Radicals in metal–organic frameworks
Thomas B. Faust
* and
Deanna M. D'Alessandro
*
School of Chemistry, The University of Sydney, New South Wales 2006, Australia. E-mail: deanna.dalessandro@sydney.edu.au; Fax: +61-2-93513329; Tel: +61-2-93513777
First published on 31st March 2014
Abstract
The burgeoning field of metal–organic frameworks (MOFs) has been marked by numerous key advances over the past two decades. An emerging theme is the incorporation of radical species which may be ligated as an integral structural component of, or simply appended to, the material, or else merely a guest within it. Radical incorporation has been shown to endow MOFs with a plethora of unique and fascinating magnetic, electronic and optical properties, paving the way towards their application as spin probes, and in magnetic/electronic devices, chemical sensing and molecular recognition. In view of the rapid growth of the literature in the area, this review highlights progress over the past three years (since 2011), and seeks to uncover promising ideas that will underscore future advancements at both the fundamental and applied levels.
 Thomas B. Faust | Thomas Faust received his Master's degree from The University of Sheffield in 2008 under Professor Mike Ward and PhD in 2011 within the Molecular Magnetism Group at The University of Manchester with Professors Richard Winpenny and Eric McInnes. His early research focussed on supramolecular assembly and magnetic exchange in multi-metallic cages and rings. Following a year as an EPSRC Doctoral Prize Fellow still in Manchester with Professor David Collison, Thomas joined the Molecular Materials Group at The University of Sydney in 2012 where his research shifted from playing with spin in discrete supramolecular structures to infinite multi-dimensional metal–organic materials. |
 Deanna M. D'Alessandro | Deanna D'Alessandro received her BSc (Honours 2001) and PhD (2006) degrees from James Cook University under the supervision of Professor Richard Keene. She held a postdoctoral position in the Molecular Electronics Group at The University of Sydney before undertaking a postdoctoral position with Professor Jeffrey Long at UC Berkeley from 2007–2009. She was appointed as a University of Sydney Postdoctoral Research Fellow in 2010 before starting her own research group in 2011 as an Australian Research Council Queen Elizabeth II Fellow. Deanna's research focuses on fundamental and applied aspects of functional inorganic materials that exhibit novel electronic and optical phenomena. |
1. Introduction
The field of metal–organic frameworks (MOFs) has been marked by astonishing developments over the past 15 years. The initial interest focussed on applications arising from their primary physical properties, namely their large accessible voids and high surface areas, which offer potential applications in gas and small molecule storage.1–3 The possibility of functionalising their cavity linings as well as tuning their topologies and pore sizes made them amenable to separation chemistry4,5 and catalysis.6 More recently attention has been drawn towards the possibility of developing more advanced properties such as magnetism, spin-state bistability, electron, hole and ion mobility, ferroelectricity, luminescence, redox and non-linear optical activity. These attributes give rise to myriad applications, not limited to but including: chemical sensing,7 small molecule sequestration (e.g., CO2)8 and release (e.g., drugs),9 electronic devices,10 magnetic refrigeration,11 second harmonic generation,12 structural determination,13 polymer templating,14 as well as multiple platforms for clean energy.15 Furthermore, the possibility of combining these elements giving rise to multi-functionality is highly sought after and difficult to induce in traditional porous materials such as zeolites.As MOF chemistry becomes more established, researchers are becoming increasingly ambitious in the selection of functional groups that are used. Whilst often seen as merely exotic curiosities, stable organic radicals have found an important place in many fields of science. Biological science has benefitted from spin labelling which provides useful insights into molecular conformation and dynamics within proteins,16,17 from in vivo imaging with radical probes,18 to spin trapping for the characterisation of usually short-lived radical intermediates in biological processes.19 Outside of biology, radicals are pivotal in the development of technologically-relevant devices such as totally organic rechargeable batteries.20
In the pursuit of new materials with interesting magnetic properties, MOFs containing paramagnetic transition metal or lanthanide ions appear ideal candidates, having regularly positioned spin centres with well-defined bridges extending infinitely in three dimensions. However for a material to be magnetic there must exist long range ordering between these spin centres. In slightly more established fields such as single molecule magnetism (SMM), order is achieved by superexchange mediated either by single atoms or very short linkers. This is juxtaposed to MOF synthesis in which metal centres are scaffolded into place by relatively long ligands such as terephthalates and bipyridines. Such ligands are often too long for magnetic ordering to exist at temperatures much above absolute zero. One solution that may be envisaged is to introduce a new spin, centred on the organic linker itself to act a conduit for the exchange process. The sign of the exchange parameter, J – i.e., whether the interaction between the metal and radical is ferromagnetic (FM) or antiferromagnetic (AFM) – is for our purposes somewhat secondary to its magnitude. If strong interaction between the radical and the adjacent metals is FM, it serves both to mediate the metal–metal exchange and to increase the overall spin present. If the radical–metal interaction is strongly AFM, we once again strengthen the metal–metal exchange (whose spins are still aligned with one another), however the radical reduces the net spin. This is a small price to pay, especially considering the spin of the radical is likely to be 1/2, whilst metal centred spins are typically much larger. Considering how difficult it can be to design a system with a particular type of exchange (FM/AFM), coupled with the iterative or serendipitous nature of many of the MOF topologies synthesised to date, we discover why this area of chemistry is still in its fledgling stage and why small steps represent significant advances in the field.
In addition to the above, radical electrons often reside over well conjugated π-systems whose resonant forms help to stabilise it. Such extended systems often span a significant proportion of the ligand's length and enhance the scope for magnetic communication along them. Moreover, the use of organic radicals offers an additional source of redox chemistry. Much manipulation of oxidation states about metal centres is concomitant with a change in coordination geometry which either alters the topology of the framework or leads to its degradation. By contrast, various oxidation states of organic radicals can often be achieved with minimal structural rearrangement and with only modest potentials. This would allow perturbation of the electronic structure of the material to be studied independent of morphological changes. The ability to switch between bistable states or turn on and off properties such as magnetism and conductivity is a current goal not just in MOF chemistry but across the molecular and nanotechnology disciplines, particularly for implementation in data storage and molecular electronic devices.
The presence of radicals and redox active moieties within MOFs can have function without possessing long range ordering. Radicals may provide a useful handle for characterisation through an ever increasing range of electron paramagnetic resonance (EPR) experiments. They may also provide a position of variable electron density which may be useful for reversibly binding small molecules upon application of an electrical potential. The intense absorption of visible light by some radicals may also lend them towards use as colourimetric sensors.
Functionalised radicals have aroused interest for their potential in magnetic materials,21 particularly within magnetically ordered organic polymers22–25 and SMMs,26–28 however their inclusion into MOFs and MOF topologies is somewhat more retarded. Theoretical studies on the adsorption of organic radicals into MOF cavities were carried out as early as 2000,29,30 but subsequent empirical studies have been limited. In this review, we aim to summarise the most recent advances (those from 2011 onwards) and provide an outlook on how the field might advance in the coming years.
