An expedient route to heterocycles through α-arylation of ketones and arylamides by microwave induced thermal SRN1 reactions†
Silvia M. Soria-Castro,
Daniel A. Caminos and
Alicia B. Peñéñory*
INFIQC, Dpto. de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, X5000HUA Córdoba, Argentina. E-mail: penenory@fcq.unc.edu.ar; Web: http://www.infiqc.fcq.edu.ar
First published on 1st April 2014
Abstract
Microwave irradiation promotes a quick aromatic nucleophilic substitution by a thermally induced electron transfer process to form new C–C bonds by the coupling of aryl radicals and enolate nucleophiles. Diverse 2-aryl-1-phenylethanones can be prepared by the direct α-arylation of acetophenone with different haloarenes. The ketone enolate anion is generated by deprotonation with tBuOK in DMSO and the reaction is carried out in a closed microwave vessel at 70–100 °C for 10 min. This simple procedure also allows the synthesis of deoxybenzoin and indole heterocycle derivatives by inter- or intra-molecular ring closure reactions, with moderate to excellent substitution yields.
Introduction
Radical reactions provide a powerful method to achieve new C–C bonds in organic synthesis.1 The process that involves an electron-transfer (ET) step to generate radical anions and radicals, as the unimolecular radical nucleophilic substitution or SRN1, has proven to be a versatile mechanism for replacing a suitable leaving group by a nucleophile at the ipso position.2 This reaction affords substitution in non-activated aromatic (ArX) compounds, with an extensive variety of nucleophiles derived from carbon, nitrogen and oxygen to form new C–C bonds. In addition, the SRN1 mechanism is an important route for the synthesis of carbocycles and heterocycles by ring-closure reactions, such as indol derivatives with interesting pharmacological properties, and for a significant number of natural products.2 On the other hand, 2-phenyl acetophenones, also known as deoxybenzoin, is a motif present in anti-inflammatory,3 anti-convulsant, analgesic,4 and antimicrobial drugs.5 These aryl acetophenone structures are also suitable precursors of other interesting compounds, such as oxazoles, pyrazoles, indoles, and fire retardants in polymeric matrixes.4,6 This deoxybenzoin (3, R = Ph) and its derivatives can be easily achieved by α-arylation of carbonyl compounds by the SRN1 reaction. For example, the reactions of non-activated haloarenes with enolate anions from both aliphatic and aromatic ketones afford substitution under photo-stimulation in DMSO or liquid ammonia as solvent (Scheme 1).7 These reactions had good yield; yet, two or more hours of photochemical induction or the use of chemical radical initiators are required.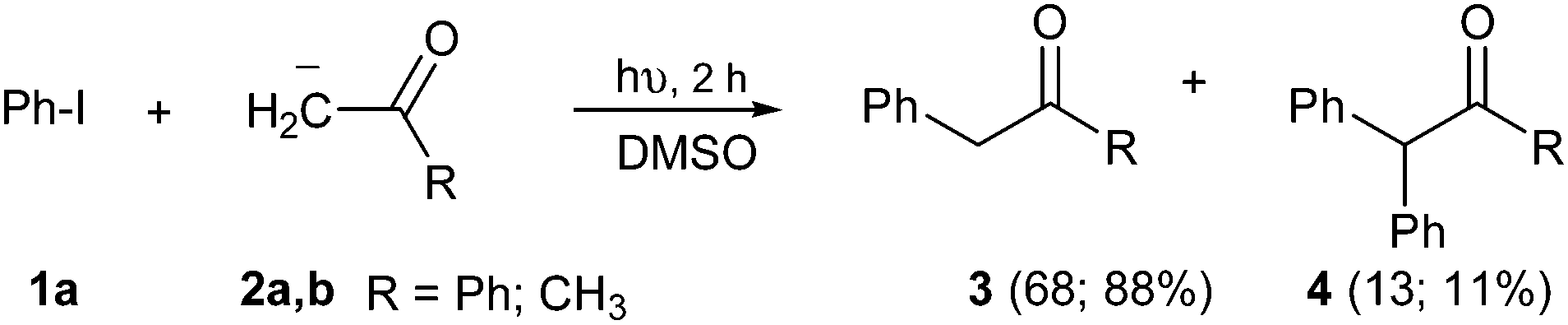 | ||
| Scheme 1 Photostimulated SRN1 α-arylation of ketone enolate anions. | ||
The selection of liquid ammonia as solvent requires distillation prior to use, which is energy costly and entails a particular careful manipulation.8 The development of new synthetic procedures, which implies simple and safer reaction conditions, is always a challenge for organic chemists. Alternatively, there are some examples of thermally induced SRN1 α-arylation of different aliphatic ketone enolate anions with ArI heating at 25 °C for 60 min in DMSO with moderate to good yield.9 Nevertheless, under these conditions the enolate anions from aromatic ketones did not react.9 In addition, thermal or spontaneous initiated reactions need about 60 min to proceed at room temperature. We consider that this synthetic pathway could be improved by microwave irradiation and be applied to heterocycles synthesis.
In the last decade microwave-assisted organic synthesis (MAOS) has proved an attractive heating method since this system reduces reaction time, increases yields, and in some cases increases selectivity in comparison with traditional oil bath heating.10 There are a few cases of thermally induced SRN1 reactions by microwave irradiation for the substitution of activated nitro benzyl derivatives. Thus, Vanelle et al. have reported that SRN1 reaction of p-nitrobenzyl chloride with 2-nitropropane anion under microwave irradiation with a domestic microwave oven affords very good coupling yields.11
This early use of a domestic microwave oven did not allow an accurate control of temperature, pressure and power applied to the sample. Today, strict reaction conditions could be set and controlled by specific laboratory microwave devices. In consequence the experiment conditions can be easily reproduced. Recently, Vanelle et al. reported the C-alkylation of 2-nitropropane anion with 4-[4-(chloromethyl)phenyl]-1,2-dimethyl-5-nitro-1H-imidazole at 140 °C (DMF) or 170 °C (DMSO) for 30 min in 25 mL using an accurate multimode microwave reactor with 200 W of applied power, with 60% yield12 (Scheme 2).
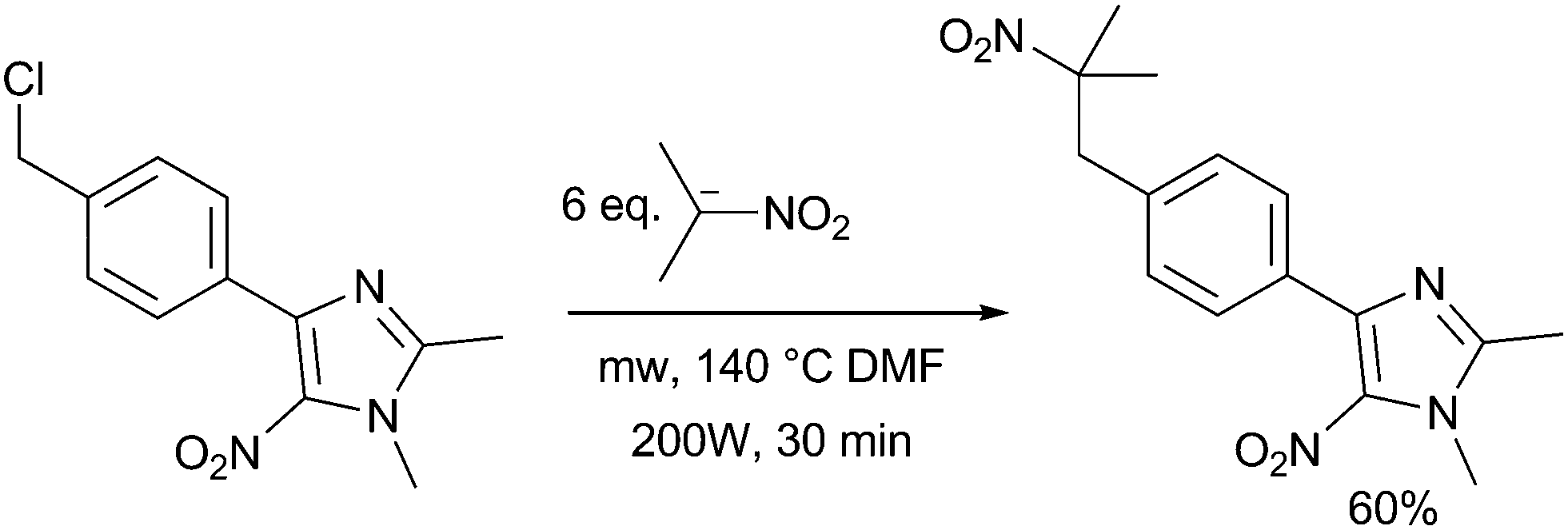 | ||
| Scheme 2 Fast thermal long distance SRN1 reaction of 4-[4-(chloromethyl)-phenyl]-1, 2-dimethyl-5-nitro-1H-imidazole with 2-nitropropane anion. | ||
In this work, we revisited the thermal initiated-SRN1 reactions of ArX with enolate nucleophiles, using for first time a controlled microwave heating to promote the reaction. Here we present a new method to produce the α-arylation of arylketones and aryl acetamides induced by thermal initiation under microwave irradiation. This methodology has also been successfully applied to the one-pot synthesis of heterocycles.
