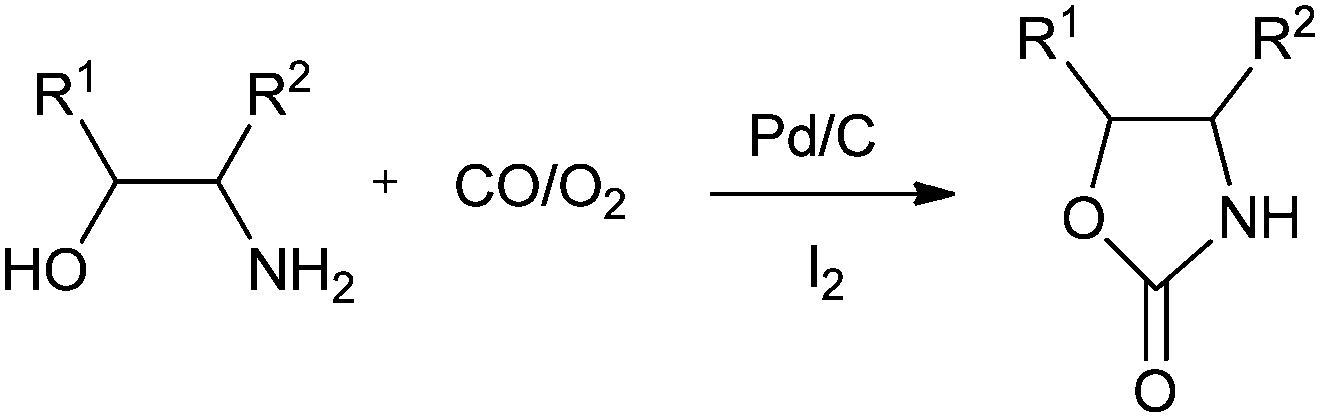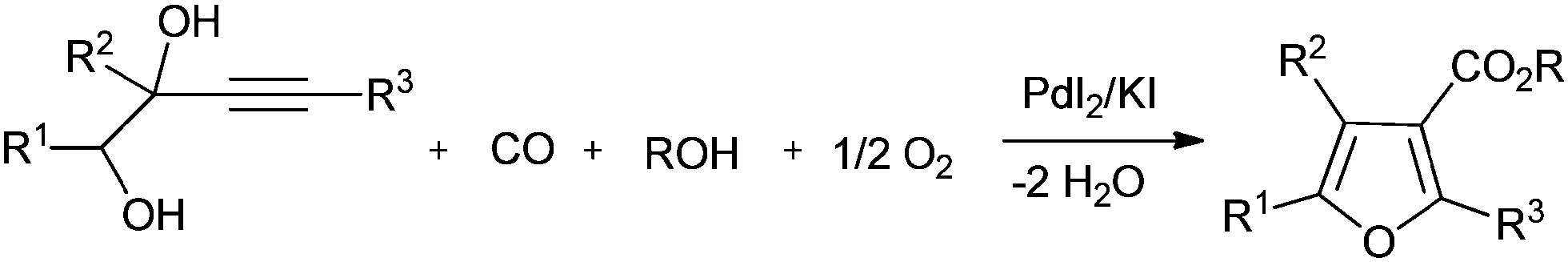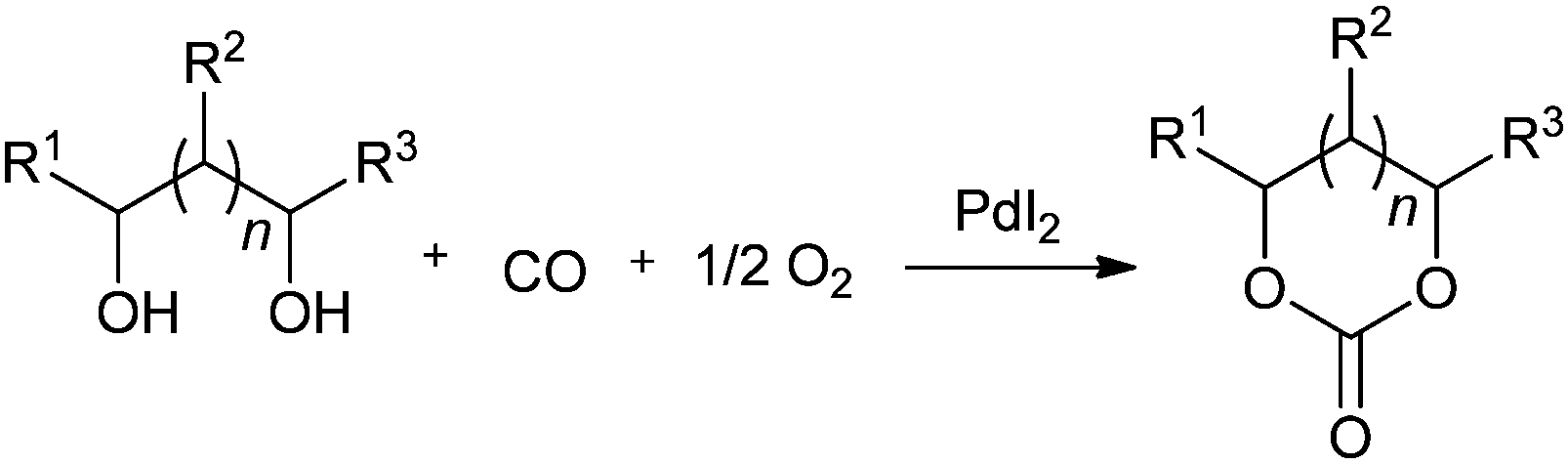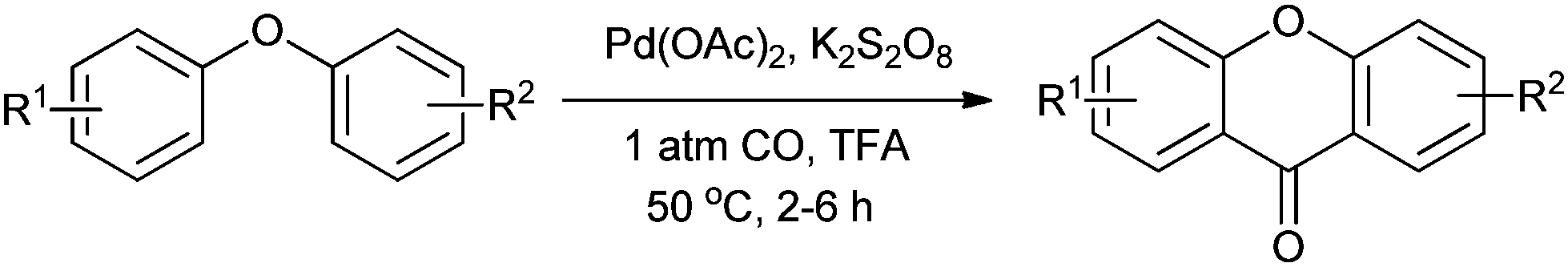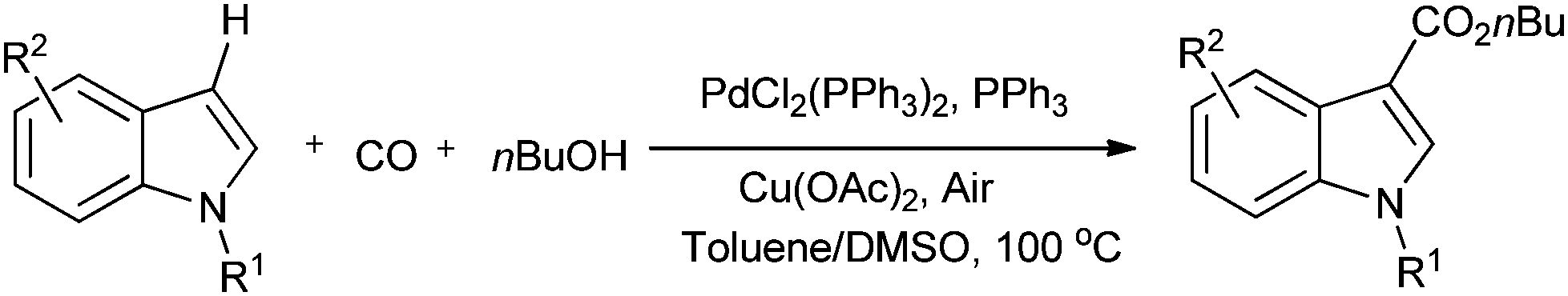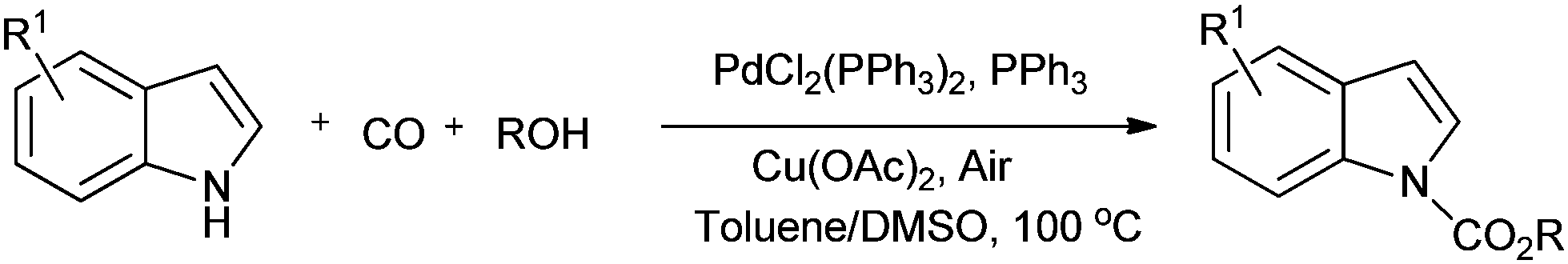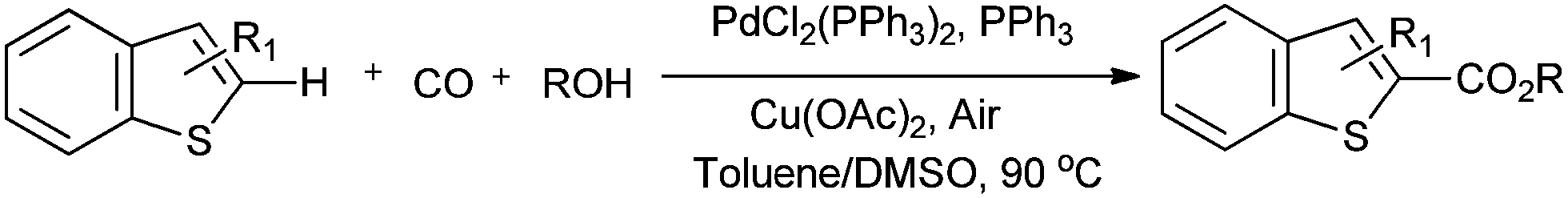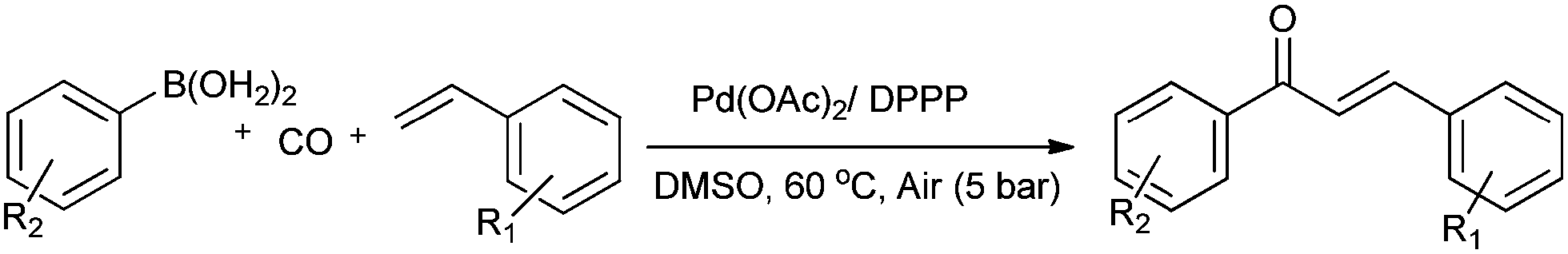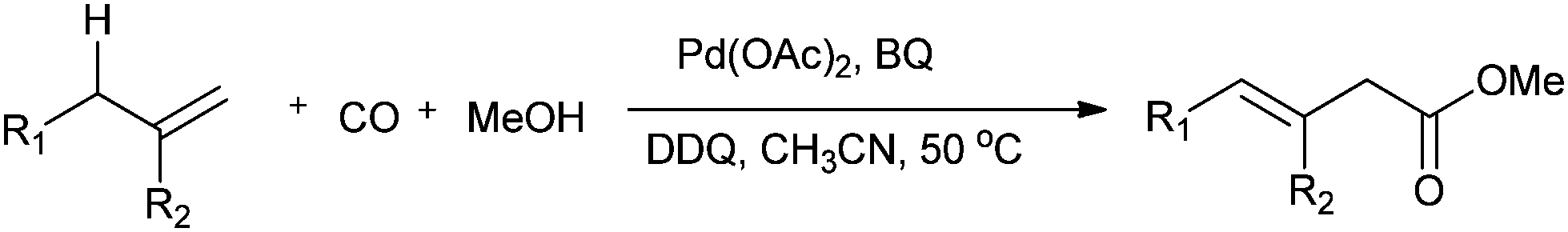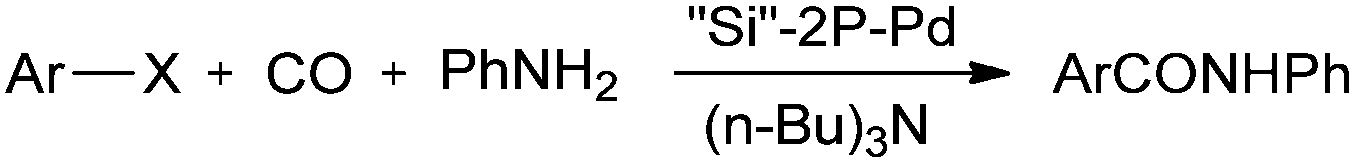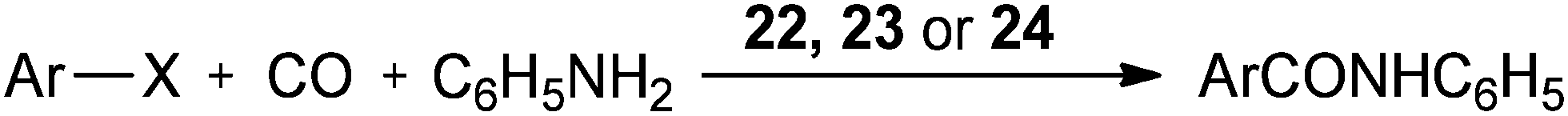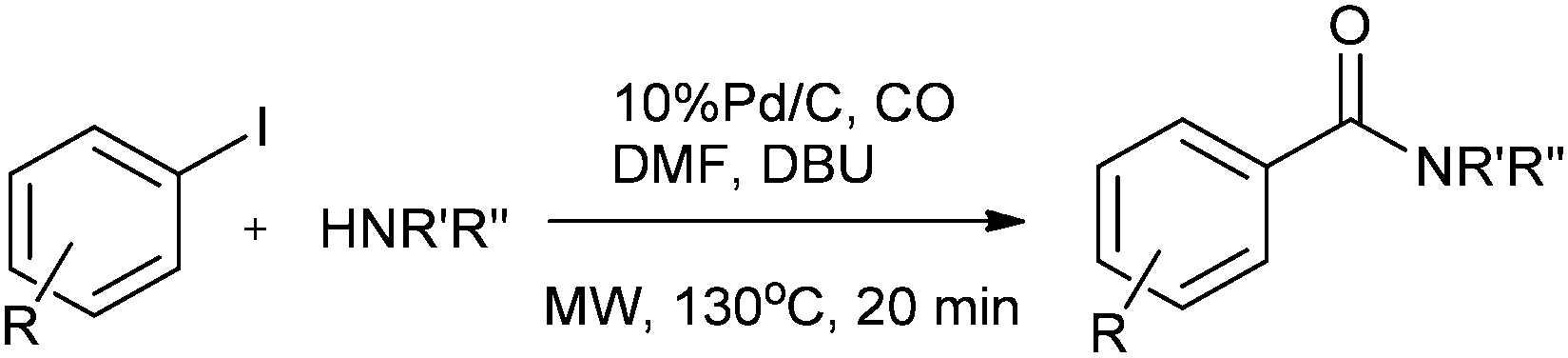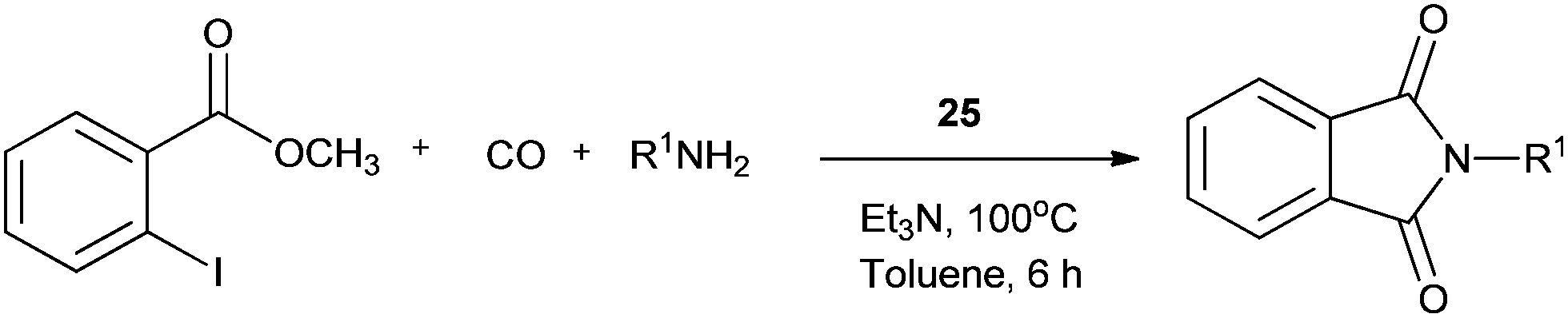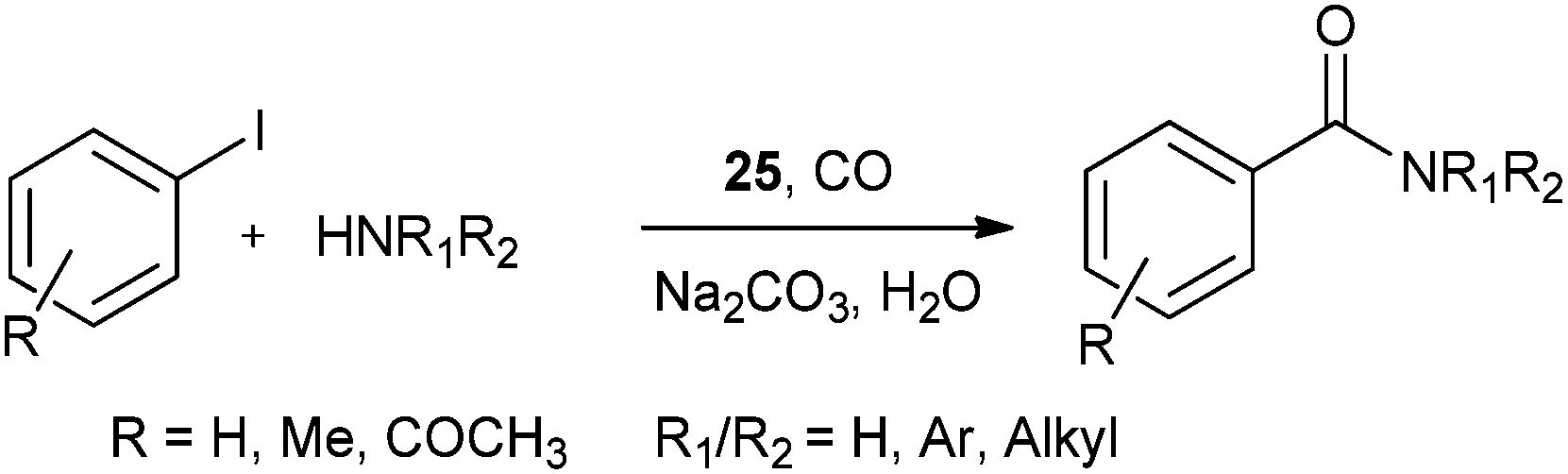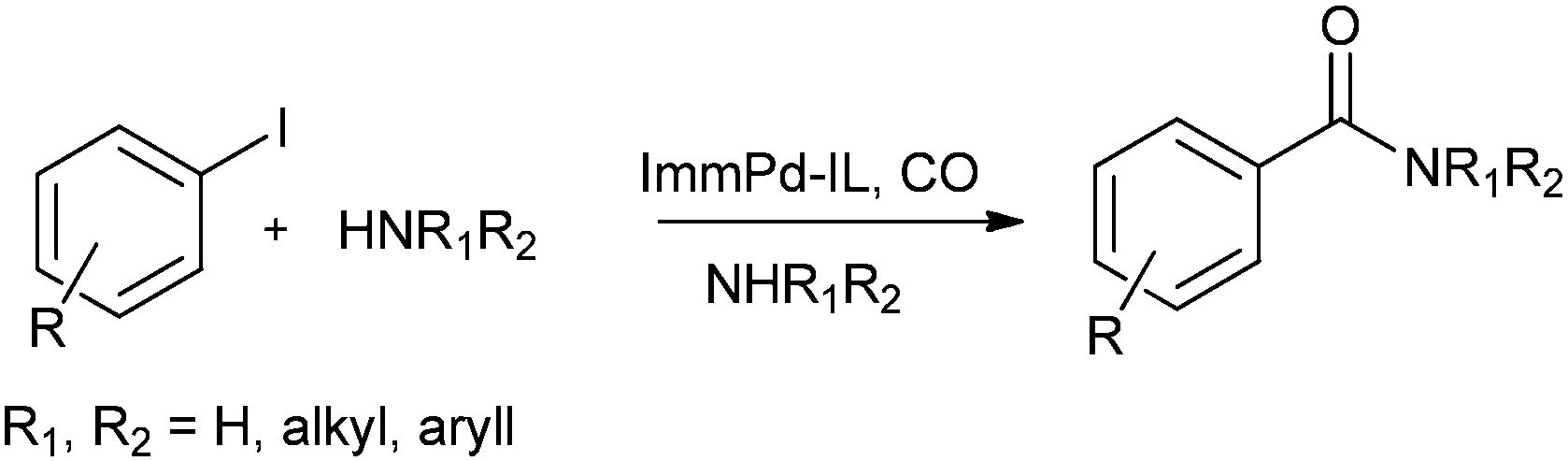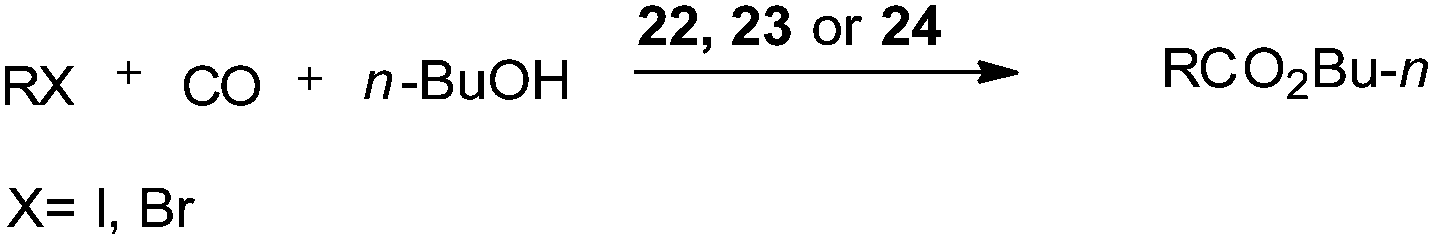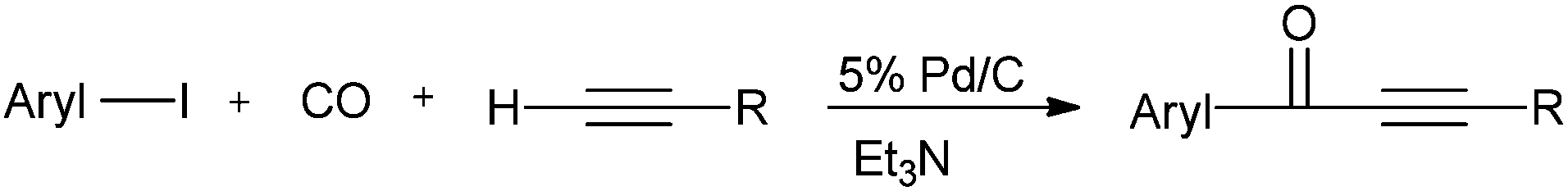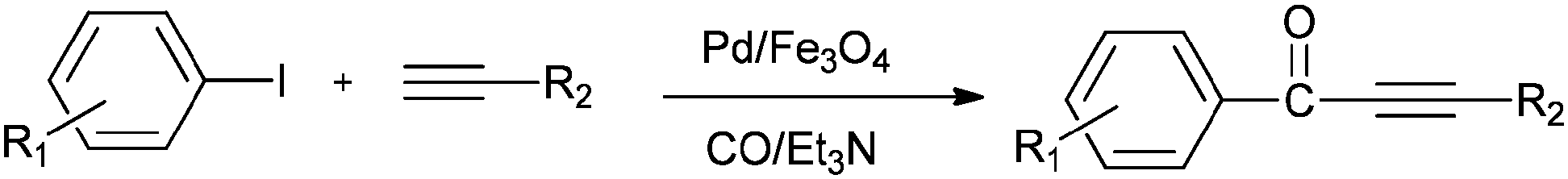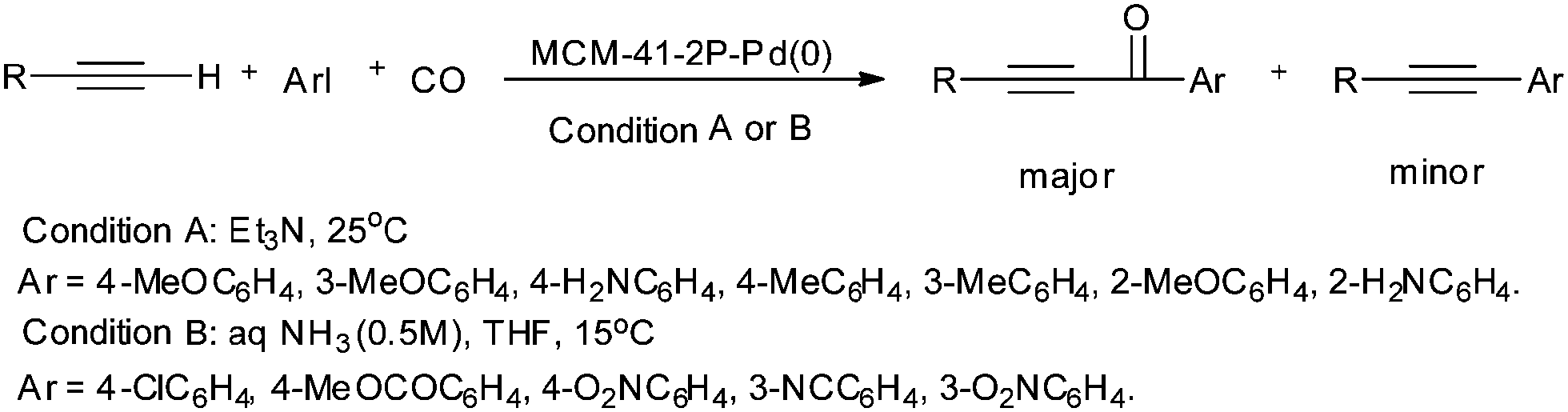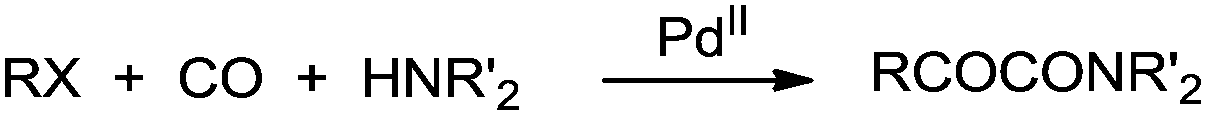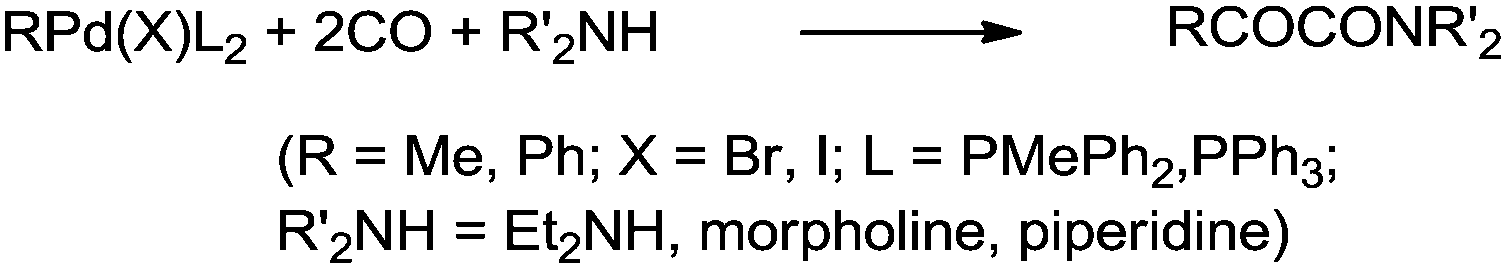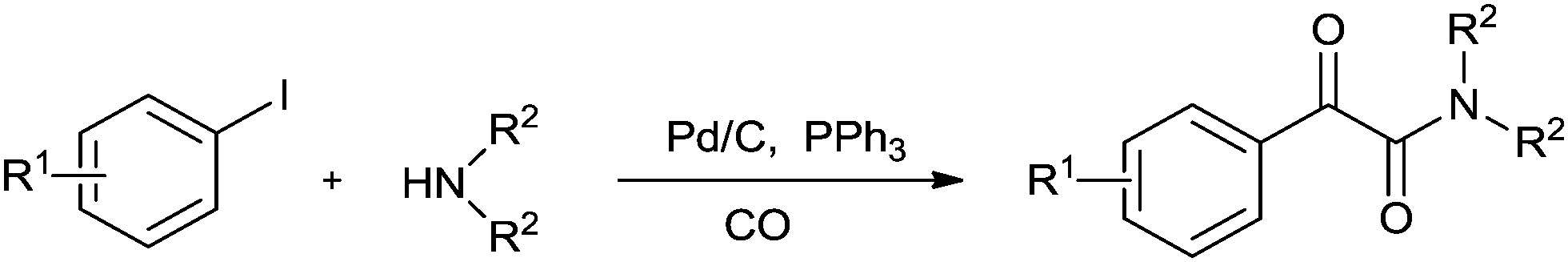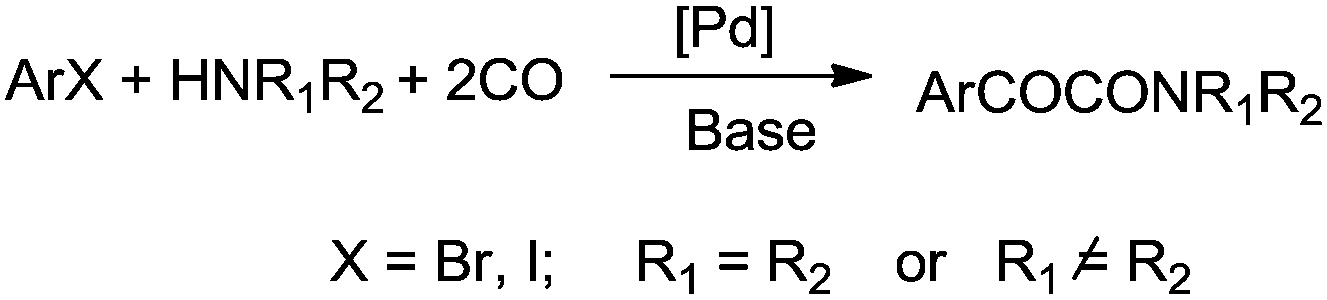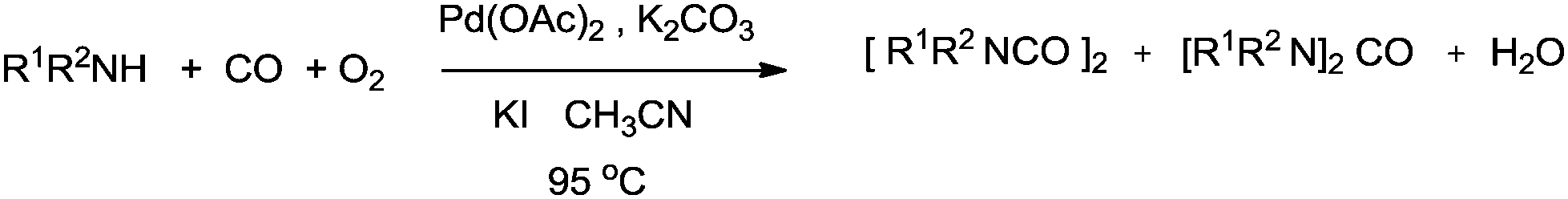DOI:
10.1039/C3RA46273K
(Review Article)
RSC Adv., 2014,
4, 10367-10389
Recent developments in palladium catalysed carbonylation reactions
Received
30th October 2013
, Accepted 19th December 2013
First published on 20th December 2013
Abstract
Recently, carbonylation reactions have gained considerable interest as they are becoming a versatile tool in the synthesis of pharmaceuticals, agrochemicals and their intermediates. Nowadays, a plethora of transition metal catalysts are available for the synthesis of various functional groups like ureas, carbamates, oxamates, oxamides, α-keto amides, ketones, esters, etc. using carbonylation methodology. Several carbonylation reactions such as aminocarbonylation, alkoxycarbonylation, double carbonylation and oxidative carbonylation, provide efficient and attractive alternatives to the conventional synthetic routes on a laboratory or industrial scale. Oxidative carbonylation is an important reaction as it allows direct carbonylative C–H bond activation. A double carbonylation reaction provides a one step alternative route for the synthesis of α-keto amides, oxamides, and oxamates. It also eliminates the use of conventional thermally unstable and toxic reagents like oxalyl chloride. Several recent studies have focused on the various aspects of these reactions, including catalyst–product separation, and catalyst recoverability and reusability. In view of this, developments in anchoring homogeneous catalysts using various techniques like biphasic catalysis and supported liquid phase catalysis are gaining importance. Carbonylation routes using these techniques are simple, efficient, economical, avoid the use of ligands, and give the desired products in excellent yields. The use of phosphine ligands is disadvantageous as it leads to air/moisture sensitivity, tedious work-up procedures and high work-up costs. Several phosphine-free carbonylation routes eliminate the use of phosphine ligands, and provide economical and simple methods for these transformations. In this review we have summarized the recent trends in carbonylative transformations, which have undergone a rapid development.