1.1 Scope of the review
As mentioned previously, the initial interest surrounding MOFs largely concerned their ability to host small molecules. Though the definition of a MOF has been debated and revised many times, one continuing theme has been the stipulation for voids or potential voids. This requirement appears to have grown out of their envisaged implementation at the time. As the field of MOFs has expanded into other areas, their usefulness is no longer solely based their capacity to uptake and store gases. Neither are properties such as interpenetration and instability to desolvation automatically negative indicators, indeed we will discover herein that densely packed MOFs can induce other desirable properties. Hence a sizable minority of the community in this area are not concerned about whether their materials meet the definitions of a MOF, since the nomenclature has little bearing on the properties. As a result in many instances a paper may not identify a MOF as such, though we might. Furthermore, some authors unfortunately identify their materials as MOFs, when this cannot be supported by even the loosest definition. This clearly makes even the most rigorous literature search fallible to some extent.We employ the IUPAC approved definition of a MOF31 as far as possible with the caveat that in many cases the presence or otherwise of potential voids is ambiguous. All materials mentioned are covalently linked to extend in at least two dimensions. If the presence of a void is not explicit, but the properties of the material complement the review, it has been included in the discussion. We do attempt to cover to the fullest extent MOFs containing radicals which have been reported in the past three years, though salient examples reported prior to 2011 may be included for perspective.
Explicitly not covered in this review are reactions within MOFs which take place via a radical pathway, for example catalysis of alkene oxidation and polymer synthesis. Also not covered are MOFs which act as radical scavenging agents, for example anti-bacterial agents and biological or industrial antioxidants.
2. Modes of radical inclusion
There are a finite number of modes for incorporation of a radical into a MOF. Radical ligands coordinated to a single metal centre can be easily identified as pendant radicals (metal appended). Radical ligands that span multiple metal centres can be classified according to the positioning of their radical moiety; those grafted onto a recognisable scaffolding ligand are also pendant radicals (ligand appended), whilst those core to the bridging ligand may be considered structurally integral. A radical moiety may be considered structurally integrated if its removal would change the network's connectivity. All non-covalently bound radicals can be considered guests within host framework. These descriptions are elucidated in Fig. 1. We prefer the term pendant over branch, as within polymer chemistry the latter term is considered to consist of repeating units.32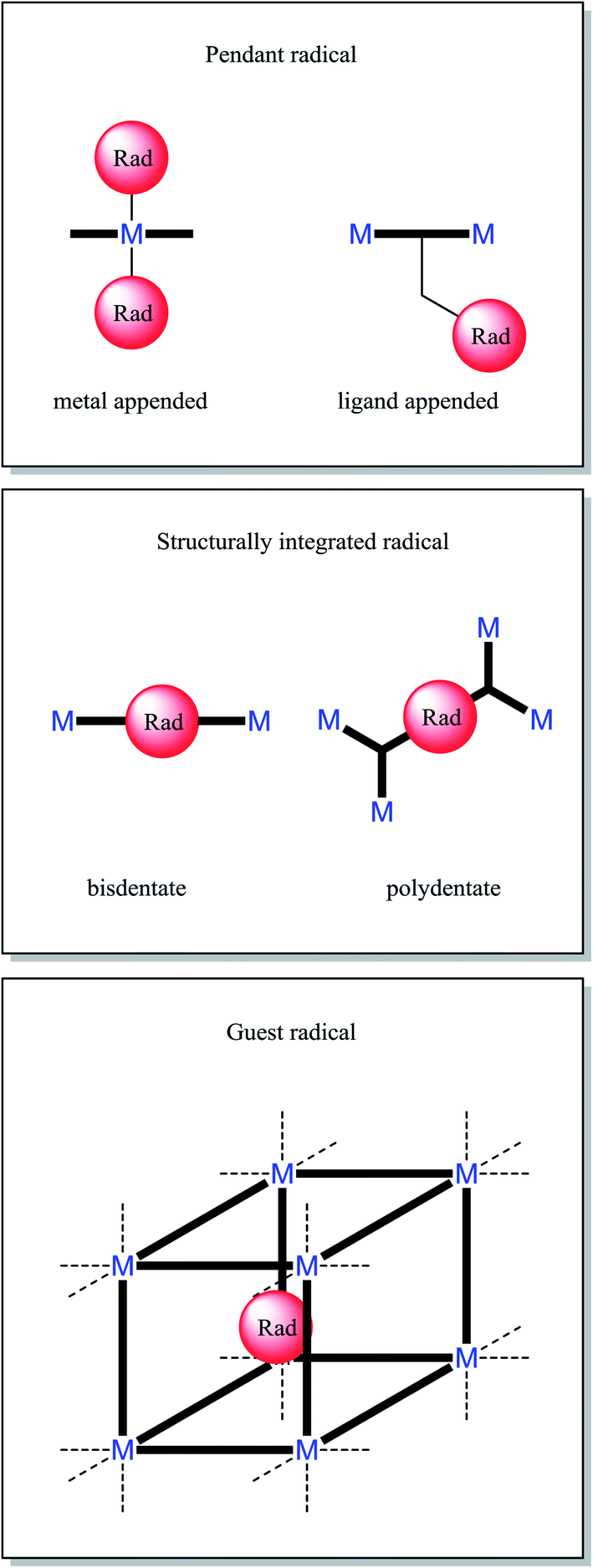 | ||
| Fig. 1 Modes of radical inclusion within MOFs. | ||
When it is desirable for continuous M–radical–M interactions, for example to engender long range magnetic ordering, it is necessary for structurally integrated radicals to be used. It is often more synthetically straightforward to adopt the pendant approach, and also allows easier synthesis of materials that are isoreticular with known frameworks.