Results and discussion
Microwave-induced reactions
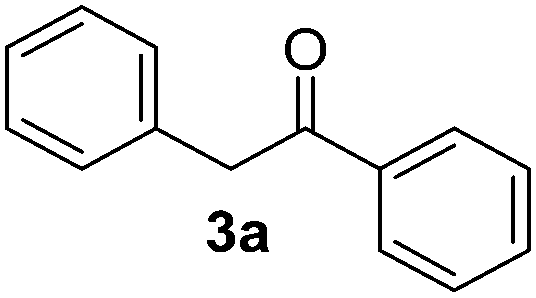
The reaction between PhI (1) and the enolate anion of acetophenone (2a) was taken as a model to explore the effectiveness of microwave irradiation as heating method for thermal initiation, as a simple alternative to photo-induction in liquid ammonia or DMSO. Table 1 shows the results yielded.
|
|||||
|---|---|---|---|---|---|
| Entry | X | Time (min) | Temp. (°C) | Product 3ab (%) | X−c (%) |
| a Reactions heated by microwave irradiation (150 Wmax) under N2 atmosphere for 10 min. Acetophenone (1.5 mmol), tBuOK (1.55 mmol), and PhI (0.5 mmol) in 2 mL of DMSO, otherwise indicated.b Quantified by NMR with internal standard.c Determined potentiometrically using a Ag/Ag(I) electrode. n.q.: no quantified.d Thermostatized in water bath, from ref. 9.e Traces (<4%) of di-arylation product 4a were observed.f DMF as solvent.g MeCN as solvent.h Without acetophenone enolate.i With 10 mol% m-dinitrobenzene.j With tBuOK as nucleophile and base.k PhOC(CH3)3 was quantified by NMR in 4% yield. | |||||
| 1d | I | 60 | 25 | <1 | <1 |
| 2 | I | 10 | 40 | Trace | n.q. |
| 3 | I | 10 | 50 | 34 | n.q. |
| 4 | I | 10 | 60 | 45e | n.q. |
| 5 | I | 10 | 70 | 55e | 78 |
| 6 | I | 5 | 70 | 49e | 70 |
| 7 | I | 30 | 70 | 54e | 72 |
| 8 | I | 10 | 100 | 58e | 92 |
| 9f | I | 10 | 70 | 7 | <10 |
| 10g | I | 10 | 70 | 8 | <10 |
| 11 | Ih | 10 | 70 | — | 0 |
| 12 | Ii | 10 | 70 | 14 | 21 |
| 13 | Ij | 10 | 70 | —k | 80 |
| 14 | Br | 10 | 70 | Trace | 4 |
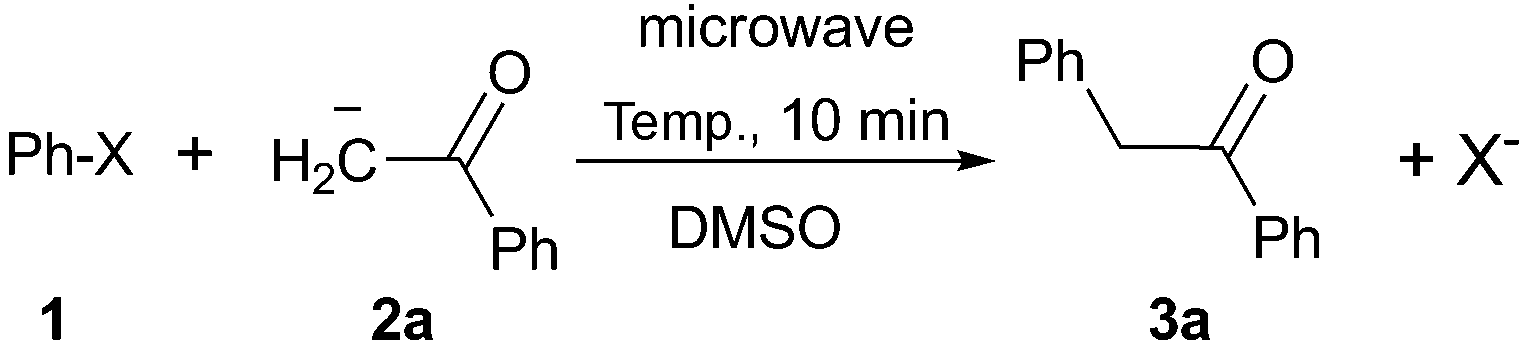
When a mixture of 1 and anion 2a was stirred at 25 °C for 60 min in DMSO, the detection of only a trace of α-arylation product 3a by GLC was reported (Table 1, entry 1).9 In view of this preliminary result we first tested the reaction between PhI with acetophenone enolate anion by heating under microwave irradiation for 10 min at different temperatures. No reaction at 40 °C under microwave irradiation was observed; however, by increasing the temperature to 50–60 °C, the substitution product 3a occurred in moderate yields and only traces (<4%) of di-arylation product 4a were observed (Table 1, entries 2–4).
Following this examination, the best result in this system was obtained at 70 °C in DMSO, irradiated with microwave; substitution product 1,2-diphenylethanone 3a was quantified in 55% yield in just 10 min, with a conversion of 78% determined by iodide ion quantification (Table 1, entry 5). A similar result was found at 5 min; yet the GC-MS analysis showed presence of unreacted PhI (Table 1, entry 6). Increasing time to 30 min, or raising temperature to 100 °C did not show a substantial rise in the yield of substitution product 3a; however, an increase in the reduction product was shown through the higher iodide ion yield (Table 1, entries 7 and 8). The 1,2-diphenylethanone 3a was stable under the reaction conditions. After GC-MS analysis it did not show any by-product produced by thermal decomposition at 70–80 °C, under microwave heating for 10 min; it was recovered in 84% isolated yield. All attempts to detect the reduced product benzene were unsuccessful.13 Another solvents such as DMF or MeCN were also tried without success (Table 1, entries 9 and 10). In addition, the main reaction by-product was the auto-condensation of acetophenone present in excess in the basic reaction media.