 Sandip T. Gadge | Sandip T. Gadge completed his M.Sc. degree in Organic Chemistry from the Department of Chemistry, University of Pune in 2009. After working as a project assistant at IISER Pune for a few months, he moved to the Institute of Chemical Technology (formerly UDCT), Mumbai in 2011 to begin his Ph.D. programme under the supervision of Prof. Bhalchandra M. Bhanage. His Ph.D. research focuses on the development of advanced methodologies for carbonylation reactions such as oxidative carbonylation, double carbonylation reactions, alkoxycarbonylation and aminocarbonylation reactions of alkynes and aryl halides. |
 Bhalchandra M. Bhanage | Bhalchandra M. Bhanage obtained his doctorate from Pune University in 1996. He spent more than one year at Tohoku University, Japan (1997–1998), and four years at Hokkaido University, Japan (2000–2003) as a postdoctoral fellow with Prof. M. Arai. In 2004, he joined the Institute of Chemical Technology, Mumbai, India as a Professor of Industrial and Engineering chemistry. Among his current interests is catalysis using CO2, CO and H2. He has worked on developing novel catalysts for various coupling reactions, carbonylation reactions, hydroformylation reactions, amination reactions, etc. He is also working on nanomaterials, enzymatic catalysis and the use of non-conventional techniques like ultrasound and microwaves for various reactions. |
1. Introduction
The development of sustainable and efficient methodologies based on catalysis and organometallic chemistry has gained considerable attention. In this context, palladium catalysed carbonylation reactions are now widely recognized as a very important tool in industrial and organic chemistry. Palladium catalysed carbonylation chemistry allows the direct synthesis of carbonyl compounds using readily available feed stocks such as carbon monoxide (CO), which is also the simplest C-1 unit and meets the requirements of “atom economy”,1 step economy2 and “green chemistry”.3 The significance of palladium catalyzed cross coupling reactions has been recognized by awarding the 2010 Nobel Prize in chemistry to Professors Heck, Negishi, and Suzuki. Nowadays, a variety of transition metal catalysts are available for the synthesis of ureas, carbamates, oxazolidinones, oxamates, oxamides, α-keto amides, ketones, esters, amides, etc. These are important intermediates in the manufacture of dyes, pharmaceuticals, agrochemicals, and other industrial products. Various homogeneous transition metals can catalyse single or double carbonylation reactions, and oxidative carbonylation methodologies have been developed for the synthesis of these compounds. Palladium catalyzed oxidative carbonylation reactions require the coupling of organic nucleophiles or electrophiles in the presence of CO and an oxidant to prepare various carbonyl-containing compounds.4 The double carbonylation methodology provides an efficient way to synthesise oxamates, oxamides and α-keto amides, which were traditionally synthesised by multi-step syntheses using thermally unstable mono esters of oxalyl chlorides. The synthesis of mono esters of oxalyl chlorides requires the combination of equimolar quantities of oxalyl chloride and the appropriate alcohol, along with a distillation setup for the isolation of the product from the reaction mixture. This is a major drawback of this method.5 Hence, the double carbonylation methodology provides an efficient alternative for the synthesis of these industrially valuable products.
Today, catalyst–product separation techniques, catalyst recoverability and catalyst reusability are the central issues to achieve an economical and environmentally friendly approach from sustainable and industrial viewpoints. Several techniques, such as biphasic catalysis,6 supported metal catalysis, polymer anchored catalysis and metal leaching re-deposition, have been developed in recent years.7 The catalysts can be easily separated, repeatedly used and, consequently, these methods provide an efficient and economical way to perform carbonylative coupling reactions. Such catalysts avoid the use of phosphines and because of this they are not air sensitive and they show moisture stability. They afford the coupling products in high yields at short reaction times. There are several reviews contributing to the area of homogeneous transition metal catalysed carbonylation reactions.4a,b,8 In this review we have described the recent developments in carbonylative transformations considering all these aspects.
2. Heterogeneous palladium-catalysed oxidative carbonylation reactions
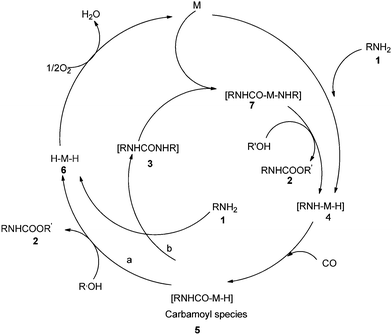
Palladium catalysed oxidative carbonylation reactions lead to carbonylated derivatives via coupling of two nucleophiles with the assistance of a suitable oxidant. The oxidant helps to oxidize the metal M(n) to M(n + 2), which promotes the catalytic cycle.4 The syntheses of urea, carbamate and oxazolidinone derivatives through the palladium catalysed oxidative carbonylation of amines and amino alcohols have been reported in several studies. Traditional synthetic methods for the synthesis of ureas use toxic and corrosive reagents, such as phosgene or isocyanates. In a typical oxidative carbonylation reaction, alcohols and amines are used as starting materials along with molecular oxygen as an oxidant in the presence of a homogeneous palladium catalyst.9 This leads to the synthesis of ureas, carbamates and oxazolidinones, and these procedures are simple and less wasteful. Later, several groups developed various palladium catalysed heterogeneous, phosphine-free protocols. They provided easy catalyst–product separation methods along with catalyst reusability and recyclability.
2.1 Heterogeneous palladium catalysed oxidative carbonylation reactions of amines for the synthesis of carbamates
Fukuoka and co-workers synthesised carbamates by the oxidative alkoxycarbonylation of amines in the presence of a platinum group metal M and an alkali metal halide or onium halide (Scheme 1).10 The reaction proceeds with various platinum group based heterogeneous catalysts and the catalyst could be recovered and reused twenty times showing excellent selectivity, yield and robustness.
 |
| | Scheme 1 Reagents and conditions: aniline (50 mmol), EtOH (50 mL), M [Pd black (0.3 mg-atom)], NaI (0.6 mmol), CO (80 kg cm−2), O2 (6 kg cm−2), temp. 160–170 °C, time 2 h. | |
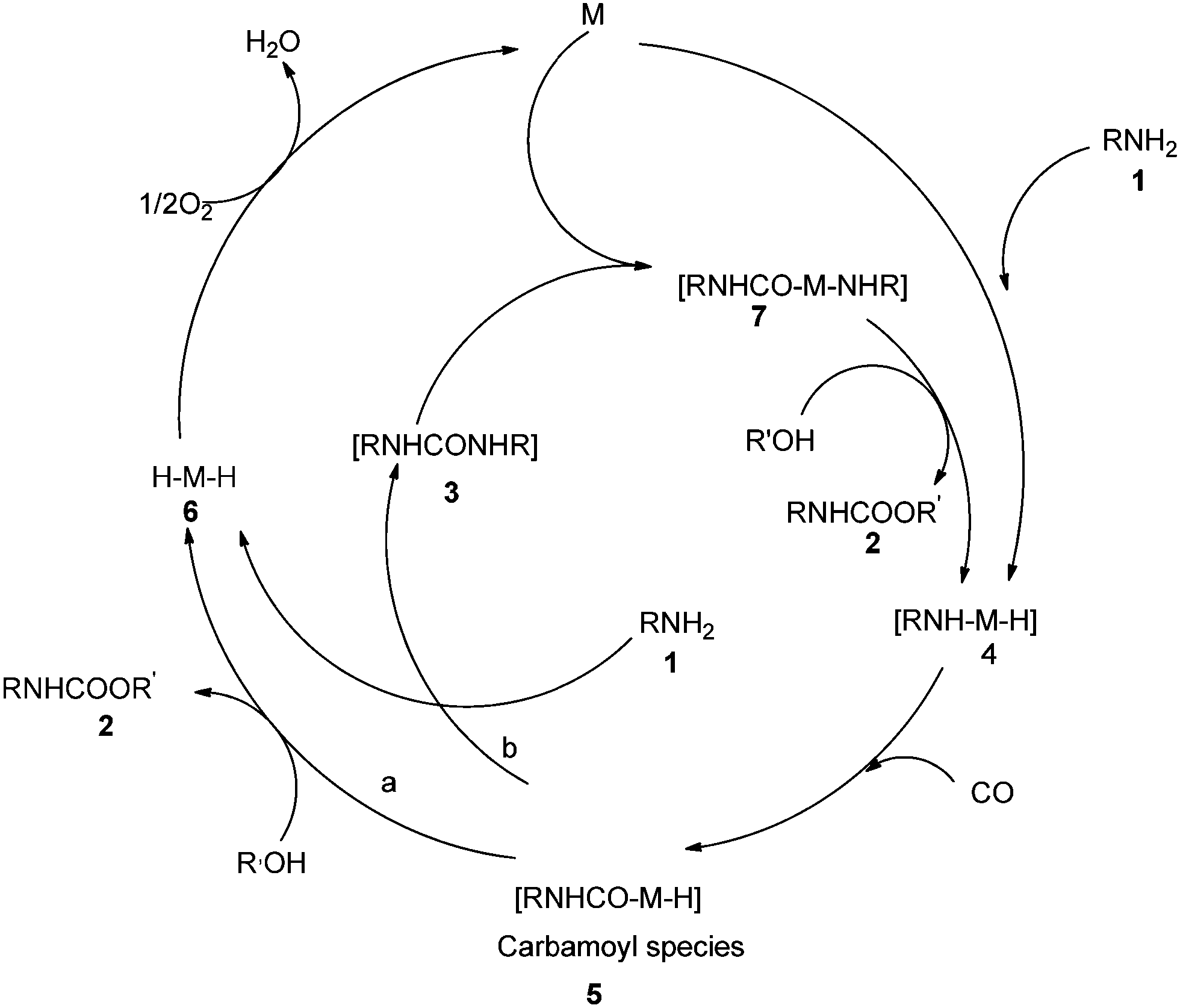
The reaction mechanism (Scheme 2) describes the oxidative addition of amine 1 to catalyst M to produce the active amino–metal species 4. The species 4 undergoes insertion of CO to produce the carbamoyl–metal species 5. The carbamate 2 was considered to be formed by two pathways, a and b. Pathway a shows the formation of the metal hydride species 6 by the attack of intermediate 5 by the alcohol. Pathway b shows the formation of urea 3 and metal hydride species 6. Further species 3 oxidatively adds to M to generate aminocarbamoyl metal species 7, which is further attacked by the alcohol to produce 2 and regenerate the active species 4.
 |
| | Scheme 2 Proposed reaction mechanism for the carbamate synthesis by oxidative carbonylation of amines. | |
In 1992, Chaudhari and co-workers studied the oxidative carbonylation of methyl amine for the synthesis of methyl N-methylcarbamate (MMC) using a palladium on carbon (Pd/C) catalyst (Scheme 3).11 The effect of various reaction parameters such as the effect of iodide promoters, the ratio of Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaI, the CO:O2 ratio, the catalyst concentration, the temperature and the effect of methyl amine concentration has been studied in detail. It was observed that at lower temperatures and higher methyl amine concentrations, N,N′-dimethylurea (DMU) was obtained as the major product. At higher temperatures and lower methyl amine concentrations, MMC was formed as the major product. At higher CO:O2 ratios, N-methylformamide (NMF) was the major product.
:NaI, the CO:O2 ratio, the catalyst concentration, the temperature and the effect of methyl amine concentration has been studied in detail. It was observed that at lower temperatures and higher methyl amine concentrations, N,N′-dimethylurea (DMU) was obtained as the major product. At higher temperatures and lower methyl amine concentrations, MMC was formed as the major product. At higher CO:O2 ratios, N-methylformamide (NMF) was the major product.
 |
| | Scheme 3 Reagents and conditions: amine (1.0 × 10−3 mol cm−3), Pd catalyst (2.35 × 10−6 mol cm−3), NaI (2.30 × 10−6 mol cm−3), solvent CH3OH, temp. 443 K, CO:O2 pressure 61 atm (CO:O2 13:1), time 120 min. | |
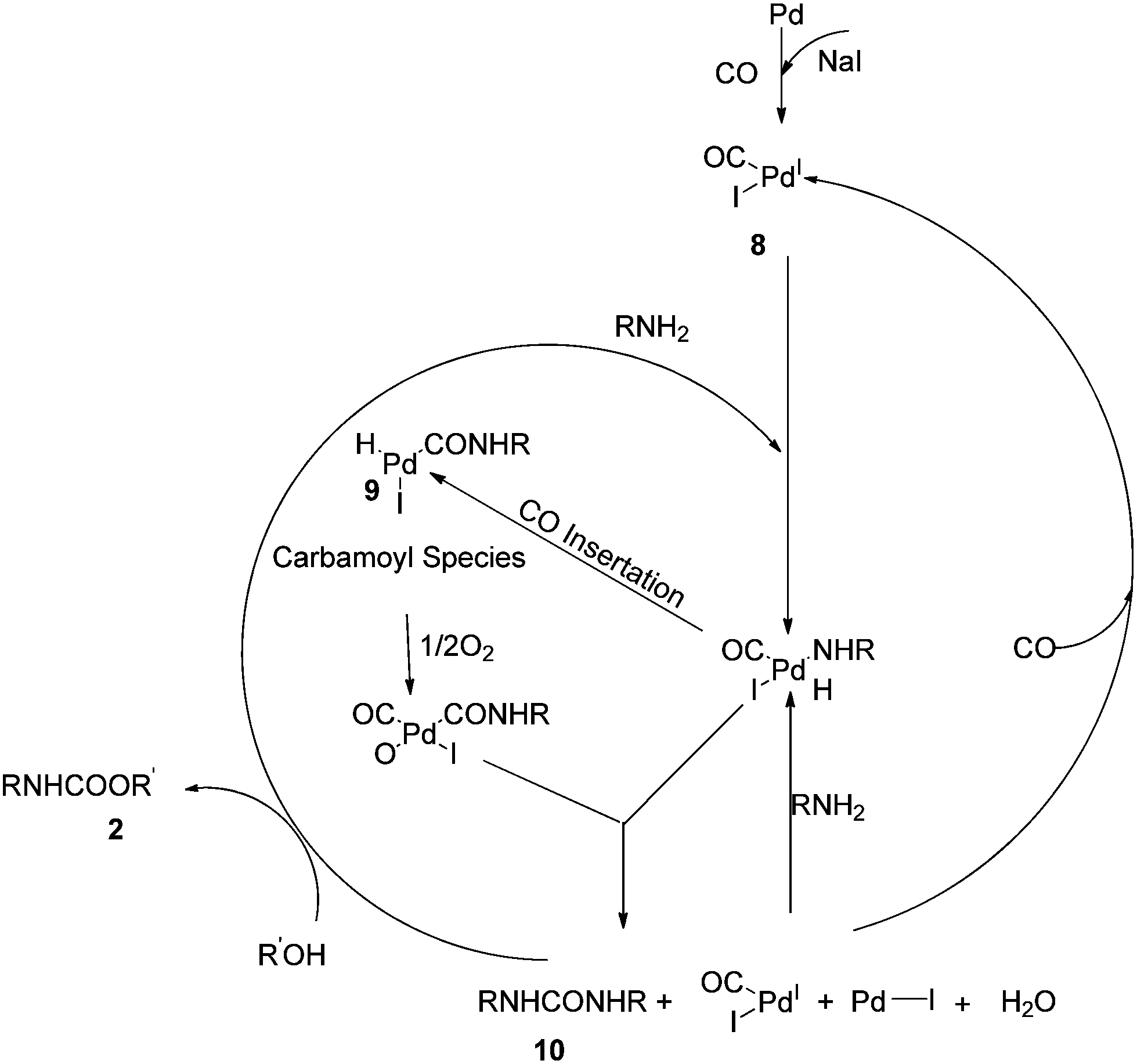
Based on these observations they proposed a reaction mechanism for carbamate synthesis (Scheme 4). DMU was found to be an intermediate product in the MMC synthesis. Fukuoka et al. proposed the Pd–carbamoyl complex intermediate 5 (ref. 10) but failed to explain the role of iodide promoters. In this mechanism, the role of the iodide promoters was clearly understood. In the absence of NaI, the reaction does not proceed, indicating Pd0 state. In the presence of iodide and oxygen, the active catalyst species 8 was observed. The formation of 2 occurs in two steps through N,N′-diarylurea 10 as an intermediate.
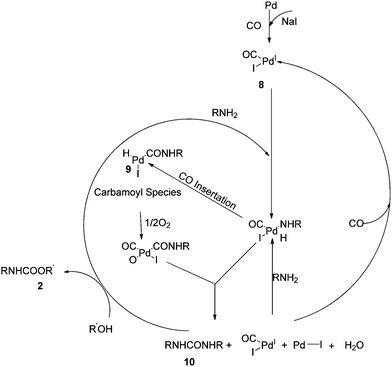 |
| | Scheme 4 Proposed mechanism for the oxidative carbonylation of amines to carbamates. | |
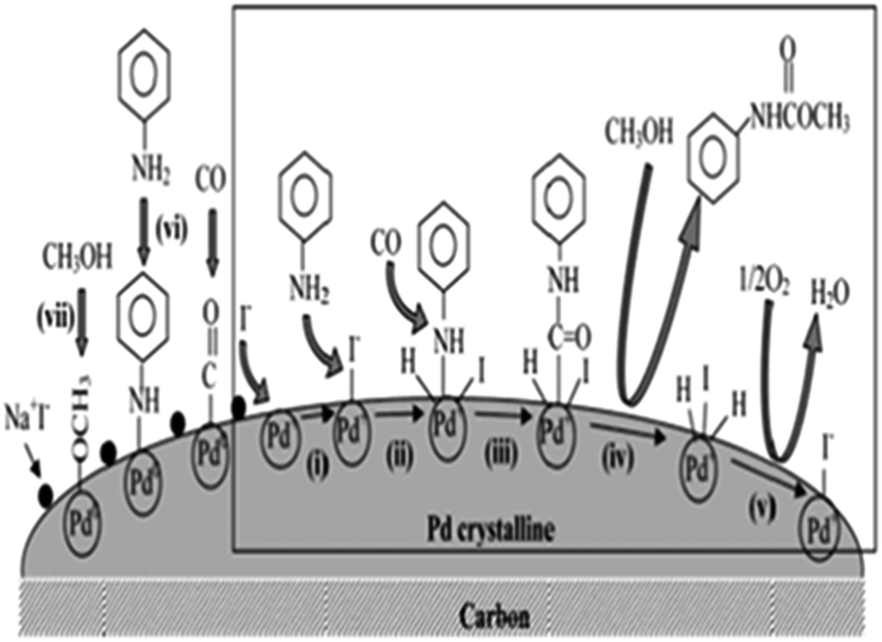
Chuang and Toochinda reported the synthesis of carbamates using a NaI–Pd/C catalytic system in slurry, gas–solid and tubular reactors.12 The oxidative carbonylation over NaI–Pd/C in a gas–solid fixed-bed reactor could provide an effective pathway for carbamate syntheses and allowed easy catalyst recovery. Atomic absorption analysis of the reactant–product mixture showed that there was no Pd loss from the carbon surface. The proposed mechanism is based on the previous studies by the Fukuoka and Chaudhari group (Fig. 1).
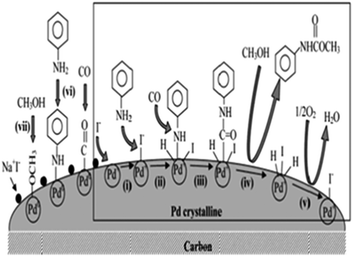 |
| | Fig. 1 Proposed mechanism for carbamate synthesis from the oxidative carbonylation over NaI–Pd/C. | |
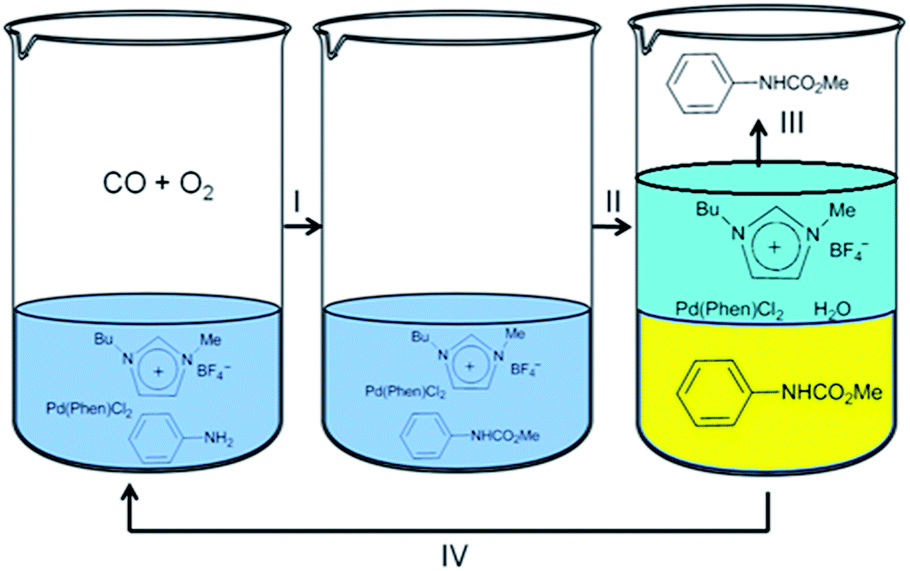
Deng and co-workers synthesised carbamates by the oxidative carbonylation of aniline with CO, O2 and methanol in the presence of a Pd(phen)Cl2/(BMImBF4) catalytic system.13 The products could be precipitated by adding water into the resulting mixture and the catalyst system could be reused. The resulting catalyst was soluble in the water phase and could be easily recycled (Fig. 2). Step I consists of the oxidative carbonylation of aniline with CO, O2 and methanol in the presence of the Pd(phen)Cl2/(BMImBF4) catalyst. A liquid mixture containing the desired product was obtained after the reaction. Step II involves the addition of 10 mL of water into the reaction mixture, which leads to the precipitation of the product. In step III the solid product is recovered. An ionic liquid containing Pd(phen)Cl2 could also be recovered and reused in step IV after the filtrate was distilled to remove the water.
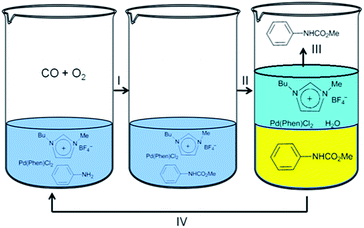 |
| | Fig. 2 The oxidative carbonylation of aniline to carbamate using a Pd(phen)Cl2/(BMImBF4) catalytic system. | |
2.2 Heterogeneous palladium catalysed oxidative carbonylation reactions of amines for the synthesis of ureas
Gupte and Chaudhari synthesised N,N′-diphenylurea (DPU) 11 by the oxidative carbonylation of aniline over a 5% Pd/C–NaI catalyst system (Scheme 5).11,14 The effects of promoters, solvents, reaction conditions, CO:O2 pressure, and pre-treatment of the catalyst with reactants have been investigated in detail. The optimum NaI/Pd ratio (molar) observed was 3.6. At higher concentrations of iodide promoters, catalyst inhibition takes place due to the strong adsorption properties of NaI, leaving behind smaller sites vacant for adsorption of CO and O2. The Pd/C–NaI catalyst system could also be reused several times without loss of activity.
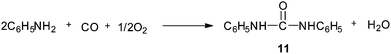 |
| | Scheme 5 Reagents and conditions: aniline (53.8 mmol), catalyst 5% Pd/C (5.0 × 10−3 g cm−3), NaI (0.87 mmol), DMF (95 cm3), CO:O2 pressure (41 bar) (CO:O2, 5:1), temp. 100 °C, time 2 h. | |
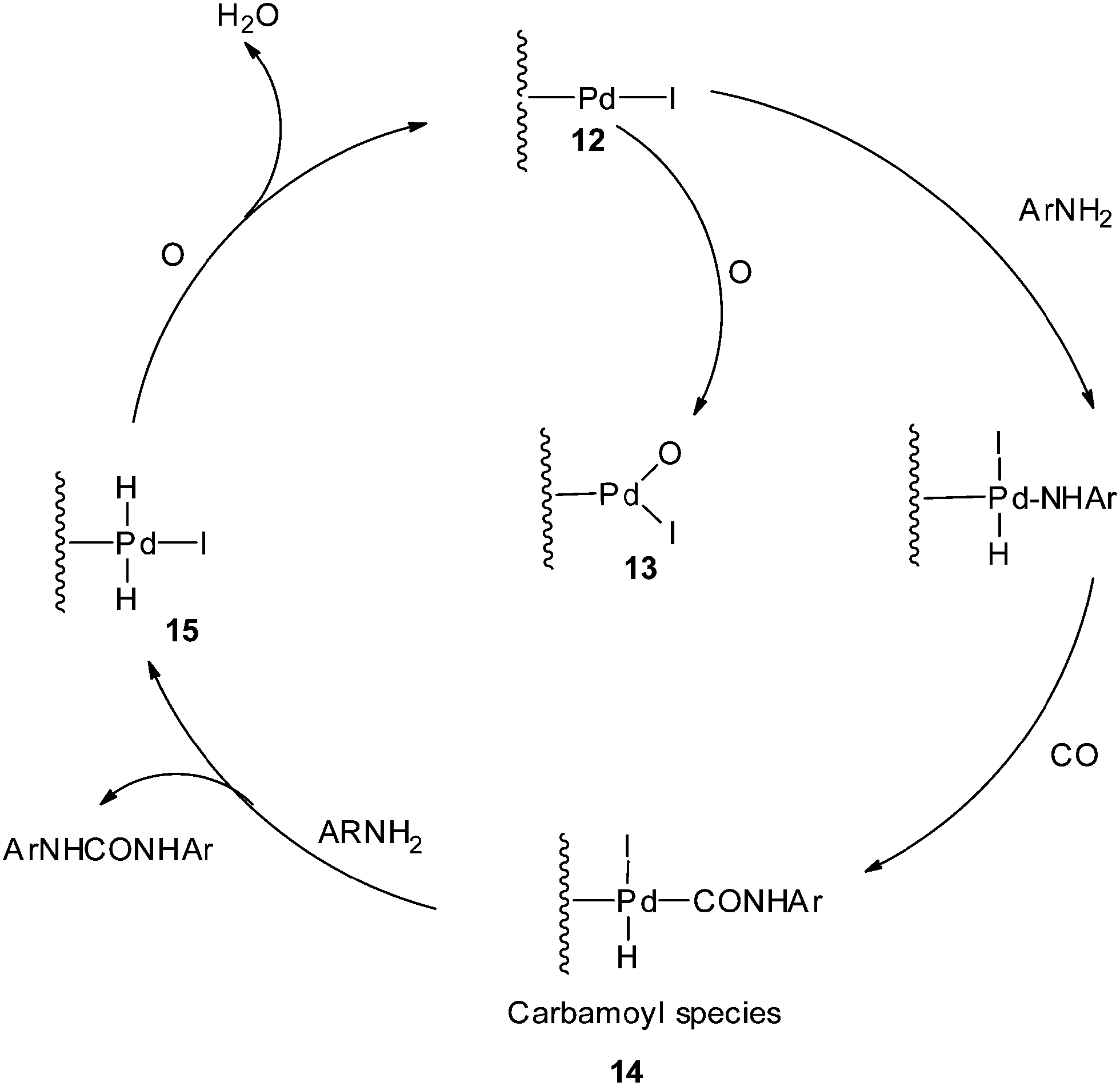
The proposed mechanism (Scheme 6) shows that the reaction proceeds through the Pd–carbamoyl complex 14 as described by Fukuoka et al..10 In the absence of iodide promoters, the reaction does not take place. In the presence of NaI, the Pd/C is modified to 12 as an active species, as proposed in Scheme 6. At a higher O2 pressure the active site may be species 13, which is inactive for oxidative carbonylation reactions, indicating inhibition of the catalyst by blocking the sites on the carbon support. The pre-treatment results also showed that there was a significant decrease in the catalytic activity at higher oxygen pressures. The carbamoyl intermediate 14 is attacked by the amine to give the urea as a product and regenerate the metal hydride species 15. The iodide promoter in the presence of oxygen plays a key role in regenerating the active catalyst by eliminating water as a by-product.