3. Radical incorporation into metal–organic frameworks
Radicals tend to be highly reactive species, susceptible to a wide range of reactions with low thermal barriers, including hydrogen abstraction, disproportionation and dimerisation. Stable radicals tend to either have localised unpaired electrons with good steric protection or else consist of conjugated systems exhibiting resonance stabilisation. The radicals tend to belong to one of a few major classes, based on their composition and we will present them according to these classifications.3.1 Nitroxides
The type of radical with which most chemists are familiar is the broad family of nitroxides. Their chemistry has received significant attention such that their use is now unremarkable. Their main attribute lies in their stability in a wide range of environments, provided they are suitably composed. Typically the aminoxyl group is flanked by quaternary carbons to prevent disproportionation to a nitrone/hydroxylamine by transfer of an α hydrogen atom. The species are resistant to dimerization due to the high degree of delocalisation within the three electron NO π-system. The best known member of this class of compound is the 2,2,6,6-tetramethylpiperidine-1-oxy radical (TEMPO, see Fig. 2). It and dozens of its derivatives are now commercially available at accessible prices (TEMPO around £3/AU$5 per gram when purchased in modest quantities).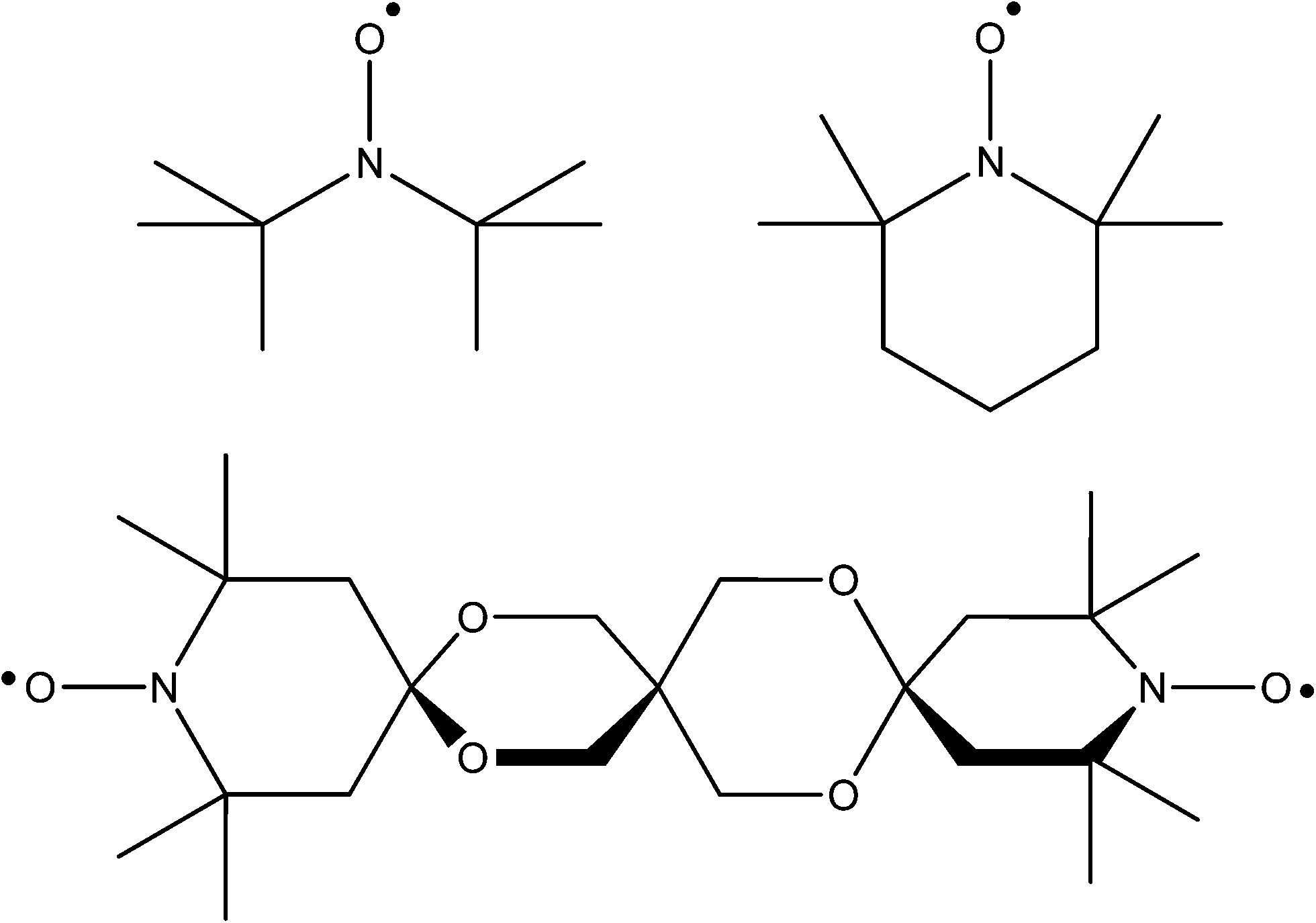 | ||
| Fig. 2 Clockwise from top left: ligands DTBN˙, TEMPO and bTbK˙˙. | ||
Pöppl and colleagues successfully demonstrated magnetic modification of an existing framework by incorporation of radical guests.33 Moreover, they employed EPR of the resulting material to shine light on possible mechanisms of ligand exchange in carboxylate MOFs. Cu3(BTC)2, or HKUST-1,34 is a MOF consisting of copper paddlewheels bridged by 1,3,5-benzene tricarboxylate (BTC) ligands to give a 3D framework. The network topology provides two large pores though which bulky molecules may pass. This has been exploited to investigate the adsorption of nitroxide radicals within the MOF cavities. HKUST-1 was dosed at liquid nitrogen temperatures with the di-tert-butyl nitroxide radical (DTBN˙, see Fig. 2). The magnetic influence upon the framework was followed by continuous wave EPR. AFM exchange within the copper paddlewheels leads to a zero ground spin state which is exclusively populated at low temperature, such that the framework is EPR silent. Upon initial exposure to DTBN˙, a low temperature EPR measurement of the framework revealed an intense signal consistent with DTBN˙ incorporation, though the lack of hyperfine structure tended to suggest pockets of radical in high concentration rather than a uniform low concentration through the material. The EPR response changed over a multi-month timescale (or through thermal acceleration) to reveal a more complicated signal which could be satisfactorily simulated with three Cu(II) species, of which the major component was square-planar. For this predominant case, it appears that the nitroxide radical binds equatorially to the copper ion. AFM exchange between this copper and the nitroxide leads to a net spin of zero which can no longer compensate for the spin of the second copper ion, resulting in an S = 1/2 ground state. Evacuation of the framework effected removal of the nitroxide and reestablishment of the copper paddlewheels, as evidenced by the disappearance of all EPR signals. This process was successfully cycled several times.
The equatorial coordination of the nitroxide clearly requires the de-coordination of at least one carboxylate oxygen from the paddlewheel. Such ligand exchange has been observed previously for HKUST-1,35,36 but the topic of post-synthetic ligand exchange has become increasingly eminent recently,37 especially as a means to synthesising both mixed ligand MOFs38 and MOFs which cannot be accessed via a direct de novo synthetic procedure.39 In this regard, handles on exchange processes such as provided by this DTBN/HKUST-1 experiment become especially pertinent.
Characterisation and chemical determination, particularly of MOFs with variable ligand ratios, is routinely performed by acid or base digestion of the MOF followed by NMR spectroscopy. This methodology is reliable but much information is lost. Solid-state NMR (ssNMR) possesses many desirable attributes for the characterisation of MOFs. It is non-destructive, can quantify the ratios of ligands within the framework, provide information about the chemical environment in which the ligands are residing, give insight to host–guest interaction and can trace the molecular dynamics within the framework.40 The relatively low-sensitivity of ssNMR often necessitates long acquisition times for acceptable signal-to-noise ratios. One method to combat this has been to induce hyperpolarisation by Dynamic Nuclear Polarisation (DNP) in which spin polarisation is transferred from electrons (which is very high due its large magnetic moment) to nuclei (which have much smaller magnetic moments). The process is driven by microwave irradiation and has the net effect of increasing the ssNMR signal intensity significantly. The biradical, bis-TEMPO-bisketal (bTbK˙˙, see Fig. 2), was first employed for this purpose in 2009.41 Other researchers have subsequently been using the bTbK˙˙ polarising agent for ssNMR, most notably for the derivatives of the MOF, MIL-68(In). Radical incorporation was achieved by incipient wetting impregnation of the dry MOF with a solution of bTbK˙˙ in 1,1,2,2-tetrachloroethane. In this case DNP was particularly useful in shortening acquisition times for experiments with low abundance isotopes, for example Cross-Polarisation Magic Angle Spinning (CPMAS) for 15N and 13C, as well as 2D Heteronuclear Correlation Spectroscopy (HETCOR). For the three derivatives studied experiment times were shortened by two orders of magnitude.42
Timofeeva and co-workers have used titration of TEMPO as a spin probe to determine the number of accessible coordinatively unsaturated metal sites in samples of MIL-96(Al), MIL-100(Al) and MIL-110(Al).43 Since TEMPO will adhere to only a single Lewis acid site, their number can be measured by the amount of TEMPO which can be added before the characteristic signal from free TEMPO appears, which is different from that of the coordinated species (see Fig. 3). The accessibility is clearly dependent on the size of the probe, which provides useful information on the accessibility for similarly sized molecules which may be employed for other purposes.