Comparison between microwave and oil bath heating
The same model reaction was tested using a sealed microwave tube on a pre-heated (to accurate temperature) oil bath, and after 10 min at 70 °C only 10% yield of product 3a was quantified by GC-MS. In addition, product 3a was obtained in 17% yield after 60 min at 70 °C. When the reaction was performed under microwave irradiation in an open vessel (at atmospheric pressure), a comparable yield of 3a and unreacted PhI were found in relation to those observed in Table 1, entry 5. Under microwave irradiation it is possible that significant and localized overheating is produced in the solution since DMSO is a strongly microwave absorbing solvent. This overheated spots, will help to accelerate reaction rate in relation to conventional heating.14 In addition, the pressure conditions of 1.7 atm did not seem to be required; however, microwave irradiation was needed to accelerate the reaction. This reaction effectively proceeds faster under microwave irradiation in an open or closed vessel; for security issues, however, the latter was adopted as a standard methodology. Further research into these microwave induced thermal ET reactions is currently in progress in our lab.Reaction mechanism
To evaluate the mechanism involved in this reaction, some experiments were performed. First we checked the presence of radical intermediates in the reaction by using 1-(allyloxy)-2-iodobenzene (5) as radical clock (Scheme 3).15 To reduce the base promoted isomerization of the double bond in 5, the reaction was conducted at 60 °C.16 Thus, the reaction of 5 with the enolate anion of acetophenone in a ratio 1 : 3, after 10 minutes under microwave irradiation at 60 °C, gave 29% and 16% of the cyclized (6) and straightforward (7) substitution products, respectively. This result clearly indicates that aryl radicals are reactive intermediates in the microwave reaction.15 Formation of product 6 must involve the intermediacy of the 2-(allyloxy)phenyl radical and its 5-exo ring closure followed by addition to the enolate anion 2a. On the other hand, direct coupling between the 2-(allyloxy)phenyl radical and the anion 2a followed by ET and isomerization of the double bond affords product 7. Isomerization of 5 prior to ET step would also account for the formation of product 7, but would not allow 5-exo cyclization to yield derivative 6 after coupling with the nucleophile. In fact, when the reaction between 5 and anion 2a was performed at 80 °C, the only product obtained was 7 in 45% yield. Moreover, PhI showed to be thermally stable under the reaction conditions. After 10 min of microwave irradiation of PhI in DMSO, either benzene or iodide ion was not detected, (Table 1, entry 11). This fact allows discarding a homolytic rupture of the CAr–I bond as the radical initiation process. Furthermore, when the reaction between PhI and the enolate anion of acetophenone was conducted in the presence of 10 mol% of m-dinitrobenzene, which is a strong electron acceptor inhibiting the ET step,2 a 14% yield of product was obtained with 21% conversion yields (Table 1, entry 12). These assays suggest that the reaction proceeds through an SRN1 mechanism thermally initiated17 with radicals and radical anions as intermediates. In this reaction, the source of initiating electrons could be the nucleophile itself, which transfers an electron to the PhI to generate Ph radical and I− by a dissociative ET.17,18 In this case, the increase in temperature or the direct microwave energy applied to the substrate could provide the necessary energy to the initial ET from the nucleophile to the ArX. Alternatively, the slightly excess of tBuO− anion could also act as the electron donor in an entrainment reaction.19 This later statement was confirmed by the reaction between PhI and tBuOK in the absence of acetophenone, which gave 80% of I− anions after 10 min of microwave irradiation (Table 1, entry 13). As in the above reactions, benzene could not be detected after microwave irradiation. In addition only a low amount of the substitution product with the tBuO− anion was observed (4%), according to the low reactivity of the alkoxide anions toward aryl radicals attributed to their very positive oxidation standard potentials, being hard nucleophiles.2,20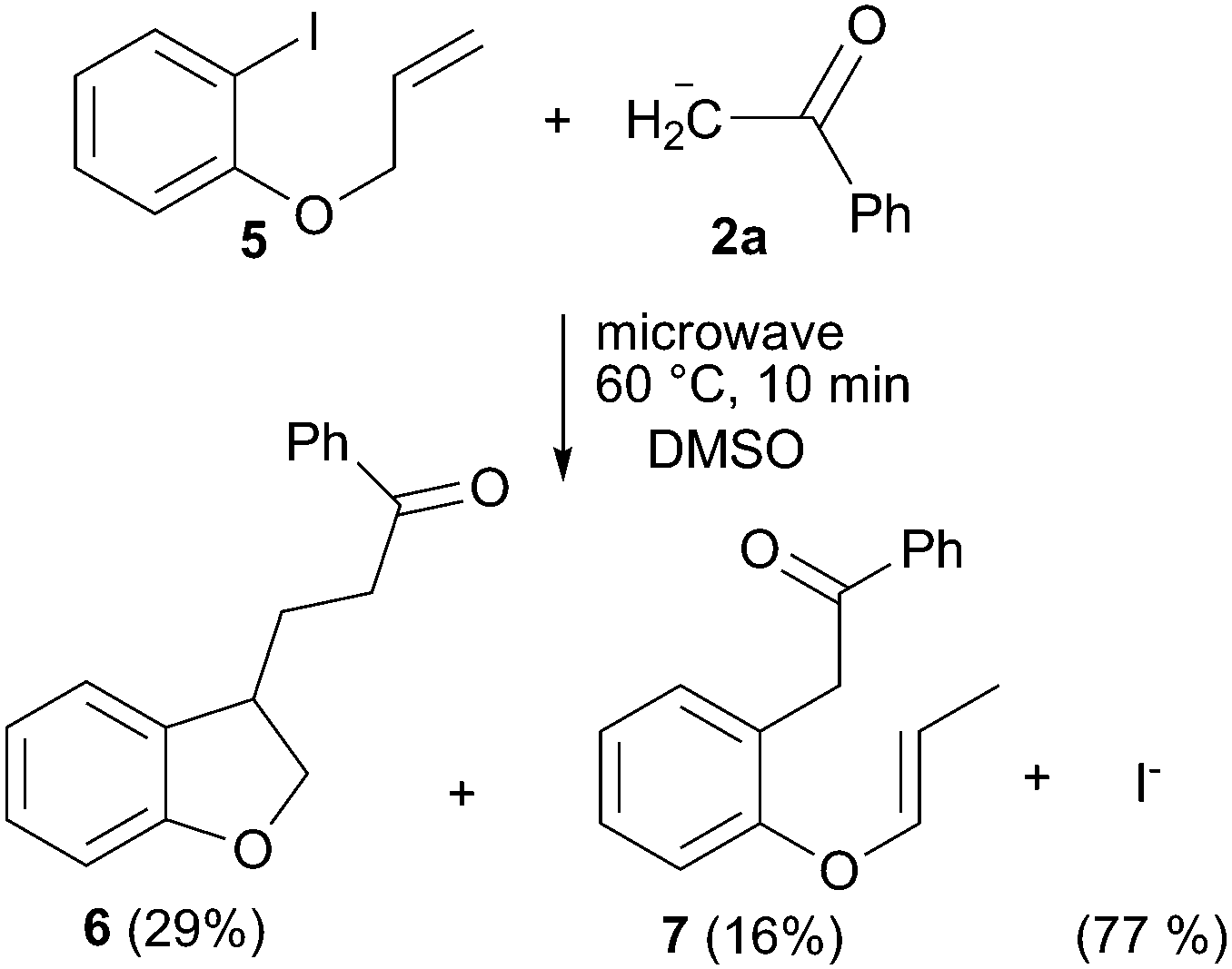 | ||
| Scheme 3 1-(Allyloxy)-2-iodobenzene as radical clock. | ||
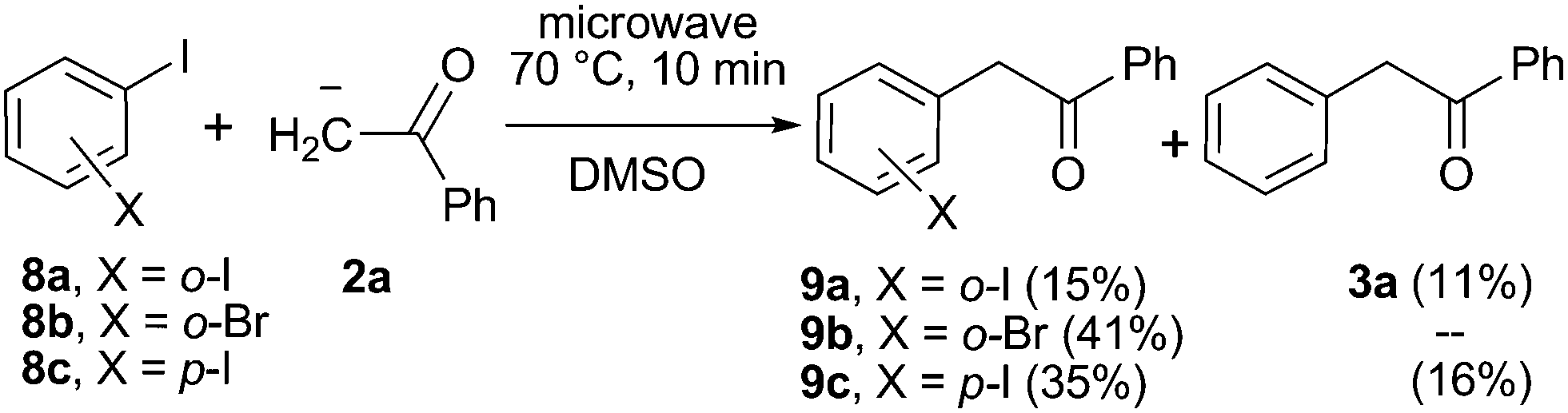 | ||
| Scheme 4 Microwave-induced reaction of acetophenone anion (2a) with ortho-haloiodobenzene (8a and b) and para-diiodobenzene (8c) in DMSO. | ||
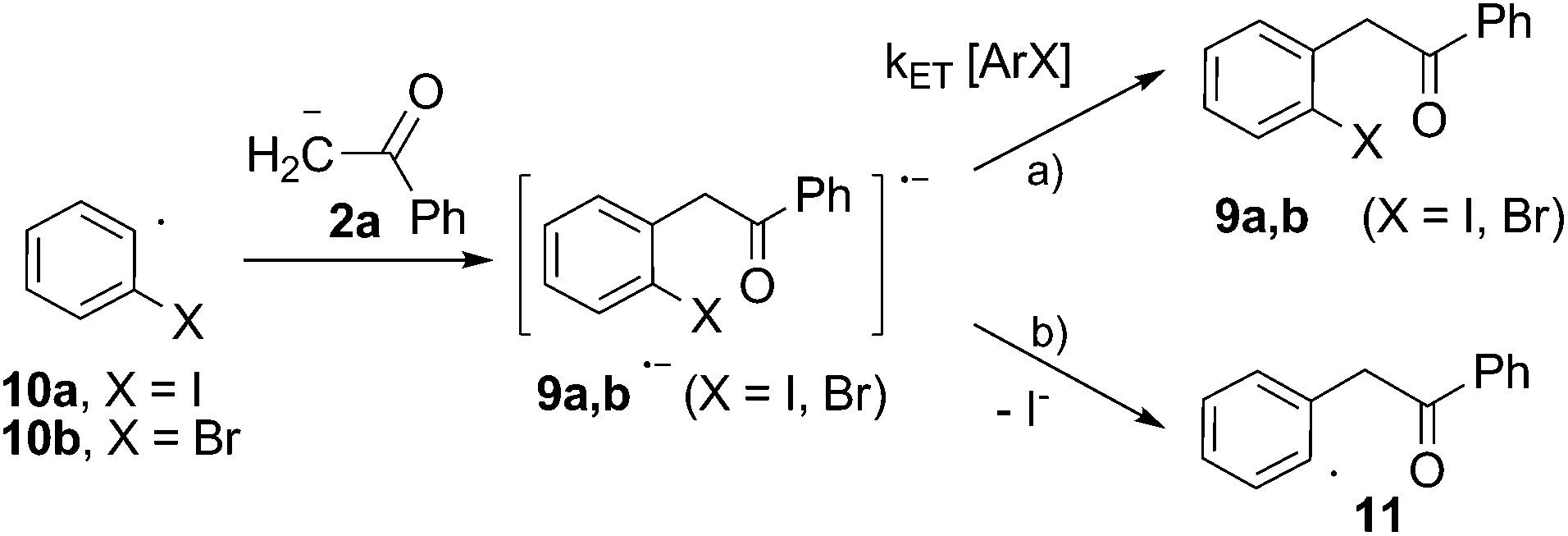 | ||
| Scheme 5 Propagation steps for the SRN1 mechanism of the reaction of acetophenone anion (2a) with ortho-haloiodobenzene (8a and b) in DMSO. | ||
Additionally, PhBr was unreactive toward the anion of acetophenone after 10 min of microwave irradiation at 70 °C, with low conversion (<5%) and traces of the substitution product 3a (Table 1, entries 14). Similar results were observed at 100 or 140 °C.
