Oxygen delignification of conventional and high alkali cooked softwood Kraft pulps, and study of the residual lignin structure†
Vahid Jafari*a,
Sara R. Labafzadehc,
Alistair Kingc,
Ilkka Kilpeläinenc,
Herbert Sixtaa and
Adriaan van Heiningenab
aDepartment of Forest Products Technology, Aalto University, FI-00076 Aalto, Finland
bDepartment of Chemical and Biological Engineering, University of Maine, 5737 Jenness Hall, Orono, ME 04469-5737, USA
cLaboratory of Organic Chemistry, Department of Chemistry, University of Helsinki, P.O. Box 55, FIN-00014, Helsinki, Finland
First published on 25th March 2014
Abstract
The selection of the optimum residual pulp lignin content in Kraft cooking has been the focus of many investigations in order to improve both the pulp yield and viscosity. Kraft cooking at a high alkali concentration (HA-Kraft) results in a higher yield compared to conventional Kraft pulping. Furthermore, the (carbohydrates yield/delignification) selectivity of oxygen delignification (O-delignification) below the fiber liberation point to a fully bleachable pulp is more selective than that of Kraft cooking. In order to obtain a high yield and preserve pulp quality, pine HA-Kraft and conventionally prepared Kraft (Ref-Kraft) pulps with kappa numbers 60 and 80 were subjected to O-delignification, during which constant but low-caustic and high-dissolved oxygen concentrations were secured by means of a flow-through (FT) reactor. The effects of operational conditions were investigated at high dissolved oxygen concentration. In addition, the chemical structure of the residual lignin of the pulps before and after O-delignification was investigated by 31P NMR. It showed that the carboxylic acid content of the residual lignin increased by 50–200% during oxygen delignification. 31P NMR data also indicated that the carboxylic acid content inside the residual lignin of oxygen-delignified pulps from Ref-Kraft samples was higher (20–70%) than that of HA-Kraft pulps. The results also demonstrated that the guaiacyl phenolic group content in the kappa range of 60 is higher compared to kappa 80 regardless of the cooking procedure that was utilized.
1. Introduction
The removal of lignin during Kraft cooking is accompanied by a severe loss of carbohydrates, primarily hemicelluloses. For example, it has been reported that Kraft pulping of Scots Pine to the bleachable grade pulp results in galactoglucomannan (GGM) losses of 75%, while the 4-O-methylglucuronoarabinoxylan (AX) and cellulose losses are 38% and 10%, respectively.1In another study, Paananen et al.2 showed that the GGM yield increased by 2.5% on oven-dry3 wood (odw) when Kraft cooking was carried out at a NaOH concentration of 1.55 mol l−1 (HA-Kraft) which is three times higher than 0.5 mol l−1 (Ref-Kraft) used in conventional Kraft cooking. The higher GGM yield can be explained by an increased stopping reaction rate at higher alkali concentration relative to the peeling reaction rate.2
Unfortunately the dissolution of AX increases with increasing alkali concentration so that at the end of a softwood Kraft cook (to 25 to 30 pulp kappa number) the overall carbohydrates yield during HA-Kraft cooking remains the same as that of Ref-Kraft cooking.2,4 However, some part of removed carbohydrates, particularly AX does not contribute into the organic acids formation and dissolved in the polymeric forms that can be retained on the fiber at the final part of cooking stage and later in O-delignification.4 Carbohydrate degradation occur in particular during the initial and continues towards the end of Kraft cook.5 The oxygen–alkali process (O-delignification) is known to be more selective (carbohydrates yield/delignification) than final phase of Kraft cooking, especially when performed at a high dissolved oxygen concentration and low but constant alkalinity.6,7 Thus, if a higher degree of delignification could be achieved with O-delignification, softwood Kraft cooking may be stopped at a higher target kappa, for example 40–80, in order to preserve more carbohydrates when oxygen delignification is continued to a kappa number of about 15 (the kappa target entering the bleach plan). In addition, it may be possible to re-precipitation a significant fraction of the AX dissolved during HA-Kraft pulping on the fiber surface during O-delignification thereby further increasing the pulp yield.