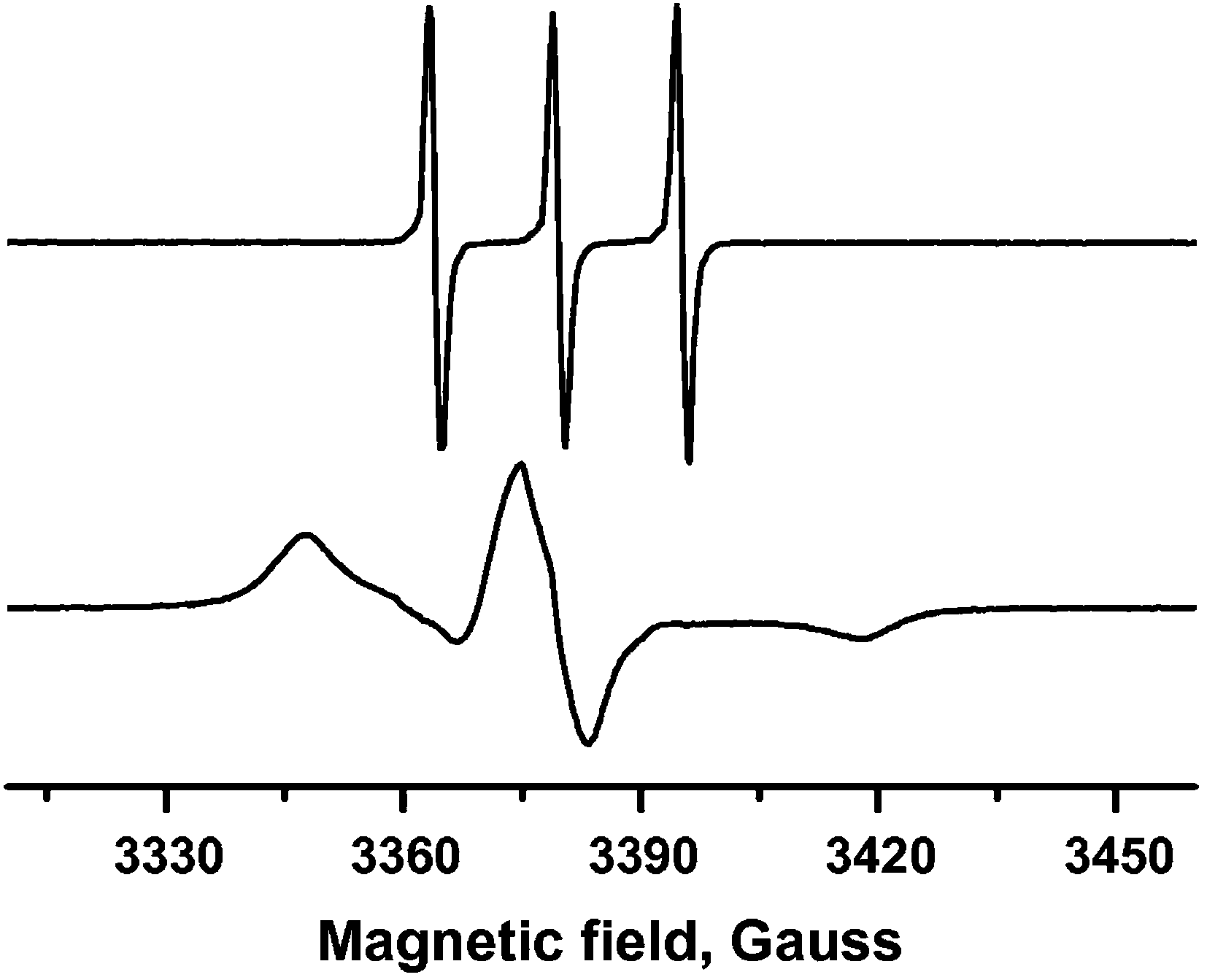 | ||
| Fig. 3 EPR spectra of (above) TEMPO in toluene and (below) TEMPO complex adsorbed on MIL-100(Al) (0.98 mmol g−1). Reprinted (adapted) with permission from ref. 43. Copyright (2014) Elsevier. | ||
TEMPO has also been used as a probe for the temperature-dependent structural rearrangement in MIL-53(Al). Guests within frameworks have a pronounced effect on the so called breathing of MOFs, but due to the high sensitivity of EPR, Bagryanskaya and co-workers have managed to incorporate very low levels (1/1000 TEMPO per unit cell) which allows investigation without perturbing the breathing process itself. MIL-53(Al) is a flexible MOF possessing pores which above ca. 350 K are large and below ca. 150 K are narrow. The transition between is these states is reversible and exhibits hysteresis such that both states are stable at 20 °C. The authors traced the EPR changes as MIL-53(Al)@TEMPO was heated/cooled to a range of temperatures, relying on the hysteresis to maintain the acquired structure upon returning to 20 °C. Upon the spatial constriction of TEMPO which is attained at low temperatures (see Fig. 4), its EPR signal is altered significantly. Multi-frequency EPR allowed calculation of the correlation times (τc) for nitroxide tumbling. At 80 K TEMPO is essentially immobilised, whilst at room temperature its τc is typical of nitroxides freely rotating in viscous solutions. It was thus possible to trace the breathing mechanism as a function of temperature. The authors were also able to elucidate aspects of the orientation of the probe and its reactivity with the framework.44
 | ||
| Fig. 4 Representation of TEMPO's confinement within MIL-53(Al); left, a large pore at high temperature and right, a narrow pore of at low temperature. Reprinted (adapted) with permission from ref. 44. Copyright (2014) American Chemical Society. | ||
These studies serve to demonstrate the increasingly routine use of radicals as diagnostic tools in MOF chemistry.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(R)N(O˙)R, see Fig. 5), are arguably the most extensively represented amongst radical containing 1D coordination polymers. One appealing property of these molecules is their propensity to ligate via both of their oxygens. In this way the radical is structurally integral to the building unit. However this binding mode has not yet been observed in higher dimensionality structures.
C(R)N(O˙)R, see Fig. 5), are arguably the most extensively represented amongst radical containing 1D coordination polymers. One appealing property of these molecules is their propensity to ligate via both of their oxygens. In this way the radical is structurally integral to the building unit. However this binding mode has not yet been observed in higher dimensionality structures.