Furthermore, the reactivity of dihalobenzenes toward the anion of acetophenone was studied under microwave induction and the results compared with the photoinduced reactions in order to get further insights into the mechanism. The microwave-irradiated reactions of ortho-haloiodobenzenes (8a and b; X = I, Br) with anion 2a gave mono-substitution with retention of halide (9a and b) and mono-substitution with reduction of the second halide (3a), the later only observed for X = I (Scheme 4). The reaction of the para-diiodobenzene (8c) with anion 2a afforded both the p-iodo substituted derivative (9c) and the mono-substituted reduced product 3a with only trace of the p-disubstituted derivative observed by GC-MS. Similar results were previously obtained in the photoinduced reactions of 8a and b with anion 2a.21 The agreement between the reactions performed under photochemical and microwave irradiation strongly supports that both reactions proceed by the same SRN1 mechanism as follows. After the initial ET, the radical anion of the substrate (8a and b˙−) fragments into iodide anion and radical 10a and b. The radical anions 9a and b˙− formed by the coupling of radical 10a and b with anion 2a can afford the mono-substituted products 9a and b after inter-ET to the substrate (Scheme 5, pathway a) or can fragment at the C–halogen bond to give radical 11 (Scheme 5, pathway b). The unpaired spin distribution in the most stable radical anion intermediate (9a and b˙−) is localized in the benzoyl moiety (π-system), which is separated from the ortho-halophenyl moiety by a sp3 carbon atom; the intra-ET is favored only when X = I to afford radical 11 after C–X fragmentation.
Finally, disubstitution was not observed, even with the ortho-diiodobenzene (8a). This can be attributed to the low excess of the acetophenone anion present in the reaction media, since di-substitution was previously observed for the ketone enolate anion under photostimulation in liquid ammonia.22 An unfavorable coupling reaction due to steric constraints can also be responsible. On the other hand, trace of disubstitution was detected by GC-MS for the reaction of para-diiodobenzene (8c) in the presence of 3 equivalents of anion 2a, under both microwave and photo-induced reactions.
Scope of the microwave induced α-arylation reaction
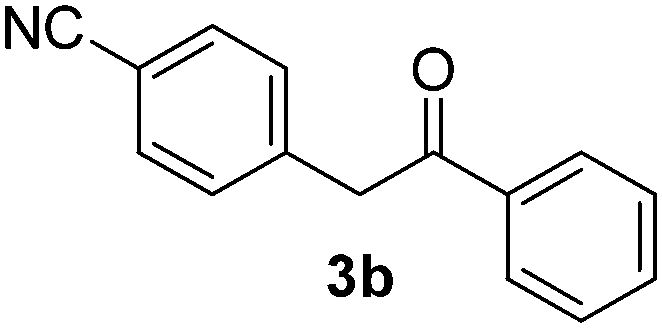
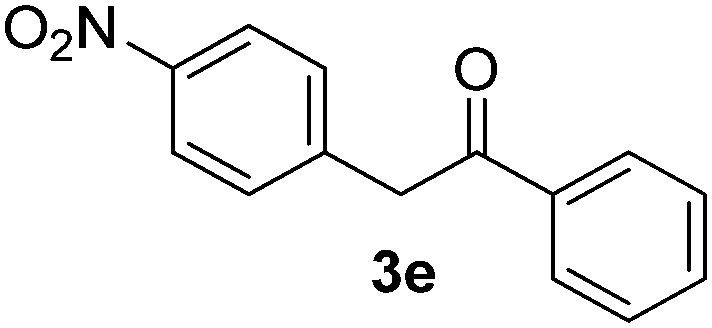
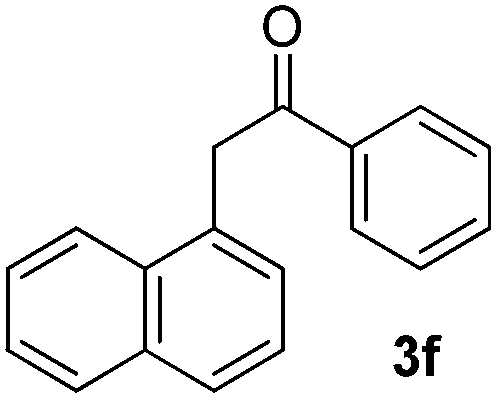
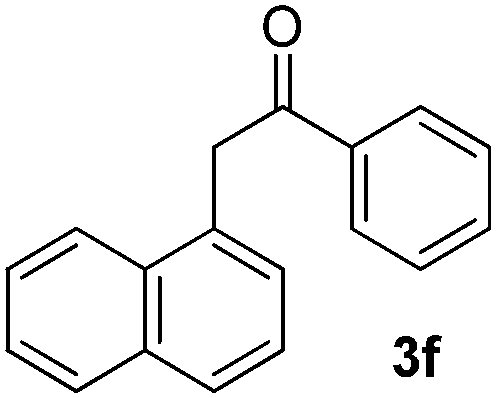
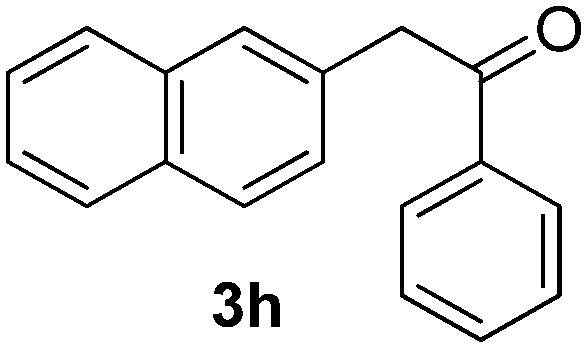
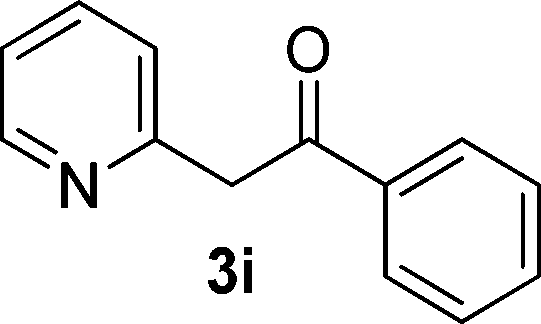
We subsequently studied the reactivity of different aryl halides to determine the scope of the microwave α-arylation reaction; results are found in Table 2. In general, the reaction of aryl iodides bearing electron-donor and -acceptor substituent gave the substitution product in moderate to good yields (46–57%, Table 2, entries 1–4). Using 4-iodobenzonitrile, anion 2a was mono-arylated in 46% yield. The α-arylation of 4-iodobenzonitrile has not been previously reported under photoinduced SRN1 conditions.2 Furthermore, attempts to α-arylation of anion 2a by Pd-catalysis failed; instead of the desired product, β-enaminones were obtained by condensation of the acetophenone with the nitrile moiety of the substrate.23 On the other hand, it is known that the fragmentation rates of the radical anions of nitro-substituted aryl halides determined electrochemically24 or by pulse radiolysis25 are slow and range from 10−3 to 102 s−1. As a consequence such substrates have not been suitable for electrochemical or photoinduced SRN1 reactions.2 In this context, it should be noted that under microwave irradiation 19% of the nitro derivate is obtained (Table 2, entry 5). Nevertheless, a classical aromatic nucleophilic substitution cannot be disregarded. In addition, 1-iodonaphtalene, 1-bromonaphtalene, 2-bromonaphtalene and 2-iodopyridine, by reaction with anion 2a gave substitution products in moderate yield (Table 2, entries 6–9).