It is well known that the chemical structure of the residual lignin is significantly affected by cooking conditions during Kraft pulping as well as by the target kappa number.8–10
Softwood Kraft pulp lignin consists of two types of lignin monomers, almost exclusively p-guaiacyl (%G) and small amount of hydroxyphenyl (% H). The model compounds investigations indicate that the removal potential for guaiacol groups is higher than their p-hydroxyphenyl counterparts during O-delignification.11,12
It was reported that, during O-delignification the phenolic groups of Kraft lignin were degraded by 41–60%. However, the degree of removal of condensed lignin is limited to only 4–29%.13–15. Another factor that contributes to lignin removal is the amount of carboxylic acid groups contained by lignin. It has been demonstrated in a study by Sun and Argyropoulos that the lignin solubilization in an oxygen–alkali stage increases at a higher level of carboxylic acid groups content.15 Besides the three-dimensional structure of lignin and content of different groups, the initial lignin content of pulp also has an effect on bleachability. High kappa number pulp is easier to delignify compared to a low kappa pulp due to more phenolic groups.5 The reactivity of residual lignin during O-delignification is also decreased due to increased bonding to hemicelluloses, as lignin–carbohydrates complexes (LCC),16,17 particularly glucomannan. The latter is more resistant to degradation and thus increases in relative content at higher degree of oxygen delignification. The elucidation of structural details of the residual lignin is therefore important for further improvements in Kraft pulping technology. Therefore the objectives of the present work are two-fold; firstly, the evaluation of O-delignification behavior of HA-Kraft and Ref-Kraft pulps in terms of yield and pulp properties, and secondly the elucidation of the chemical structure of the residual lignin isolated from the pulps before and after O-delignification by acidolysis by means of 31P NMR spectroscopy.18
2. Experimental section
2.1. Pulping
Laboratory cooking trials were conducted in a 30 l displacement digester. Approximately 4 kg of oven-dry screened pine (Pinus sylvestris) chips were placed in the digester. The digester was connected to a tank farm consisting of separate vessels for the impregnation and the cooking liquors. The alkali concentration i.e. effective alkali, the sulfidity, and the temperature of both liquors were adjusted in both liquor tanks to the targeted conditions in the digester. Both the impregnation and cooking liquors were pumped through the digester from the bottom to the top and followed the targeted cooking schedule. Together with a high liquor-to-wood ratio i.e. ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the concentration profile of the active cooking chemicals was kept fairly constant. The alkali concentration of the reference or low alkalinity (LA) cook was 0.5 mol l−1, and the sulfidity was 33%. For the high alkalinity (HA) Kraft cook, the alkali concentration was adjusted to 1.55 mol l−1, while the sulfidity remained unchanged at 33%. The alkali concentration of the impregnation liquor of HA Kraft cooking was adjusted to 2.0 mol l−1 to ensure a minimum hydroxide ion concentration of 1.55 mol l−1 throughout the cooking. The target kappa numbers of approximately 60 and 80 were adjusted by H-factor control. The cooking conditions and resulting pulp properties are summarized in Table 1.
:1, the concentration profile of the active cooking chemicals was kept fairly constant. The alkali concentration of the reference or low alkalinity (LA) cook was 0.5 mol l−1, and the sulfidity was 33%. For the high alkalinity (HA) Kraft cook, the alkali concentration was adjusted to 1.55 mol l−1, while the sulfidity remained unchanged at 33%. The alkali concentration of the impregnation liquor of HA Kraft cooking was adjusted to 2.0 mol l−1 to ensure a minimum hydroxide ion concentration of 1.55 mol l−1 throughout the cooking. The target kappa numbers of approximately 60 and 80 were adjusted by H-factor control. The cooking conditions and resulting pulp properties are summarized in Table 1.
| Pulp | H-factor | T (°C) | Time (min) | Alkalinity (%) [OH−] mol l−1 | Yield on wood (%) | Reject from screening (%) | Kappa | Viscosity (mL g−1) | Lignin (%) | Cella (%) | GGM (%) | AX (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Cellulose, initial amount of wood components is: lignin = 26.6%, cell = 42.5%, GGM = 16.9% and AX = 8.2%. | ||||||||||||
| HA80 | 262 | 155 | 55 | 2 | 58 | 0.22 | 79 | 1310 | 6.9 | 41.6 | 5.8 | 3.8 |
| LA80 | 650 | 165 | 50 | 0.5 | 58 | 0.35 | 79 | 1477 | 6.8 | 41.1 | 5.2 | 5.0 |
| HA60 | 310 | 150 | 110 | 2 | 52 | 2.4 | 64 | 1300 | 5.1 | 38.1 | 5.7 | 3.1 |
| LA60 | 1199 | 160 | 180 | 0.5 | 52 | 0.06 | 56 | 1430 | 4.7 | 38.6 | 4.4 | 4.2 |
2.2. O-delignification
O-delignification was carried out in an FT reactor (commercially called Berty reactor) with 280 ml nominal volume containing a 100 ml stationary basket which holds the pulp suspension in a medium consistency (10–12% od pulp in the fibrous suspension). A constant flow of an aqueous solution containing 1–4 g NaOH per l is fed to the reactor continuously, thus the pH value is always constant at 12.5–13. The solution is pre-saturated with oxygen at room temperature and 10 bar. During an experiment, the oxygenated solution is pushed through a preheater into the reactor at 90–100 °C using a pressure of 20 bar to avoid the degassing of the solution at the reaction temperature.19 The reactor operation was validated by showing that the O-delignification kinetics are not affected by the amount of pulp inside the basket (3–5 g od pulp) or the mixer speed (≥1200 rpm), which forces the flow through the basket and creates uniform conditions within the reactor and the feed flow rate (70–130 ml min−1).Therefore, the stirring speed, the weight of the pulp, and the flow rate were kept constant at 1200 rpm, 4 g, and 100 ml min−1, respectively. The process scheme of the FT reactor setup is shown in Fig. 1.