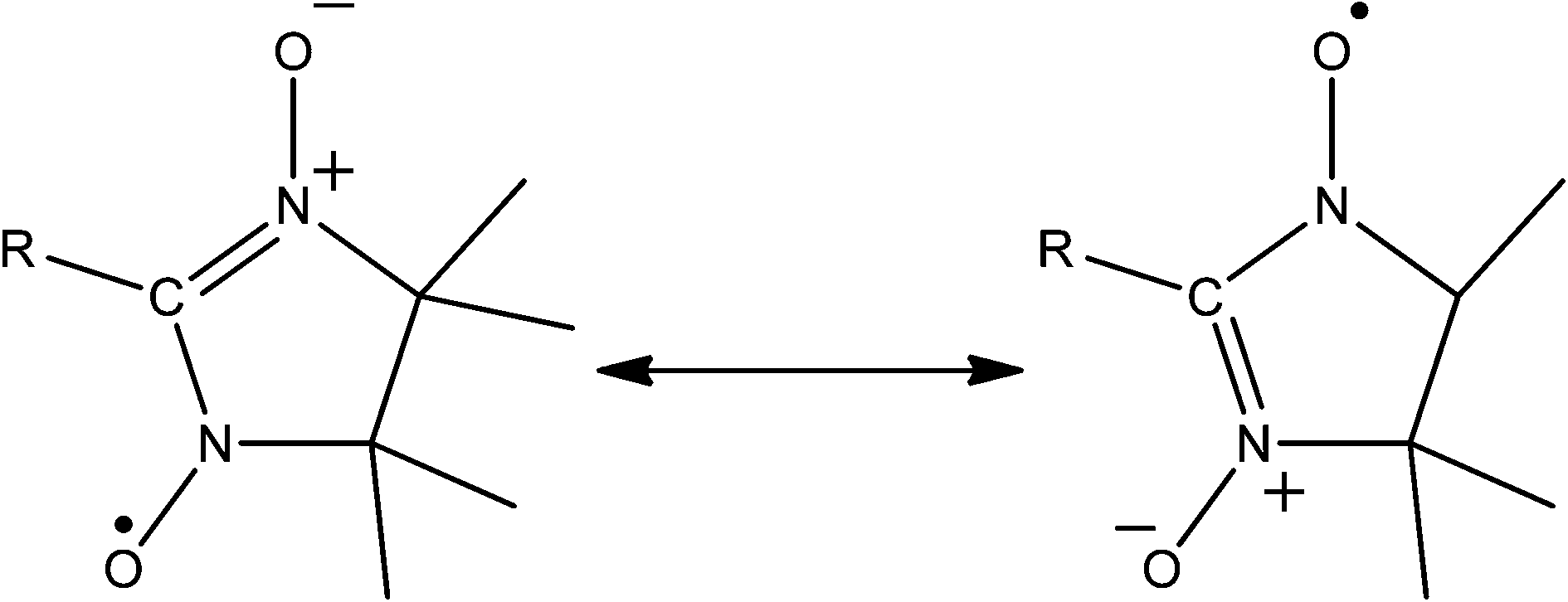 | ||
| Fig. 5 Resonance forms of a generic nitronyl-nitroxide. | ||
The only examples of nitronyl-nitroxides inclusion are as a metal appended pendants. For instance, in 2003 Mohanta and Wei reported a 3D coordination polymer, [Mn(II)(NIT-TZ˙)(DCA)2], where NIT-TZ˙ is the chelating ligand 2-(2-thiazole)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol-1-oxy-3-oxide radical (see Fig. 6) and DCA is the dicyanamide anion. The material exhibited strong metal–ligand AFM exchange and much weaker AFM metal–metal exchange through the DCA, as might be expected.45 More recently Kitazawa took advantage of a mineralomimetic aqua cadmium cyanide framework, into which 2-(4′-pyridyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (NIT4PY˙, see Fig. 6) was incorporated.46 The room temperature magnetic susceptibility revealed that the spins were essentially isolated from one another. Reducing the temperature revealed Curie–Weiss behaviour with a small negative Weiss constant consistent with weak anti-ferromagnetism which can be ascribed to interaction through the hydrogen bonded network.
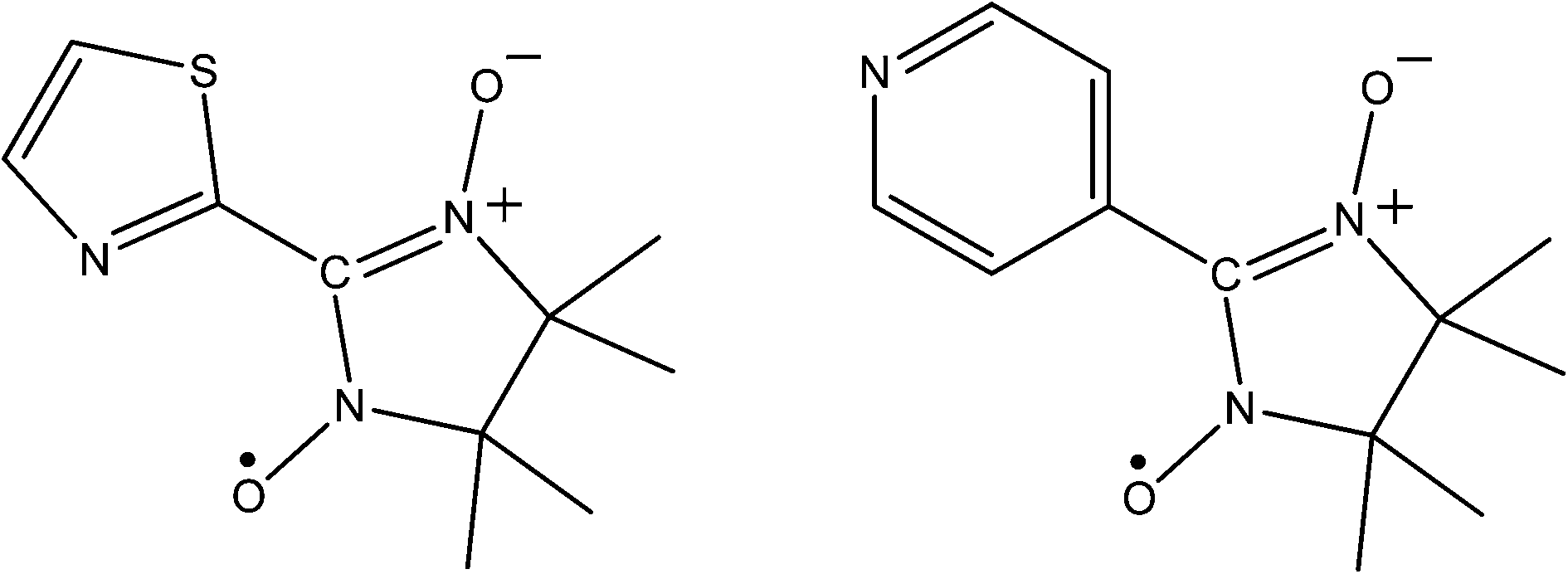 | ||
| Fig. 6 The radical ligands NIT-TZ˙ and NIT4PY˙. | ||
3.2 Organonitriles
Despite the initial discovery some two centuries ago of the first cyanide-based framework known as Prussian blue, coordination polymers based on cyanide ligands are the subject of ongoing scientific curiosity.47 Organonitrile ligands such as DCA (dicyanamide), TCM (tricyanomethanide), [N(CN)2]− and [C(CN)3]− have been used since the 1960's for the construction of network systems, underscoring the significant historical importance of the broad class of organonitriles in the development of the field.48 Organonitrile radical anions have also been extensively employed to develop 1D, 2D and 3D coordination polymers which have been of particular interest for fundamental studies of magnetically-coupled pathways.49Within the scope of this review which covers advances in the field over the last three years, the radical anion ligands of relevance are tetracyanoethylene (TCNE˙−) and 7,7,8,8-tetracyano-p-quinodimethane (TCNQ˙−), as shown in Fig. 7. These highly non-innocent ligands are capable of bridging up to four metal centres, and may exist in neutral π-acceptor form, as stable monoanionic radicals, or as dianions. The interconversion between the three redox states is facile and occurs in solution with a small reorganisation energy, such that these electron acceptors often acquire electrons from coordinating metal centres.