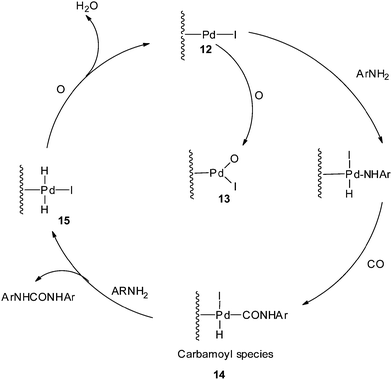 |
| | Scheme 6 Proposed mechanism for the Pd/C catalysed oxidative carbonylation of aniline to diphenylurea. | |
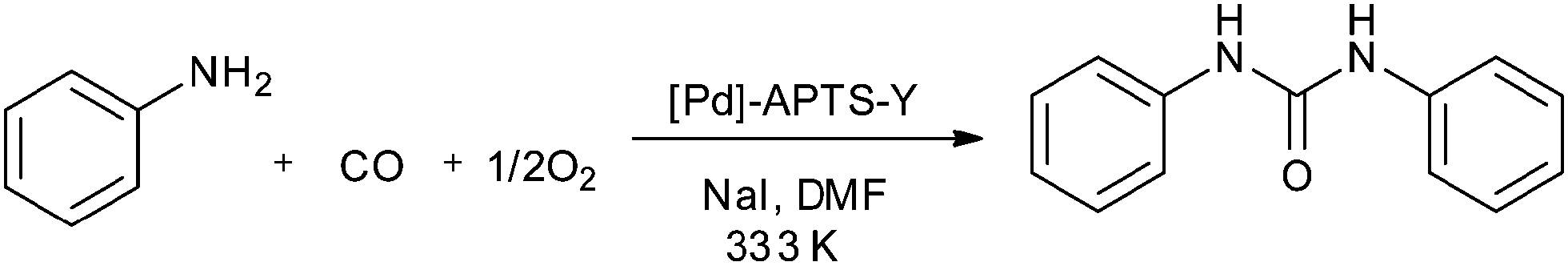
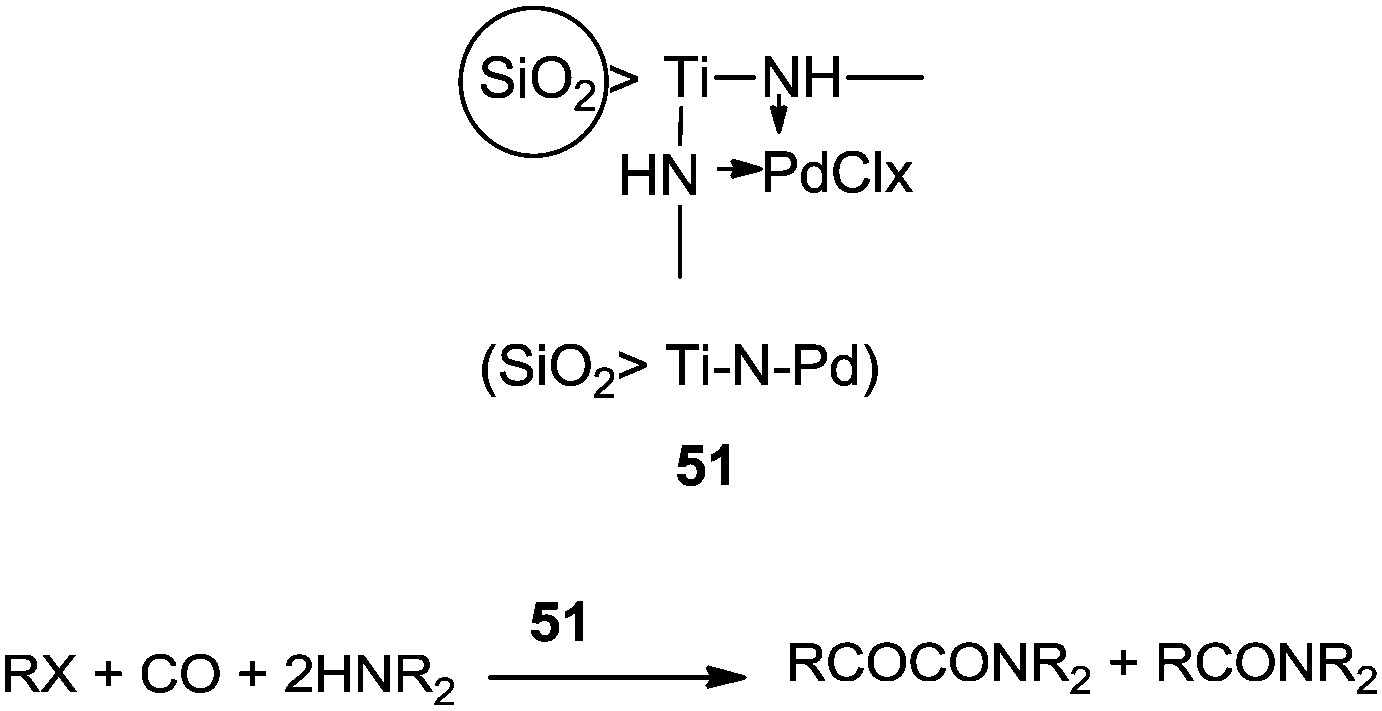
In 2010, Didgikar et al. reported palladium nanoparticles immobilized on a NaY zeolite support through 3-aminopropyl-trimethoxysilane (APTS) as an anchoring agent ([Pd]–APTS–Y). The [Pd]–APTS–Y catalyst along with NaI promoter showed excellent selectivity towards the desired urea products with a wide range of amine compounds (Scheme 7).15 The previously reported 5% Pd/C catalyst had a very poor TOF of 23.6 h−1 compared to the [Pd]–APTS–Y catalyst (TOF of 157.5 h−1). The [Pd]–APTS–Y catalyst had a high activity (TOF of 157 h−1) even at lower temperatures and pressures compared to the catalysts reported previously. The catalyst was easily separated and recycled several times without any loss of activity (Fig. 3).15
 |
| | Scheme 7 Reagents and conditions: amine 0.80 kmol m−3, [Pd]–APTS–Y (1.854 kg m−3), NaI (1.24 × 10−3 kmol m−3), pressure (CO:O2) 50 psi, DMF, time 8 h, temp. 333 K. | |
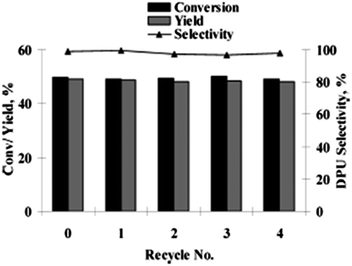 |
| | Fig. 3 Recyclability studies of the [Pd]–APTS–Y catalyst for the oxidative carbonylation of aniline. Reagents and conditions: aniline (0.82 kmol m−3), [Pd]–APTS–Y (1.854 kg m−3), NaI (1.24 × 10−3 kmol m−3), pressure (CO:O2) 50 psi, temp. 333 K, DMF, time 4 h. | |
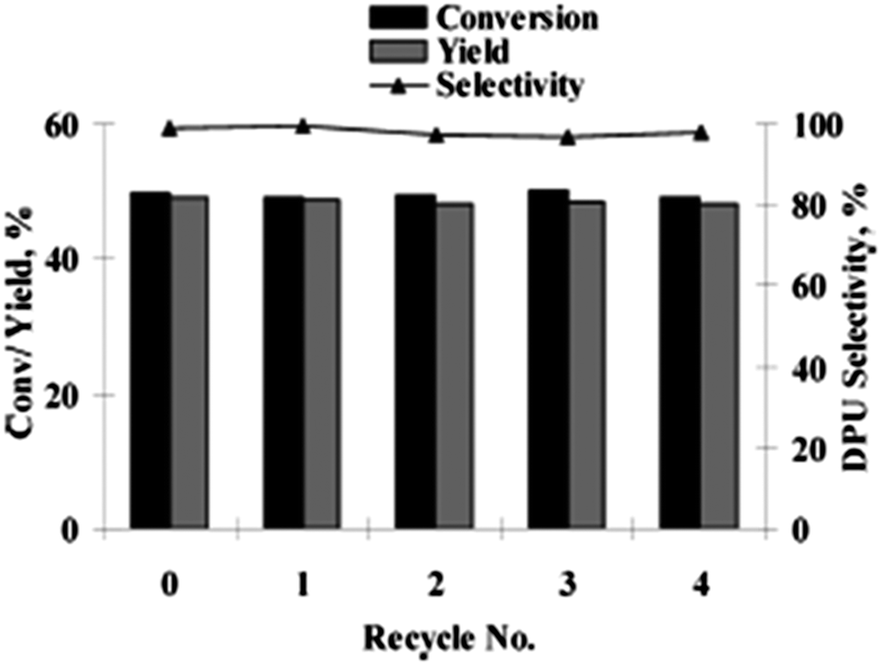
Deng and co-workers reported a sulfate modified zirconia supported palladium catalyst (Pd/ZrO2–SO42−) for the synthesis of disubstituted ureas by the oxidative carbonylation of a series of aliphatic amines (Scheme 8).16
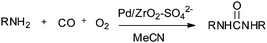 |
| | Scheme 8 Reagents and conditions: amine (2 mL) (liquid substrate) or 1.5 g (solid substrate), (Pd/ZrO2–SO42−) 35 mg, CH3CN (15 mL), CO (3.5 MPa), O2 (0.5 MPa), temperature 135 °C, time 1 h. (Unsymmetrical ureas synthesised by directly using aniline as a solvent in the presence of aliphatic amines.) | |
The combination of aliphatic amines in aniline as a solvent led to various unsymmetrical ureas. The catalyst could be easily separated and recovered from the reaction mixture. Afterwards the same group developed a homogeneous catalyst system consisting of a palladium complex and an ionic liquid (Pd(phen)Cl2/BMImBF4)13 for the synthesis of ureas. The reaction occurs in a homogeneous phase and at the end of the reaction, water is added to the reaction mixture, which creates biphasic conditions. In this case the catalyst components separated into the aqueous phase, while the product (and unconverted amine, etc.) remained in the organic phase. In 2011 Gupte and co-workers reported the synthesis of 11 by the oxidative carbonylation of an amine using water-soluble palladium catalysts in a biphasic media (Scheme 9).17 This methodology overcomes the catalyst–product separation drawbacks of the previously reported method by Deng and co-workers.13
 |
| | Scheme 9 Reagents and conditions: amine (74 mmol), Pd(OAc)2(BipyDS) (0.078 mmol), water (10 mL), toluene (50 mL), NaI (0.90 mmol), pressure (68.9 bar), CO:O2 (13:1), agitation (16.66 Hz), temp. 423 K, time 2 h. | |
The aqueous–organic biphasic catalytic system involving a water-soluble catalyst can be of great advantage in terms of catalyst and product separation and easy recyclability.
Water soluble ligands like sulfonated N-containing ligands, e.g. disodium 2,2′-bipyridine-4,4′-disulfonate (Bipy-DS), were found to be highly active and stable under the reaction conditions. The urea product could be precipitated out from the reaction medium and thus the soluble catalyst could be recycled. The oxidative carbonylation of aniline in a biphasic catalytic system using the Pd(BipyDS)Pd(OAc)2 catalyst gave a TOF of ∼210 h−1 with 97% conversion and 91% selectivity for 11. The aliphatic amines were found to be unreactive under such reaction conditions. Comparison of the XPS patterns of the recovered and fresh complexes also confirmed that the molecular nature of the complex and more importantly the oxidation state of palladium remained unchanged after the reaction.
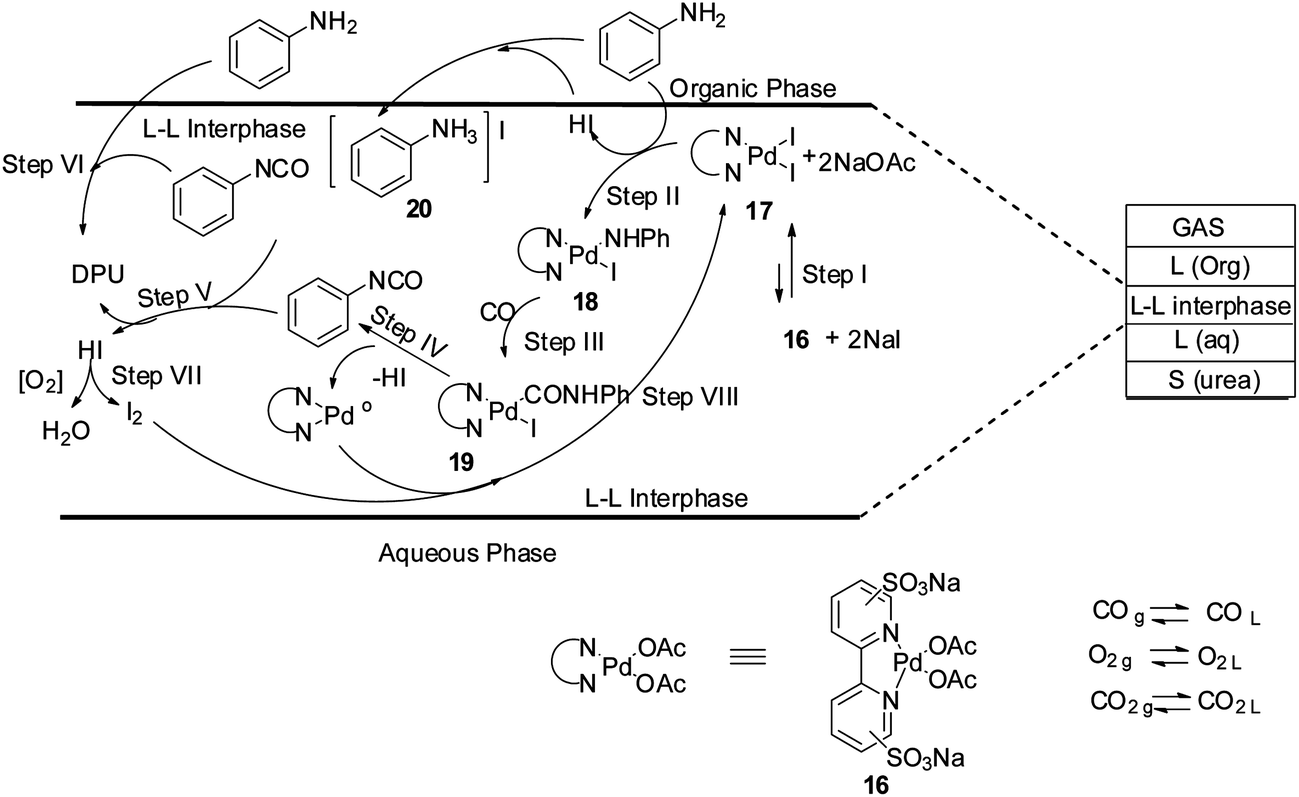
Plausible reaction mechanisms for the oxidative carbonylation reaction has been reviewed by various groups, e.g. Gabriele et al.,18 McElwee-White and co-workers19 and Ragaini.20 Based on these observations, a reaction mechanism has been proposed for the oxidative carbonylation in the liquid–liquid interphase of a biphasic medium (Scheme 10). The reaction mechanism highlights the role of the iodide promoter. Step I shows that complex 16 and NaI promoters leads to the formation of the active catalytic intermediate species 17. In step II the oxidative addition of aniline to 17 generates the species 18 which undergoes CO insertion in step III to give the Pd–carbamoyl intermediate 19. It is postulated that phenyl isocyanate is formed at the interface in step IV, which is highly reactive and is attacked by 20 to generate DPU (steps V and VI).
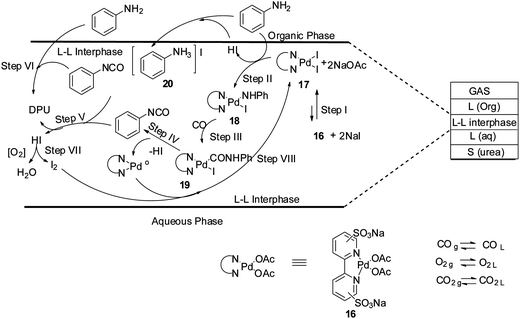 |
| | Scheme 10 Proposed mechanism for urea synthesis in a biphasic system. | |
2.3 Palladium catalysed oxidative carbonylation reactions of amino alcohols, alcohols and diols
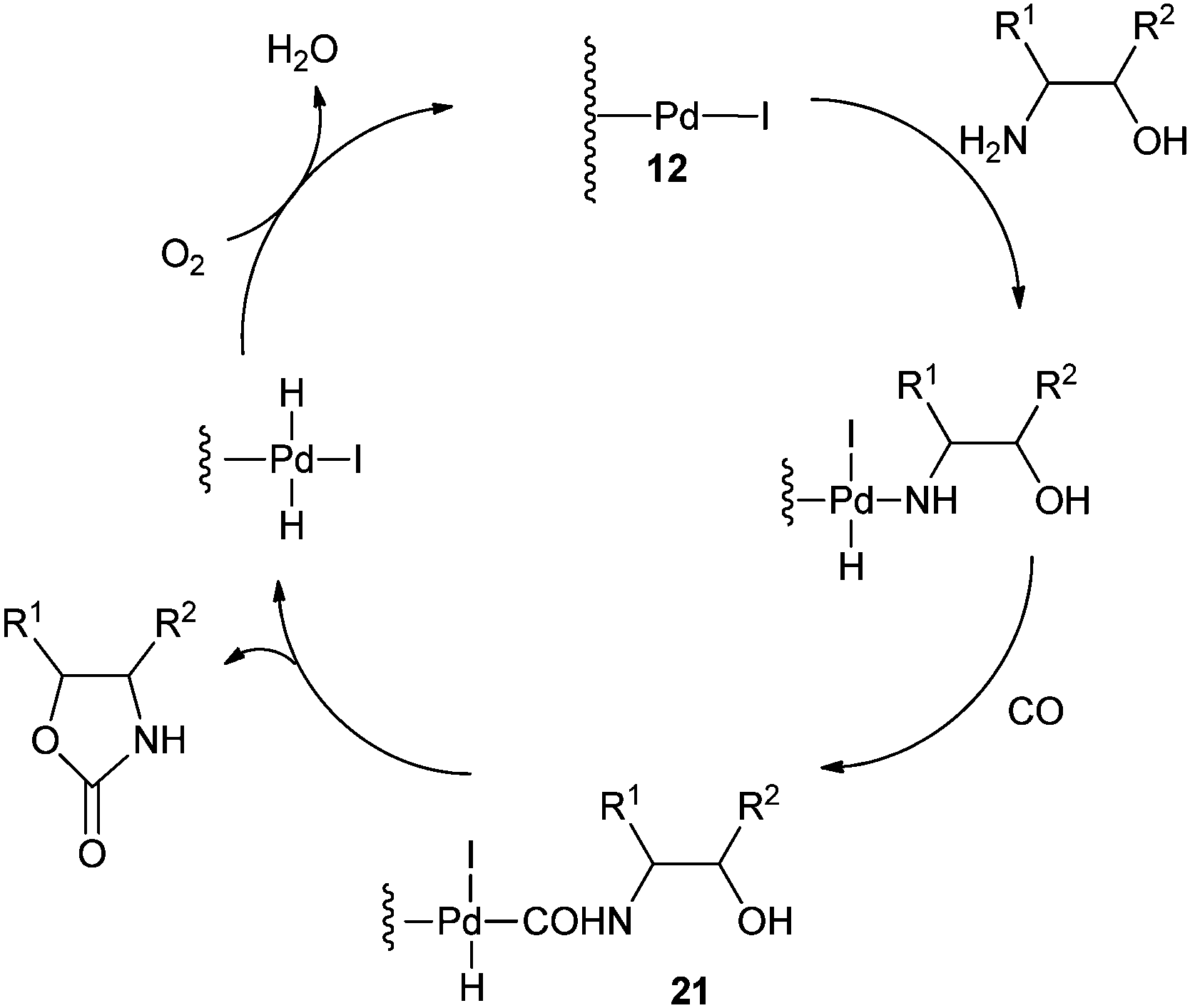
Xia and co-workers have synthesised oxazolidinones using a Pd/C–I2 catalytic system by the oxidative cyclocarbonylation of amino alcohols (Scheme 11).21 The catalyst could be reused five times without significant loss in the catalytic activity and selectivity. The catalyst system not only solves the basic problems of catalyst separation and recovery but also avoids the use of phosphine ligands. The catalyst gave high TOF values, which are 15 times larger than the best results previously reported using homogeneous Pd catalyst systems. The 2-aminophenols are also reactive under such catalytic conditions.
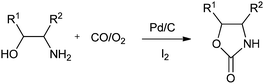 |
| | Scheme 11 Reagents and conditions: ethanolamine (10 mmol), Pd/C (0.01 mmol), promoter (0.036 mmol), temp. 100 °C, CO (1.0 MPa), O2 (0.25 MPa), DME (8 mL), time 1 h. | |
A plausible mechanism shows that in the presence of iodide promoters, Pd/C is modified to 12 (Scheme 12). The active species 12 reacts with the –NH2 group of the β-aminoalcohols and CO to produce the cabamoyl-type complexes 21, which further react with the –OH group of the aminoalcohols to produce the oxazolidinones products.
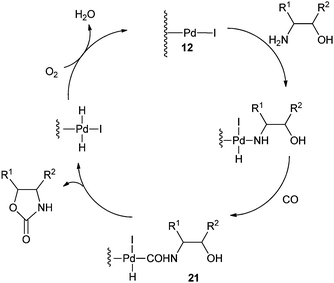 |
| | Scheme 12 Proposed mechanism for the oxazolidinone synthesis by the Pd/C catalysed oxidative carbonylation reaction. | |
In recent years, synthesis methods for carbonates, cyclic carbonates and furan-3-carboxylic esters have been developed using homogeneous palladium catalysed oxidative carbonylation reactions of alcohols and diols.22 Gabriele and co-workers synthesised furan-3-carboxylic esters by the heterocyclodehydration–alkoxycarbonylation of readily available 3-yne-1,2-diol derivatives using the PdI2/KI catalytic system and air as an oxidant (Scheme 13).22a Bell and co-workers investigated the oxidative carbonylation of ethanol to diethyl carbonate. The catalyst was prepared by dispersing CuCl2 and PdCl2 on activated carbon and carbon nanofibers.22b The catalysts prepared with CuCl2 alone were also active. A fivefold increase in activity was realized using a PdCl2/CuCl2 catalytic system. It was proposed that the Pd2+ cations interact with the [CuCl2]− anions to form Pd[CuCl2]2 complexes that are stabilized through dative bonds formed with the oxygen groups present at the edges of the graphene sheets of the support.
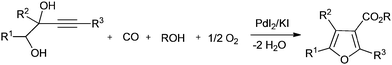 |
| | Scheme 13 Reagents and conditions: alcohol (0.05 mmol of 3-yne-1,2-diol per mL of alcohol), PdI2 (2 mol%), KI (KI/PdI2 molar ratio of 5), CO [40 atm (4/1 mixture of CO and air)], temp. 100 °C, time 2 h. | |
Yang et al. prepared palladium catalysts supported on Pb-cation-doped manganese oxide octahedral molecular sieves and used them for the heterogeneous oxidative carbonylation of phenol to diphenyl carbonate (DPC).22c The DPC yield increased to 18.1% after the doping of Pb. This suggested that the new support of Pb2–xMn8O16 was responsible for the enhanced catalytic performance. The syntheses of cyclic carbonates have been described by various groups using the homogeneous palladium catalysed oxidative carbonylation of diols. Gabriele and co-workers presented an efficient approach for 5- and 6-membered organic carbonates using the PdI2-catalyzed direct oxidative carbonylation of 1,2- and 1,3-diols (Scheme 14).22d The use of an excess of a dehydrating agent, such as trimethyl orthoacetate, was needed to achieve appreciable results. Li and co-workers showed various catalytic systems for the synthesis of glycerol carbonate using the oxidative carbonylation of glycerol. Such catalytic systems are PdCl2(phen)/KI, PdCl2(phen) with the aid of CuI, and the zeolite–Y confined Pd catalyst (PdCl2(phen)@Y) (Scheme 15).22e,f The zeolite based catalyst showed comparable activity to its homogeneous counterpart and could be reused five times without any significant decrease in activity.
 |
| | Scheme 14 Reagents and conditions: DMA (0.5 mmol of diol/mL of DMA, 4 mmol scale based on diol), KI/PdI2 molar ratio of 10, CO (20 atm, 4:1 CO/air mixture), temp. 100 °C. | |
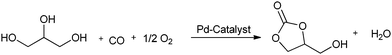 |
| | Scheme 15 Oxidative carbonylation of glycerol to glycerol carbonate using a Pd-catalyst. | |
Muller and co-workers synthesised cyclic carbonates by the oxidative carbonylation of polyols. Polyols are easily available from bio-feedstock and can be converted efficiently into their corresponding cyclic carbonates by using a Wacker-type Pd/Mn catalyst system.22g Waymouth and co-workers synthesised 5- and 6-membered cyclic carbonates.22h The sterically uncongested 1,2- and 1,3-diols were found to be reactive substrates for the catalyst.
2.4 Miscellaneous oxidative carbonylation reactions
The oxidative carbonylation reaction is an efficient methodology for the direct insertion of the carbonylation functionality by activation of the C–H bond. Various homogeneous palladium catalysts along with an oxidant are used for the carbonylative synthesis of xanthones, phenanthridinones, chalcones, esters, acids, etc.23 Classically, esters, acids and amides were synthesised using aryl halides. These oxidative carbonylation methods carbonylatively activate the C–H bond of arenes and heteroarenes, and eliminate the need for aryl halides, providing an environmentally benign protocol for the synthesis of valuable chemicals.
During the past decade, increased attention has focused on the Pd-catalyzed aromatic C–H functionalization/carbonylation. Lei and co-workers synthesised xanthones by the Pd-catalyzed double C–H functionalization/carbonylation of diaryl ethers (Scheme 16).23a
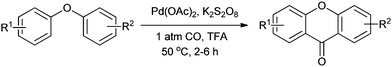 |
| | Scheme 16 Reagents and conditions: diaryl ether (0.2 mmol), Pd(OAc)2 (2.5 mol%), K2S2O8 (2 equiv.), 1 atm CO, TFA (1.0 mL), temp. 50 °C, time 2–6 h. | |
Chuang and co-workers synthesised phenanthridinone derivatives by the palladium catalyzed oxidative carbonylation of N-sulfonyl-2-aminobiaryls through C–H bond activation and C–C, C–N bond formation under TFA-free conditions (Scheme 17).23b
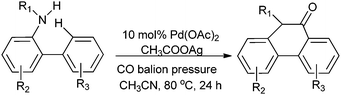 |
| | Scheme 17 Reagents and conditions: N-sulfonyl-2-aminobiaryls (50.0 mg), Pd(OAc)2 (10 mol%), CH3COOAg (5 equiv.), 4 mL of anhydrous CH3CN, CO balloon, temp. 80 °C, time 24 h. | |
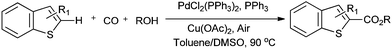
Lei and co-workers showed the regioselective aerobic oxidative C–H carbonylation of heteroarenes using a balloon pressure of CO (Schemes 18–20).23c This oxidative carbonylation catalytic system was also applied to the aerobic N–H carbonylation of N-free indoles to afford various indole carbamates. Later on, Lang et al. reported the oxidative iodination and subsequent Pd0-catalyzed carbonylation of heteroarenes.23d Wrigglesworth et al. showed the heteroannulation reactions of N-alkoxybenzamides by a Pd(II) catalyzed carbonylative C–H activation reaction.23e Beller and co-workers synthesised chalcones by palladium catalyzed oxidative carbonylative coupling reactions of arylboronic acids with styrenes (Scheme 21).23f This reaction proceeds under mild conditions using air as an oxidant.
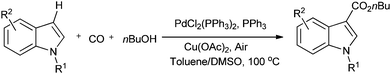 |
| | Scheme 18 Reagents and conditions: indole (0.2 mmol), PdCl2(PPh3)2 (2.5 mol%), PPh3 (5 mol%), Cu(OAc)2 (10 mol%), 1 atm CO/air (7:1), nBuOH (0.2 mL), toluene/DMSO (1.5 mL/0.1 mL), temp. 100 °C, time 36 h. | |
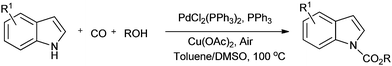 |
| | Scheme 19 Reagents and Conditions: indole (0.2 mmol), PdCl2(PPh3)2 (2.5 mol%), PPh3 (5 mol%), Cu(OAc)2 (10 mol%), 1 atm CO/air (7:1), nBuOH (0.2 mL), toluene/DMSO (1.5 mL/0.1 mL), temp. 100 °C, time 36 h. | |
 |
| | Scheme 20 Reagents and conditions: benzothiopene (0.2 mmol), PdCl2(PPh3)2 (2.5 mol%), PPh3 (5 mol%), Cu(OAc)2 (10 mol%), 1 atm CO/air (7:1), alcohol (0.2 mL), toluene/DMSO (1.5 mL/0.1 mL), temp. 90 °C, time 36 h. | |
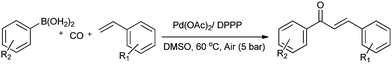 |
| | Scheme 21 Reagents and conditions: ArB(OH)2 (1 mmol), styrene (1 mmol), DMSO (2 mL), Pd(OAc)2 (2 mol%), DPPP (2 mol%), air (5 bar), CO (5 bar), temp. 60 °C, time 20 h. | |
Bell and co-workers showed the mechanism and kinetics of the liquid-phase oxidative carbonylation of toluene to toluic acid over Pd(II) in the presence of trifluoroacetic acid, trifluoroacetic anhydride, and molecular oxygen.23g Elman synthesised 2,6-naphthalenedicarboxylic acid via the liquid-phase oxidative carbonylation of naphthalene using a Pd(OAc)2/K2S2O8 catalytic system.23h The reaction goes via the formation of naphthalic anhydride (NAn) by oxidative carbonylation, subsequent alkaline hydrolysis of NAn and isomerization giving 1,8-naphthalenedicarboxylic acid, which is known to lead to 2,6-naphthalenedicarboxylic acid. Sarkar and Chaudhari synthesised a Pd(pyca)(PPh3)(OTs) complex anchored on different mesoporous matrices, such as MCM-41, MCM-48 and SBA-15. Such anchored Pd-complex catalysts were found to be effective for the hydrocarboxylation of alkenes or alcohols to acids.23i The materials showed excellent activity, regioselectivity, stability and recyclability for the low-pressure carbonylation of olefins and alcohols.
Lei and co-workers synthesised aromatic esters by the oxidative carbonylation of arylboronic acid derivatives under balloon pressure of CO with air as the oxidant using [PdCl2(PPh3)2] as the catalyst precursor (Scheme 22).23j
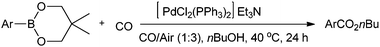 |
| | Scheme 22 Reagents and conditions: arylboronic acid (0.25 mmol), nBuOH (2 mL), Et3N (0.5 mmol), [PdCl2(PPh3)2] (0.0125 mmol), CO/air (1:3) at 1 atm pressure. | |
Jiang and co-workers synthesised β-enoic acid esters by the palladium-catalysed direct oxidative carbonylation of allylic C–H bonds with carbon monoxide (Scheme 23).23k
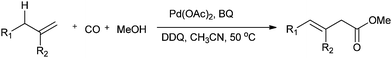 |
| | Scheme 23 Reagents and conditions: alkene (0.3 mmol), Pd(OAc)2 (10 mol%), oxidant (BQ: 2 equiv.; DDQ: 0.5 equiv.), MeCN (2 mL), MeOH (0.5 mL), CO (1 atm), temp. 50 °C, time 24 h. | |
Huang and co-workers developed an efficient Pd-catalyzed carbonylation of benzylic C–H bonds with CO through nondirected C(sp3)–H bond activation.23l This process represents a practical and efficient methodology for the synthesis of substituted phenylacetic acid esters from simple toluene.