 | ||
| Fig. 1 Process flow schematic for the flow-through reactor. | ||
The UV absorption at 280 nm in a downstream flow cell is used for the quantification of the dissolved lignin since absorption at this wavelength is high and not affected by the presence of sodium hydroxide. An extinction coefficient of 22 l g−1 cm−1 was used based on Indulin AT as the reference lignin material in order to better predict the experimentally determined final kappa number for different initial kappa numbers range of ∼60–80. Each reaction was repeated three times and the results (kappa, viscosity, delignification rates) indicate the satisfying reproducibility, with standard deviation in the 4–8% range. The average error in the prediction of the final kappa number based on this extinction coefficient is ±1.5 kappa units.
2.3. Data reduction for FT-reactor
The rate of delignification, r(t), in mg lignin per g pulp per min, can be obtained from a mass balance for the dissolved lignin concentration C(t) within the reactor boundaries, according to eqn (1) and (2)| Inflow − outflow + dissolved = accumulated |
| φv × C(t + td) × dt + r(t) × mp × dt = VrdC(t + td) | (1) |
 | (2) |
The amount of lignin removed is calculated by mathematical integration from eqn (2) as:
 | (3) |
 | (4) |
The value of 0.15 approximately relates the kappa number and lignin content in the softwood Kraft pulp.6
2.4. Woodchips and pulp analysis
The kappa number and viscosity of the treated pulps were measured based on Scan-C 1:00 and Scan-CM 15:99, respectively. Pulps with a kappa number higher than 35 were subjected to chlorite treatment before viscosity determination (5 g pulp in 200 ml water + 5 g NaClO2 + 2 ml acetic acid at 70 °C for 5 min).20 Anion exchange chromatography, Dionex HPAEC was used to quantify the sugar and lignin content of the woodchips, pulps and isolated lignin based on NREL/TP-510-42618 procedure. The polysaccharide composition can be determined from the monosaccharide quantities according to Janson.212.5. Lignin isolation and purification
Pulp samples at different kappa numbers before and after O-delignification in the reference conditions (2.2 g NaOH per l, 95 °C and 10 bar) subjected to acetone extraction (SCAN-CM 49:03), with a liquid to solid ratio (L/S) of 10. Lignin was isolated using multistep acidolysis according to Evtuguin et al.'s approach.22 The purity and degraded carbohydrates in the lignin sample are listed in Table 2. The purity of lignin is defined as a percentage of lignin in the isolated lignin sample and measured in the same way that the pulp sample compounds were determined. The yield of lignin isolation is also reported in Table 2, where the initial kappa was compared with the kappa value of the sample after acidolysis treatment.| HA64 | O2-HA64 | LA56 | O2-LA56 | HA80 | O2-HA80 | LA80 | O2-LA80 | |
|---|---|---|---|---|---|---|---|---|
| Yield (in pulp lignin %) | 65 | 61 | 70 | 65 | 68 | 63 | 61 | 63 |
| Lignin average (in lignin sample %) | 96 | 94 | 97 | 96 | 93 | 94.8 | 94 | 95.2 |
| Total carbohydrate average (in lignin sample %) | 4 | 6 | 3 | 4 | 7 | 5.2 | 6 | 4.8 |
2.6. 31P NMR measurement for lignin characterization
Lignin samples were analyzed using quantitative 31P NMR spectroscopy on a Varian Unity INOVA 600 spectrometer (600 MHz proton frequency), which was equipped with a 5 mm direct detection broadband probe-head at 27 °C. The quantitative 31P NMR spectra were collected with 1000 transients, a 75° pulse flip angel, 5 seconds relaxation delay, and 1 s acquisition time.The samples were prepared using a modified technique based on previously published methods.23,24 Pyridine (100 μl) and chloroform (500 μl) were added to 25 mg of lignin, and the mixture was agitated. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP, 100 μl, 0.63 mmol) was added and vortex-mixed until it was completely dissolved. Finally, endo-N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide solution (e-HNDI, 200 μl, 121.5 mM in Pyr–CDCl3/3:2, 0.0243 mmol) and Cr(acac)3 (500 μl, 0.08 M in CDCl3, 0.04 mmol) were added to serve as the internal standard and relaxation agent, respectively. The 31P NMR spectra were recorded with 700 μl samples in a 5 mm diameter NMR tube. The spectra were calibrated with TMDP-anhydride at 132.2 ppm, and the amounts of the different functional groups were calculated from their integration values against the phosphitylated internal standard.