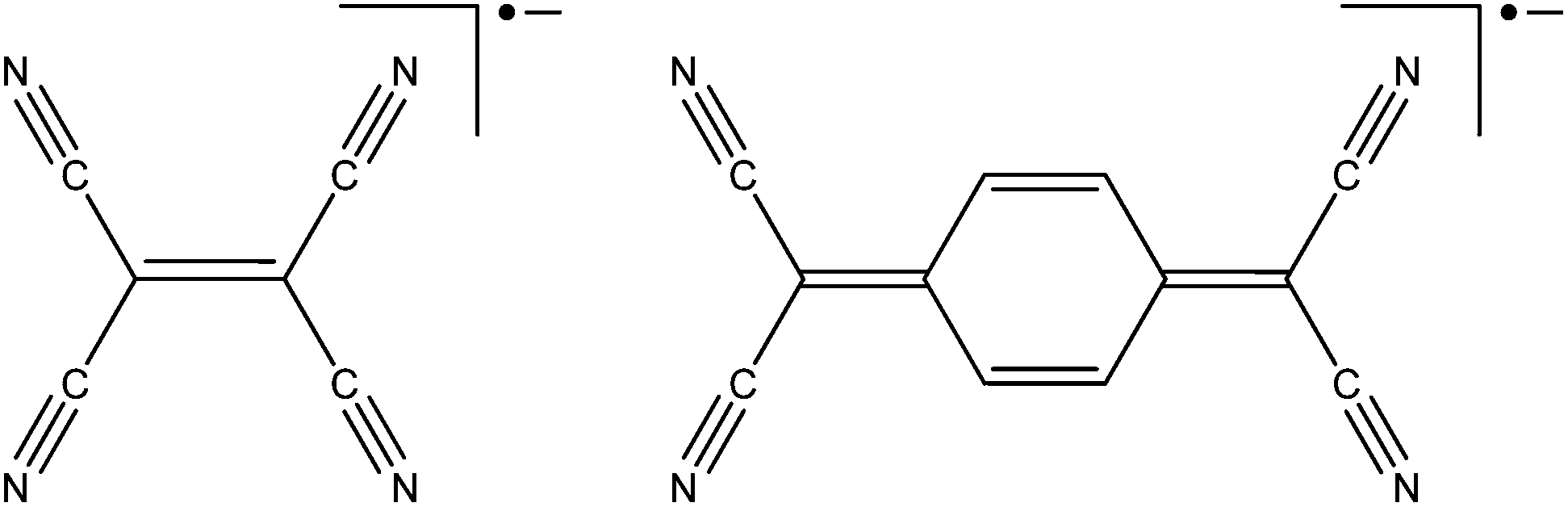 | ||
| Fig. 7 The TCNE˙− and TCNQ˙− radical anions. | ||
While numerous coordination polymers comprising the TCNE and TCNQ ligands in their various redox states have been realised, our focus here is on MOFs containing the radical states. Indeed, these radical anions have been intensively investigated as components of conducting organic charge transfer salts, particularly with TTF derivatives, by virtue of their propensity to undergo π–π stacking. In the same way that the physical properties of these purely organic systems have garnered significant decades-long attention, a growing number of studies are recognising the potential for incorporation of organonitrile radical anion ligands into MOFs. Investigations of the electrical conductivities, magnetic coupling and long-wavelength optical absorption features of such materials will underscore critical advances at the fundamental and applied levels. Importantly, there is potential to exploit the porosities of MOFs to engender multifunctionality beyond that achievable in purely organic charge transfer systems.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe).50 Two major families of 3D networks containing the radical anion ligand have been intensively investigated in recent years by Miller and co-workers, namely MII(TCNE)[C4(CN)8]1/2·zCH2Cl2 (M = Mn, Fe)51,52 and MnII(TCNE)3/2(I3)1/2·zTHF.53 In the former case, corrugated layers of MII bonded to four μ4-[TCNE˙−] (S = 1/2) are bridged by the diamagnetic μ4-[C4(CN)8]2− dimer (S = 0) to yield a structure commonly formulated as MII[μ4-[TCNE]˙−(μ4-[C4(CN)8]2−)1/2. Initial studies indicated that the isostructural Fe and Mn frameworks exhibited an unusual AFM ground state by virtue of direct AFM exchange coupling between μ4-[TCNE˙−] (S = 1/2) and MII (S = 5/2 for MnII and S = 2 for FeII) leading to 2D FM layers which couple AFM via conjugated –N
000 Oe).50 Two major families of 3D networks containing the radical anion ligand have been intensively investigated in recent years by Miller and co-workers, namely MII(TCNE)[C4(CN)8]1/2·zCH2Cl2 (M = Mn, Fe)51,52 and MnII(TCNE)3/2(I3)1/2·zTHF.53 In the former case, corrugated layers of MII bonded to four μ4-[TCNE˙−] (S = 1/2) are bridged by the diamagnetic μ4-[C4(CN)8]2− dimer (S = 0) to yield a structure commonly formulated as MII[μ4-[TCNE]˙−(μ4-[C4(CN)8]2−)1/2. Initial studies indicated that the isostructural Fe and Mn frameworks exhibited an unusual AFM ground state by virtue of direct AFM exchange coupling between μ4-[TCNE˙−] (S = 1/2) and MII (S = 5/2 for MnII and S = 2 for FeII) leading to 2D FM layers which couple AFM via conjugated –N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C–CN– superexchange pathways. It was recognised that their description differed from classical antiferromagnets in which a spin centre (or lattice) is adjacent to identical spins that couple antiferromagnetically, thus cancelling one another.51 A ferrimagnetic state should result if two adjacent spins differ in magnitude but do not cancel leading to a net magnetic moment. While MII(TCNE)[C4(CN)8]1/2·zCH2Cl2 is akin to a ferrimagnet in respect to its two different spin sites, the unusual antiferromagnetic ground state has led to its description as a compensated ferrimagnet.51 The magnetic behaviour was recently reassessed, and it was recognised that the FeII analogue exhibited even greater complexity than the MnII analogue. The observation of metamagnetism in the former was attributed to the single-ion anisotropy of high spin FeII (S = 2), in contrast to the isotropic high spin MnII ion (S = 5/2).