3. Heterogeneous palladium catalysed aminocarbonylation and alkoxycarbonylation reactions
Palladium catalysed aminocarbonylation and alkoxycarbonylation reactions of aryl halides and alkynes are efficient methods for the synthesis of amides and esters.8d,f,24 Various homogeneous and heterogeneous palladium catalysed protocols have been developed for the synthesis of these valuable products.8d,f
3.1 Heterogeneous palladium catalysed aminocarbonylation reactions of aryl halides
Cai and co-workers reported a silica-supported bidentate phosphine palladium complex (abbreviated as ‘Si’–2P–Pd) for the Heck carbonylation of aryl halides (Scheme 24).25 The catalytic activity of ‘Si’–2P–Pd was comparable to that of the analogous homogeneous (Pd(PPh3)2Cl2) catalyst. The catalyst could be separated from the product by a simple filtration and could be reused without any noticeable deactivation.
 |
| | Scheme 24 Reagents and conditions: aryl halide (5 mmol), aniline (8 mmol), tri-n-butylamine (7 mmol), ‘Si’–2P–Pd (180 mg, 0.075 mmol Pd), atmospheric pressure of CO. | |
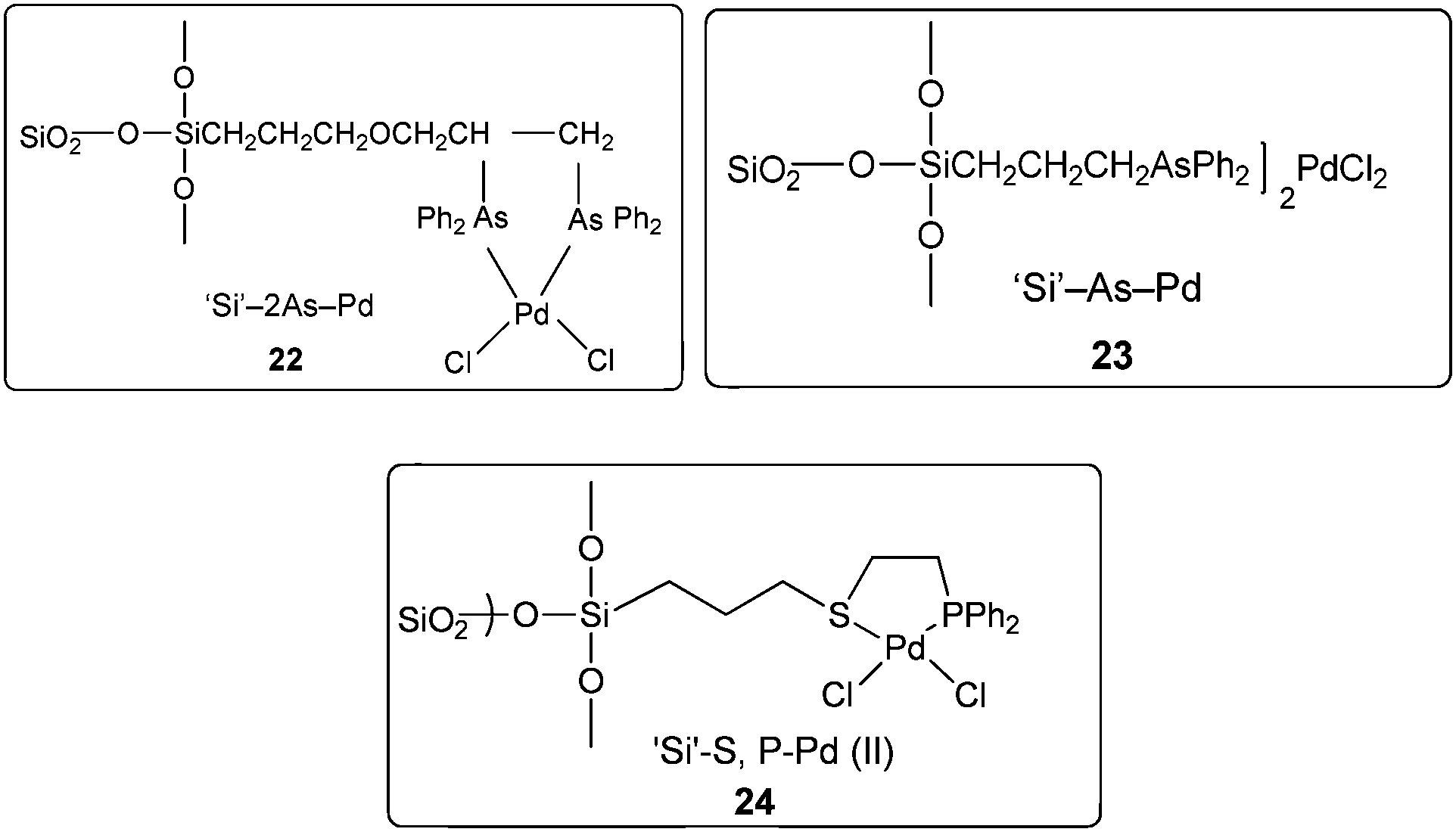
In 2004 the same group reported a silica-supported bidentate arsine palladium complex 22 (abbreviated as ‘Si’–2As–Pd),26 a silica-supported poly-γ-diphenylarsinopropylsiloxane palladium complex 23 (abbreviated as ‘Si’–As–Pd)27 and a silica-supported sulfur and phosphine mixed bidentate palladium complex 24 (abbreviated as ‘Si’–S,P–Pd(II))28 as heterogeneous and reusable catalysts for the aminocarbonylation of aryl halides (Scheme 25). Both aryl iodides and activated aryl bromides under atmospheric pressure of CO gave aminocarbonylation and the catalyst could be recovered and reused.
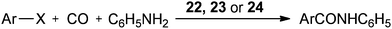 |
| | Scheme 25 Reagents and conditions: aryl halide (5 mmol), aniline (8 mmol), n-Bu3N (7 mmol), CO (1 atm), 22 (0.08 mmol) or 23 (0.078 mmol) or 24 (0.08 mmol). | |
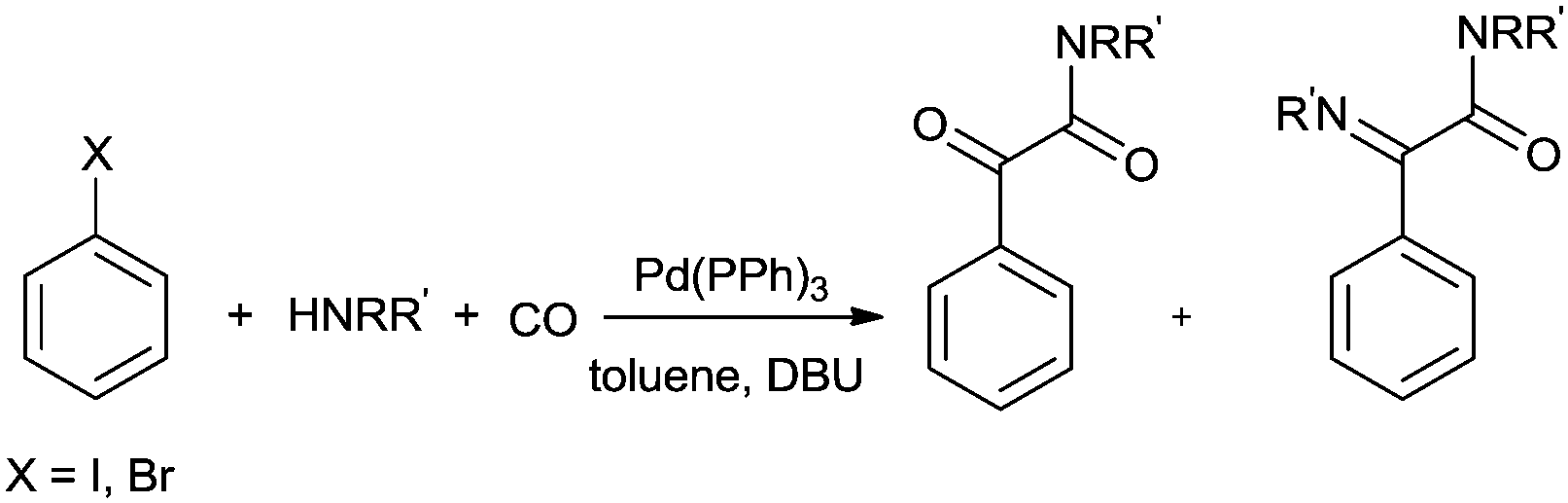
Csajági and co-workers reported the aminocarbonylation of iodobenzoic acids in a micro/meso fluidic continuous flow reactor using a polymer-supported tetrakis(triphenylphosphine) palladium catalyst.29 The method gave improved results over comparable batch techniques. Petricci and co-workers described the microwave-assisted aminocarbonylation of aryl iodides in the presence of a Pd/C catalyst at 10 bar and 130 °C (Scheme 26).30 The catalyst could be recycled for up to two runs without considerable differences in the reaction yields.
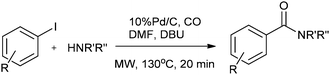 |
| | Scheme 26 Reagents and conditions: aryl iodide (0.50 mmol), amine (0.50 mmol), DMF (1 mL), DBU (224 μL, 1.50 mmol), Pd/C 10% (10 mg, 0.01 mmol), CO (130 psi), time 20 min, temp. 130 °C at 150 W. | |
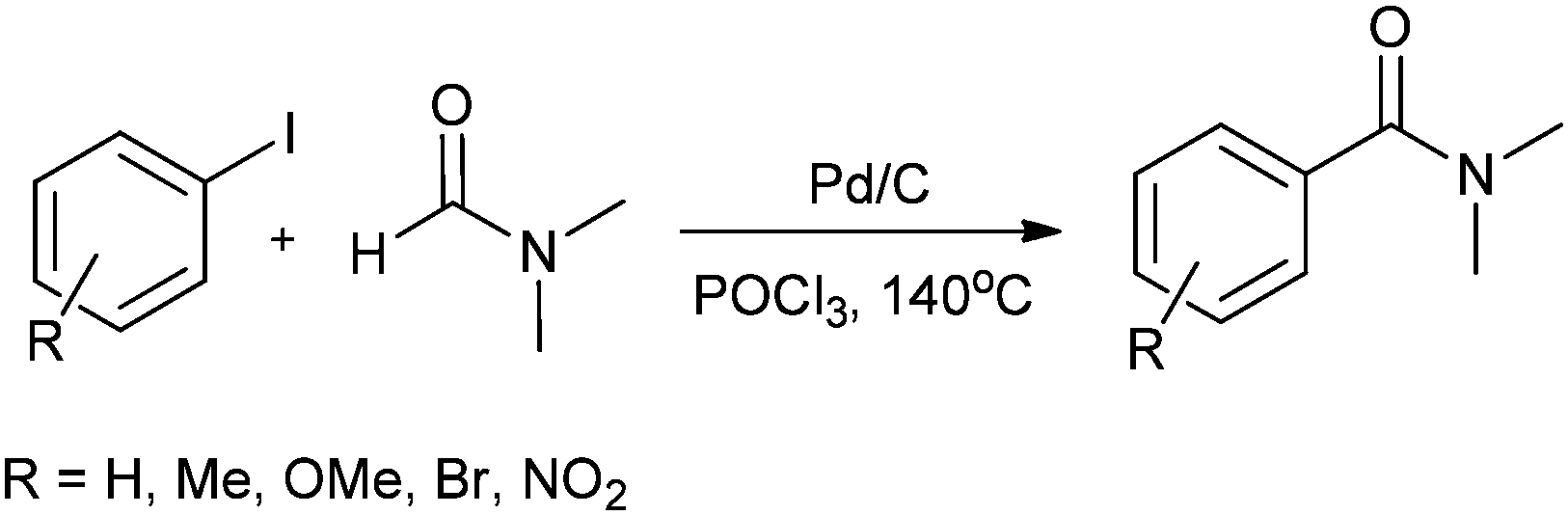
In 2008, Bhanage and co-workers reported the aminocarbonylation of aryl iodides by N,N′-dimethylformamide (DMF) as a non-gaseous CO source using Pd/C as a heterogeneous catalyst (Scheme 27).31 The catalyst showed excellent reusability. Pd leaching was examined and effective redeposition of Pd metal on the support was carried out using a thermal deposition technique.
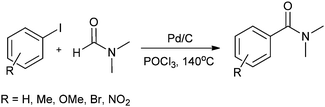 |
| | Scheme 27 Reagents and conditions: aryl iodide (1 mmol), Pd/C (0.1 mmol), POCl3 (2 mmol), DMF (10 mL), temp. 140 °C, time 24 h. | |
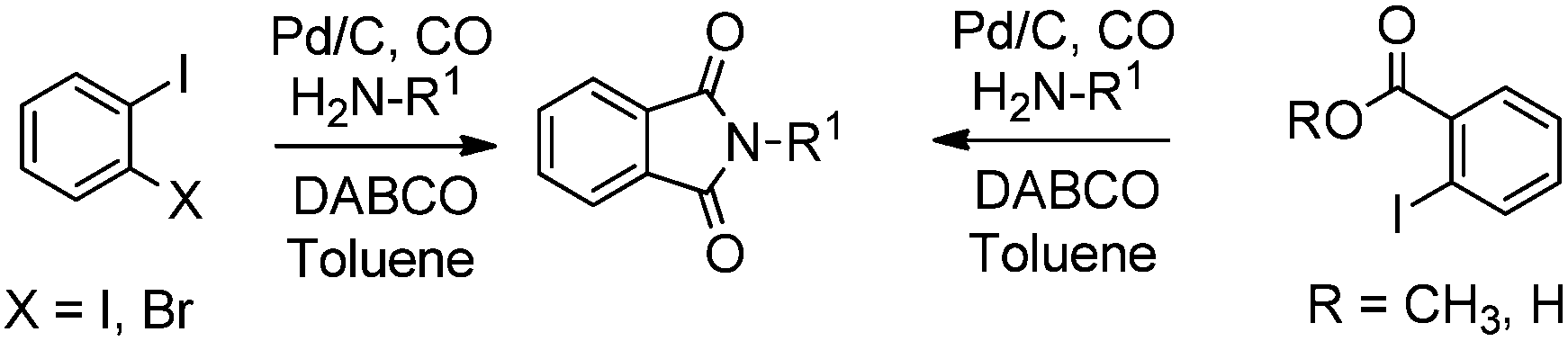
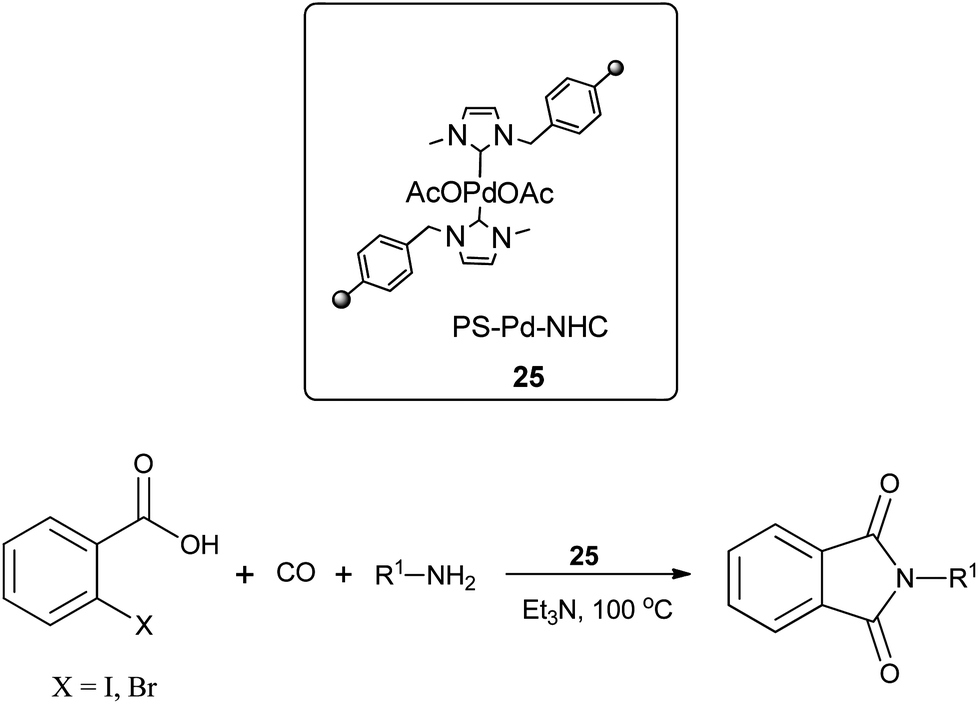
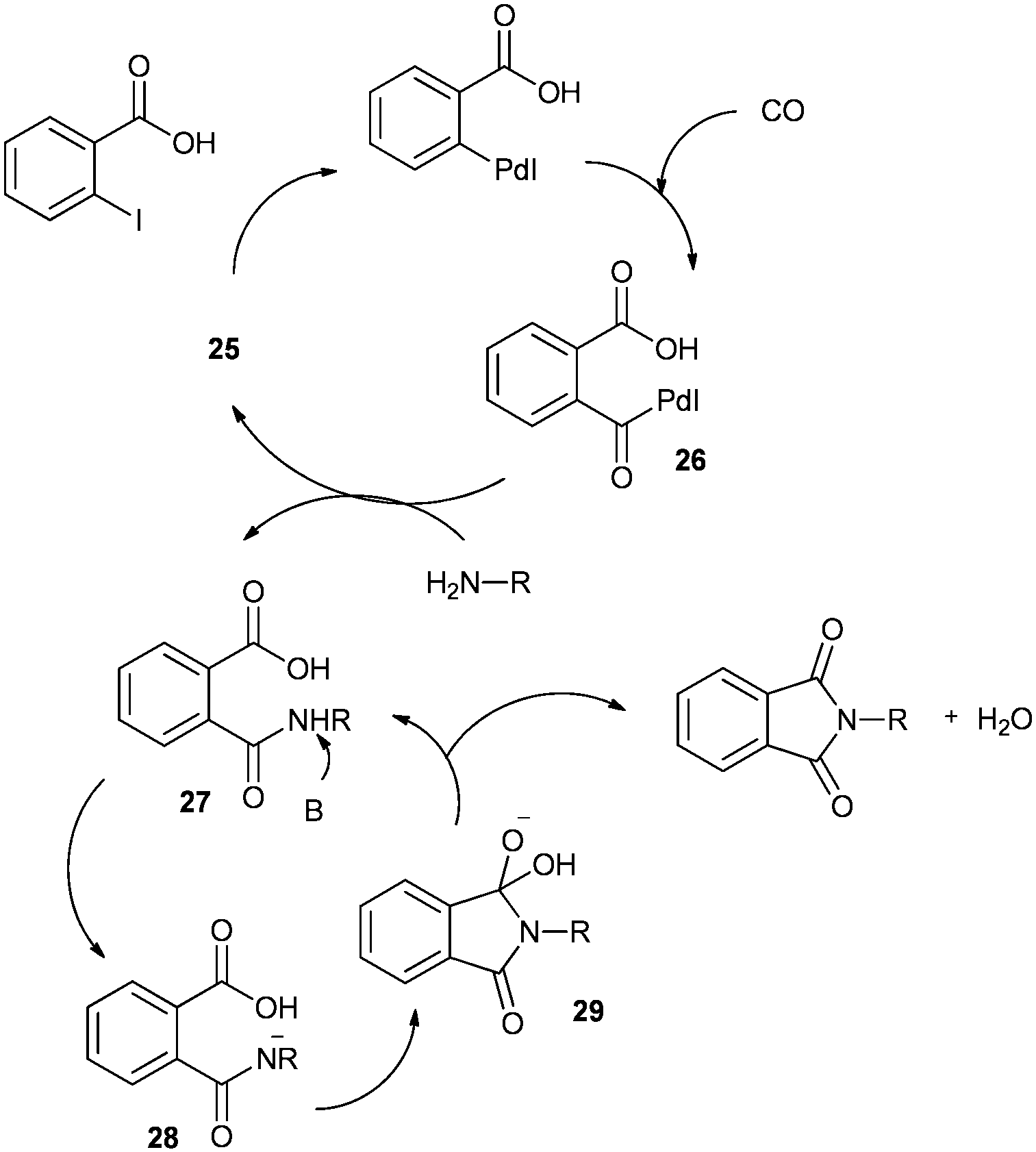
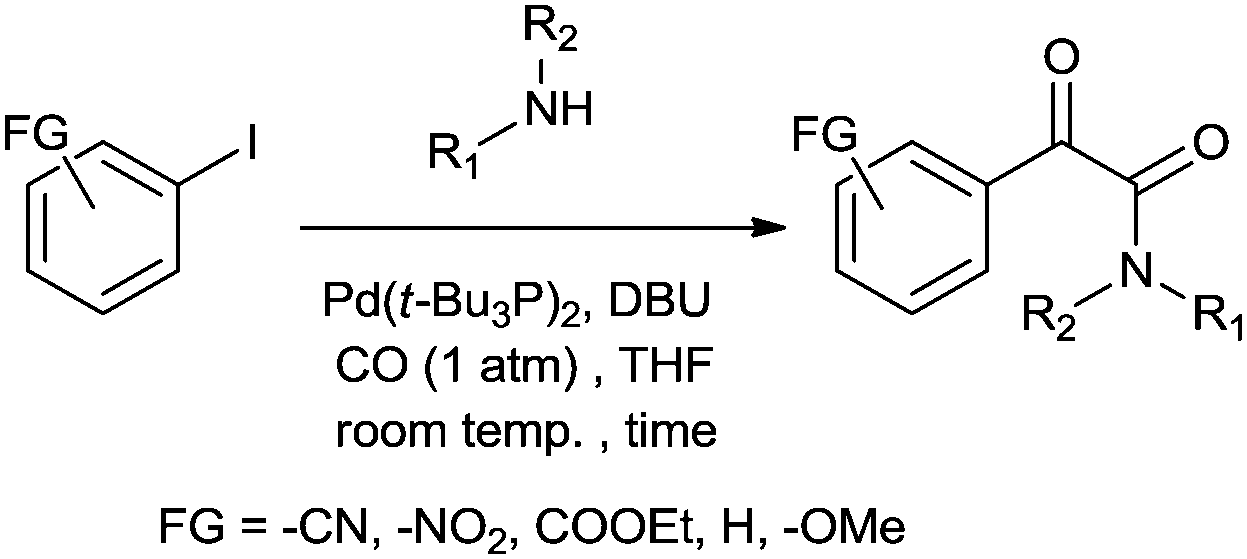
Redeposition of the catalyst was monitored using ICP-AES analysis. At 140 °C, efficient redeposition of Pd took place and the Pd content was found to be 4.45 ppm, which was in agreement with a literature report for a similar catalyst system used for the Heck reaction.32 Afterwards, the same group reported Pd/C and a polymer-supported palladium-N-heterocyclic carbene complex (PS–Pd–NHC) 25 as heterogeneous, phosphine free and recyclable catalysts for the synthesis of N-substituted phthalimides using a variety of substrates such as o-dihaloaryls, o-halobenzoate and o-halobenzoic acid (Schemes 28–30).33 Using Pd/C, they examined the ICP-AES analysis of the 1st and 8th recycle run, where the palladium in solution was estimated below 0.01 ppm and thus no significant leaching of the palladium catalyst was observed. The catalyst 25 was prepared by the reported literature procedure.34 It was found that 25 was also an effective catalytic system for the aminocarbonylation of aryl halides (Scheme 31).35 Various aliphatic and aromatic amines undergo carbonylation with aromatic and heteroaromatic halides to provide an excellent yield of the amides. The polymer supported catalyst was found to show remarkable recyclability without loss in catalytic activity and selectivity.
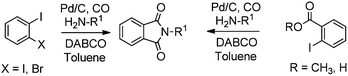 |
| | Scheme 28 Reagents and conditions: o-dihaloaryl/o-halobenzoate/o-halobenzoic acid (0.5 mmol), amine (0.7 mmol), 10% Pd/C (2.5 mol%), DABCO (1.5 mmol), toluene (7 mL), CO (90 psi), temp. 120 °C, time 2 h. | |
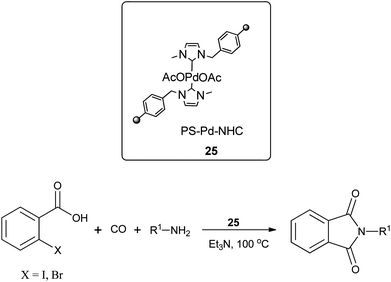 |
| | Scheme 29 Reagents and conditions: o-halobenzoic acid (1 mmol), amine (1.5 mmol), 25 (1 mol%), Et3N (2 mmol), toluene (10 mL), CO (1 atm), temp. 100 °C, time 4 h. | |
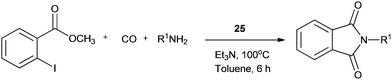 |
| | Scheme 30 Reagents and conditions: o-halobenzoate (1 mmol), amine (1.5 mmol), 25 (1 mol%), Et3N (2 mmol), toluene (10 mL), CO (1 atm), temp. 100 °C, time 6 h. | |
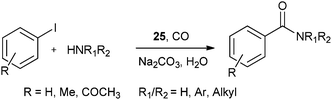 |
| | Scheme 31 Reagents and conditions: aryl iodide (1 mmol), amine (2 mmol), Na2CO3 (2 mmol), 25 (50 mg, 14.2 μmol), water (10 mL), CO pressure (100 psi), temp. 100 °C, time 8 h. | |
A plausible mechanism for the synthesis of N-substituted phthalimides (Scheme 32) shows that the reaction goes by an acylpalladium complex 26. Complex 26 then reacts with the amine to give intermediate 27, which, in the presence of base, gives intermediate 28. Species 28 is then cyclised to produce intermediate 29, which can reductively eliminate to give the corresponding N-substituted phthalimide product.
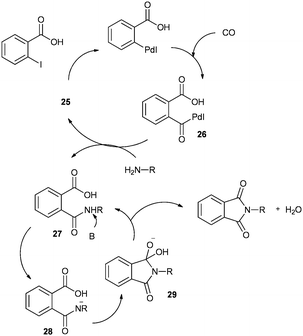 |
| | Scheme 32 Possible mechanism for the PS–Pd–NHC catalysed synthesis of N-substituted phthalimides. | |
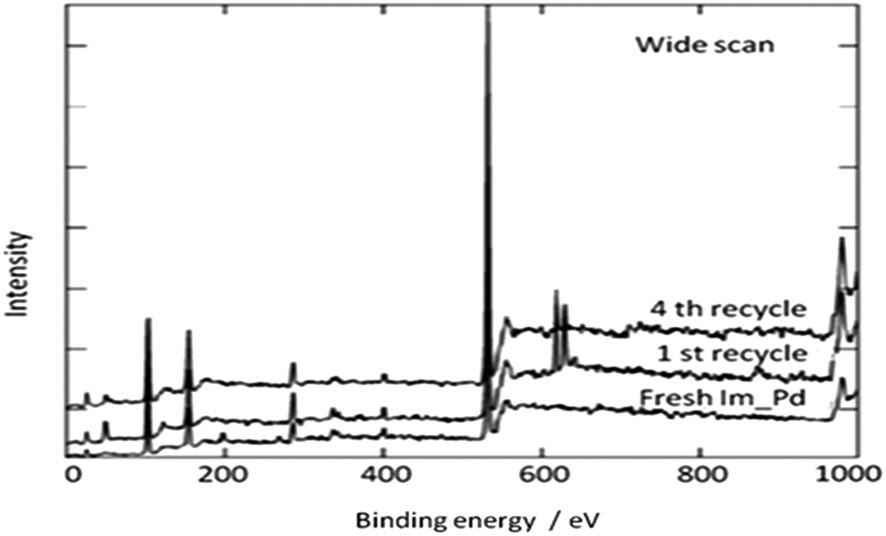
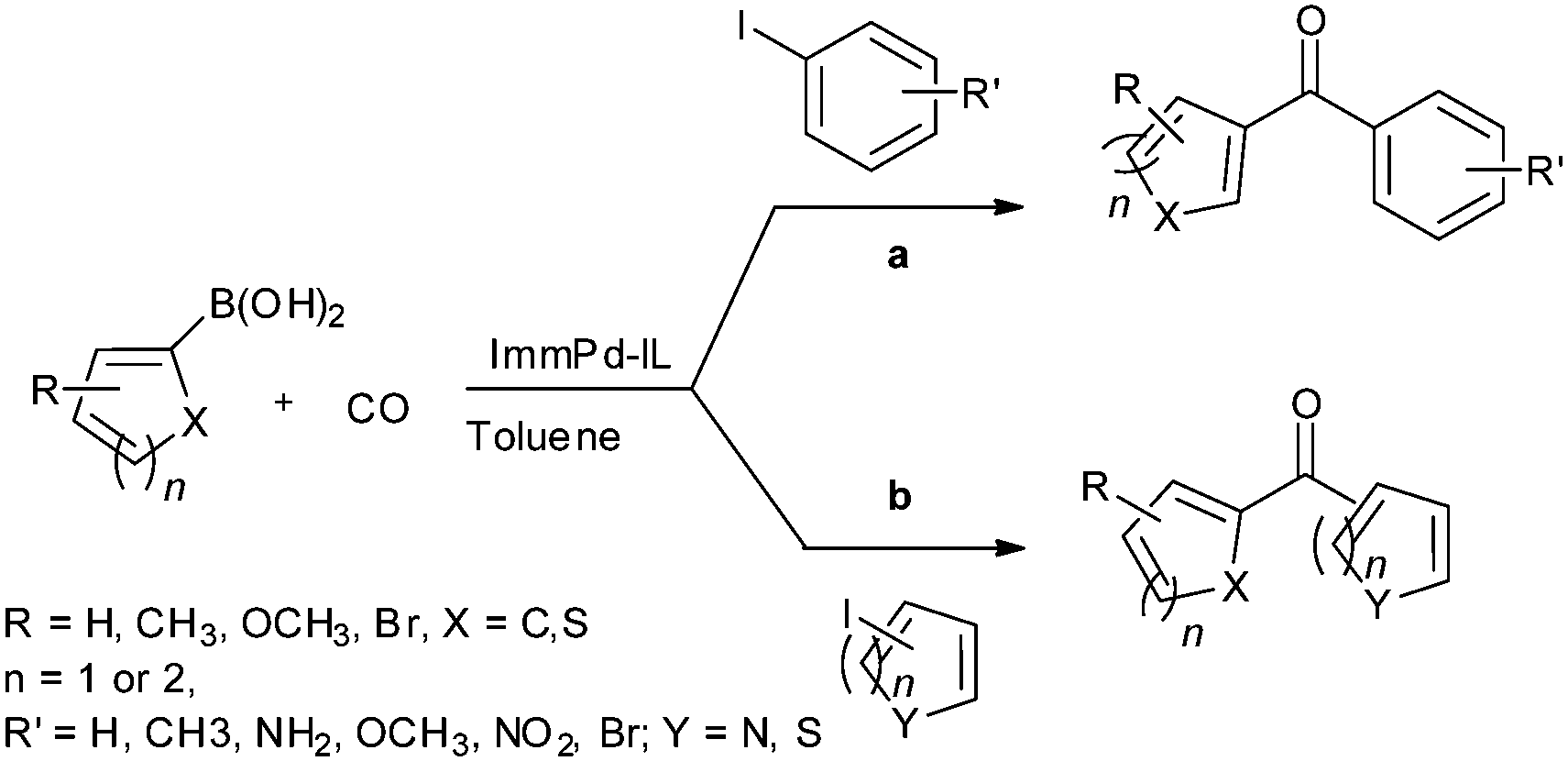
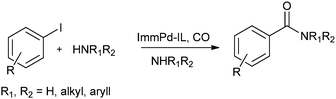
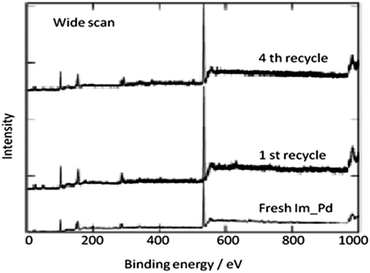
In 2013 the same group reported an immobilized palladium metal-containing ionic liquid (ImmPd-IL), which is a structurally well-defined transition metal complex, as a heterogeneous, phosphine-free catalytic system for aminocarbonylation of aryl halides (Scheme 33).36 The ImmPd-IL catalyst could be easily recovered and reused for up to four consecutive cycles. The recycled catalyst was characterized using XPS analysis, which indicated that after the fourth recycle run, reduction of the Pd2+ species to the Pd0 species takes place (Fig. 4).
 |
| | Scheme 33 Reagents and conditions: aryl iodide (1 mmol), amine (2 mmol), ImmPd-IL (2 mol%), Et3N (3 mmol), toluene (10 mL), CO pressure (1 MPa), temp. 100 °C, time 8 h. | |
 |
| | Fig. 4 XPS of the fresh and recycled ImmPd-IL catalyst. | |
Seayad and co-workers reported Pd nanoparticles supported on a zeolitic imidazole framework (ZIF-8) as a robust and efficient catalyst for the aminocarbonylation of both aryl bromides and iodides (Scheme 34).37 There are several advantages to using ZIF-8 as a support. It has a high surface area, is easy to prepare, has high stability in water, and has high temperature tolerance in the presence of methanol and acids/bases. The catalyst is recyclable and a TON of 2540 was achieved in a single batch reaction.