| Entry | ArX | Product 3 | % Yieldb |
|---|---|---|---|
| a Reactions heated to 70 °C by microwave irradiation (150 Wmax) under N2 atmosphere for 10 min. Acetophenone (1.5 mmol), tBuOK (1.55 mmol), and ArX (0.5 mmol) in 2 mL of DMSO, otherwise indicated.b Quantified by NMR with internal standard.c Isolated yield from a preparative scale reaction: acetophenone (7.5 mmol), tBuOK (7.75 mmol), and PhI (2.5 mmol) in 10 mL of DMSO. | |||
| 1 |  |
 |
55 (53)c |
| 2 | 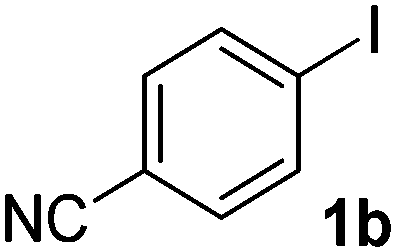 |
 |
46 |
| 3 | 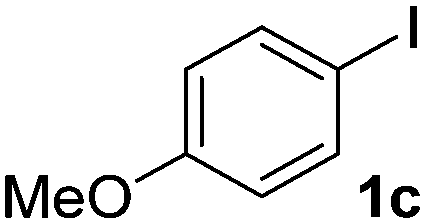 |
 |
54 |
| 4 | 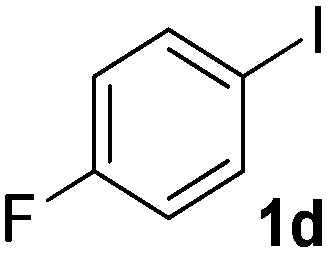 |
 |
57 |
| 5 |  |
 |
19 |
| 6 |  |
 |
59 |
| 7 | 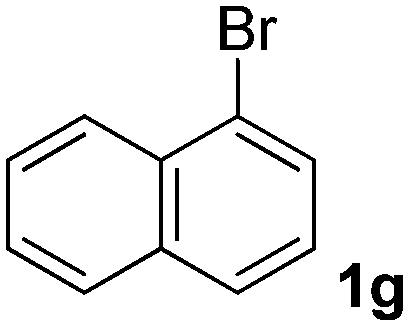 |
 |
55 |
| 8 |  |
 |
35 |
| 9 | 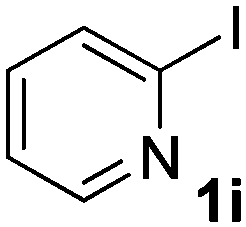 |
 |
44 |
Besides, p-substituted acetophenone enolate anions (2j–m) were also investigated as nucleophiles in the reaction with PhI (Table 3). As expected for an ET-initiated reaction, the presence of an electron donor group such as CH3 in anion 2j afforded comparable yield of substitution product relative to anion 2a (Table 3, entries 1 and 2). The amino group is also deprotonated in the reaction media and the yield of α-arylation decreased (Table 3, entry 3). On the other hand the presence of electron withdrawing groups such as Cl and NO2 in the enolate anion strongly inhibits or avoids ET pathways (Table 3, entries 4 and 5). The formation of 3a in the α-arylation of anion 2m supports the participation of 3m˙− as intermediate (Table 3, entry 5).
| Entry | Anion | Product | %Yieldb |
|---|---|---|---|
| a Reactions heated to 70 °C by microwave irradiation (150 Wmax) under N2 atmosphere for 10 min. Ketone (1.5 mmol), tBuOK (1.55 mmol), and PhI (0.5 mmol) in 2 mL of DMSO.b Quantified by NMR with internal standard.c Isolated yield from a preparative scale reaction.d Together with 7% of 3a. | |||
| 1 | 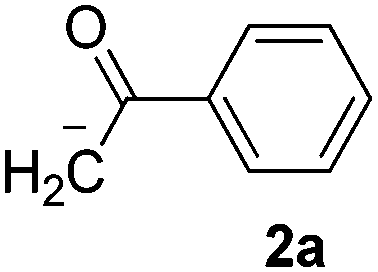 |
 |
55 (53)c |
| 2 |  |
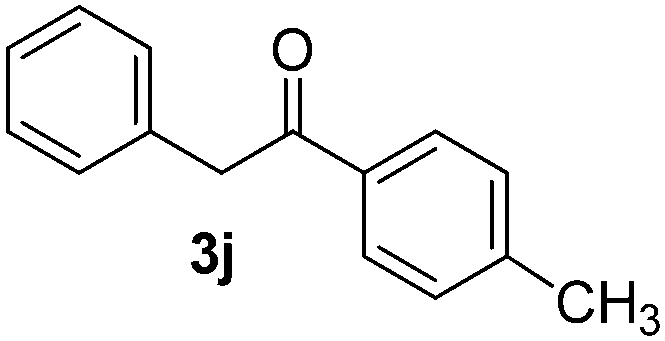 |
40 |
| 3 | 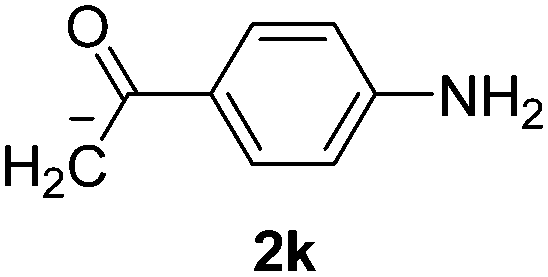 |
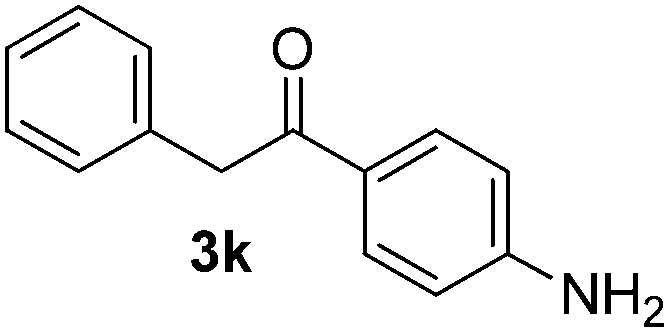 |
34 |
| 4 |  |
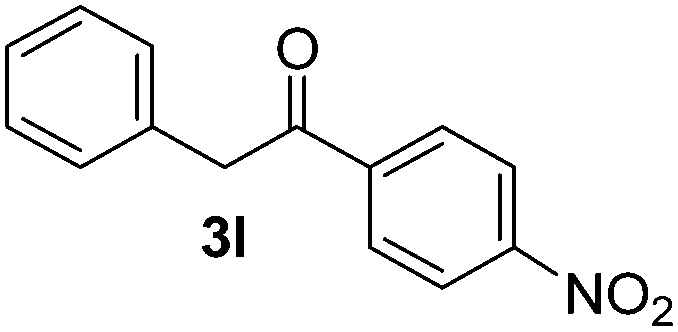 |
0 |
| 5 | 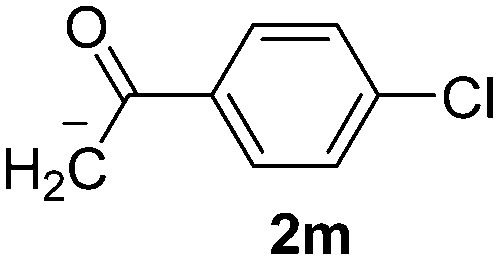 |
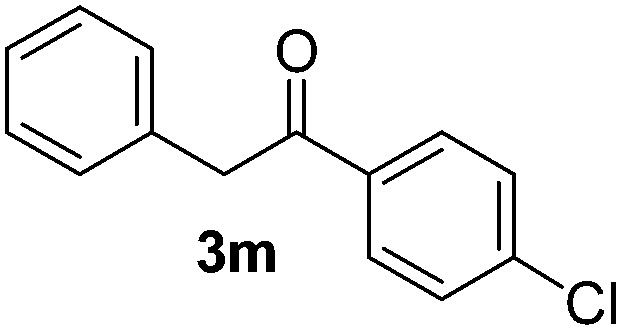 |
14d |
| 6 | 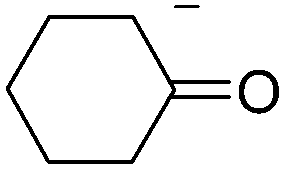 |
 |
10 |
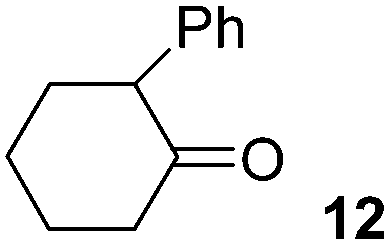
In addition, we have performed some experiments using the enolate anion of cyclohexanone as nucleophile under microwave irradiation to compare with the thermal induced reaction previously reported.9 To our surprise at 70 °C only 10% of the α-arylated product, 2-phenylcyclohexanone (12) was observed and most IPh remained unreacted (Table 3, entry 6). The yield of 12 was not improved by increasing the temperature to 100 °C, except for the ketone condensation product. Further attempts to reproduce the previous reported results9 were unsuccessful. In our hands, IPh did not react with the enolate anion of cyclohexanone in DMSO after 60 min heating at 25 °C.