C–C–CN– superexchange pathways. It was recognised that their description differed from classical antiferromagnets in which a spin centre (or lattice) is adjacent to identical spins that couple antiferromagnetically, thus cancelling one another.51 A ferrimagnetic state should result if two adjacent spins differ in magnitude but do not cancel leading to a net magnetic moment. While MII(TCNE)[C4(CN)8]1/2·zCH2Cl2 is akin to a ferrimagnet in respect to its two different spin sites, the unusual antiferromagnetic ground state has led to its description as a compensated ferrimagnet.51 The magnetic behaviour was recently reassessed, and it was recognised that the FeII analogue exhibited even greater complexity than the MnII analogue. The observation of metamagnetism in the former was attributed to the single-ion anisotropy of high spin FeII (S = 2), in contrast to the isotropic high spin MnII ion (S = 5/2).In view of the technological importance of magnetic switching as a useful property to endow multifunctional behaviour, a number of studies have explored the application of external stimuli such as light and pressure to alter the magnetic ordering of materials for applications in sensors, transducers and actuators.50 Recent work by Miller and co-workers on MnII(TCNE)[C4(CN)8]1/2·zCH2Cl2 demonstrated that the material exhibits a reversible pressure-induced piezomagnetic transition under an applied hydrostatic pressure.52 Specifically, the AFM ground state was transformed to an induced ferrimagnetic state with an enhanced TC up to 97 K. The origin of the switching was ascribed to structural changes in the framework above an applied pressure of 0.5 ± 0.15 kbar due either to the formation of cis-μ4-[TCNE˙−] between the layers, or incomplete cancellation of the moments due to spin canting.
The related framework MnII(TCNE)3/2(I3)1/2·zTHF has also been reported to exhibit a pressure-induced enhancement in the magnetic properties.53 The structure consists of layers of octahedrally-coordinated MnII ions that are bound to four μ4-[TCNE˙−] ions to form corrugated layers; each MnII is bound to two additional μ4-[TCNE˙−] that bridge the layers in a planar fashion, giving rise to a 3D ferromagnetic structure with one-half of a I3− counter anion per MnII residing in the pores. Interestingly, a 2D analogue of the structure, namely MnII(TCNE)I(OH)2 which exhibits MnII ions bound to four μ4-[TCNE˙−] ligands that does not, however, display interconnectivity between the layers, was characterised by a more marked pressure dependence up to 14 kbar.53 While the behaviour was attributed to the reduction in interlayer spacing upon pressurisation, the nature of the coupling mechanism requires further investigation. These results suggest that 2D coordination polymers, or 3D frameworks with flexible bridges between 2D layers may also be of great interest for future exploration of materials with piezomagnetic transitions.
In addition to coordination polymers containing TCNE˙−, the radical anion is well-known to undergo dimerisation in the presence of alkali cations to form π-[TNCE2]2−. A notable anomaly was recently reported in which the structure of Li+[TCNE˙−] was elucidated using Rietveld refinement.53,54 The material was shown to contain the radical anion form of the ligand μ4-[TCNE˙−], which was bound to four tetrahedral Li+ ions. The extended 3D diamondoid topology exhibits 2-fold interpenetration, and magnetic studies suggested that the spins on the two magnetic sub-lattices are slightly canted, giving rise to a transition from a paramagnetic state to a weak FM (canted antiferromagnetic) ground state.
Two important themes of emerging research interest are TCNQ-based network structures incorporating main group metals59,60 and electrosynthesised TCNQ-based phases incorporating transition metal ions.61–63 These materials offer noteworthy examples of semiconducting materials containing TCNQ˙− that can be fabricated as macroscopic structures. Critically, these networks often exist in different phases which exhibit markedly different magnetic and electronic properties, highlighting the significant potential that exists for the incorporation of these radical ligands into frameworks with diverse and potentially exotic physical properties.
3.3 Tetrathiafulvalene
Tetrathiafulvalene (TTF) is an unsaturated but non-aromatic organosulfur bis-heterocyclic moiety, notable for its easily accessible redox chemistry. In its charge neutral state it is diamagnetic; upon one electron oxidation it becomes partially aromatised and is stabled as a radical cation. Loss of a second electron provides access to the fully aromatised diamagnetic dication as shown in Fig. 8. Charge transfer complexes containing TTF derivatives have been the target of intense interest since the report of metallic conductivity in the aforementioned TTF-TCNQ charge transfer salt in 1973.56 It has since developed to include many other organic and inorganic TTF salts, most notably the family of Bechgaard salts which exhibit superconductivity at low temperatures. A good account of these materials is given in the review by Coronado and Day64 and a concise chronological account of TTF development is given by Martín.65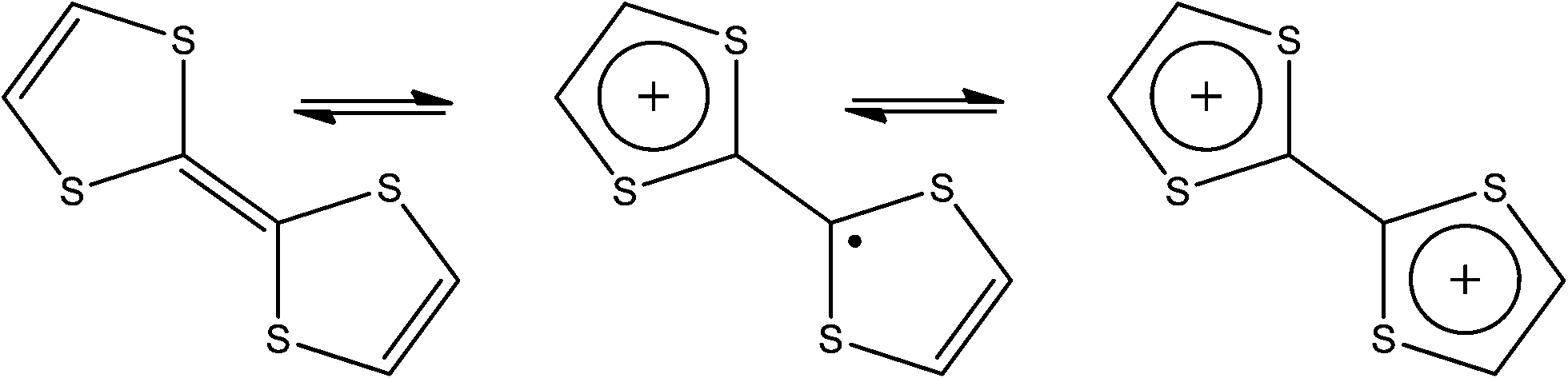 | ||
| Fig. 8 Oxidation states of TTF, from left to right; neutral species, radical cation, diamagnetic dication. | ||
Incorporation of TTF as a guest within a framework can be relatively facile as demonstrated by Fujita and co-workers in 2010.66 The diffusion of TTF in toluene into the pores of single crystals of (Co(SCN)2)3(TPT)4, (TPT = 2,4,6-tris(4-pyridyl)-1,3,5-triazine) was concomitant with a colour change from orange to black as a result of a new absorption band at 500–900 nm caused by a broad charge-transfer band. The authors suggest that guest recognition occurs through π-stacking and electrostatic interactions between the electron rich TTF and highly electron deficient TPT. The study is nicely complemented by analogous studies with a discrete cage complex possessing a void similar to one existing within the 3D framework.