 |
| | Scheme 34 Reagents and conditions: ArBr (1 mmol, 1 equiv.), CO (4 bar), NHR1R2 (1.5 equiv.), Pd/ZIF-8 (24 mg, 0.21 mol% Pd), dpePhos (1 mol%), K2CO3 (1.5 equiv.), toluene (3 mL). | |
3.2 Heterogeneous palladium catalysed alkoxycarbonylation reactions of aryl halides
Reddy et al. reported a polymer-anchored phosphine palladium complex catalysed ethoxycarbonylation of organic halides in ethanol.38 The activity of the catalyst was moderate and decreased gradually with repeated use. Huang reported a silica-supported poly[3-(2-cyanoethylsulfanyl)propylsiloxane palladium] complex (abbreviated as ‘Si’–S–Pd) as an efficient catalyst for the carbonylation of aryl bromides/iodides using carbon monoxide and n-butyl alcohol (Scheme 35).39 The ‘Si’–S–Pd catalyst was separated from the mixture by filtration and reused.
 |
| | Scheme 35 Reagents and conditions: ‘Si’–S–Pd (0.01 g, 0.04 mmol), iodobenzene (0.61 g, 3 mmol), n-Bu3N (0.65 g, 3.5 mmol), n-butyl alcohol (2 cm3), CO stream, temp. 100 °C, time 25 h. | |
Cai and co-workers described 22,26 2327 and 2428 as heterogeneous and reusable catalysts for the butoxycarbonylation of aryl iodides and activated aryl bromides (Scheme 36).
 |
| | Scheme 36 Reagents and conditions: (a) aryl halide (5 mmol), n-butyl alcohol (4 mL), n-Bu3N (7 mmol), CO (1 atm), 22 (0.08 mmol) or 23 (0.078 mmol) or 24 (0.80 mmol). | |
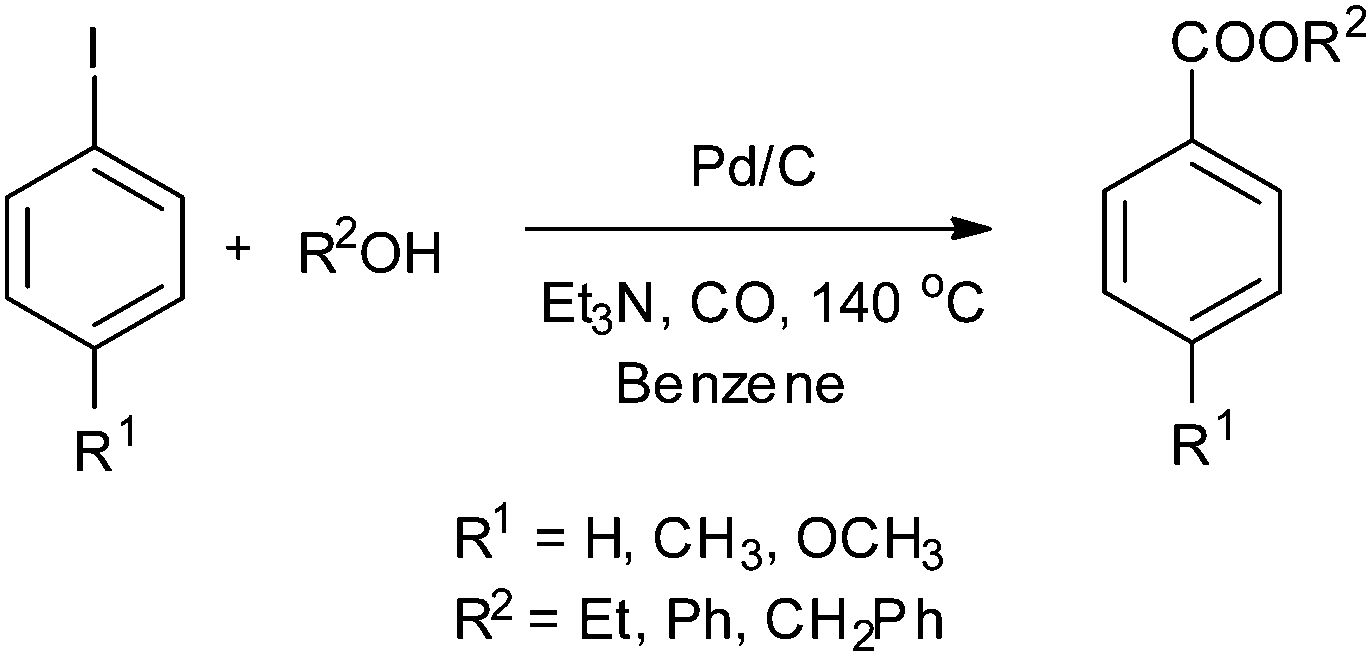
In 2003, Sugi and co-workers reported the Pd/C catalysed alkoxycarbonylation of aryl iodides (Scheme 37).40 The Pd/C catalyst could be recovered after the reaction, and reused for further reactions. Chen and Xia reported the alkoxy and phenoxycarbonylation of aryl halides using Pd/C as a novel, efficient, versatile, recyclable, and phosphine-free catalytic system (Scheme 38).41 The Pd/C catalyst was reused several times with no significant loss in the catalytic activity and selectivity.
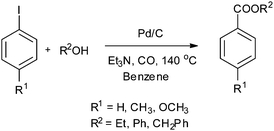 |
| | Scheme 37 Reagents and conditions: aryl iodide (2.5 mmol), alcohol (2.5–3.0 mmol), Et3N (3 mmol), 5% Pd/C (50 mg), benzene (5 mL), carbon monoxide (1 MPa), temp. 140 °C, time 6 h. | |
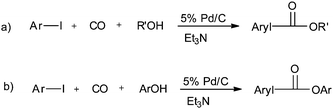 |
| | Scheme 38 Reagents and conditions: (a) iodobenzene (2.5 mmol), 5% Pd/C (5 mg), Et3N (7.2 mmol), alcohol (4 mL), CO (0.5 MPa), temp. 130 °C, time 2 h; (b) iodobenzene (2.5 mmol), ArOH (3 mmol), 5% Pd/C (30 mg), Et3N (7.2 mmol), toluene (4 mL), CO (2.0 MPa), temp. 130 °C, time 8 h. | |
Petricci and co-workers reported the alkoxycarbonylation of aryl iodides using Pd/C as a heterogeneous catalytic system under microwave dielectric heating conditions (Scheme 39).30 Various alcohols such as aliphatic, alicyclic, chiral alcohols, and phenols showed excellent reactivity towards carbonylation with aryl iodides. The Pd/C catalyst was recycled for up to two runs without a considerable decrease in the yield of the products.
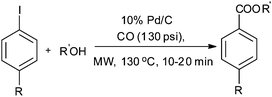 |
| | Scheme 39 Reagents and conditions: aryl iodide (0.50 mmol), alcohol (0.50 mmol), DMF (1 mL), DBU (224 μL, 1.50 mmol), Pd/C 10% (10 mg, 0.01 mmol), CO (130 psi), temp. MW, 130 °C at 150 W, time 20 min. | |
Beletskaya and Ganina reported palladium nanoparticles immobilised on poly(N-vinylimidazole-co-N-vinylcaprolactam) (PdNPs/PVI-PVC) in methanol for the alkoxycarbonylation of aryl iodides (Scheme 40).42 The PdNPs/PVI-PVC catalyst could be recycled several times without loss of activity.
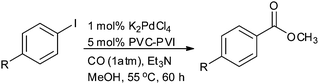 |
| | Scheme 40 Reagents and conditions: aryl iodide (0.2 mmol), Et3N (0.4 mmol), K2PdCl4 (1 mol%), PVI-PVC (70:30) (5 mol%), methanol (2 mL), temp. 55 °C, time 60 h. | |
In 2013 Bhanage and co-workers reported the alkoxy and phenoxycarbonylation of aryl iodides using an immobilized palladium metal-containing ionic liquid (ImmPd-IL) as a heterogeneous and phosphine-free catalytic system (Scheme 41).36 The reaction avoids the use of phosphine ligands and the precious palladium metal catalyst could be recovered. The catalyst showed excellent recyclability for up to four consecutive cycles of the methoxycarbonylation reaction.
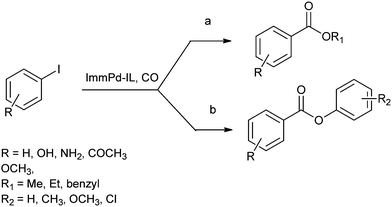 |
| | Scheme 41 Reagents and conditions: (a) aryl iodide (1 mmol), alcohol (5 mL), ImmPd-IL (2 mol%), Et3N (3 mmol), CO pressure (0.5 MPa), temp. 80 °C, time 3 h; (b) aryl iodide (1 mmol), phenol (2 mmol), ImmPd-IL (2 mol%), Et3N (3 mmol), toluene (10 mL), CO pressure (1 MPa), temp. 100 °C, time 8 h. | |
3.3 Heterogeneous palladium catalysed amino and alkoxycarbonylation reactions of terminal alkynes
In 2012, Gadge et al. synthesised alk-2-ynamides by the oxidative aminocarbonylation of terminal alkynes with secondary amines using Pd/C as a heterogeneous and reusable catalyst (Scheme 42).43 Various aromatic, heteroaromatic, and aliphatic terminal alkynes underwent aminocarbonylation reactions with excellent product yields. Only secondary amines were found to be effective. Primary amines were found to give the urea as the major product. The developed methodology avoids the use of phosphine ligands and bases, and has the additional advantages that the palladium catalyst can be recovered and reused for up to four consecutive cycles. The ICP-AES analysis of the reaction mixture on a larger scale (4 mmol of alk-1-yne) showed that the palladium content was below the detectable level, indicating no significant leaching.
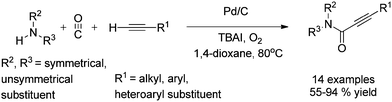 |
| | Scheme 42 Reagents and conditions: alk-1-yne (1 mmol), secondary amine (1.5 mmol), 10% Pd/C (8 mol%), CO/O2 pressure (5/1 atm), TBAI (0.90 mmol), 1,4-dioxane (10 mL), temperature 80 °C, time 14 h. | |
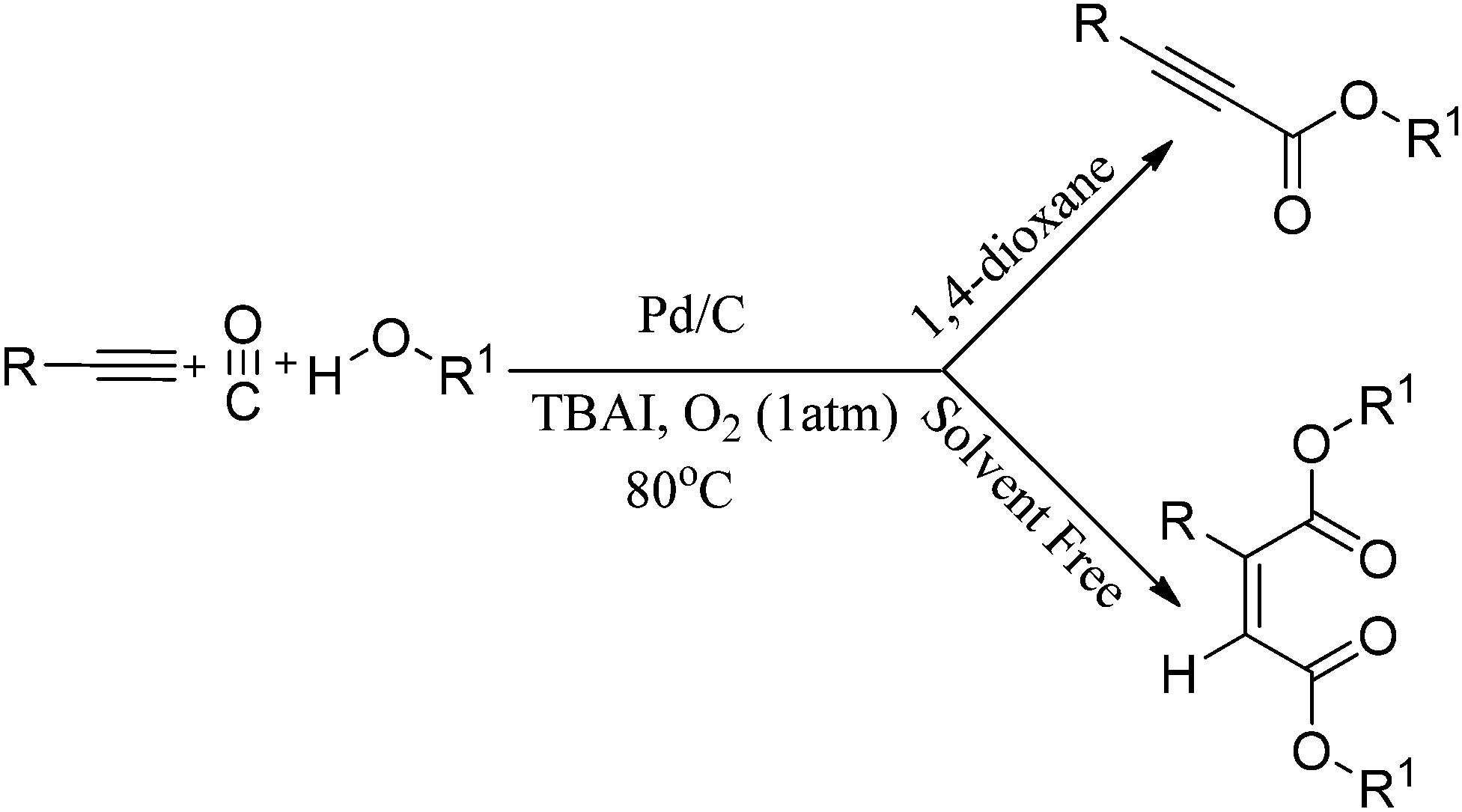
Instead of using amines as nucleophiles, the same group demonstrated the phosphine-free oxidative alkoxycarbonylation of terminal alkynes using alcohols in the presence of Pd/C as a heterogeneous catalyst (Scheme 43).44 In the presence of 1,4-dioxane solvent, the reaction provides α,β-alkynyl esters. However, with only ethanol, double carbonylation takes place to produce selectively unsymmetrical maleate esters. Various aromatic, heteroaromatic and aliphatic terminal alkynes resulted in excellent yields of the α,β-alkynyl esters and unsymmetrical maleate esters. The catalytic system has competitive advantages since it eliminates the use of phosphine ligands, bases and acids. In addition to this, the catalyst can be easily separated and recovered, allowing it to be reused several times without significant loss in the catalytic activity and selectivity.
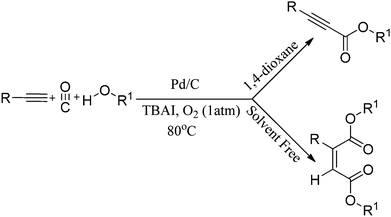 |
| | Scheme 43 Reagents and conditions: alk-1-yne (1 mmol), alcohol (0.5 mL), 10% Pd/C (10 mol%), CO/O2 (5:1 atm), TBAI (0.6 mmol), 1,4-dioxane (10 mL), temp. 80 °C, time 8 h. (The unsymmetrical maleate esters were formed directly by using the alcohol as the solvent under the same reaction conditions). | |
4. Phosphine-free carbonylative Suzuki and Sonogashira coupling reactions
Biaryl ketones and α,β-alkynyl ketones are important moieties in many biologically active molecules, natural products, and pharmaceuticals. Palladium catalysed carbonylative Suzuki and Sonogashira reactions have provided attractive routes for the synthesis of these ketones using aryl halides, gaseous CO and one of the partner boronic acids or terminal alkynes respectively.45 Various homogeneous palladium catalysts, along with N/P-containing ligands have been developed for these reactions.46 Heterogeneous and ligand-free palladium catalysed reactions for the synthesis of these biaryl ketones and α,β-alkynyl ketones would be more desirable from economical and environmental points of view. The carbonylative Sonogashira coupling reaction is generally reported using CuI as a co-catalyst, but recently new catalytic systems have investigated eliminating the co-catalyst. The high cost of transition metal catalysts and the toxic effects associated with them has led to an increased interest in immobilizing catalysts onto a support. Such heterogeneous and phosphine ligand-free cross-coupling reactions have been enthusiastically investigated because of their current toxicity and cost burden, and the difficulty in purifying the product from the reaction mixture.
4.1 Phosphine-free palladium-catalysed carbonylative Suzuki coupling reactions
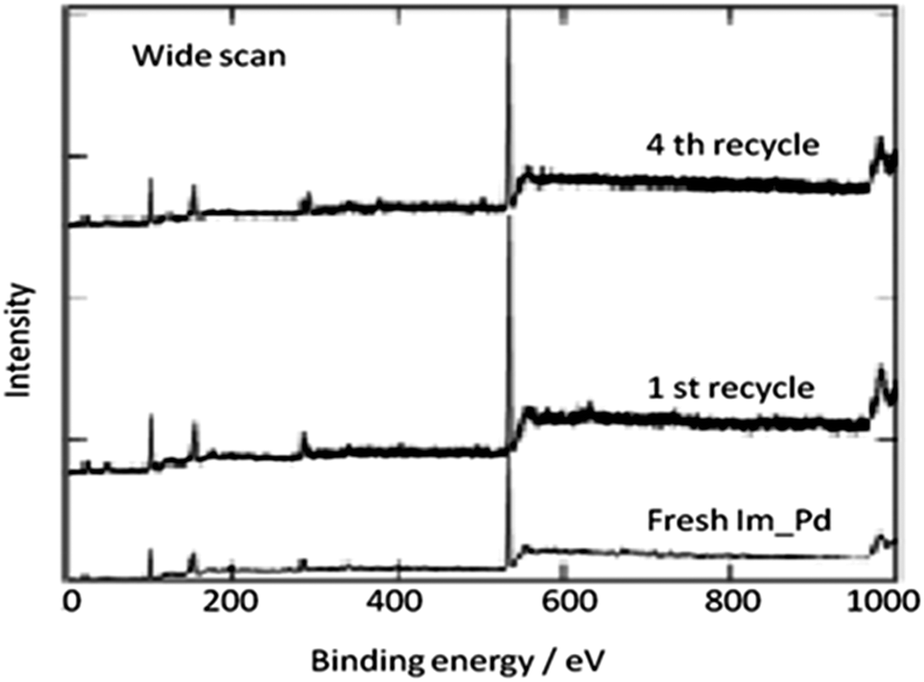
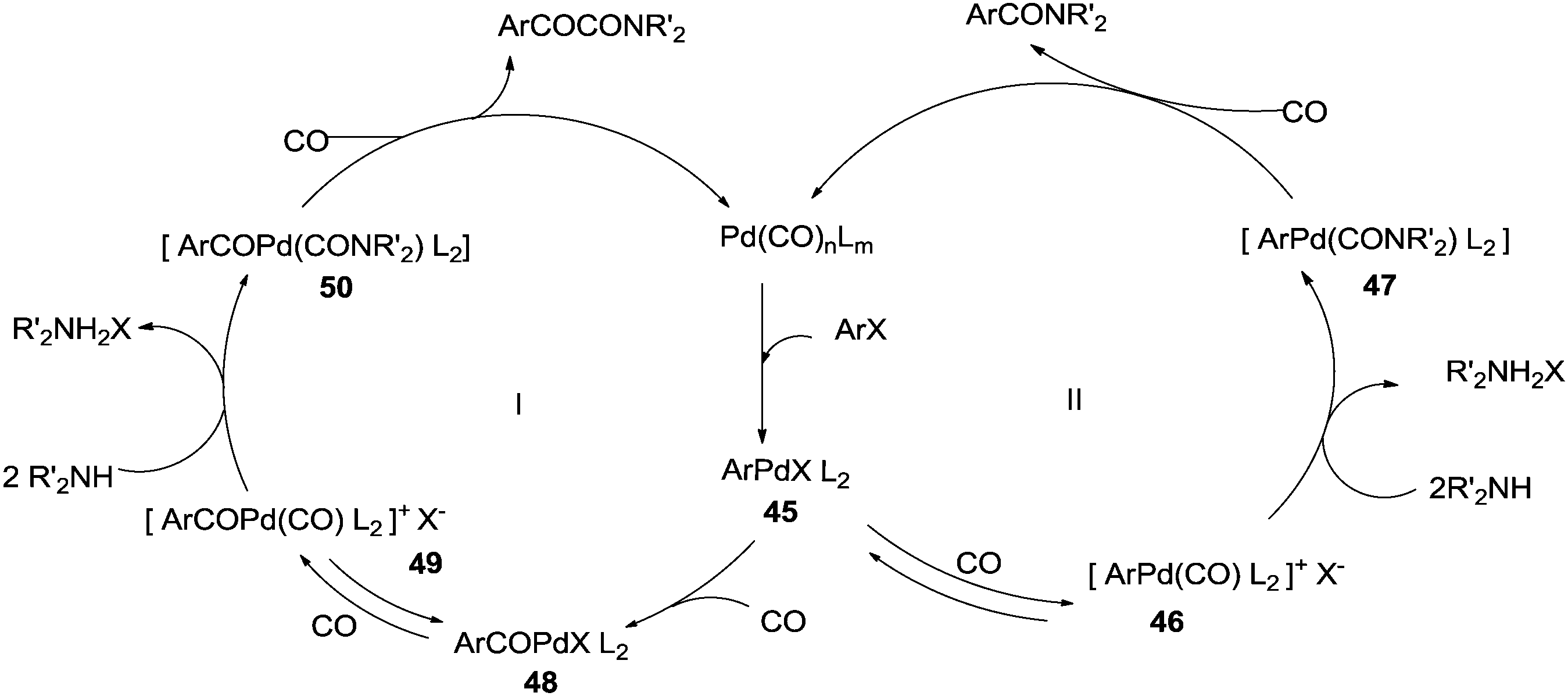
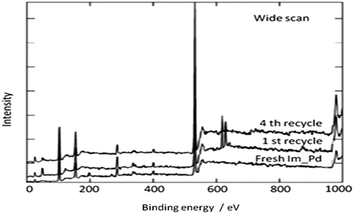
In 2009, Bhanage and co-workers discovered Pd(tmhd)2/Pd(OAc)2 as a phosphane-free catalyst for the carbonylative Suzuki reaction for biaryl ketones synthesis (Scheme 44).47 The developed protocol was applicable for both aryl and heteroaryl iodides with various boronic acids using phosphane-free homogeneous palladium complexes. The loss of the precious palladium metal catalyst was the main issue under such reaction conditions. Afterwards various heterogeneous supported palladium catalysts were investigated for such reactions, which includes Pd/C (Scheme 45),48 PS–Pd–NHC (Scheme 46)49 and ImmPd-IL catalysts (Scheme 47).50 The advantages of all these methods are: the easy separation of the catalyst and product, catalyst recoverability, catalyst recyclability, and avoiding the use of expensive phosphine ligands. XPS spectra were measured for the fresh ImmPd-IL catayst, the catalyst after the 1st recycle and the catalyst after the 4th recycle (Fig. 5). The wide scan showed that the fresh and recycled catalysts do not differ much. All these methods were applicable to aryl/heteroaryl iodides and resulted in excellent product yields.
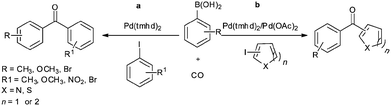 |
| | Scheme 44 (a) Reagents and conditions: aryl iodide (1 mmol), arylboronic acid (1.1 mmol), Pd(tmhd)2 (2 mol%), K2CO3 (3 mmol), anisole (10 mL), 6 h, 80 °C, CO (100 psi); (b) heteroaryl iodide (1 mmol), arylboronic acid (1.2 mmol), Pd(tmhd)2 (2.5 mol%), K2CO3 (3 mmol), toluene (10 mL), time 12 h, 100 °C, CO (200 psi). | |
 |
| | Scheme 45 Reagents and conditions: (a) aryl iodide (1 mmol), aryl boronic acid (1.2 mmol), CO (200 psi), 10% Pd/C (2 mol%), K2CO3 (3 mmol), anisole (10 mL), 100 °C, 8 h; (b) heteroaryl iodide (1 mmol), aryl boronic acid (1.2 mmol), CO (200 psi), 10% Pd/C (2.5 mol%), K2CO3 (3 mmL), toluene (10 mL), time 10 h, temp. 100 °C. | |
 |
| | Scheme 46 Reagents and conditions: (a) aryl iodide (1 mmol), arylboronic acid (1.2 mmol), 25 (14.5 mmol), K2CO3 (3 mmol), toluene (10 mL), 100 °C, CO (100 psi), 10 h; (b) aryl iodide (1 mmol), arylboronic acid (1.2 mmol), 25 (14.5 mmol), K2CO3 (3 mmol), toluene (10 mL), 100 °C, CO (100 psi), 10 h. | |
 |
| | Scheme 47 Reagents and conditions: (a) aryl iodide (1 mmol), aryl boronic acid (1.2 mmol), CO pressure (1 MPa), ImmPd-IL (2 mol%), K2CO3 (3 mmol), toluene (10 mL), 100 °C, 8 h; (b) hetero aryl iodide (1 mmol), hetero aryl boronic acid (1.2 mmol), CO pressure (1 MPa), ImmPd-IL (2.5 mol%), K2CO3 (3 mmol), toluene (10 mL), 100 °C, 10 h. | |
 |
| | Fig. 5 XPS spectra of the fresh and recycled ImmPd-IL catalysts. | |
Yang and Xue reported Pd2(dba)3 as a ligand-free catalyst under atmospheric pressure of carbon monoxide with high TONs up to 1700 (Scheme 48).51 The catalytic system was also applicable to a broad range of heteroaryl iodides with heteroaryl boronic acids (Scheme 49). The catalyst was recovered simply by filtration and it was found that the activity of the catalyst remained after four consecutive runs.
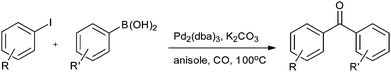 |
| | Scheme 48 Reagents and conditions: aryl iodide (0.2 mmol), aryl boronic acid (0.2 mmol), Pd2(dba)3 (1 mol%), K2CO3 (3 equiv.), CO (balloon pressure), anisole (2 mL), temp. 100 °C, time 20 h. | |
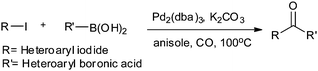 |
| | Scheme 49 Reagents and conditions: heteroaryl iodide (0.2 mmol), heteroaryl boronic acid (0.2 mmol), Pd2(dba)3 (1 mol%), K2CO3 (3 equiv.), CO (balloon pressure), anisole (2 mL), temp. 100 °C, time 20 h. | |
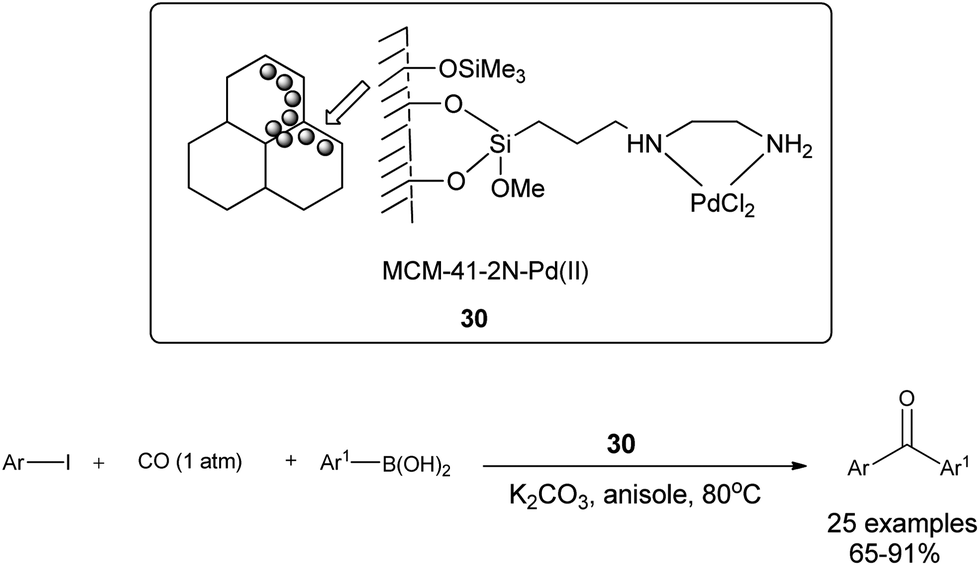
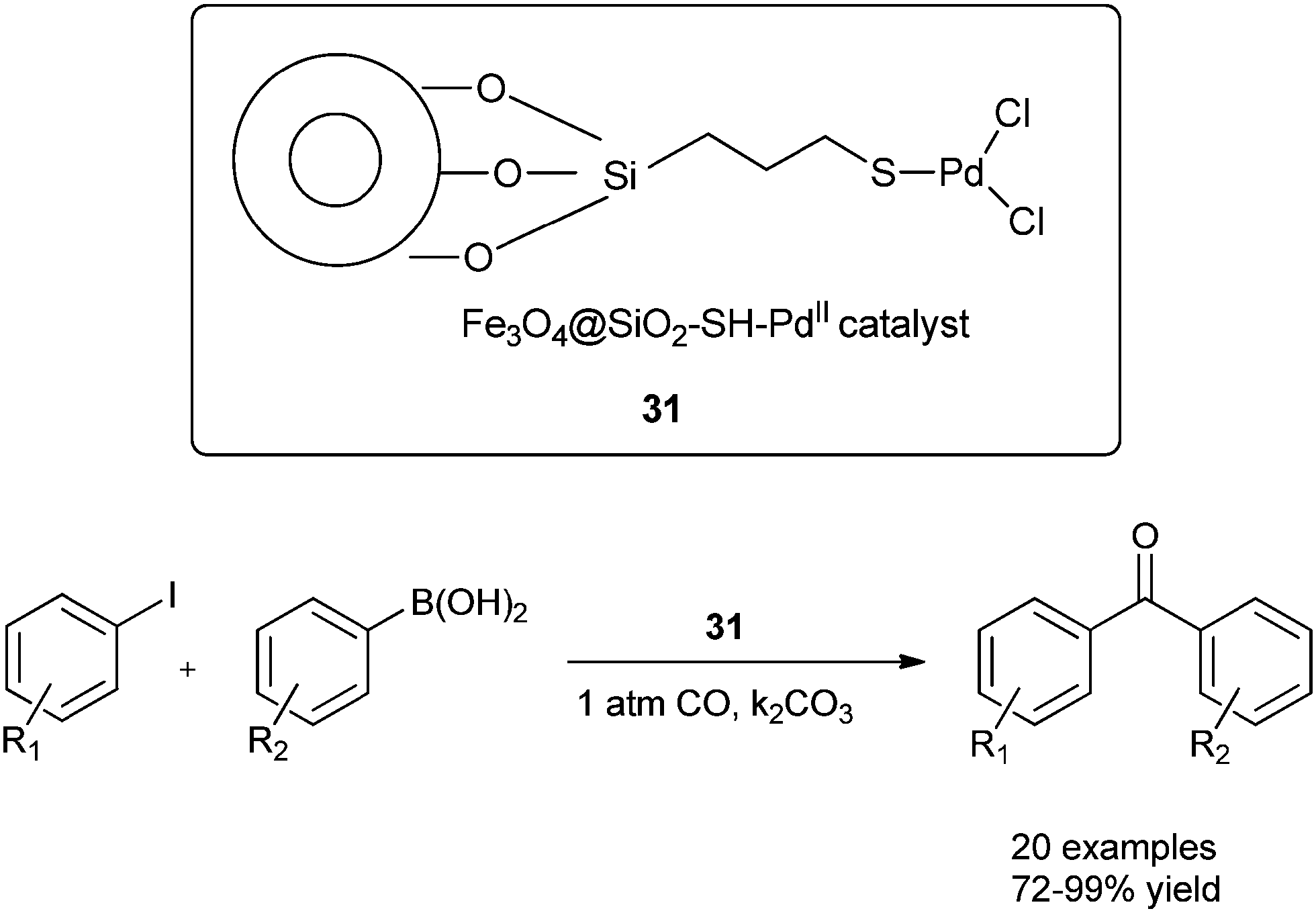
Cai and co-workers discovered a 3-(2-aminoethylamino) propyl-functionalized MCM-41-immobilized palladium(II) complex 30 as a phosphine-free heterogeneous catalyst for the carbonylative cross-coupling of aryl iodides with aryl boronic acids under an atmospheric pressure of carbon monoxide (Scheme 50).52 The MCM-41 support has a high surface area and large pore size, and the catalytic palladium species is anchored on the inner surface of the mesopores of the MCM-41 support and exhibit a high activity and good reusability. The catalyst and product could be separated by a simple filtration procedure and the catalyst was recycled up to ten consecutive trials without any decrease in activity. Niu et al. discovered magnetically separable mercaptopropyl-modified silica-coated palladium nanoparticles (Fe3O4@SiO2–SH–PdII) 31 and used them for the Suzuki carbonylative cross-coupling reaction (Scheme 51).53 The catalyst could be magnetically separated from the solution phase and reused five times without any significant loss in the catalytic activity and selectivity.
 |
| | Scheme 50 Reagents and conditions: arylboronic acid (1.1 mmol), aryl iodide (1.0 mmol), CO (1 atm), K2CO3 (3 mmol), 30 (2 mol%), anisole (5 mL), temp. 80 °C. | |
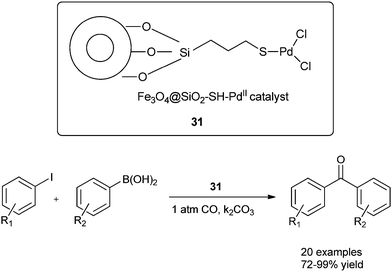 |
| | Scheme 51 Reagents and conditions: aryl iodide (0.5 mmol), arylboronic acid (0.6 mmol), CO (1 atm), K2CO3 (1.5 mmol), 31 (1 mol%), anisole (5 mL), temp. 80 °C. | |
4.2 Phosphine-free palladium-catalysed carbonylative Sonogashira coupling reactions
Xia and co-workers reported Pd/C as an efficient, versatile, recyclable, and phosphine-free catalytic system for the carbonylative Sonogashira coupling reaction of aryl iodides with terminal alkynes for the first time (Scheme 52).54
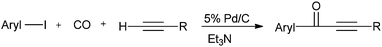 |
| | Scheme 52 Reagents and conditions: aryl iodide (2.5 mmol), alkyne (3 mmol), 5% Pd/C (10 mg), Et3N (7.2 mmol), toluene (4 mL), CO (2.0 MPa), temp. 130 °C, time 4 h. | |
The reaction avoids the use of CuI as a co-catalyst. This catalytic system is advantageous as there is no need to add phosphine ligands and the catalyst can be easily recycled, allowing it to be reused several times.