This procedure was used for isolated lignin from pulps with kappa number 60. Since lignins from kappa 80 pulps were found to be only partially soluble in the pyridine–CDCl3–2-Cl-TMDP system, a new solvent was used in this effort. Lignin (25 mg) was heated in N,N-dimethyl acetamide (DMA, 250 μl) for 10 min at 100 °C. Pyridine (100 μl) and CDCl3 (500 μl) were then added, and the samples were phosphitylated with 2-Cl-TMDP (100 μl). Finally, e-HNDI (200 μl) and Cr(acac)3 (500 μl) were added, the mixture agitated and the 31P NMR spectra collected.
3. Results and discussion
3.1. O-delignification of kappa number 80 and 60 pulp
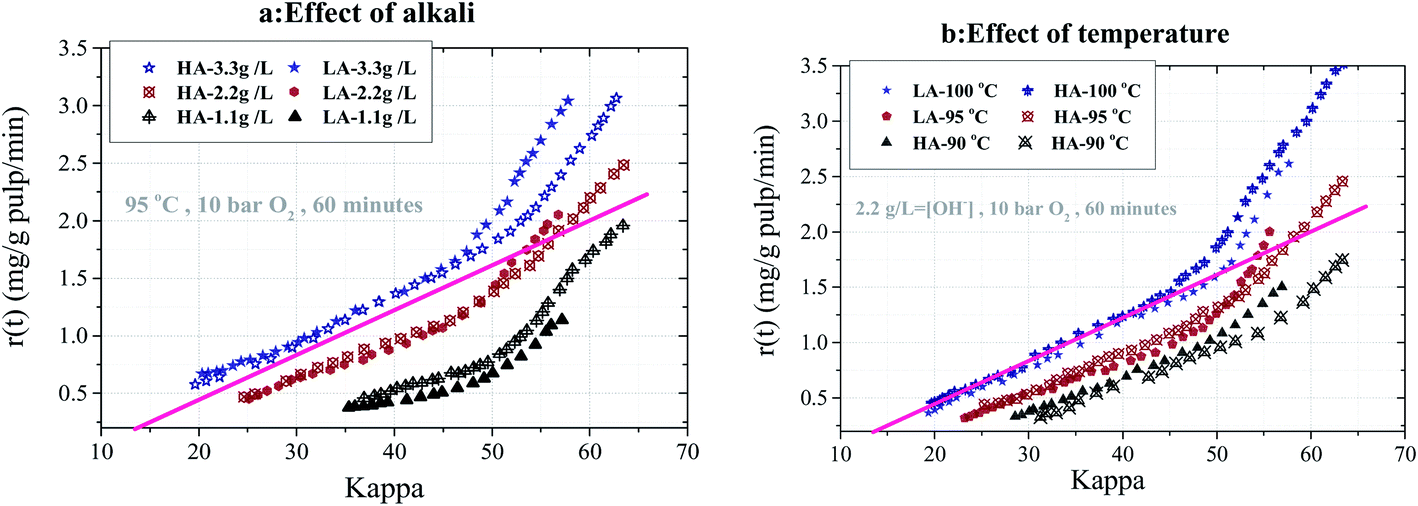
HA and LA Kraft pulps with a kappa number of approximately 80 and 60 were oxygen-delignified in the flow through reactor for 60 minutes. The experiments were carried out at different alkali concentrations (1.1, 2.2 and 3.3 g l−1) at 95 °C and (90, 95, 100 °C) at 2.2 g l−1 NaOH, where the oxygen pressure was kept constant at 10 bar (room temperature). The development of the delignification rate for kappa 80 pulps is shown in Fig. 2a and b, respectively. It is evident that a significantly higher delignification rate can be achieved at a higher alkali charge or higher temperature. The results also show that the rate of delignification at the same kappa number is lower for the HA pulp compared to the LA pulp. | ||
| Fig. 2 Delignification rate vs. kappa for HA80 and LA80 pulp at (a) different alkali concentrations at 95 °C and (b) at different temperatures at 2.2 g l−1 NaOH. | ||
The rate difference increases at milder O-delignification conditions, such as at lower temperature and alkali concentration, for example. Another interesting observation is that, with the exception of the highest concentration (3.3 g l−1 NaOH in Fig. 2a) and the highest temperature (100 °C in Fig. 2b), the delignification rate displays two phases. The initial rapid phase is followed by a second slower phase where the delignification rate decreases roughly linearly with the decreasing kappa number. It can also be observed that the initial delignification rate is not affected significantly by increasing temperature or alkali concentration above 95 °C and 2.2 g l−1 NaOH. One might speculate that the rate of reaction is not controlled by chemical kinetics but by diffusion since the oxygen supply at these conditions (about 40 mg min−1) is at least four times larger than the maximum oxygen requirement. The rate of oxygen consumption is estimated by multiplying the rate of delignification (in mg lignin per g pulp per min) by the pulp weight and the stoichiometric constant for the oxygen–lignin reaction (g O2 consumed per g lignin removed).7 The latter coefficient is taken from literature as 1.0 g g−1, which is in agreement with 0.14% oxygen consumption per kappa unit.25,26
Fig. 3a and b show the selectivity plots based on the measured kappa numbers and intrinsic viscosities of the final pulps. These samples were removed from the reactor after O-delignification of the HA80 and LA80 pulps at the different alkali concentrations and temperatures, respectively. The degree of delignification is less for the HA pulp, and the difference between delignification rate development increases with decreasing alkali concentration and temperature.