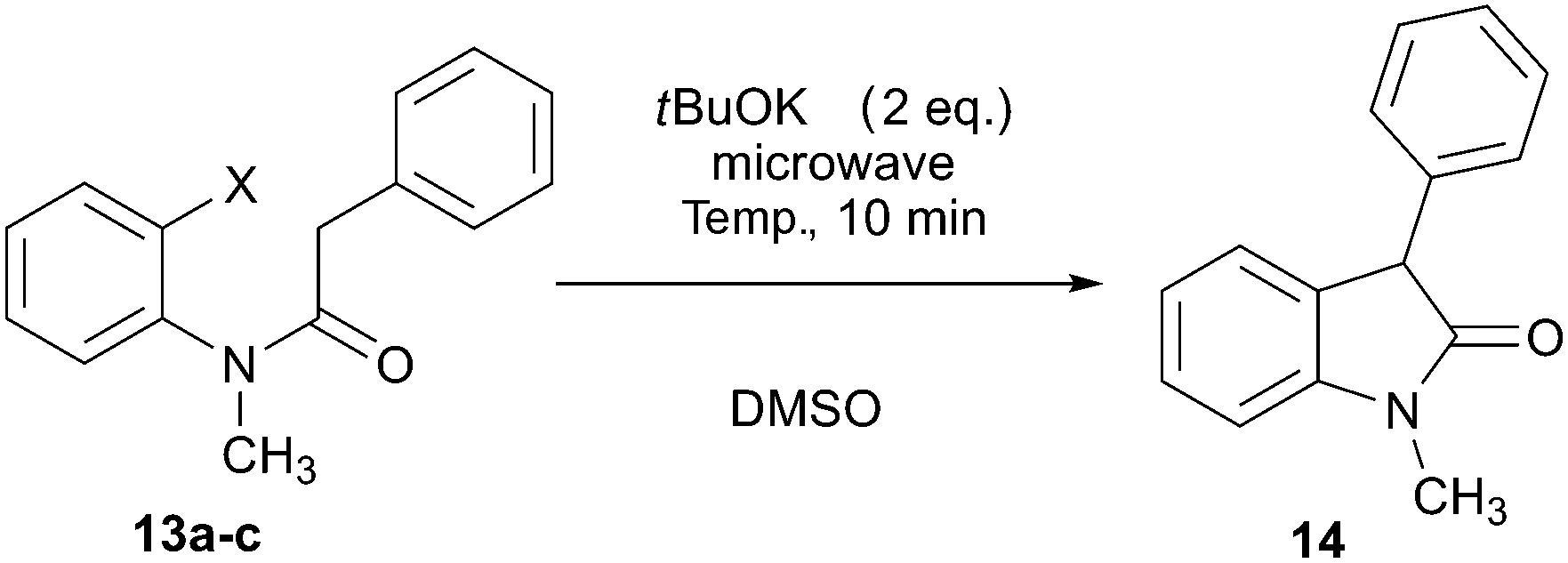
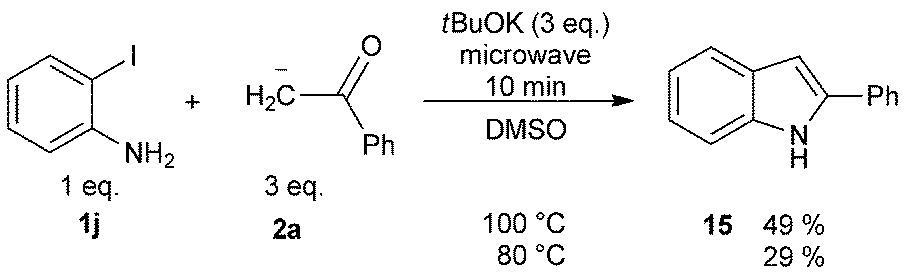
As it has been well established, the photoinduced SRN1 reaction provides a good alternative to the synthesis of heterocycles.2 Therefore, two different approaches were explored to achieve target compounds under microwave irradiation: (1) intramolecular SRN1 reaction and (2) intermolecular SRN1 reaction, followed by a polar ring closure reaction. Initially, the synthesis of indolinone derivatives was performed by intramolecular cyclization of N-(2-halophenyl)phenylacetamides (13) under microwave irradiation (Table 4). Thus, the reaction of the iodo derivative 13a in the presence of tBuOK gave 90% yield of the cyclized compound 1-methyl-3-phenylindolin-2-one (14) and 10% of the reduced product N-methyl-N,2-diphenylacetamide at 70 °C in DMSO (Table 4, entry 1). Quantitative yield was achieved by increasing temperature to 100 °C (Table 4, entry 2). The bromo derivative 13b, afforded 80% of substitution at 100 °C, while the chloro derivative 13c required increasing temperature to 130 °C to observe only 14% of product 14 (Table 4, entries 3–6). The reactivity order I > Br > Cl is according to the reduction potential of the aryl halides and with the average fragmentation rate constant for their radical anion intermediates.2 Finally, this methodology was extended to the “one-pot” synthesis of indole derivatives (15) by two consecutives reactions, an intermolecular nucleophilic substitution of ortho-iodo aniline with the enolate anion 2a induced by microwave irradiation followed by a condensation. The best isolated yield (49%) of indole 15 was obtained at 100 °C (Scheme 6). This result agrees with that found in the previous examples in Table 2, for intermolecular process.
 | ||
| Scheme 6 “One-pot” synthesis of 2-phenylindole (15). | ||
Conclusions
The use of modern microwave heating devices proves valuable in the fine chemist and pharmaceutical chemistry offering a faster and safer synthetic methodology. In this report new C–C bonds were obtained by α-arylation of aromatic ketones and acetamides by a thermally induced SRN1 reaction by microwave irradiation at moderate temperatures. This is the first report of microwave-induced SRN1 reaction in the aromatic system. The initiation step is proposed to take place by a microwave-induced thermal ET.By comparison with the photoinduced procedures, the main disadvantage of the microwave-initiated reaction is the competitive reduction observed for intermolecular process, consequently the best substitution yields is about 60%. On the other hand, the microwave-induced reaction shows as many advantages as simplicity, short times (10 min of microwave irradiation compared with 120 min of photoirradiation), compatibility with substituents like NO2, CN, F, Br, and a better performance in intramolecular reactions.
Thus, this process allows the synthesis of 2-aryl-1-phenyletanones (3a–k and 3m) by α-arylation of the enolate anion of acetophenones (2) with different haloarenes (1a–i). In addition, this methodology provides a simple way to achieve heterocycles as 1-methyl-3-phenylindolin-2-one (14) and 2-phenylindol (15) by intra- or intermolecular SRN1 reactions, respectively, in a very fast reaction.