A possible reaction mechanism (Scheme 53) shows that the carbonylative coupling product was obtained through 33 via the electron deficient acylpalladium complex 32. An alternative route is the insertion of an arylalkyne into the 32 species to give 34, followed by β-hydrogen elimination. The same group then reported two different catalytic systems for the carbonylative Sonogashira reaction. The first one is based on a magnetically separable palladium catalyst (Pd/Fe3O4) (Scheme 54)55 and the second one is based on a cross-linked polymer-supported ionic liquid immobilized palladium catalyst (P(DVB-IL)–Pd) (Scheme 55).56 The magnetically separable Pd/Fe3O4 catalyst was prepared by the wet impregnation of Fe3O4 with Na2[PdCl4], followed by reduction with KBH4, and it consists of Pd particles of 5 nm in diameter entrapped in iron oxide nanoparticles. The Pd/Fe3O4 catalyst was recycled seven times after magnetic separation without loss in activity and selectivity (Fig. 6). Both catalytic systems not only solve the basic problem of palladium metal recovery, but also avoid the use of toxic and expensive phosphine ligands.
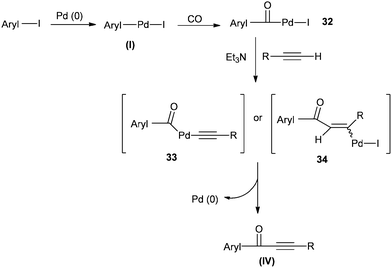 |
| | Scheme 53 Possible mechanism for the Pd/C catalysed carbonylative Sonogashira coupling reaction. | |
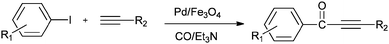 |
| | Scheme 54 Reagents and conditions: aryl iodides (2.5 mmol), phenylacetylene alkynes (3.0 mmol), Pd/Fe3O4 catalyst (Pd, 1.04 wt%), 50 mg, Et3N (7.2 mmol), toluene (5.0 mL), CO (2.0 MPa), temp. 130 °C, time 4 h. | |
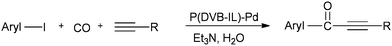 |
| | Scheme 55 Reagents and conditions: aryl iodide (1 mmol), terminal alkyne (1.2 mmol), P(DVB-IL)–Pd (20 mg, 0.005 mmol Pd), Et3N (0.4 mL), CO pressure (3.0 MPa), distilled water (5.0 mL), temperature 130 °C, time 6 h. | |
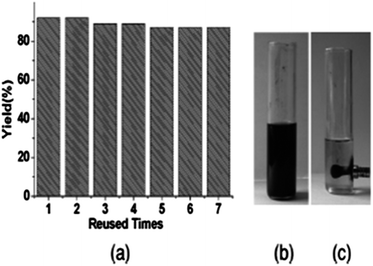 |
| | Fig. 6 Recyclability of the Pd/Fe3O4 catalyst. | |
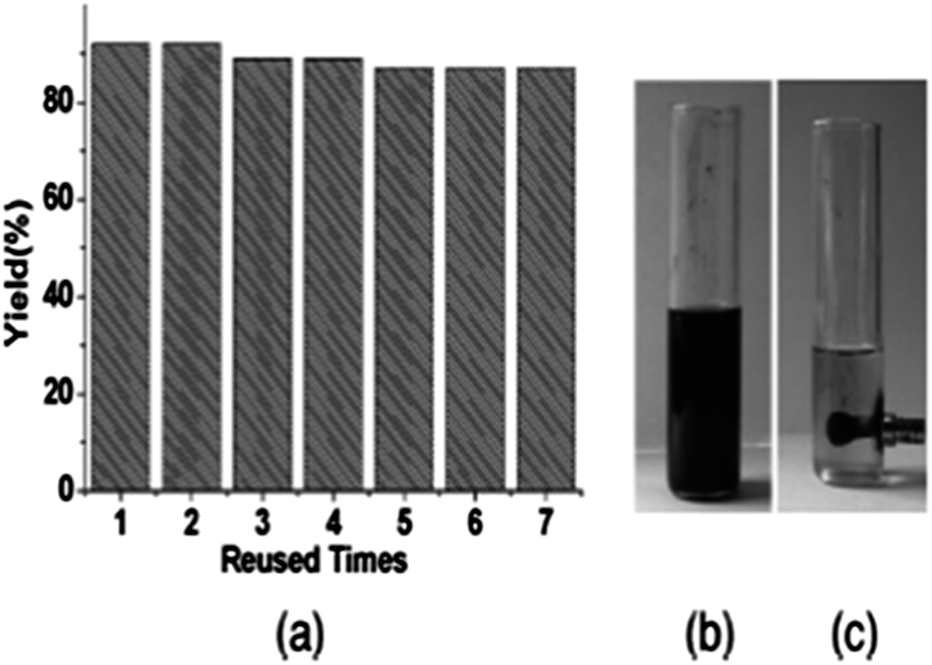
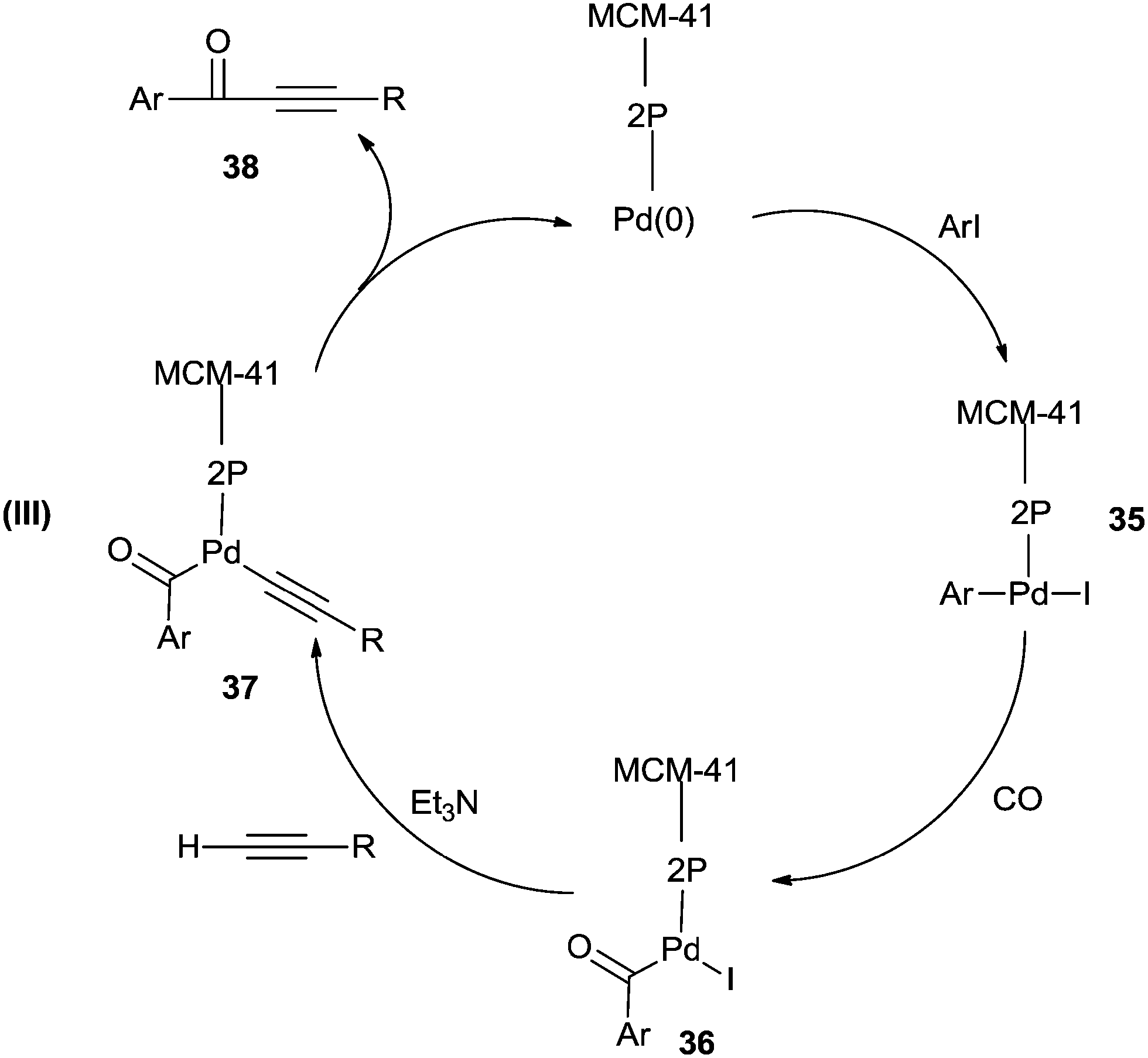
Cai and co-workers reported a heterogeneous carbonylative Sonogashira coupling reaction using a MCM-41-supported bidentate phosphine based palladium(0) complex catalyst [MCM-41–2P–Pd(0)] (Scheme 56).57a This polymeric palladium catalyst could be reused at least ten times without any decrease in activity. A plausible reaction mechanism is shown in Scheme 57. When an electron-rich aryl iodide is used as a substrate, the insertion reaction of CO to the less reactive arylpalladium complex 35 can proceed smoothly, giving the corresponding acylpalladium complex 36. Complex 36 reacts with a terminal alkyne in the presence of Et3N to afford 37 and the carbonylative coupling product 38, respectively. When an electron-deficient aryl iodide is used as the substrate in the presence of Et3N, the electron-deficient arylpalladium complex 35 reacts too rapidly with a terminal alkyne without insertion of CO, affording the direct Sonogashira coupling products in good yields. Djakovitch and co-workers synthesised several organophosphino palladium complexes grafted on SBA-15 silica or incorporated within the walls of SBA-3 silica, and applied them to the carbonylative Sonogashira reaction, particularly concerning the leaching/redeposition phenomena.57b The catalyst showed successive recycling runs.
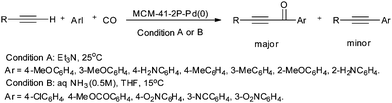 |
| | Scheme 56 Reagents and conditions: terminal alkyne (1.2 mmol), aryl iodide (1 mmol), CO (1 atm), [MCM-41–2P–Pd(0)] (0.05 mmol); conditions A: Et3N (3 mL), 25 °C; conditions B: aq. NH3 (0.5 M, 4 mL), THF (8 mL), 15 °C. | |
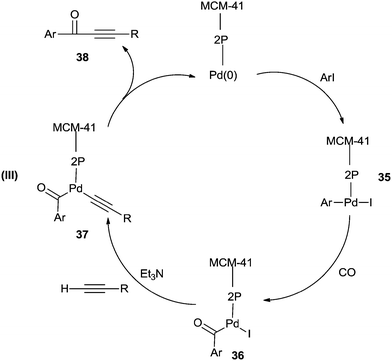 |
| | Scheme 57 A plausible mechanism for the MCM-41–2P–Pd(0) catalysed carbonylative Sonogashira coupling of aryl iodides with terminal alkynes. | |
5. Palladium-catalysed double carbonylation reactions of aryl halides and amines
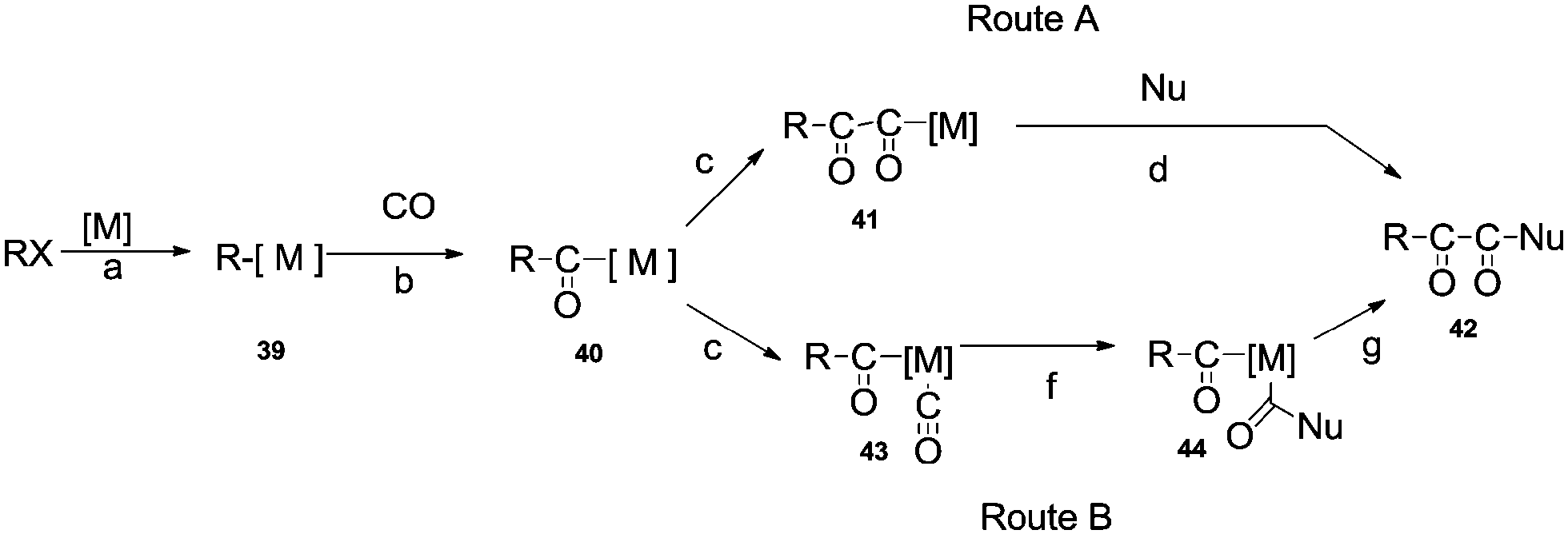
Palladium catalysed double carbonylation of amines and aryl halides is an important reaction for the synthesis of α-keto amides, oxamides and oxamates. Previous routes for the synthesis of these products involves the use of thermally unstable oxalyl chloride based chemicals such as the monoester of oxalyl chloride or diesters of oxalic acid.58 The synthesis of these starting materials involves a multistep synthesis and isolation procedures require a distillation setup.58a The alternative to these routes is the double carbonylation reaction. Various palladium metal salts and phosphine ligands have been developed to achieve these valuable products using the double carbonylation technique. Two basic routes have been proposed for the double carbonylation of aryl halides to α-keto amides (Scheme 58).59 The first step is an oxidative addition of the aryl halide on the transition metal catalyst to produce 39 (step a). An oxidative addition followed by the migratory insertion of CO (step b), gives the acylmetal complex 40. Then, two routes may be possible. Route A proposes a further migratory insertion of CO to 40, creating the (–CO–CO–) chaining species 41 (step c), followed by a nucleophilic attack on the α-carbonyl of the formed ligand to liberate the double carbonylated product 42 (step d). Route B proposes a nucleophilic attack on a terminal CO of 43 (step f), then a carbon–carbon coupling of complex 44 would afford 42 (step g).
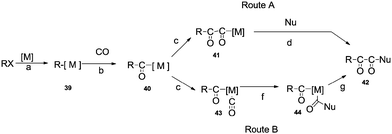 |
| | Scheme 58 General proposed mechanism for the double carbonylation of an aryl halide. | |
For the synthesis of oxamides and oxamates, a palladium catalysed oxidative carbonylation methodology has been developed. The palladium catalysed double carbonylation methodology has provided an atom economical, environmentally benign protocol for the synthesis of these valuable products.
5.1 Palladium-catalysed double carbonylation reactions of aryl halides for the synthesis of α-keto amides
In 1982, Tanaka demonstrated the synthesis of α-keto amides by the double carbonylation of aryl and heteroaryl halides using the dichloro[l,4-bis(diphenylphosphino) butane]palladium complex, [PdCl2(dppb)], under a CO atmosphere (Scheme 59).60 Various secondary amines gave excellent yields of the desired products, however with primary amines the imine of the α-keto amide was obtained as the main product.
 |
| | Scheme 59 Reagents and conditions: aryl halide (4.0 mmol), amine (3 mL), PdCl2(dppb) (1.88 × 10−2 mmol), CO (40 atm), temp. 60 °C, time 4 h. | |
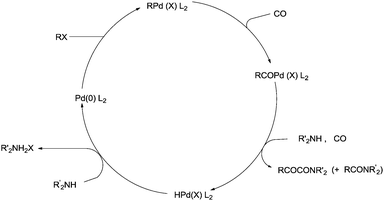
Yamamoto and co-workers reported the double carbonylation of aryl iodides, bromides and chlorides using PdC12(PMePh2)2 as a catalytic system for the synthesis of α-keto amides (Scheme 60).61 A reaction mechanism for the double carbonylation reaction has been proposed in Scheme 61. It is undecided whether the double carbonylation reaction proceeds by a double CO insertion into a Pd–C bond giving a RCOCO–Pd species to be trapped by an amine or by a single CO insertion giving a CO-coordinated acylpalladium species RCO–Pd(CO), followed by attack of the amine on the coordinated CO to give acyl-carbamoyl species RCO–Pd–CONŔ2, which reductively eliminates to form RCOCONŔ2.
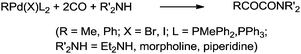 |
| | Scheme 60 Reagents and conditions: aryl halide (1 mL, 9.6 mmol), amine (3 mL, 29 mmol), PdC12(PMePh2)2, (49 mg, 0.085 mmol), CO (10 atm), temp. 100 °C, time 40 h. | |
 |
| | Scheme 61 Proposed mechanism for the double carbonylation of aryl halides. | |
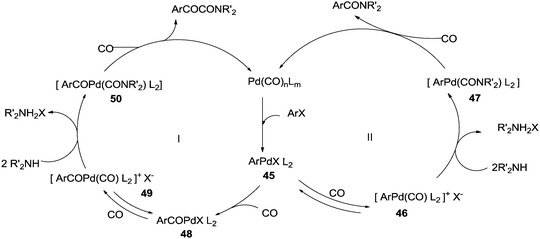
In 1985 the same research group studied the detailed mechanism of the diphenylmethylphosphine palladium complex catalysed double carbonylation of aryl halides (Scheme 62).62
 |
| | Scheme 62 Mechanistic studies of the double carbonylation of aryl iodides. | |
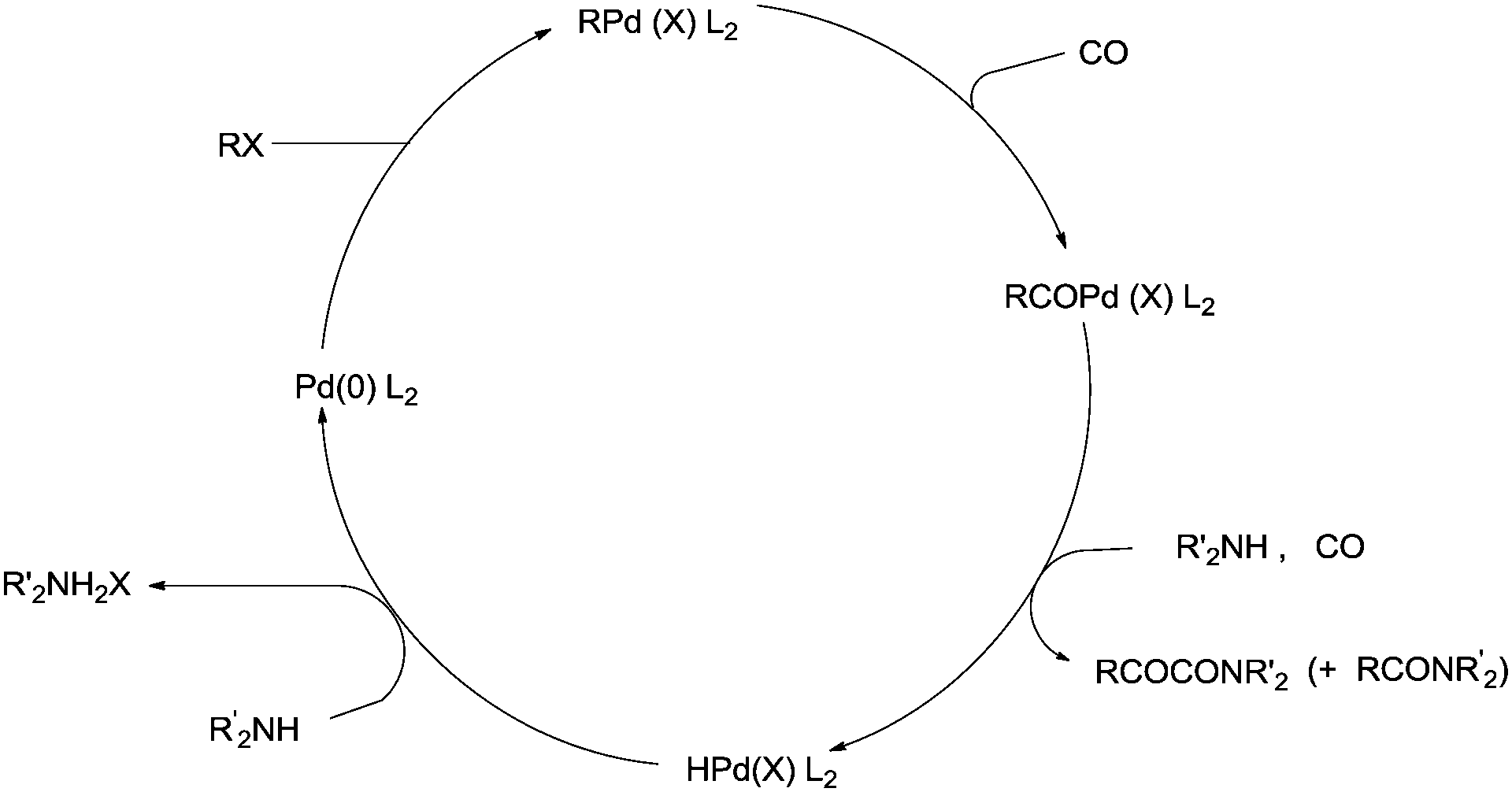
The reactivity of the phenyl halides decreases in the order PhI > PhBr ≫ PhCl. Highly basic secondary amines were found to be the best substrates for the α-keto amide synthesis, while the selectivity for the α-keto amide formation decreased with decreasing steric bulk. Primary amides are not suitable for the double carbonylation reaction. The mechanism consists of two catalytic cycles to produce α-keto amides and amides. Cycle I produces the α-keto amide whereas cycle II yields the amide. The reaction goes via an oxidative addition of the aryl halide to a zero-valent palladium species to give the common intermediate arylpalladium complex 45 for both cycles. A highly reactive amine attacks the coordinated CO in the arylcarbonylpalladium species 46 to give the arylcarbamoyl species 47. After reductive elimination the amide is generated as well as the zero-valent palladium species. When a less reactive amine was used, the intermediate 46 converts to the aroylpalladium species 48. After CO insertion the ionic species is generated (49) and this is attacked by the amine to generate the aroylcarbamoyl species 50. Finally, a reductive elimination step gives the α-keto amide. The smooth formation of the aroylpalladium intermediate 48 is key to the successful double carbonylation reaction of the aryl halide. The migration step, forming 48 from the intermediate 46, is greatly influenced by the electron density of the aryl moiety. Aryl halides with electron withdrawing groups have been regarded as unfavourable substrates and the protocol is only applicable to those aryl halides without electron withdrawing groups. Miura et al. demonstrated the formation of α-keto amides at atmospheric pressure of CO.63 By adding CuI as a co-catalyst, the uptake of CO could be promoted and the rate of reaction is enhanced. Yan and co-workers prepared silica-supported polytitazane–palladium complex 51 and used it to catalyse the double carbonylation reaction of phenyl halides (Scheme 63).64 It was found that the reactivity of the phenyl halides decreased in the order of PhI > PhBr ≫ PhCl. The catalyst could be reused ten times without any noticeable decrease in the catalytic activity and selectivity. In 1999, Carpentier and Castanet used the double carbonylation method for the synthesis of α-keto amides using 4-iodopyridines, CO and diethylamine in the presence of a catalytic amount of Pd(OAc)2(PCy3) complex catalyst (Scheme 64).65
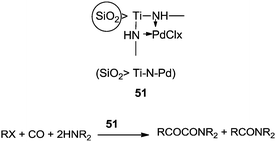 |
| | Scheme 63 Reagents and conditions: 51/PhI (0.5%), phenyl halide (5.1 mmol), E2NH (100 mmol), temp. 100 °C, CO pressure (3.0 MPa), time 6–24 h. | |
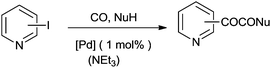 |
| | Scheme 64 Reagents and conditions: 4-iodopyridine (0.50 g, 2.44 mmol), HNEt2 (1.2 mL, 12 mmol), Pd(OAc)2 (5.5 mg, 0.024 mmol) and PCy3 (19.2 mg, 0.072 mmol), CH2C12 (30 mL), CO (60 bar), temp. 50 °C, time 6 h. | |
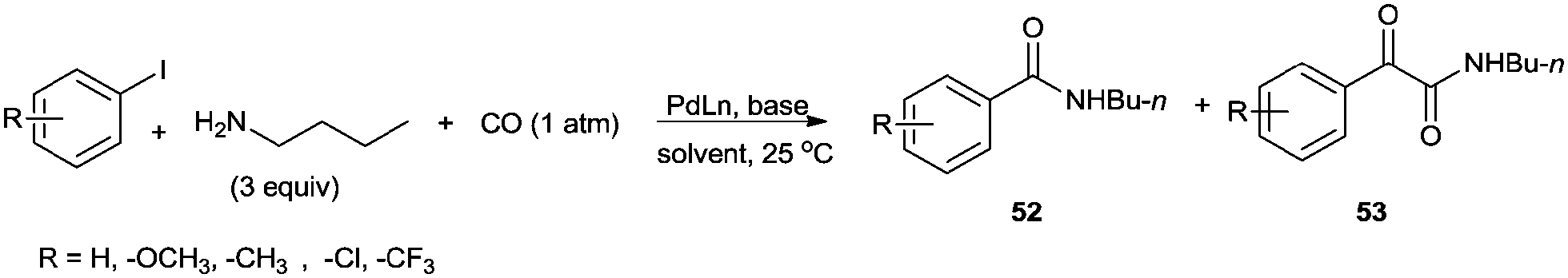
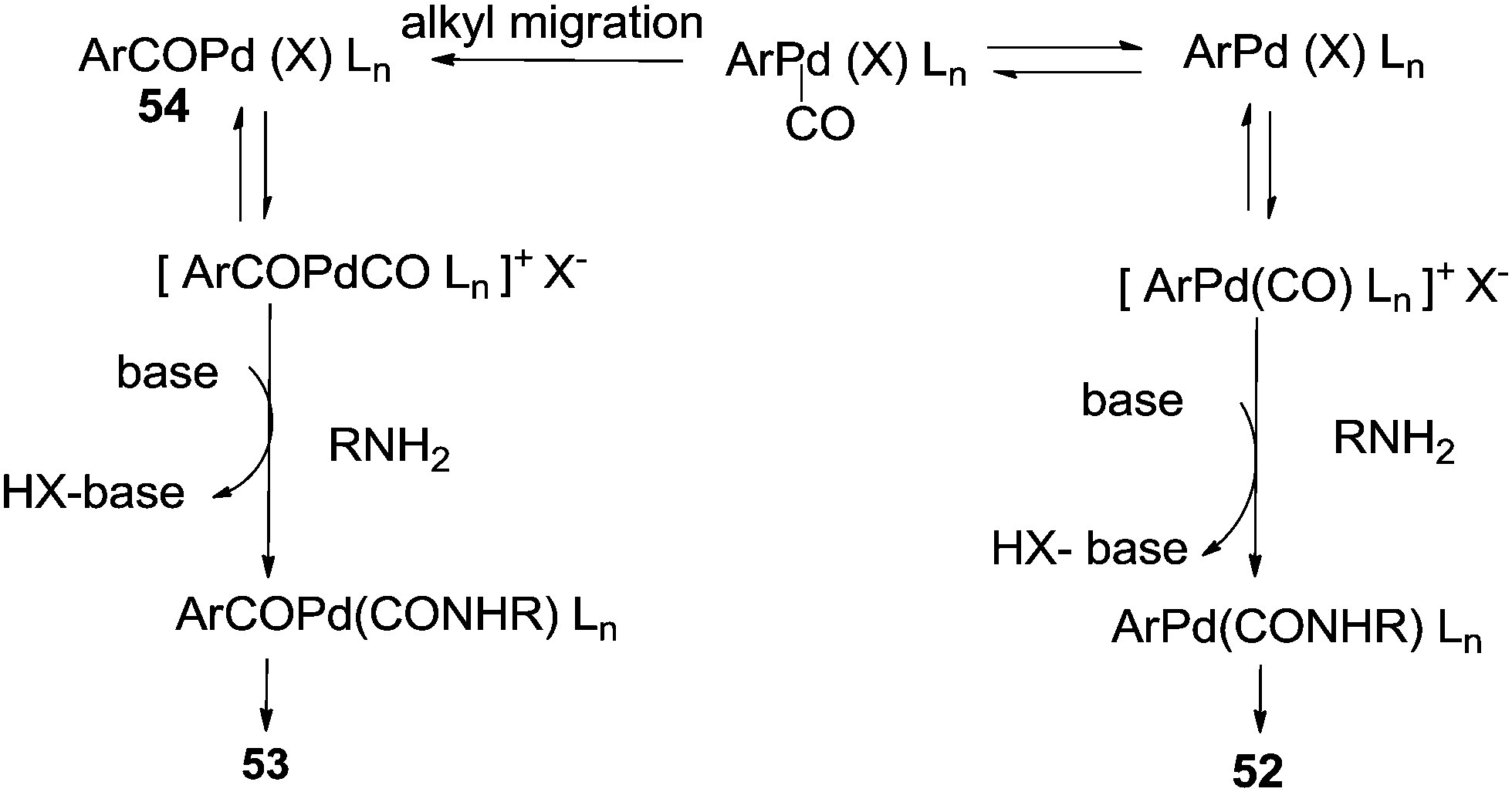
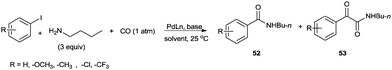
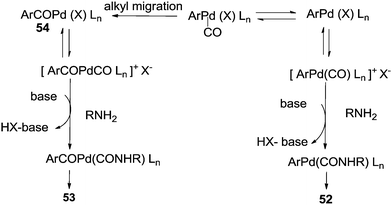
The reaction resulted in more than 90% yield of the α-keto amides under mild conditions. Uozumi and co-workers synthesised α-keto amides by the double carbonylation of aryl iodides with primary amines under atmospheric pressure and ambient temperature conditions using the Pd/PPh3/DABCO/THF system (Scheme 65).66 High selectivity for the double carbonylation reaction was obtained using DABCO as a base. In the presence of other bases such as Et3N, pyridine and DBU, the selectivity decreased. The role of DABCO is not properly understood in the double carbonylation reaction. The reaction mechanism shows that the irreversible alkyl migration step forming 54 is the divergence of the reaction pathway towards the double carbonylation reaction for the synthesis of 53 (Scheme 66). The protocol is only applicable to those aryl iodides without electron withdrawing groups.