 | ||
| Fig. 3 Viscosity and kappa number development of HA80 and LA80 pulps at different operating conditions. | ||
This is in agreement with the results shown in Fig. 2. Fig. 3 reveals that the selectivity of O-delignification of the HA Kraft pulp is basically not dependent on the operating conditions. For the LA Kraft pulp, the selectivity of O-delignification is initially poor but improves significantly during the final delignification stages. Since the HA Kraft pulp contains less AX but more GGM compared to the LA Kraft pulp, a possible explanation for the slower delignification of the former may be related to the presence of lignin–carbohydrate complexes (LCCs), which impair delignification.27 Lawoko et al. reported that galactan–lignin structures in the fiber wall lead to poorer lignin accessibility.27,28 The results for HA and LA kappa 60 indicate that the differences in the delignification rate between the LA and HA pulps are generally small, which is contrary to what was observed for the HA80 and LA80 pulps in Fig. 2. Surprisingly, the initial delignification rates for the HA64 and LA56 pulps at 3.3 g l−1 NaOH shown in Fig. 4a are higher than the initial delignification rates of the 80 kappa pulps in Fig. 2 despite the lower lignin content of the former pulps.
 | ||
| Fig. 4 Delignification rate vs. kappa for HA64 and LA56 pulp at (a) different alkali concentrations at 95 °C and (b) at different temperatures at 2.2 g l−1 NaOH. | ||
The lower initial delignification rates for the kappa 80 pulps may be related to the poor accessibility of the lignin in these pulps, as proposed in the previous section. It is also noticeable that for HA 80, the effect of caustic concentration on the delignification rate is much larger than that of temperature. Finally, from Fig. 4a and b, it can be noted that during the slow delignification phase, the rates decrease linearly with decreasing kappa and extrapolate to a limiting kappa number of about 14. A similar behavior was also observed by Ji et al.29 However, in the latter study the final kappa could decreases further due to the lower starting kappa number (kappa 24).7
The results in Fig. 5 show that the delignification-cellulose degradation relationship is comparable for the HA64 and LA56 pulps. This is different from what was indicated by the kappa 80 pulps in Fig. 3, where the HA80 pulp initially demonstrated a much lower selectivity than the LA80 pulp. Therefore, for the kappa number level of about 60, there is no significant difference in rate and selectivity of O-delignification for the HA and LA pulps. Lawoko et al.28 found that most of the residual lignin in kappa-50 softwood Kraft pulp was linked to xylan, while most of the residual lignin in a kappa-20 softwood Kraft pulp, received by applying a higher H-factor, was linked to glucomannan. Similarly, upon subsequent O-delignification, a further relative increase in glucomannan-linked LCC was observed.
 | ||
| Fig. 5 Viscosity and kappa number of HA64 and LA56 pulps at different operating conditions. | ||
Based on these findings, it can be concluded that the significant difference in xylan and glucomannan content of the two pulps, HA64 and LA56, may not be related to different LCC contents.
3.2. Effect of initial kappa number on delignification
In Fig. 6, the delignification rate at 2.2 g l−1 NaOH and 95 °C of the pulps with the two different initial kappa number levels is plotted against the kappa numbers in the course of O-delignification. It can be noted that the delignification rates of kappa 60 and 80 pulps develop largely in the same manner. The delignification rates of the kappa number 80 pulps follow a parallel pattern until reaching the lowest kappa number, keeping the differences largely constant. This is different for the kappa number 60 pulps, where the delignification rates between the HA and the LA-Kraft pulps are quite comparable with a slight advantage for the HA-Kraft pulp at very low kappa. | ||
| Fig. 6 Delignification rate versus kappa for different HA and LA kappa pulps at 95 °C and 2.2 g l−1 NaOH. | ||
From the results, it can be concluded that the kappa number target of 15 after O-delignification can be achieved for the Kraft pulps with a kappa number of about 60, while this goal cannot be achieved for the kappa number 80 pulps. This raises the question about the structural changes in lignin during Kraft cooking as well as how the target kappa number facilitates dissolution of lignin during following O-delignification stages. Analysis by 31P NMR of isolated Kraft pulp lignins, as well as the analysis of samples after the oxygen stage, gives additional information for behavior of lignin towards oxygen–alkali stage.