Experimental section
Chemicals
Potassium tert-butoxide (tBuOK), iodobenzene, bromobenzene, cyclohexanone, acetophenone, 1-p-tolylethanone, 1-(4-aminophenyl)ethanone, 1-(4-nitrophenyl)ethanone, 1-(4-chlorophenyl)ethanone, 1-iodonaphthalene, 1-bromonaphthalene, 2-bromonaphtalene, 2-iodopyridine, 2-iodoaniline 1,2-diiodobenzene, 1,4-diiodobenzene, 1,2-bromo-iodobenzene, 1,4-fluoro-iodobenzene, 1,4-cyano-iodobenzene, 1,4-methoxy-iodobenzene, and 1,4-iodo-nitrobenzene were all high purity commercial samples used without further purification.DMSO absolute grade was used without further purification and stored over molecular sieves (4 Å). N-Methylanilides 13a–c were synthesized by standard procedures from the reaction between commercial 2-haloanilines and the corresponding acyl chloride in CH2Cl2 in the presence of pyridine.26 N-(2-iodophenyl)-N-methyl-2-phenylacetamide (13a),27 N-(2-bromophenyl)-N-methyl-2-phenylacetamide (13b),28 and N-(2-chlorophenyl)-N-methyl-2-phenylacetamide (13c)27 present spectral data in good agreement with the literature.
The ketone enolate anion or the anion of N-methylanilides was generated in situ by acid–base deprotonation using tBuOK.
General methods
1H and 13C NMR spectra were recorded at 400.16 and 100.62 MHz respectively on a 400 spectrometer, and all spectra were reported in δ (ppm) relative to Me4Si, with CDCl3 as solvent. Gas chromatographic analyses were performed with a flame-ionization detector, on 30 m capillary column of a 0.32 mm × 0.25 μm film thickness, with a 5% phenylpolysiloxane phase. GC-MS analyses were performed employing a 25 m × 0.2 mm × 0.33 μm with a 5% phenylpolysiloxane phase column. HRMS spectra were recorded on a GCT Premie orthogonal acceleration time-of-flight (oa-TOF) GC mass spectrometer. Ionization was achieved by electronic impact (70 eV) and detection set up positive mode.Representative experimental procedure
The reactions were carried out in a 10 mL CEM Discover microwave glass vessel, filled with nitrogen and with a magnetic stirrer. The tube was dried under vacuum, filled with nitrogen, and then charged with dried DMSO (2 mL) and degassed. Next, for the α-arylation of the haloarene, tBuOK (179 mg, 1.55 mmol), acetophenone (1.5 mmol), and aryl halide (0.5 mmol) were added to the degassed solvent under nitrogen. For the intramolecular cyclization reaction halophenylacetamide (0.5 mmol) and tBuOK (112 mg, 1.0 mmol) were added to the degassed solvent under nitrogen. Then, the reaction tube was heated by microwave irradiation.Microwave-induced reactions were performed in a single mode instrument equipped with a noncontact infrared temperature sensor, direct pressure control system for measuring the pressure of the reaction vessel contents and a cooling system by compressed air. The sample vessels reach the selected temperature in about 30 s (∼2.5 °C s−1). Although, the maximum microwave power was set at 150 W, after the initial heating pulse of maximum 100 W for 30 s, the average applied power was about 1 W to keep the selected temperature (see ESI† for irradiation details). After 10 min of irradiation the device cooled the tube to 50 °C with compressed air above 1 min (−0.5 °C s−1).
The average pressure in the vessel was 1.7 atm during the reaction time. After completion of the reaction, the vessel was removed from the microwave cavity and opened to the atmosphere. The reaction was subsequently quenched by addition of water (30 mL) and NH4NO3 excess, and the mixture was extracted with methylene chloride (3 × 20 mL). The combined organic extract was dried over anhydrous CaCl2, and the products were quantified by GC or NMR by the internal standard method or isolated by silica gel chromatography from the crude product reaction mixture. Water layer was recovered to quantify the halide ions by potentiometric titration with an AgNO3 standard solution.
Product isolation from a preparative scale reaction
This reaction was performed following the representative experimental procedure using acetophenone (7.5 mmol), tBuOK (7.75 mmol), and PhI (2.5 mmol) in 10 mL of DMSO. After reaction and usual workup, the combined dried extract was first chromatographed on a silica gel short-column eluted first with n-pentane and second with n-pentane : ethyl ether (90 : 10). After evaporation the oily residue was distilled in a kügelrohr to separate the product 1,2-diphenylethanone (3a) from acetophenone. The substitution product 3a crystallizes at 0 °C with a small amount of n-hexane: 260 mg (53%); mp 53–55 °C.Products characterization
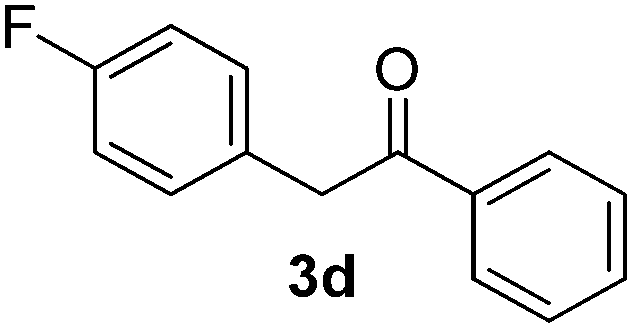
All the products present in Table 2 were obtained following the general procedure, quantified by NMR or GC. Products 3a–k, 3m, 9a–c, 12, 14, and 15 are known compounds and present spectral data as shown in the literature, in agreement with the proposed structures. 1,2-Diphenylethanone (3a),29 4-(2-oxo-2-phenylethyl)benzonitrile (3b),30 2-(4-methoxyphenyl)-1-phenylethanone (3c),31 2-(4-fluorophenyl)-1-phenylethanone (3d),31 2-(4-nitrophenyl)-1-phenylethanone (3e),32 2-(naphthalen-1-yl)-1-phenylethanone (3f),33 2-(naphthalen-2-yl)-1-phenylethanone (3h),34 1-phenyl-2-(pyridin-2-yl)ethanone (3i),35 1-(4-methylphenyl)-2-phenylethanone (3j),36 1-(4-aminophenyl)-2-phenylethanone (3k),37 1-(4-chlorophenyl)-2-phenylethanone (3m),36 1-(allyloxy)-2-iodobenzene (5),16 2-(2-iodophenyl)-1-phenylethanone (9a),21 2-(2-bromophenyl)-1-phenylethanone (9b),38 2-(4-iodophenyl)-1-phenylethanone (9c),21 2-phenylcyclohexanone (12),39 1-methyl-3-phenylindolin-2-one (14),28 and 2-phenyl-1H-indole (15).323-(2,3-Dihydrobenzofuran-3-yl)-1-phenylpropan-1-one (6)
Following the general procedure, using tBuOK (179 mg, 0.75 mmol), acetophenone (0.75 mmol), and aryl halide 5 (0.25 mmol), microwave irradiation at 60 °C for 10 min and then purification by radial chromatography (petroleum/ethyl ether, 8/2) provided 6 as light yellow crystals (18 mg, 29% yield).1H NMR (400 MHz, CDCl3) δ 7.94–7.92 (m, 2H), 7.57 (tt, 7.3, 1.3 Hz, 1H), 7.46 (tb, J = 7.6 Hz, 2H), 7.21 (db, J = 7.3 Hz, 1H), 7.14 (tb, J = 7.7 Hz, 1H), 6.87 (td, J = 7.4, 1 Hz, 1H), 6.81 (d, J = 8 Hz, 1H), 4.66 (t, J = 8.9 Hz, 1H), 4.28 (dd, J = 8.9, 5.8 Hz, 1H), 3.61-3.54 (m, 1H), 3.11-2.95 (m, 2H), 2.24-2.16 (m, 1H), 2.11-2.02 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 199.5, 160.0, 136.8, 133.2, 130.1, 128.7, 128.4, 128.0, 124.5, 120.5, 109.7, 41.1, 35.3, 28.9. HRMS ESI+ [M + Na+] calcd for C17H16O2Na: 275.1043, found 275.1056.
2-(2-(Prop-1-enyloxy)phenyl)-1-phenylethanone (7)
Following the general procedure, using tBuOK (179 mg, 0.75 mmol), acetophenone (0.75 mmol), and aryl halide 5 (0.25 mmol), microwave irradiation at 60 °C for 10 min and then purification by radial chromatography (petroleum/ethyl ether, 8/2) provided 7 as light brown crystals, (10 mg, 16% yield).1H NMR (400 MHz, CDCl3) δ 8.08–8.06 (m, 2H), 7.56 (tt, J = 7, 1 Hz, 1H), 7.56 (tt, J = 7, 1 Hz), 7.46 (tb, J = 7.5 Hz, 1H), 7.27–7.23 (m, 2H), 7.02 (td, J = 7.5, 1 Hz, 1H), 6.96 (dd, J = 8, 1 Hz, 1H), 6.34 (dc, J = 6, 1.7 Hz, 1H), 4.85 (cd, J = 6.8, 6 Hz, 1H), 4.35 (s, 2H), 1.60 (dd, J = 6.8, 1.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 197.7, 155.2, 140.9, 136.9, 133.0, 131.3, 128.6, 128.5, 124.5, 122.6, 114.8, 107.6, 39.9, 9.3. HRMS ESI+ [M + Na+] calcd for C17H16O2Na: 275.1043, found 275.1047.