 |
| | Scheme 65 Reagents and conditions: iodobenzene (170 mg, 0.83 mmol), n-butylamine (183 mg, 2.5 mmol), [PdCl(n3-C3H5)]2 (4.6 mg, 0.0125 mmol), PPh3 (13.1 mg, 0.05 mmol), THF (8.3 mL), DABCO (280 mg, 2.5 mmol), temp. 25 °C, CO (1 atm), time 12 h. | |
 |
| | Scheme 66 Reaction mechanism for the synthesis of 53 by the double carbonylation reaction of aryl iodides. | |
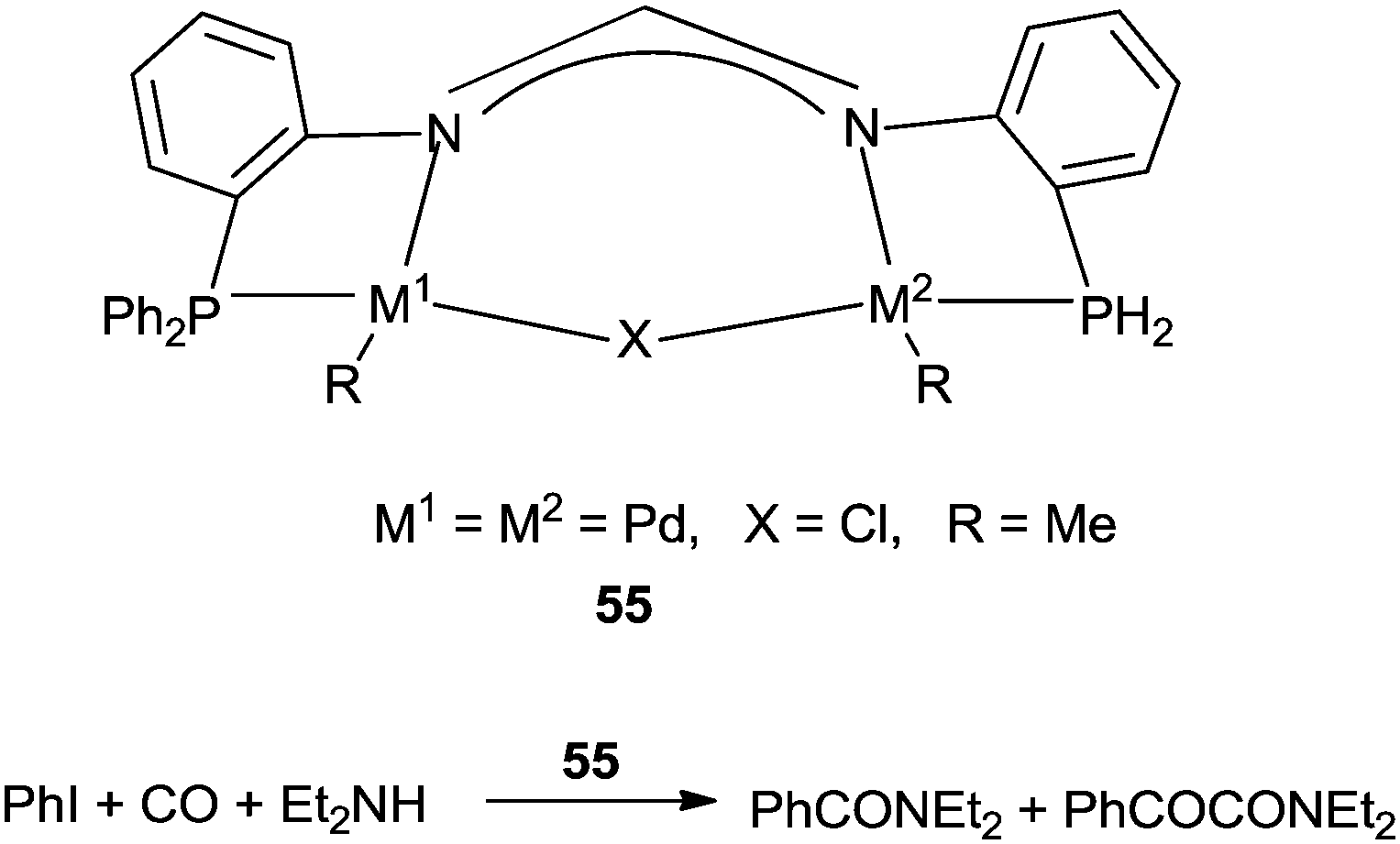
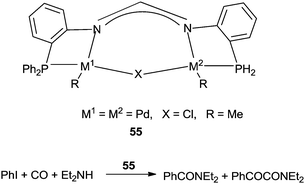
Inoue and co-workers reported the dinuclear palladium complexes 55 bridged by a novel PNNP ligand (dpfam) for the double carbonylation of iodobenzene (Scheme 67).67 The catalytic system allowed the double carbonylation reaction to proceed with a TON up to 105 and a selectivity of 96%.
 |
| | Scheme 67 Reagents and conditions: iodobenzene (5.0 mmol), amine (7.5 mmol), K3PO4 (5.0 mmol), 1,4-dioxane (3 mL), 55 (0.01 mol%), CO (10 atm), temp. 100 °C, time 15 h. | |
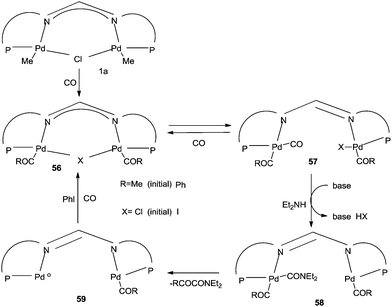
A plausible mechanism proposed for the 55-catalysed double carbonylation reaction is shown in Scheme 68. Insertion of CO gives the acetyl complex 56, which was confirmed by NMR analysis. Coordination of a second molecule of CO to species 56 generates intermediate 57. Nucleophilic attack of the amine generates the acyl-carbamoyl species 58, which on reductive elimination generates the Pd0PdII bimetallic species 59. The intermediate 59 undergoes oxidative addition with iodobenzene and continues the cycle by CO insertion through the acyl species 56.
 |
| | Scheme 68 Double carbonylation of aryl iodide using a dinuclear palladium complex catalyst. | |
Iizuka and Kondo showed that the use of t-Bu3P as a ligand dramatically improved the generality of the double carbonylation reaction of aryl iodides (Scheme 69).68 Using t-Bu3P as a ligand resulted in almost complete selectivity for the α-keto amide at room temperature and atmospheric pressure of CO in THF.
 |
| | Scheme 69 Double carbonylation of aryl iodides catalysed by a Pd(t-Bu3P)2 catalyst. | |
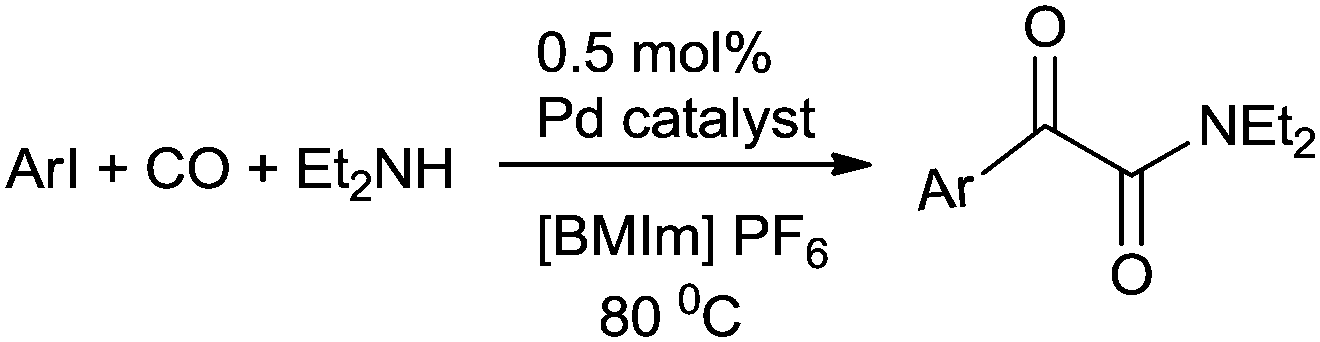
Moreover, this altered protocol gave access to the double carbonylation reaction of electron-deficient aryl iodides. Rahman et al. developed a low pressure microflow system for the palladium catalyzed double carbonylation of aryl iodides in an ionic liquid (Scheme 70).69 In comparison with a conventional batch system, the microflow system resulted in superior selectivity and higher yields for the α-keto amide synthesis (Scheme 70). Murphy et al. developed a microreactor system for the double aminocarbonylation reaction of aryl iodides.70 Microreactors have enhanced heat and mass transfers, reduced reaction volumes, and the ability to run several experiments within a sealed system. They also have the additional advantage of continuous flow. Here both electron donating and electron withdrawing substituents on aryl bromide showed remarkable activity for the double carbonylation reaction (Scheme 71).
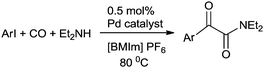 |
| | Scheme 70 Reagents and conditions: microflow system: aryl iodide (14.0 mmol), Et2NH (70 mmol, 5.0 equiv.), CO (20 or 15 atm), Pd cat. (0.5 mol%) in [BMIm]PF6 (17 mL); flow rates: mixture of aryl iodide and Et2NH (0.04 mL min−1), Pd/[BMIm]PF6 (0.14 mL min−1), CO (0.25 mL min−1), temp. 80 °C, residence time 34 min; conventional batch system: aryl iodide (2.66 mmol), Et2NH (5.0 equiv.), Pd cat. (0.5 mol%), [BMIm]PF6 (2.0 mL), CO (20 or 15 atm), 80 °C, time 5 h. | |
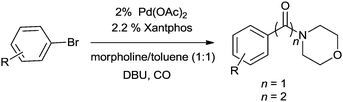 |
| | Scheme 71 Double carbonylation of aryl bromides in a micro reactor system. | |
Balogh et al. showed the double carbonylation reaction of iodobenzene in the presence of various amines with high selectivity in a microfluidic-based flow reactor (X-CubeTM)29,71 using an immobilized Pd(PPh3)4 catalyst (Scheme 72).72 The double carbonylation reaction observed selectively at 80 °C using DBU as the base. At higher temperatures the imine was observed as a side product.
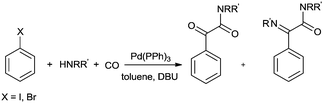 |
| | Scheme 72 Reagents and conditions: aryl halide (0.05 M), amine (0.05 M or 0.1 M), DBU (0.1 M), immobilized Pd(PPh3)4 (0.4 g), flow rates between 0.2 and 0.5 mL min−1, CO (40 bar), temp. 70–100 °C. | |
In 2009 Xia and co-workers used Pd/C–PPh3 as a simple and efficient catalyst system for the double carbonylation reaction of aryl iodides.73 The reaction proceeded under homogeneous conditions through a leached palladium species from the Pd/C source (Scheme 73). Skrydstrup discovered the palladium-catalyzed double carbonylation reaction of aryl iodides with amine nucleophiles using a stoichiometric amount of CO for α-keto amide syntheses (Scheme 74).74 9-Methylfluorenecarbonyl chloride (COgen) acts as a solid source of CO in a sealed two-chamber glass reactor. Various aryl and heteroaryl iodides showed excellent reactivity and selectivity towards the double carbonylation reaction with various amines, and excellent yields of the α-keto amide derivatives were achieved.
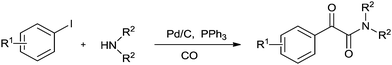 |
| | Scheme 73 Reagents and conditions: aryl iodide (1.0 mmol), amine (1.5 mmol), DABCO (2.0 mmol), the molar ratio of catalyst/PPh3/substrate = 1/4/100, solvent (6.0 mL), CO (4.0 MPa), temp. 60 °C, temp. 20 h. | |
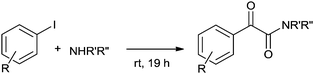 |
| | Scheme 74 Reagents and conditions: aryl iodide (0.5 mmol), amine (2 equiv.), room temperature, time 19 h; chamber 1: Pd(dba)2 (2 mol%), HBF4P(tBu)3 (4 mol%), DBU (2 equiv.), THF; chamber 2: COgen (1.5 mmol), Pd(dba)2 (5 mol%), P(tBu)3 (10 mol%), Cy2NMe (2 equiv.), DMF. | |
Djakovitch and Dufaud synthesised α-keto amides using covalently immobilized palladium complexes on mesostructured SBA-15 type silica [PdCl2(PPh2)2@SBA-15](Scheme 75).75 It was found that the PdCl2(PPh2)2@SBA-15 catalyst could be reused for up to three cycles without affecting the catalyst performance.
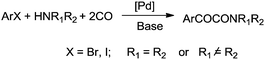 |
| | Scheme 75 Reagents and conditions: aryl iodide (1 mmol), amine (2 mmol), K2CO3 (2 mmol), PdCl2(PPh2)2@SBA-15 (1 mol% [Pd]), MEK (5 mL), temp. 60 °C, CO 40 bar, time 48 h. | |
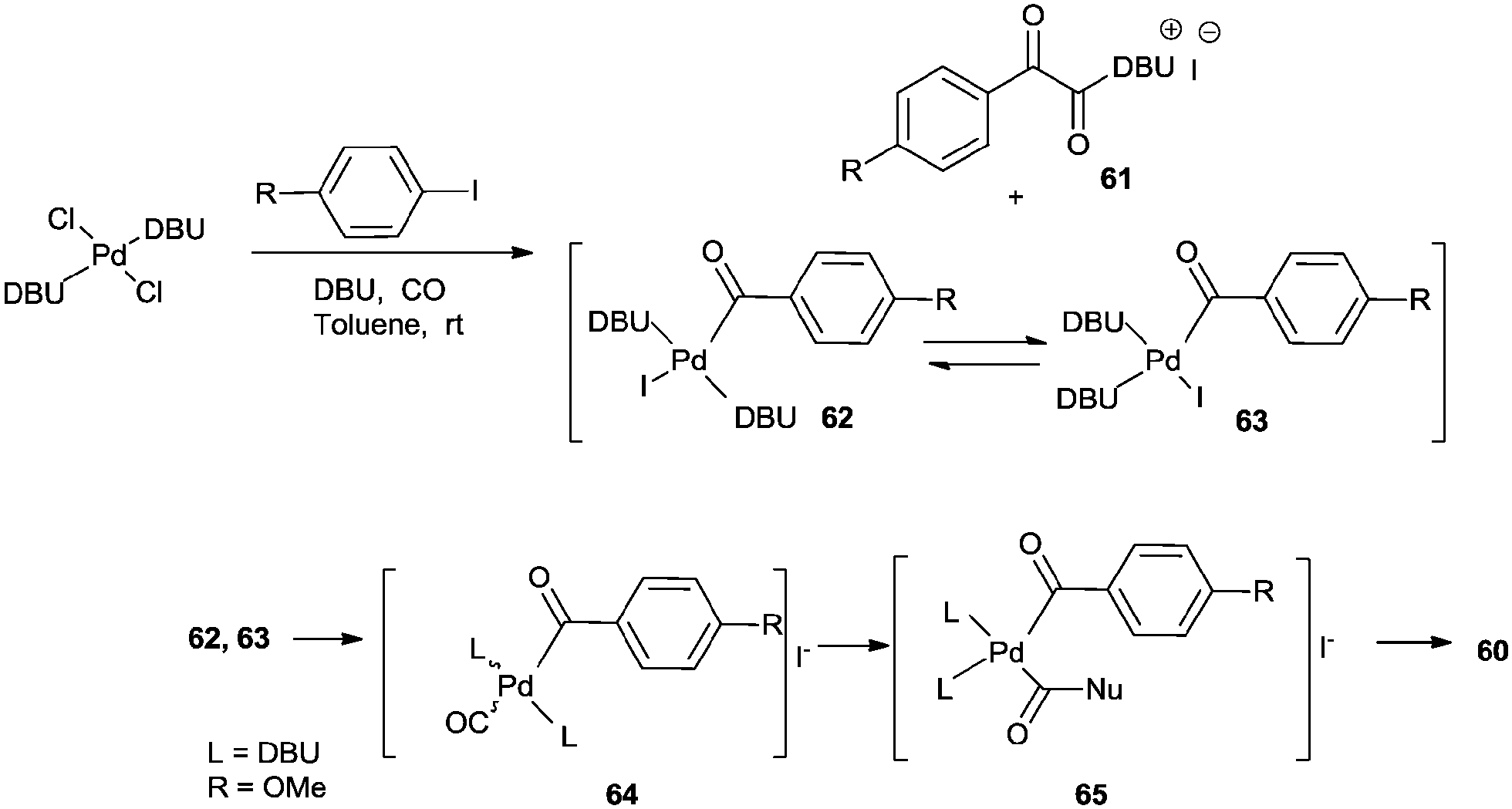
Recently Castillo'n reported the first phosphine-free Pd-catalysed double carbonylation reaction of aryl iodide using Pd/DBU as a catalytic system under atmospheric CO pressure (Scheme 76).76 They proved the key roles of DBU were a nucleophile (or acyl transfer agent) and a base. As shown in Scheme 77, in the absence of an amine nucleophile, three new products 61, 62 and 63 were formed and detected by NMR and MS analysis. These products were the α-keto amide 61 containing a charged DBU unit, and the cis and trans isomers of the Pd-acyl complex 62, 63, respectively. This experiment clearly demonstrated that DBU acts as a nucleophile under these conditions.
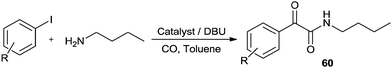 |
| | Scheme 76 Reagents and conditions: aryl iodide (0.5 mmol), amine (1.2 mmol), catalyst [Pd(n3-C3H5)(μ-Cl)]2 (0.005 mmol), DBU (1 mmol), toluene (5 mL), CO (1 bar). | |
 |
| | Scheme 77 Proposed mechanism for the double carbonylation reaction of aryl halides in the presence of DBU. | |
Intermediates 62 and 63 can react with a second molecule of CO to form a cationic species 64, containing a terminal CO group and an acyl moiety with iodide as the counter-ion. At this stage, nucleophilic attack at the terminal CO could occur from the amine nucleophile to directly form Pd–acyl–amide species 65. Reductive elimination of the complex 65 regenerates the Pd0 species and 60. Papp and Skoda-Földes reported various phosphine-free silica supported palladium catalysts (SILP–Pd) for the double carbonylation reaction of iodobenzene in the presence of secondary amines (Scheme 78).77 The catalyst was prepared by impregnating silica with a mixture of a palladium precursor and an ionic liquid (Scheme 79). The catalyst could be recycled at least six times and resulted in excellent yields of the α-keto amides.
 |
| | Scheme 78 Reagents and conditions: iodobenzene (0.2 mmol), secondary amine (0.5 mmol), Et3N (0.25 mmol), catalyst (3.6 μmol Pd-content), DMF 1 mL, CO 30 bar, temp. 100 °C, time 3 h. | |
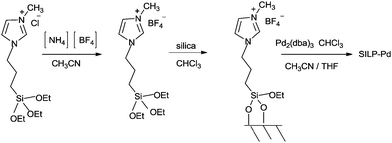 |
| | Scheme 79 Preparation of the silica supported phosphine-free catalyst (SILP–Pd). | |
5.2 Palladium-catalysed double carbonylation reactions of amines for the synthesis of oxamides
In 1966, Tsuji synthesised oxamides by a palladium catalysed double carbonylation reaction of primary amines for the first time (Scheme 80).78 The by-product of hydrogen was confirmed by gas chromatography.
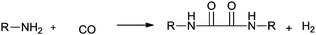 |
| | Scheme 80 Reagents and conditions: n-decylamine (7.9 g), PdCl2 (0.5 g), benzene (30 mL), CO (100 kg cm−2), temp. 180 °C, time 20 h. | |
Alper and co-workers have shown palladium catalysed oxidative coupling reactions of amines and carbon monoxide for the synthesis of oxamides and ureas (Scheme 81).79 Oxamides were formed in excellent yields when secondary amines were carbonylated in the presence of iodide as a promoter and oxygen. When using primary amides, the selectivity was towards the urea.
 |
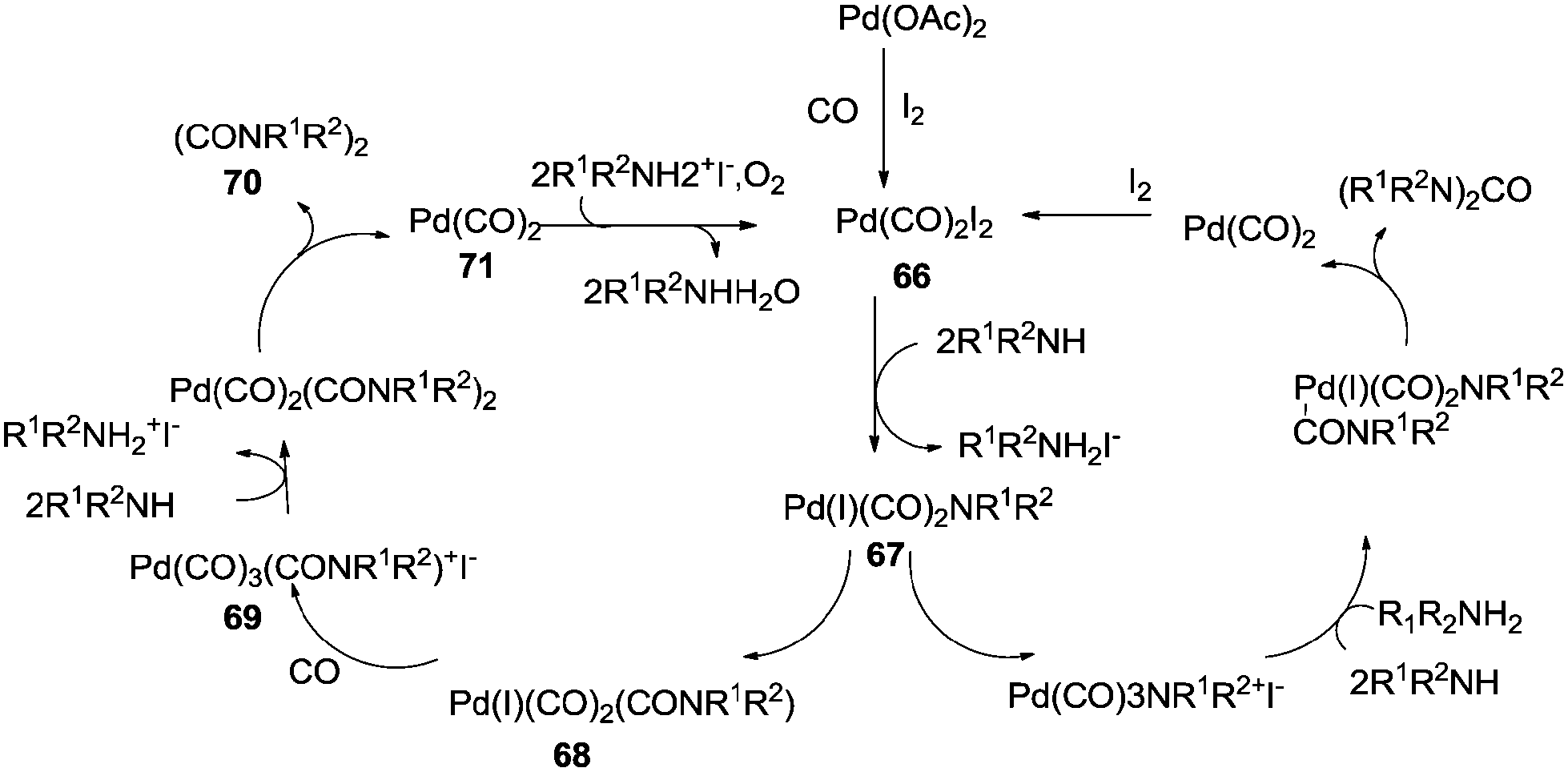
| | Scheme 81 Reagents and conditions: amine (1.5 mmol), Pd(OAc)2 (0.022g, 0.10 mmol), KI or (C4H9)N+I− (0.2–0.8 mmol), K2CO3 (1.5 mmol), THF (10 mL), CO (2.7 atm), O2 (1 atm), temp. 95 °C, time 3 h. | |
The reaction mechanism showed that there were two independent reaction pathways using iodide ligands for the synthesis of oxamides and ureas, respectively (Scheme 82). Palladium acetate can convert to the diiododicarbonyl palladium compound 66 using iodide and carbon monoxide. Displacement of iodide by the amine gives 67. With complex 67 ligand migration followed by CO insertion takes place to generate species 68. Addition of another carbon monoxide molecule generates the ionic species 69. At this stage amine addition, followed by reductive elimination generates the oxamide 70. Reoxidation of 71 in the presence of oxygen and the iodide promoter regenerates the active species 66.
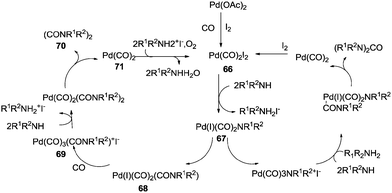 |
| | Scheme 82 Proposed reaction mechanism for the oxamide synthesis. | |
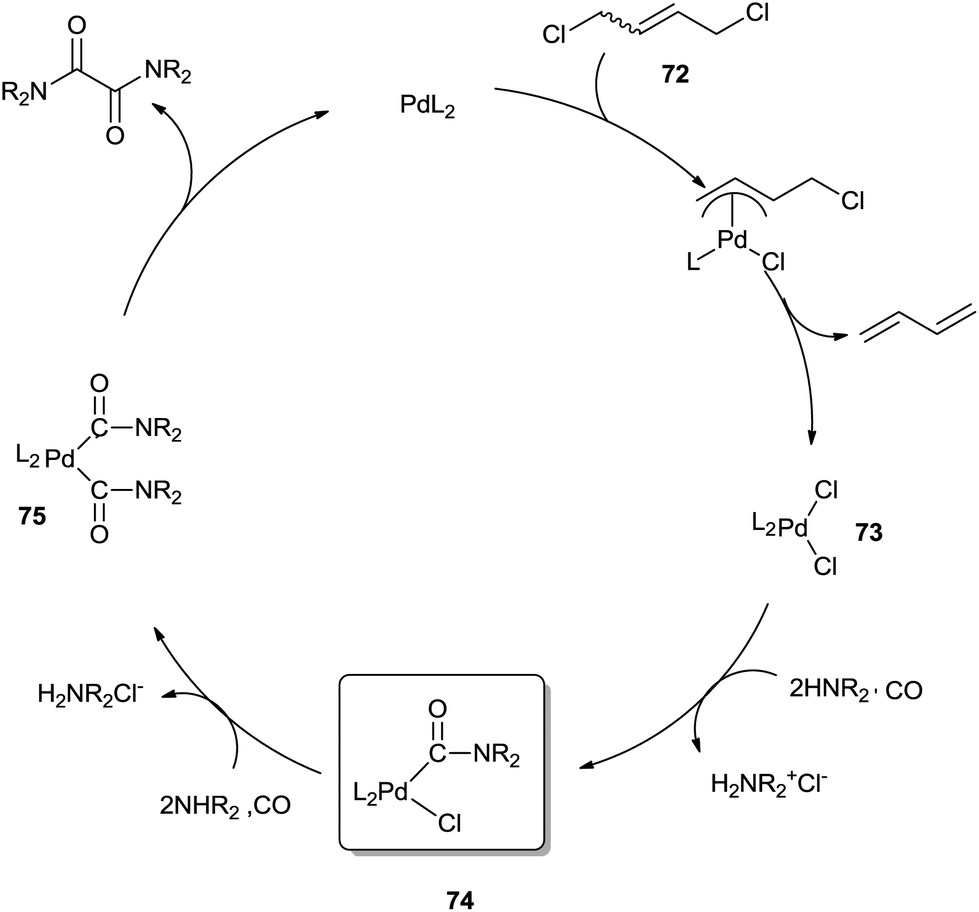
Hiwatari et al. developed a new process for converting secondary amines into oxamides under CO pressure, catalyzed by homogeneous palladium complexes in the presence of 1,4-dichloro-2-butene (DCB) 72 as an oxidant (Scheme 83).80
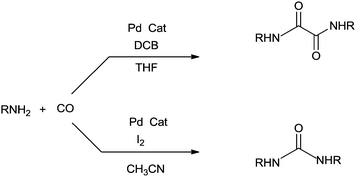 |
| | Scheme 83 Reagents and conditions: diethylamine (2.07 mL, 20.0 mmol), trans-[PdCl2(PPh3)2] (17.5 mg, 0.0250 mmol), THF (3 mL), 1,4-dichloro-2-butene (0.540 mL, 5.00 mmol), carbon monoxide (1 to 100 atm), rt, time 3–17 h. | |
The mechanism (Scheme 84) proposes that in the presence of 72, oxidation of the Pd(0) species readily gives dichloropalladium(II) complex 73. Complex 73, upon treatment with CO and diethylamine, forms the monocarbamoylpalladium intermediate 74. Bis(carbamoyl)palladium complex 75, generated by another CO molecule and amine molecule insertion to species 74, undergoes reductive elimination to produce the oxamide and regenerate Pd0.