3.3. Lignin structural information based on 31P NMR
Quantitative 31P NMR is used to characterize the structure of isolated lignins. Lignin samples were isolated from the pulps of different kappa number before and after oxygen delignification at reference operational conditions (2.2 g NaOH per l, 95 °C and 10 bar oxygen). The typical reactive groups present in lignin samples in this study (phenolic hydroxyls) are illustrated in Fig. 7, while Fig. 8 shows the 31P NMR spectra of HA60 and O-delignified HA60, which represents signal assignments according to previous studies.24,30 Further information concerning the NMR data can be found in the ESI.† | ||
| Fig. 7 Typical phenolic structures present in the lignin samples in this study. | ||
 | ||
| Fig. 8 31P NMR spectra of HA60 and HA60-OD (G: Guaiacyl phenolic OH, H: p-hydroxyphenolic OH). | ||
It is evident from Table 3 that the amount of carboxylate groups increased by 48–159% through O-delignification, depending on the pulp sample.
| Lignin sample | Carboxyl OHa | p-Hydroxy-phenolic OHb | Guaiacyl OHc | Total condensed phenolic OHd | 5–5′ condensed OHe | Aliphatic OHf | Total free phenolic OHg |
|---|---|---|---|---|---|---|---|
| a 133–137 ppm.b 137–138.6 ppm.c 138.6–140.2 ppm.d 140.2–145.2 ppm.e 140.2–141.4 ppm.f 145.2–151.4 ppm.g Sum of b, c & d. | |||||||
| HA60 | 0.58 | 0.21 | 1.25 | 1.32 | 0.4 | 2.97 | 2.78 |
| HA60-O | 0.86 | 0.24 | 0.75 | 1.20 | 0.57 | 2.77 | 2.19 |
| LA60 | 0.46 | 0.16 | 1.12 | 1.40 | 0.37 | 2.86 | 2.68 |
| LA60-O | 1.19 | 0.21 | 0.58 | 0.96 | 0.38 | 2.96 | 1.74 |
| HA80 | 0.43 | 0.15 | 0.96 | 0.95 | 0.30 | 3.08 | 2.06 |
| HA80-O | 0.79 | 0.17 | 0.58 | 0.86 | 0.35 | 3.03 | 1.62 |
| LA80 | 0.36 | 0.15 | 1.02 | 1.12 | 0.43 | 2.88 | 2.29 |
| LA80-O | 0.79 | 0.12 | 0.59 | 0.80 | 0.40 | 2.79 | 1.51 |
The magnitude of the increase is generally higher for the lignin samples isolated from the LA-Kraft pulp than for those isolated from the HA-Kraft pulp. On the contrary, the quantity of aliphatic hydroxyl groups decreased under O-delignification (Table 3). The degradation of condensed phenolic structures, and thus the formation of carboxylic acids, are known to be pathways for lignin dissolution during the oxygen–alkali stage, according to a previous study.31
The free phenolic hydroxyl groups attract more attention amongst all of the other functional groups due to their higher reactivity compared to non-phenolic structures. Early study suggested that a very low content of free phenolic groups represent a very low reactivity towards O-delignification.3 The reactivity of guaiacol groups is known to be higher than their p-hydroxyphenyl counterparts during O-delignification.11,12
Table 3 shows that the quantity of free phenolic hydroxyls within residual Kraft lignin decreased by 19–34% after O-delignification. In addition, the degree of elimination of such groups is higher in low alkali pulps than high alkali samples for both pulp samples, with kappa 60 and 80 (Fig. 9a and b). It should also be noted that the amount of these groups are higher in kappa 60 pulps than for kappa 80 pulps.
 | ||
| Fig. 9 The amounts (mmol g−1 of 100% lignin) of the various hydroxyls in lignin samples isolated from softwood Kraft pulp, with kappa number 60 and 80, before and after O-delignification. | ||
Amongst phenolic structures, non-condensed phenolic hydroxyls require more attention because the most significant changes in lignin structure are due to a loss of such groups. These moieties possess excellent reactivity towards O-delignification compared to condensed phenolics, and therefore their content decreased as delignification proceeded.32 As it is obvious from Table 3, Fig. 9a and b, guaiacyl phenolic hydroxyl groups decreased over a range of 39–48% for the analyzed lignins that is in agreement with the earlier studies.13–15 The magnitude of the decrease is also higher in low alkali pulps than for high alkali samples, regardless of the pulp kappa number.