Acknowledgements
This work was supported by Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and Agencia Nacional de Promoción Científica y Técnica (ANPCyT), Ministerio de Ciencia y Tecnología de la Provincia de Córdoba, Argentina and SECyT-UNC. A.B.P. and D.A.C. are scientific members from CONICET, S.S.C. gratefully acknowledges the receipt of a fellowship from CONICET.Notes and references
- (a) Radicals in Organic Synthesis, ed. P. Renaud and M. Sibi, Wiley-VCH, Weinheim, vol. 1 and 2, 2001 Search PubMed; (b) R. A. Rossi and A. B. Peñéñory, Curr. Org. Chem., 2006, 3, 437–451 Search PubMed.
- (a) R. A. Rossi, A. B. Pierini and A. B. Peñéñory, Chem. Rev., 2003, 103, 71–167 CrossRef CAS PubMed; (b) A. B. Peñéñory, J. E. Argüello, Aromatic and Heteroaromatic Substitution by SRN1 and SN1 Reactions, in Handbook of Synthetic Photochemistry, ed. A. Albini and M. Fagnoni, Wiley-VCH, Weinheim, 2010, ch. 10, pp. 319–346 Search PubMed; (c) R. A. Rossi and A. B. Peñéñory, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. M. Horspool and F. Lenci, CRC Press Inc., Boca Raton, 2nd edn, 2003, ch. 47, pp. 47-1–47-24 Search PubMed; (d) R. A. Rossi, A. B. Pierini and A. N. Santiago, in Organic Reactions, ed. L. A. Paquette and R. Bittman, Wiley & Sons, 1999, pp. 1–271 Search PubMed.
- H.-Q. Li, Y. Luo, P. C. Lv, L. Shi, C.-H. Liu and H.-L. Zhu, Bioorg. Med. Chem. Lett., 2010, 20, 2025–2028 CrossRef CAS PubMed.
- J. Sävmarker, J. Lindh and P. Nilson, Tetrahedron Lett., 2012, 51, 6886–6889 CrossRef PubMed.
- H.-Q. Li, J.-Y. Xue, L. Shi, S.-Y. Gui and H.-L. Zhu, Eur. J. Med. Chem., 2008, 43, 662–667 CrossRef CAS PubMed.
- B.-Y. Ryu, S. Moon, I. Kosif, T. Ranganathan, R. J. Farris and T. Emrick, Polymer, 2009, 50, 767–774 CrossRef CAS PubMed.
- (a) G. L. Borosky, A. B. Pierini and R. A. Rossi, J. Org. Chem., 1992, 57, 247–252 CrossRef CAS; (b) M. F. Semmelhack and T. Bargar, J. Am. Chem. Soc., 1980, 102, 7765–7774 CrossRef CAS; (c) R. Beugelmans and M. Bois-Choussy, Heterocycles, 1987, 26, 1863–1871 CrossRef CAS.
- Liquid ammonia has a boiling point of −33 °C and is extremely basic and corrosive. Inhalation may cause asphyxia.
- R. G. Scamehorn, J. M. Hardacre, J. M. Lukanich and L. R. Sharpe, J. Org. Chem., 1984, 49, 4881–4883 CrossRef CAS.
- C. O. Kappe and D. Dallinger, Nat. Rev. Drug Discovery, 2006, 5, 51–63 CrossRef CAS PubMed.
- P. Vanelle, A. Gellis, M. Kaafarani, J. Maldonado and M. P. Crozet, Tetrahedron Lett., 1999, 40, 4343–4346 CrossRef CAS.
- L. Zink, M. D. Crozet, T. Terme and P. Vanelle, Tetrahedron Lett., 2011, 52, 6991–6996 CrossRef CAS PubMed.
- After microwave irradiation the reaction vessel reached 1.7 atm of pressure. To condense the possible gaseous reaction products, a tube was connected from the vessel to a nitrogen liquid tramp for the reaction of 1-iodonaphthalene with anion 2a. Nevertheless, after releasing pressure only traces of naphthalene were detected in the tramp by GC-MS.
- C. O. Kappe, B. Pieber and D. Dallinger, Angew. Chem., Int. Ed., 2013, 52, 1088–1094 CrossRef CAS PubMed.
- J. I. Bardagí, S. E. Vaillard and R. Rossi, Tetrahedron Lett., 2006, 47, 3149–3152 CrossRef PubMed.
- S. E. Vaillard, A. Postigo and R. A. Rossi, J. Org. Chem., 2002, 67, 8500–8506 CrossRef CAS PubMed.
- C. Costentin, P. Hapiot, M. Médebielle and J. M. Savéant, J. Am. Chem. Soc., 1999, 121, 4451–4460 CrossRef CAS.
- C. Costentin, P. Hapiot, M. Médebielle and J.-M. Savéant, J. Am. Chem. Soc., 2000, 122, 5623–5635 CrossRef CAS.
- L. C. Schmidt, J. E. Argüello and A. B. Peñéñory, J. Org. Chem., 2007, 72, 2936–2944 CrossRef CAS PubMed.
- J.-M. Savéant, J. Phys. Chem., 1994, 98, 3716–3724 CrossRef.
- M. T. Baumgartner, L. B. Jiménez, A. B. Pierini and R. A. Rossi, J. Chem. Soc., Perkin Trans. 2, 2002, 1092–1097 RSC.
- J. F. Bunnett and P. Singh, J. Org. Chem., 1981, 46, 5022–5025 CrossRef CAS.
- S. M. Crawford, P. G. Alsabeh and M. Stradiotto, Eur. J. Org. Chem., 2012, 6042–6050 CrossRef CAS.
- R. G. Compton, R. A. W. Dryfe, J. C. Eklund, S. D. Page, J. Hirst, L. B. Nei, G. W. J. Fleet, K. Y. Hsia, D. Bethell and L. J. Martingale, J. Chem. Soc., Perkin Trans. 2, 1995, 1673–1677 RSC , and references therein reported.
- M. Meot-Ner, P. Neta, R. K. Norris and K. Wilson, J. Phys. Chem., 1986, 90, 168–173 CrossRef CAS.
- A. R. Fersht and W. P. Jencks, J. Am. Chem. Soc., 1970, 92, 5432–5442 CrossRef CAS.
- V. Rey, A. B. Pierini and A. B. Peñéñory, J. Org. Chem., 2009, 74, 1223–1230 CrossRef CAS PubMed.
- R. R. Goehring, Y. P. Sachdeva, J. S. Pisipati, M. C. Sleevi and J. F. Wolfe, J. Am. Chem. Soc., 1985, 107, 435–443 CrossRef CAS.
- J.-C. Hsieh, Y.-C. Chen, A.-Y. Cheng and H.-C. Tseng, Org. Lett., 2012, 14, 1282–1285 CrossRef CAS PubMed.
- A. Padwa, T. Brookhart, D. Dehm and G. Wubbels, J. Am. Chem. Soc., 1978, 100, 8247–8259 CrossRef CAS.
- K. Huang, G. Li, W.-P. Huang, D.-G. Yu and Z.-J. Shi, Chem. Commun., 2011, 47, 7224–7226 RSC.
- T. Hering, D. P. Hari and B. König, J. Org. Chem., 2012, 77, 10347–10352 CrossRef CAS PubMed.
- A. Odedra, C.-J. Wu, T. B. Pratap, C.-W. Huang, Y.-F. Ran and R.-S. Liu, J. Am. Chem. Soc., 2005, 127, 3406–3412 CrossRef CAS PubMed.
- J. McEwen and K. Yates, J. Am. Chem. Soc., 1987, 109, 5800–5808 CrossRef CAS.
- M. A. Nazareno and R. A. Rossi, Tetrahedron Lett., 1994, 35, 5185–5188 CrossRef CAS.
- T. Miao and G.-W. Wang, Chem. Commun., 2011, 47, 9501–9503 RSC.
- I. Ivanov, S. Nikolova and S. Statkova-Abeghe, Synth. Commun., 2006, 36, 1405–1411 CrossRef CAS.
- L. Ackermann and L. T. Kaspar, J. Org. Chem., 2007, 72, 6149–6153 CrossRef CAS PubMed.
- L. Li, P. Cai, Q. Guo and S. Xue, J. Org. Chem., 2008, 73, 3516–3522 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Typical reaction profile under microwave irradiation and spectra (1H and 13C NMR) for products 6 and 7. See DOI: 10.1039/c4ra00120f |
| This journal is © The Royal Society of Chemistry 2014 |