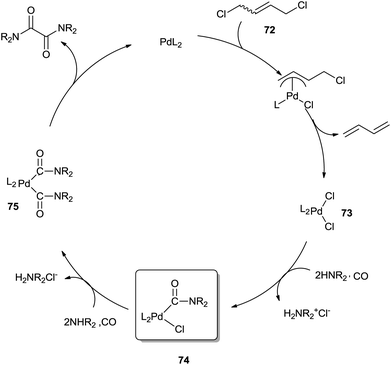 |
| | Scheme 84 Proposed mechanism for the catalytic carbonylation of a secondary amine to form an oxamide with DCB as an oxidant. | |
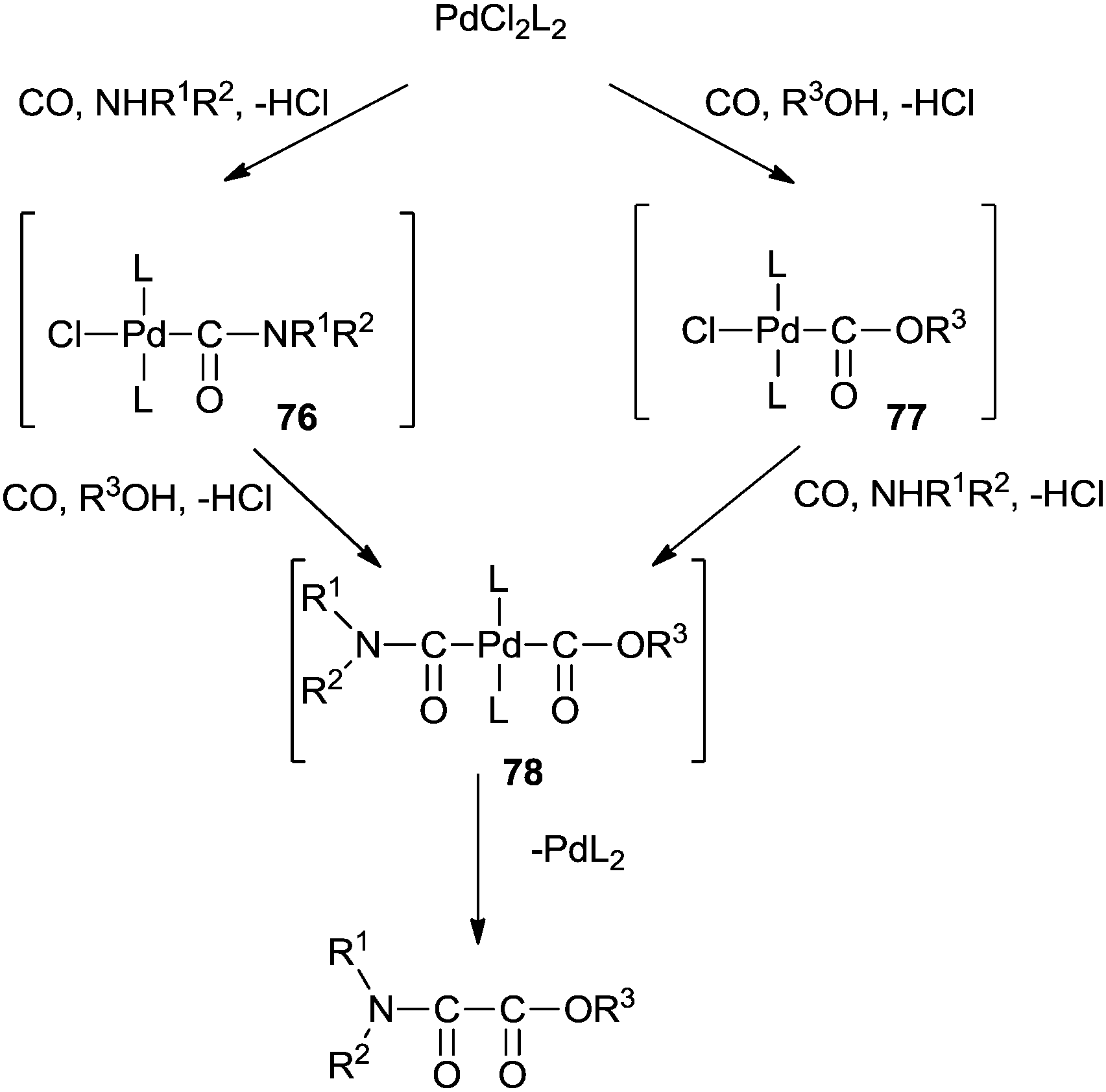
5.3 Palladium-catalysed double carbonylation reactions of amines for the synthesis of oxamates
In 1987 Murahashi and co-workers synthesised oxamates by oxidative cross double carbonylation of amines and alcohols using a PdC12(MeCN)2/CuI catalytic system (Scheme 85).81 The reaction required CuI as a co-catalyst and the dehydrating reagent CH(OMe)3 under a very high pressure of CO (80 kg cm−2) and O2 (5 kg cm−2). In the case of β-aminoalcohols, intramolecular double carbonylation proceeds highly efficiently, giving the corresponding cyclic oxamates (Scheme 86). In this case, addition of the dehydrating reagent CH(OMe)3 was not necessary.
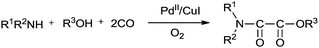 |
| | Scheme 85 Reagents and conditions: amine (1.0 mmol), PdC12(MeCN)2 (0.05 mmol), CuI (0.15 mmol), alcohol (50 mmol), dehydrating reagent CH(OMe)3 (2 mmol), CO (80 kg cm−2), O2 (5 kg cm−2). | |
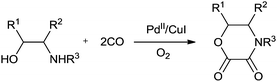 |
| | Scheme 86 Reagents and conditions: amino alcohol (1 mmol), PdC12(MeCN)2 (0.05 mmol), CuI (0.25 mmol), acetonitrile (2 mL), CO (80 kg cm−2) and O2 (5 kg cm−2), rt, time 20 h. | |
The mechanism shows that the double carbonylation reaction proceeds via the carbamoylpalladium complex 76 or the alkoxycarbonylpalladium complex 77 (Scheme 87). Further co-ordination of carbon monoxide to 76 or 77 and the subsequent nucleophilic attack of the alcohol or amine gives the alkoxycarbonylcarbamoylpalladium complex 78. Reductive elimination of 78 gives the oxamate and PdL2. PdL2 is readily oxidized with O2–CuI–HCl to continue the catalytic cycle.
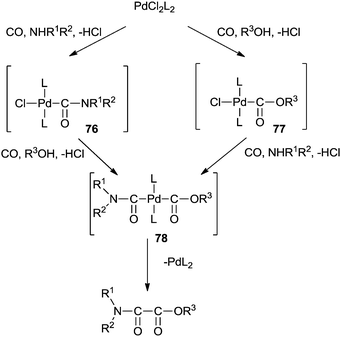 |
| | Scheme 87 A plausible reaction mechanism for the cross double carbonylation reaction of an amine and an alcohol for the synthesis of an oxamate. | |
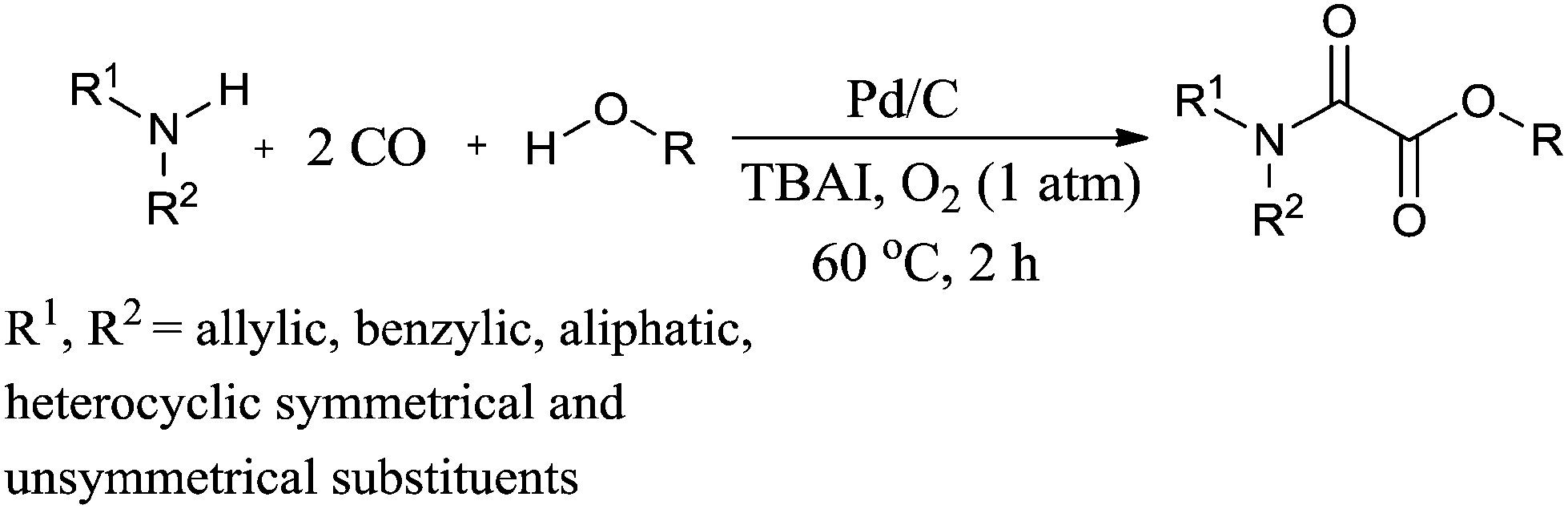
Recently Gadge and Bhanage used Pd/C as a heterogeneous catalytic system for the synthesis of oxamates (Scheme 88).82 The reaction proceeds under a low pressure of CO/O2 (6:1 atm), a short reaction time, and does not require a co-catalyst, base, dehydrating agent or ligand. A variety of amines, such as allylic, benzylic, aliphatic, symmetrical and unsymmetrical amines, underwent cross double carbonylation reactions with alcohols, providing excellent yields of the desired products. Moreover the catalyst was recovered by a simple filtration and reused for up to six consecutive cycles.
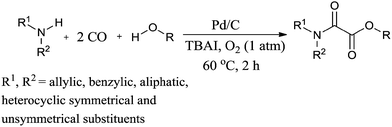 |
| | Scheme 88 Reagents and conditions: amine (1 mmol), alcohol (10 mL), 10% Pd/C (8 mol%), TBAI (0.2 mmol), CO/O2 (6:1 atm), temp. 60 °C, time 2 h. | |
6. Conclusions
To summarize, we have reviewed various palladium catalysed carbonylation reactions such as oxidative carbonylation, double carbonylation and several other carbonylative coupling reactions. These reactions provide a novel and efficient tool for constructing various carbonylative compounds. It is necessary to conduct research towards the synthesis of various heterogeneous catalysts and the development of economical procedures for the synthesis of these heterogeneous catalysts, as this will result in a higher efficiency, selectivity, and a larger substrate scope. Many of these reactions work at high pressure, limiting their application. Future effort needs to be directed towards developing a catalyst which works at atmospheric pressures. There is also a requirement for the deeper understanding of the fundamental aspects of the mechanistic studies of heterogeneous catalysed carbonylation reactions. The catalyst–product separation and the catalyst recyclability in homogeneous catalysis is a major problem and there is a need for the development of various novel strategies to solve this problem. The invention of novel catalyst–product separation techniques and the recovery of precious metal catalysts are current challenges. The development of various biphasic catalysis systems, supported liquid phase catalysis systems, polymer anchored catalysis systems and metal leaching re-deposition techniques for carbonylation reactions have provided attractive ways for process economy and for the overall development of truly ecofriendly reaction methodologies.
Abbreviations
| MMC | Methyl N-methylcarbamate |
| DMU | N,N′-dimethylurea |
| NMF | N-methylformamide |
| DPU | N,N′-diphenylurea |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| TBAI | Tetrabutylammonium iodide |
| DMF | Dimethylformamide |
| MW | Microwave |
| DCB | 1,4-Dichloro-2-butene |
| TOF | Turnover frequency |
| TON | Turnover number |
| DME | Dimethoxyethane |
| dpfam | [N,N′-bis[(2-diphenylphosphino)phenylformamidinate] |
| DMA | Dimethylacetamide |
| phen | 1,10-Phenanthroline |
| DMSO | Dimethyl sulfoxide |
| dppp | 1,3-Bis(diphenylphosphino)propane |
| dppb | 1,4-Bis(diphenylphosphino)butane |
| pyca | 2-Picolinate |
| BQ | 1,4-Benzoquinone |
| DDQ | 2,3-Dichloro-5,6-dicyanobenzoquinone |
Acknowledgements
S.T.G. is thankful to CSIR (Council of Scientific and Industrial Research), New Delhi, India for providing a senior research fellowship.
Notes and references
-
(a) B. M. Trost, Science, 1991, 254, 1471–1477 CAS;
(b) B. M. Trost, Angew. Chem., 1995, 107, 285 (Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281) CrossRef;
(c) B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS PubMed;
(d) L. P. Kollar, Modern Carbonylation Methods, Willey-VCH, Weinheim, 2008 Search PubMed.
- P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS PubMed.
-
(a) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC;
(b) J. J. Song, J. T. Reeves, D. R. Fandrick, Z. Tan, N. K. Yee and C. H. Senanayake, Green Chem. Lett. Rev., 2008, 1, 141–148 CrossRef CAS;
(c) S. Tang, R. Bourne, R. Smith and M. Poliakoff, Green Chem., 2008, 10, 268–269 RSC;
(d) R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC;
(e) R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC;
(f) I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 107, 2169–2173 CrossRef PubMed;
(g) J. A. Dahl, B. L. S. Maddux and J. E. Hutchison, Chem. Rev., 2007, 107, 2228–2269 CrossRef CAS PubMed;
(h) P. T. Anastas and M. M. Kirchoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
-
(a) X.-F. Wu, H. Neumann and M. Beller, ChemSusChem, 2013, 6, 229–241 CrossRef CAS PubMed;
(b) Q. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788–10799 CrossRef CAS PubMed.
- S. J. Rhoads and R. E. Michel, J. Am. Chem. Soc., 1963, 85, 585–591 CrossRef CAS.
-
(a) R. V. Chaudhari, B. M. Bhanage, R. M. Deshpande and H. Delmas, Nature, 1995, 373, 501–503 CrossRef CAS;
(b) B. M. Bhanage and M. Arai, Catal. Rev.: Sci. Eng., 2001, 43, 315–344 CrossRef CAS PubMed.
- A. Molnar, Chem. Rev., 2011, 111, 2251–2320 CrossRef CAS PubMed.
-
(a) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC;
(b) X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1–35 CrossRef CAS PubMed;
(c) B. Gabriele, R. Mancuso and G. Salerno, Eur. J. Org. Chem., 2012, 6825–6839 CrossRef CAS;
(d) A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed;
(e) R. Grigg and S. P. Mutton, Tetrahedron, 2010, 66, 5515–5548 CrossRef CAS PubMed;
(f) S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867–9923 CrossRef CAS PubMed.
-
(a) D. J. Díaz, A. K. Darko and L. McElwee-White, Eur. J. Org. Chem., 2007, 4453–4465 CrossRef;
(b) B. Gabriele, G. Salerno, R. Mancuso and M. Costa, J. Org. Chem., 2004, 69, 4741–4750 CrossRef CAS PubMed;
(c) P. Giannoccaro, J. Organomet. Chem., 1987, 336, 271–278 CrossRef CAS;
(d) N. D. Ca', P. Bottarelli, A. Dibenedetto, M. Aresta, B. Gabriele, G. Salerno and M. Costa, J. Catal., 2011, 282, 120–127 CrossRef PubMed;
(e) K. Orito, M. Miyazawa, T. Nakamura, A. Horibata, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita, T. Yamazaki and M. Tokuda, J. Org. Chem., 2006, 71, 5951–5958 CrossRef CAS PubMed;
(f) B. Gabriele, R. Mancuso, G. Salerno and M. Costa, Chem. Commun., 2003, 486–487 RSC;
(g) H. Alper and F. W. Hartstock, J. Chem. Soc., Chem. Commun., 1985, 1141–1142 RSC;
(h) W. Tam, J. Org. Chem., 1986, 51, 2977–2981 CrossRef CAS;
(i) B. Gabriele, R. Mancuso, G. Salerno and M. Costa, J. Org. Chem., 2003, 68, 601–604 CrossRef CAS PubMed;
(j) B. Gabriele, G. Salerno, D. Brindisi, M. Costa and G. P. Chiusoli, Org. Lett., 2000, 2, 625–627 CrossRef CAS PubMed;
(k) Z.-H. Guan, H. Lei, M. Chen, Z.-H. Ren, Y. Bai and Y.-Y. Wang, Adv. Synth. Catal., 2012, 354, 489–496 CrossRef CAS.
-
(a) S. Fukuoka, M. Chono and M. Kohno, J. Chem. Soc., Chem. Commun., 1984, 399–400 RSC;
(b) S. Fukuoka, M. Chono and M. Kohno, J. Org. Chem., 1984, 49, 1458–1460 CrossRef CAS.
- A. A. Kelkar, D. S. Kolhe, S. Kanagasabapathy and R. V. Chaudhari, Ind. Eng. Chem. Res., 1992, 31, 172–176 CrossRef CAS.
- P. Toochinda and S. S. C. Chuang, Ind. Eng. Chem. Res., 2004, 43, 1192–1199 CrossRef CAS.
- F. Shi, J. Peng and Y. Deng, J. Catal., 2003, 219, 372–375 CrossRef CAS.
-
(a) S. P. Gupte and R. V. Chaudhari, J. Catal., 1988, 114, 246–258 CrossRef CAS;
(b) S. P. Gupte and R. V. Chaudhari, Ind. Eng. Chem. Res., 1992, 31, 2069–2074 CrossRef CAS.
- M. R. Didgikar, D. Roy, S. P. Gupte, S. S. Joshi and R. V. Chaudhari, Ind. Eng. Chem. Res., 2010, 49, 1027–1032 CrossRef CAS.
- F. Shi, Y. Deng, T. SiMa and H. Yang, Tetrahedron Lett., 2001, 42, 2161–2163 CrossRef CAS.
- M. R. Didgikar, S. S. Joshi, S. P. Gupte, M. M. Diwakar, R. M. Deshpande and R. V. Chaudhari, J. Mol. Catal. A: Chem., 2011, 334, 20–28 CrossRef CAS PubMed.
- B. Gabriele, R. Mancuso, G. Salerno and M. Costa, Top. Organomet. Chem., 2006, 18, 239–271 CrossRef CAS.
- D. J. Diaz, A. K. Darko and L. McElwee-White, Eur. J. Org. Chem., 2007, 4453–4465 CrossRef CAS.
- F. Ragaini, Dalton Trans., 2009, 6251–6266 RSC.
- F. Li and C. Xia, J. Catal., 2004, 227, 542–546 CrossRef CAS PubMed.
-
(a) B. Gabriele, R. Mancuso, V. Maltese, L. Veltri and G. Salerno, J. Org. Chem., 2012, 77, 8657–8668 CrossRef CAS PubMed;
(b) D. N. Briggs, G. Bong, E. Leong, K. Oei, G. Lestari and A. T. Bell, J. Catal., 2010, 276, 215–228 CrossRef CAS PubMed;
(c) X. Yang, J. Han, Z. Du, H. Yuan, F. Jin and Y. Wu, Catal. Commun., 2010, 11, 643–646 CrossRef CAS PubMed;
(d) B. Gabriele, R. Mancuso, G. Salerno, L. Veltri, M. Costa and A. Dibenedetto, ChemSusChem, 2011, 4, 1778–1786 CrossRef CAS PubMed;
(e) J. Hu, J. Li, Y. Gu, Z. Guan, W. Mo, Y. Ni, T. Li and G. Li, Appl. Catal., A, 2010, 386, 188–193 CrossRef CAS PubMed;
(f) J. Hu, Y. Gu, Z. Guan, J. Li, W. Mo, T. Li and G. Li, ChemSusChem, 2011, 4, 1767–1772 CrossRef CAS PubMed;
(g) F. Doro, P. Winnertz, W. Leitner, A. Prokofieva and T. E. Muller, Green Chem., 2011, 13, 292–295 RSC;
(h) D. M. Pearson, N. R. Conley and R. M. Waymouth, Adv. Synth. Catal., 2011, 353, 3007–3013 CrossRef CAS.
-
(a) H. Zhang, R. Shi, P. Gan, C. Liu, A. Ding, Q. Wang and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 5204–5207 CrossRef CAS PubMed;
(b) V. Rajeshkumar, T.-H. Lee and S.-C. Chuang, Org. Lett., 2013, 15, 1468–1471 CrossRef CAS PubMed;
(c) H. Zhang, D. Liu, C. Chen, C. Liu and A. Lei, Chem.–Eur. J., 2011, 17, 9581–9585 CrossRef CAS PubMed;
(d) R. Lang, L. Shi, D. Li, C. Xia and F. Li, Org. Lett., 2012, 14, 4130–4133 CrossRef CAS PubMed;
(e) J. W. Wrigglesworth, B. Cox, G. C. Lloyd-Jones and K. I. Booker-Milburn, Org. Lett., 2011, 13, 5326–5329 CrossRef CAS PubMed;
(f) X.-F. Wu, H. Neumann and M. Beller, Chem.–Asian J., 2012, 7, 282–285 CrossRef CAS PubMed;
(g) A. Behn, J. Zakzeski, M. Head-Gordon and A. T. Bell, J. Mol. Catal. A: Chem., 2012, 361–362, 91–97 CrossRef CAS PubMed;
(h) A. R. Elman, Tetrahedron Lett., 2013, 54, 5527–5531 CrossRef CAS PubMed;
(i) B. R. Sarkar and R. V. Chaudhari, Catal. Today, 2012, 198, 154–173 CrossRef CAS PubMed;
(j) Q. Liu, G. Li, J. He, J. Liu, P. Li and A. Lei, Angew. Chem., Int. Ed., 2010, 49, 3371–3374 CrossRef CAS PubMed;
(k) H. Chen, C. Cai, X. Liu, X. Li and H. Jiang, Chem. Commun., 2011, 47, 12224–12226 RSC;
(l) P. Xie, Y. Xie, B. Qian, H. Zhou, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 9902–9905 CrossRef CAS PubMed.
-
(a) P. Mathur, R. K. Joshi, B. Jha, A. K. Singha and S. M. Mobin, J. Organomet. Chem., 2010, 695, 2687–2694 CrossRef CAS PubMed;
(b) B. El Ali, A. El-Ghanam, M. Fettouhi and J. Tijani, Tetrahedron Lett., 2000, 41, 5761–5764 CrossRef CAS;
(c) Y. Izawa, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2004, 77, 2033–2045 CrossRef CAS;
(d) B. Gabriele, G. Salerno, L. Veltri and M. Costa, J. Organomet. Chem., 2001, 622, 84–88 CrossRef CAS.
- M.-Z. Cai, C.-S. Song and X. Huang, Synth. Commun., 1997, 27, 361–366 CrossRef.
- M. Cai, Y. Huang, R. Hu and C. Song, J. Mol. Catal. A: Chem., 2004, 212, 151–154 CrossRef CAS PubMed.
- M. Cai, Y. Huang, R. Hu and C. Song, J. Mol. Catal. A: Chem., 2004, 208, 17–20 CrossRef CAS.
- M. Cai, H. Zhao and Y. Huang, J. Mol. Catal. A: Chem., 2005, 238, 41–45 CrossRef CAS PubMed.
- C. Csajági, B. Borcsek, K. Niesz, I. Kovacs, Z. Szekelyhidi, Z. Bajko, L. Urge and F. Darvas, Org. Lett., 2008, 10, 1589–1592 CrossRef PubMed.
- J. Salvadori, E. Balducci, S. Zaza, E. Petricci and M. Taddei, J. Org. Chem., 2010, 75, 1841–1847 CrossRef CAS PubMed.
- P. J. Tambade, Y. P. Patil, M. J. Bhanushali and B. M. Bhanage, Tetrahedron Lett., 2008, 49, 2221–2224 CrossRef CAS PubMed.
- F. Zhao, B. M. Bhanage, M. Shirai and M. Arai, Chem.–Eur. J., 2000, 6, 843–848 CrossRef CAS.
-
(a) M. V. Khedkar, S. R. Khan, D. N. Sawant, D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2011, 353, 3415 CrossRef CAS;
(b) M. V. Khedkar, S. R. Khan, K. P. Dhake and B. M. Bhanage, Synthesis, 2012, 44, 2623 CrossRef CAS PubMed.
-
(a) J. Byun and Y. Lee, Tetrahedron Lett., 2004, 45, 1837 CrossRef CAS PubMed;
(b) J. Kim, B. Jun, J. Byun and Y. Lee, Tetrahedron Lett., 2004, 45, 5827 CrossRef CAS PubMed.
- Z. S. Qureshi, S. A. Revankar, M. V. Khedkar and B. M. Bhanage, Catal. Today, 2012, 198, 148–153 CrossRef CAS PubMed.
- M. V. Khedkar, T. Sasaki and B. M. Bhanage, ACS Catal., 2013, 3, 287–293 CrossRef CAS.
- T. T. Dang, Y. Zhu, J. S. Y. Ngiam, S. C. Ghosh, A. Chen and A. M. Seayad, ACS Catal., 2013, 3, 1406–1410 CrossRef CAS.
- N. P. Reddy, M. L. Kantam and B. M. Choudary, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1989, 28, 105 Search PubMed.
- M.-Z. Cai, C.-S. Song and X. Huang, J. Chem. Soc., Perkin Trans. 1, 1997, 2273–2274 RSC.
- C. Ramesh, R. Nakamura, Y. Kubota, M. Miwa and Y. Sugi, Synthesis, 2003, 4, 501–504 Search PubMed.
- J. Liu, J. Chen and C. Xia, J. Catal., 2008, 253, 50–56 CrossRef CAS PubMed.
- I. P. Beletskaya and O. G. Ganina, React. Kinet., Mech. Catal., 2010, 99, 1–4 CAS.
- S. T. Gadge, M. V. Khedkar, S. R. Lanke and B. M. Bhanage, Adv. Synth. Catal., 2012, 354, 2049–2056 CrossRef CAS.
- S. T. Gadge and B. M. Bhanage, Synlett, 2013, 24, 0981–0986 CrossRef CAS PubMed.
-
(a) T. Ishiyama, H. Kizaki, T. Hayashi, A. Suzuki and N. Miyaura, J. Org. Chem., 1998, 63, 4726–4731 CrossRef CAS;
(b) S. Shi and Y. Zhang, Synlett, 2007, 1843 CAS;
(c) P.-H. Li and L. Wang, Adv. Synth.
Catal., 2006, 348–681 Search PubMed.
-
(a) M. S. M. Ahmed and A. Mori, Org. Lett., 2003, 5, 3057–3060 CrossRef CAS PubMed;
(b) S. K. Kang, K. H. Lim, P. S. Ho and W. Y. Kim, Synthesis, 1997, 874 CrossRef CAS PubMed;
(c) T. Kobayashi and M. J. Tanaka, J. Chem. Soc., Chem. Commun., 1981, 333 RSC;
(d) S. Bonnaire, J. F. Carpentier, A. Mortreux and Y. Castanet, Tetrahedron Lett., 2001, 42, 3689–3691 CrossRef;
(e) E. Maerten, F. Hassouna, S. Bonnaire, A. Mortreux, J. F. Carpentier and Y. Castanet, Synlett, 2003, 12, 1874–1876 Search PubMed.
- P. J. Tambade, Y. P. Patil, A. G. Panda and B. M. Bhanage, Eur. J. Org. Chem., 2009, 3022–3025 CrossRef CAS.
- M. V. Khedkar, P. J. Tambade, Z. S. Qureshi and B. M. Bhanage, Eur. J. Org. Chem., 2010, 6981–6986 CrossRef CAS.
- Z. Qureshi, K. M. Deshmukh, P. J. Tambade and B. M. Bhanage, Synthesis, 2011, 2, 243–250 Search PubMed.
- M. V. Khedkar, T. Sasaki and B. M. Bhanage, RSC Adv., 2013, 3, 7791–7797 RSC.
- H. Li, M. Yang, Y. Qi and J. Xue, Eur. J. Org. Chem., 2011, 2662–2667 CrossRef.
- M. Cai, J. Peng, W. Hao and G. Ding, Green Chem., 2011, 13, 190–196 RSC.
- J.-R. Niu, X. Huo, F.-W. Zhang, H.-B. Wang, P. Zhao, W.-Q. Hu, J. Ma and R. Li, ChemCatChem, 2013, 5, 349–354 CrossRef CAS.
- J. Liu, J. Chen and C. Xia, J. Catal., 2008, 253, 50–56 CrossRef CAS PubMed.
- J. Liu, X. Peng, W. Sun, Y. Zhao and C. Xia, Org. Lett., 2008, 10, 3933–3936 CrossRef CAS PubMed.
- Y. Wang, J. Liu and C. Xia, Tetrahedron Lett., 2011, 52, 1587–1591 CrossRef CAS PubMed.
-
(a) W. Hao, J. Sha, S. Sheng and M. Cai, J. Mol. Catal. A: Chem., 2009, 298, 94–98 CrossRef CAS PubMed;
(b) M. Genelot, V. Dufaud and L. Djakovitch, Adv. Synth. Catal., 2013, 355, 2604–2616 CrossRef CAS.
-
(a) S. J. Rhoads and R. E. Michel, J. Am. Chem. Soc., 1963, 85, 585–591 CrossRef CAS;
(b) E. P. Burrows and D. H. Rosenblatt, J. Org. Chem., 1982, 47, 892–893 CrossRef CAS;
(c) R. Peters, M. Althaus and A.-L. Nagy, Org. Biomol. Chem., 2006, 4, 498–509 RSC;
(d) F. A. Cotton, C. Y. Liu, C. A. Murillo, D. Villagran and X. Wang, J. Am. Chem. Soc., 2003, 125, 13564–13575 CrossRef CAS PubMed.
- J. T. Chen and A. Shen, J. Am. Chem. Soc., 1984, 106, 1506 CrossRef CAS.
- T. Kobayashi and M. Tanaka, J. Organomet. Chem., 1982, 233, C64–C66 CrossRef CAS.
- F. Ozawa, H. Soyama, T. Yamamoto and A. Yamamoto, Tetrahedron Lett., 1982, 23, 3383–3386 CrossRef CAS.
-
(a) F. Ozawa, H. Soyama, H. Yanagihara, I. Aoyama, H. Takino, K. Izawa, T. Yamamoto and A. Yamamoto, J. Am. Chem. Soc., 1985, 107, 3235–3245 CrossRef CAS;
(b) F. Ozawa, H. Yanagihara and A. Yamamoto, J. Org. Chem., 1986, 51, 415–417 CrossRef CAS.
- T. Satoh, K. Kokubo, M. Miura and M. Nomura, Organometallics, 1994, 13, 4431 CrossRef CAS.
- Y.-Y. Yan, H.-P. Zuo and Z.-L. Jin, React. Funct. Polym., 1997, 32, 21–24 CrossRef CAS.
- S. Couve-Bonnaire, J.-F. Carpentier, Y. Castanet and A. Mortreux, Tetrahedron Lett., 1999, 40, 3717–3718 CrossRef CAS.
- Y. Uozumi, T. Arii and T. Watanabe, J. Org. Chem., 2001, 66, 5272–5274 CrossRef CAS.
- N. Tsukada, Y. Ohba and Y. Inoue, J. Organomet. Chem., 2003, 687, 436–443 CrossRef CAS PubMed.
- M. Iizuka and Y. Kondo, Chem. Commun., 2006, 1739–1741 RSC.
- Md. T. Rahman, T. Fukuyama, N. Kamata, M. Sato and I. Ryu, Chem. Commun., 2006, 2236–2238 RSC.
- E. R. Murphy, J. R. Martinelli, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 1734–1737 CrossRef CAS PubMed.
- C. Csajági, G. Szatzker, E. R. Toke, L. Ürge, F. Darvas and L. Poppe, Tetrahedron: Asymmetry, 2008, 19, 237–246 CrossRef PubMed.
- J. Balogh, Á. Kuik, L. Ürge, F. Darvas, J. Bakos and R. Skoda-Földes, J. Mol. Catal. A: Chem., 2009, 302, 76–79 CrossRef CAS PubMed.
- J. Liu, S. Zheng, W. Sun and C. Xia, Chin. J. Chem., 2009, 27, 623–627 CrossRef CAS.
- D. U. Nielsen, K. Neumann, R. H. Taaning, A. T. Lindhardt, A. Modvig and T. Skrydstrup, J. Org. Chem., 2012, 77, 6155–6165 CrossRef CAS PubMed.
- M. Genelot, N. Villandier, A. Bendjeriou, P. Jaithong, L. Djakovitch and V. Dufaud, Catal. Sci. Technol., 2012, 2, 1886–1893 CAS.
- V. de la Fuente, C. Godard, E. Zangrando, C. Claver and S. Castillo'n, Chem. Commun., 2012, 48, 1695–1697 RSC.
- M. Papp and R. Skoda-Földes, J. Mol. Catal. A: Chem., 2013, 378, 193–199 CrossRef CAS PubMed.
- J. Tsuji and N. Iwamoto, Chem Commun., 1966, 380 RSC.
- I. Pri-Bar and H. Alper, Can. J. Chem., 1990, 68, 1544–1547 CrossRef CAS.
- K. Hiwatari, Y. Kayaki, K. Okita, T. Ukai, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2004, 77, 2237–2250 CrossRef CAS.
-
(a) S.-l. Murahashi, Y. Mitsue and K. Ike, J. Chem. Soc., Chem. Commun., 1987, 125–127 RSC;
(b) Y. Imada, Y. Mitsue, K. Ike, K.-I. Washizuka and S.-I. Murahashi, Bull. Chem. Soc. Jpn., 1996, 69, 2079–2090 CrossRef CAS.
- S. T. Gadge and B. M. Bhanage, J. Org. Chem., 2013, 78, 6793–6797 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.