It has been demonstrated that condensed phenolic units containing β-5, 4–O–5 and 5–5′ biphenyl structures are fairly resistant to oxidative degradation even at higher temperatures. Therefore, the degradation of such compounds is less than for non-condensed guaiacyl units.3 Our observations are in agreement with earlier studies showing that condensed structures decreased by 6–31% after O-delignification with the highest decrease for LA cooked pulps. 5–5′ Biphenolic moieties are the most unreactive units among the condensed structures. Table 2 shows that such structures basically accumulate during O-delignification.
The presence of p-hydroxyphenyl units was also quantified by 31P NMR. Our findings illustrate that these groups increase somewhat during delignification for all examined samples, while for the LA80 pulps, they decrease slightly. However, this is not very significant. In addition, these functional groups were found to be fairly stable under alkali–oxygen conditions.
A comparison of the two categories of pulps, such as 60 kappa and 80 kappa pulps, clearly indicate that there is a higher amount of total free phenolic units, especially guaiacyl structures in lignin for kappa 60 pulps compared to kappa 80 pulps. It indicates an increase in the reactive structure of residual lignin as the cook proceeds. One of the main fragmentation reactions in the cooking stage is the cleavage of the β-aryl ether linkage in β–O–4 structures. This reaction breaks down the lignin polymer by the simultaneous liberation of a new phenolic hydroxyl group and the generation of coniferyl alcohol. Gellerstedt et al. have shown that increased dissolution of cross-linked lignin groups takes place mainly at the end of the cooking process.14 The latter study indicates that cleavage of α-aryl ether structures still occurs, even at the end of pulping stage in the lignin fibers, but not necessarily with simultaneous lignin dissolution. It may be concluded that the interruption of cooking at kappa 80 impairs the fragmentation of lignin. Therefore, the variety of lignin structure remains intact or in larger polymeric forms, which makes it resistant to the oxygen–alkali reaction.
3.4. Effect of initial kappa number on lignin-free pulp yield (based on original wood)
Fig. 10 illustrates the lignin-free yield (based on original wood) versus kappa number at reaction conditions of 95 °C and 2.2 g l−1 NaOH. It can be noted that the lignin-free yield (i.e., carbohydrate yield) loss is significantly smaller for O-delignification than for Kraft cooking. The yield after O2-delignification was found to be slightly higher for the LA pulp compared to the corresponding HA pulp. Fig. 10 shows a 2.5% higher carbohydrate yield after O-delignification of the LA80 pulp compared to an oxygen-delignified LA60 pulp at a given kappa number. Thus, termination of cooking at high kappa numbers followed by extended O-delignification may substantially increase the lignin-free yield, due to smaller carbohydrate losses during O-delignification. | ||
| Fig. 10 Lignin-free yield versus kappa during O-delignification of different pulps cooked at HA and LA charge (95 °C, 2.2 g l−1 NaOH and 10 bar oxygen pressure for 60 min). | ||
However, the present results also show that it is very difficult to reach a bleachable grade pulp of kappa 15 (kappa that is introduced to the bleaching sequences) by O-delignification when starting at kappa numbers of about 80. As previously stated, this may be due to the interruption of cooking at a high kappa number that attributes to low lignin polymer fragmentation and thus the poor liberation of a new active phenolic hydroxyl group.
4. Conclusion
It was found that the rate of O-delignification for HA-80 is lower than that of a LA-80. The rate difference also increases at milder O-delignification conditions, i.e. lower temperature and alkali concentration. This difference between HA60 and LA60 pulps, however, was not observed. The results of the present study suggest that the delignification in the kappa 80 pulps is limited by both accessibility and the limited amount of free phenolic units in lignin compared to kappa 60 pulps. The lower amount of free phenolic groups is attributed to the interruption of cooking at a higher kappa number that, in turn results to lower polymeric lignin fragment masses. The lignin-free yield decrease is significantly smaller for O-delignification than for HA or LA Kraft pulping. However, owing to the lower selectivity (chain scission/removed kappa number) of O-delignification than Kraft cooking, the kappa number level entering an oxygen–alkali stage is not only selected by the yield advantage but is also limited by the minimal viscosity requirements for subsequence bleachable grade pulps of about 850 ml g−1. Our future focus will be centered on study of lignin structure during O-delignification of HK pulp, where the operational conditions are intensified for example by increasing temperature and using an appropriate cellulose-protecting additive. The higher temperature will allow for more efficient removal of the condensed lignin, while the presence of additive will maintain viscosity above the acceptable threshold value.Abbreviations
| GGM | Galactoglucomannan |
| AX | Arabinoxylan |
| OD | Oven-dry |
| HA | High alkalinity |
| LA | Low alkalinity |
| O-delignification | Oxygen delignification |
| FT reactor | Flow through reactor |
Acknowledgements
The authors would like to acknowledge the Finnish Bioeconomy Cluster Ltd (FIBIC) for financial support. We would also like to give special thanks to Terhi Toivari for their high-quality work to make the pulps.References
- E. Sjostrom, Tappi J., 1977, 60, 151–154 CAS.
- M. Paananen, T. Tamminen, K. Nieminen and H. Sixta, Holzforschung, 2010, 64, 683–692 CrossRef CAS.
- L. Akim, J. Colodette and D. S. Argyropoulos, Can. J. Chem., 2001, 79, 201–210 CAS.
- M. Paananen, T. Liitia and H. Sixta, Ind. Eng. Chem. Res., 2013, 52, 12777–12784 CrossRef CAS.
- B. Parsad, J. Gratzl, A. Kirkman, H. Jameel, T. Rost and V. Magnotta, Tappi J., 1994, 77, 135–147 CAS.
- A. van Heiningen and Y. Ji, Tappi J., 2012, 11, 9–18 Search PubMed.
- V. Jafari, H. Sixta and A. van Heiningen, Industrial & Engineering Chemistry Research Journal, 2014 Search PubMed , submitted.
- M. Backstrom and A. Jensen, Appita Annu. Gen. Conf. Proc., 1999, 53rd, 101–109 CAS.
- G. Gellerstedt, K. Gustafsson and R. A. Northey, Nord. Pulp Pap. Res. J., 1988, 3, 87–94 CAS.
- E. W. Rutkowska, P. Wollboldt, G. Zuckerstatter, H. K. Weber and H. Sixta, BioResources, 2009, 4, 172–193 CAS.
- S. Fu and L. Lucia, Ind. Eng. Chem. Res., 2003, 42, 4269–4276 CrossRef CAS.
- L. A. Lucia, A. J. Ragauskas and F. S. Chakar, Ind. Eng. Chem. Res., 2002, 41, 5171–5180 CrossRef CAS.
- G. Gellerstedt, K. Gustafsson and E. L. Lindfors, Nord. Pulp Pap. Res. J., 1986, 1, 14–17 CAS.
- G. Gellerstedt and E. L. Lindfors, Holzforschung, 1984, 38, 151–158 CrossRef CAS.
- Y. Sun and D. S. Argyropoulos, J. Pulp Pap. Sci., 1995, 21, J185–J190 CAS.
- S. Antonsson, M. E. Lindstroem and M. Ragnar, Nord. Pulp Pap. Res. J., 2003, 18, 388–394 CAS.
- H. Sixta, Book of Abstracts, 211th ACS National Meeting, New Orleans, LA, March 24–28 1996 Search PubMed.
- E. Johansson and S. Ljunggren, J. Wood Chem. Technol., 1994, 14, 507–525 CrossRef CAS.
- D. Tromans, Hydrometallurgy, 1998, 48, 327–342 CrossRef CAS.
- V. Jafari, H. Sixta and A. van Heiningen, Holzforschung, 2013 DOI:10.1515/hf-2013-0148.
- J. Janson, Pap. Puu, 1970, 52, 323–326 CAS , 328–329.
- D. V. Evtuguin, C. P. Neto, A. M. S. Silva, P. M. Domingues, F. M. L. Amado, D. Robert and O. Faix, J. Agric. Food Chem., 2001, 49, 4252–4261 CrossRef CAS PubMed.
- A. W. T. King, J. Jalomaki, M. Granstrom, D. S. Argyropoulos, S. Heikkinen and I. Kilpelainen, Anal. Methods, 2010, 2, 1499–1505 RSC.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- C. P. J. Bennington and I. Pineault, Pulp Pap. Can., 1999, 100, 123–131 CAS.
- A. van Heiningen, D. Krothapalli, J. Genco and A. Justason, Pulp Pap. Can., 2003, 104, 96–101 CAS.
- C. T. Laine and B. H. Tamminen, Holzforschung, 2004, 58, P611–P621 Search PubMed.
- M. Lawoko, R. Berggren, F. Berthold, G. Henriksson and G. Gellerstedt, Holzforschung, 2004, 58, P603–P610 CrossRef.
- Y. Ji, E. Vanska and A. van Heiningen, Holzforschung, 2009, 63, 264–271 CrossRef CAS.
- Z. Jiang, D. S. Argyropoulos and A. Granata, Magn. Reson. Chem., 1995, 33, 375–382 CrossRef CAS.
- T. Elder, Holzforschung, 1997, 51, 47–56 CrossRef CAS.
- H. Sixta, H. U. Süss, A. Potthast, M. Schwanninger and A. W. Krotscheck, in Handbook of Pulp, Wiley-VCH Verlag GmbH, 2008, pp. 609–708 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00115j |
| This journal is © The Royal Society of Chemistry 2014 |