Advancements in the synthetic expertise of TTF derivative preparation have allowed a broad spectrum of polyfunctionalisation which may permit it to exist as a structurally integrated or pendant radical.67,68 As such their inclusion might be envisioned in a large number of MOFs, and yet there exists a paucity of such materials in the literature.
TTF-tetracarboxylic acid (TTF-TC)H4 (see Fig. 9) is somewhat reminiscent of 3,3′,5,5′-biphenyltetracarboxylic acid (BPTC)H4 used in MOF-501 and MOF-502.69 Its use is limited to a very small number of frameworks probably due to its low thermal and chemical stability, however Devic and co-workers did report the successful synthesis of a range of non-porous 3D coordination polymers with alkali metals in the series MIL-132 to MIL-134 incorporating (TTF-TC)H22− at modest temperatures, as well MIL-135 containing the oxidised (TTF-TC˙)H24−, which was synthesised under electro-(sub)hydrothermal synthetic conditions.70 None of these frameworks possess potential voids nor good TTF π-stacking which is required for charge mobility.
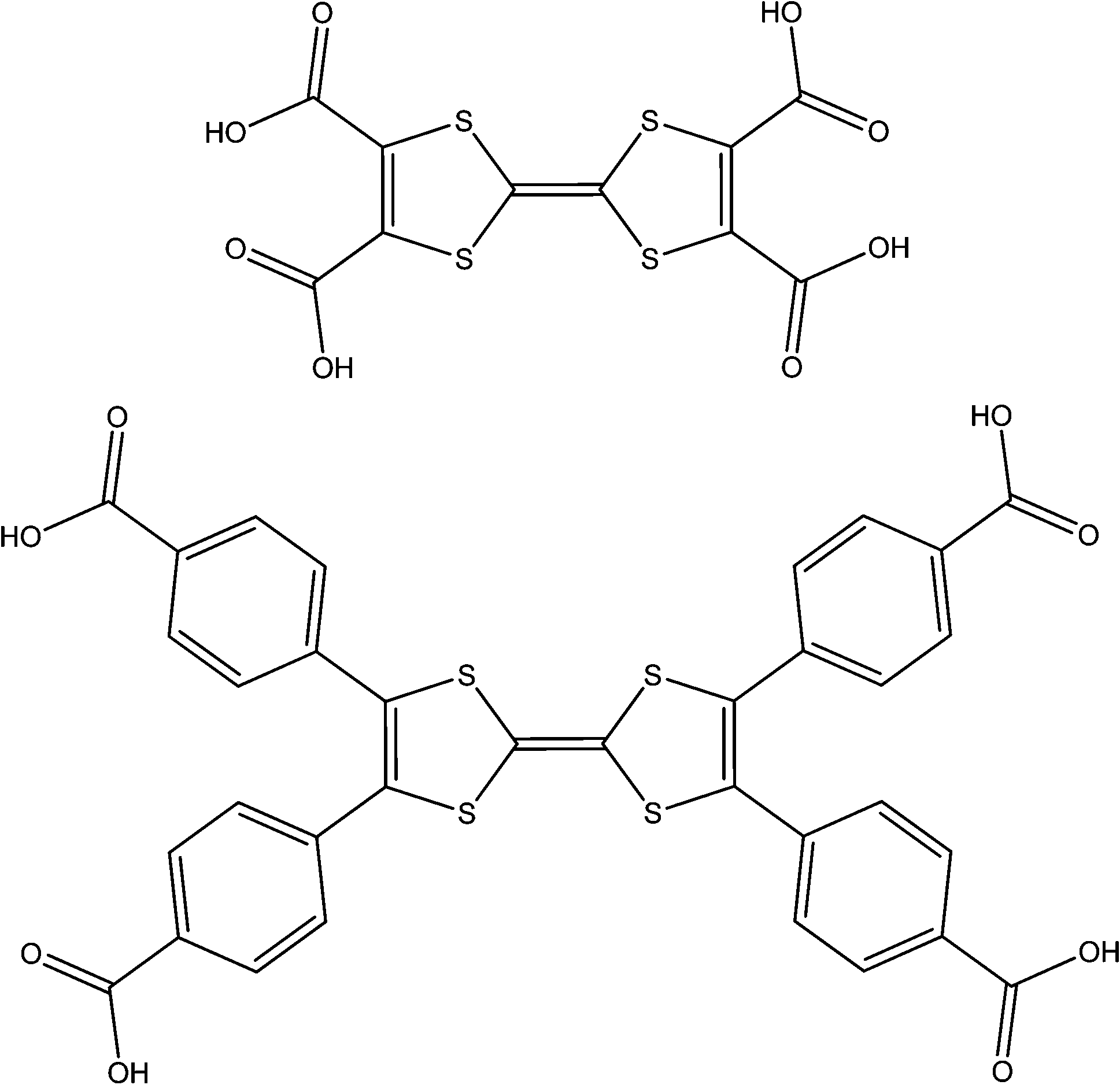 | ||
| Fig. 9 The ligands (TTF-TC)H4 and (TTF-TB)H4. | ||
Zhu and Dai and co-workers also synthesised a 2D framework Mn(TTF-TC)0.5(PHEN)(H2O)2·nH2O and 3D framework, Mn(TTF-TC)0.5(BIPY)·nH2O, at room temperature. For both structures the TTF exists in it diamagnetic neutral state but can undergo oxidation in two steps as evidenced by cyclic voltammetry, however no magnetic data was reported for the oxidised frameworks.71 Similar frameworks also exist with different arms, including ones with 2-fold (rather than 4-fold) symmetry.72,73 While the redox-activity of these systems is generally demonstrated, their radical states are neither measured magnetically nor exploited for other purposes.
One 2D framework, presented by Golhen and Ouahab and coworkers, is reported as being in the oxidised state as synthesised, despite appearing that the radical does not contribute to the magnetic response. However spectroscopic data does support the notion, and furthermore the cobalt analogue of the series, Co(TCE-TTF˙+)2Co(H2O)4](BF4)6·2H2O, (TCE = 2,3,6,7-tetrakis(2-cyanoethylthio)), exhibits semiconductor behaviour (though experimental evidence to support this statement is absent).74
One strategy to increase framework stability is by spatially separating the redox active moiety from the carboxylic acids as achieved by Dincă and colleagues in the synthesis of TTF-tetrabenzoic acid (TTF-TB)H4 (see Fig. 9). The extra aryl groups also expand the ligand such that, in the resulting framework, permanent porosity is achieved. The crystal structure revealed infinite helical stacks of TTF-TB with short interlayer distances (shortest interlayer S⋯S = 3.80 Å) (see Fig. 10). Existing as TTF-TB4−, the linker is diamagnetic but EPR revealed a sharp signal consistent with inherent partial radical doping. Both of these characteristics are positive markers for good charge mobility and indeed measurement revealed an excellent intrinsic charge mobility of 0.2 cm2 V−1 s−1.75
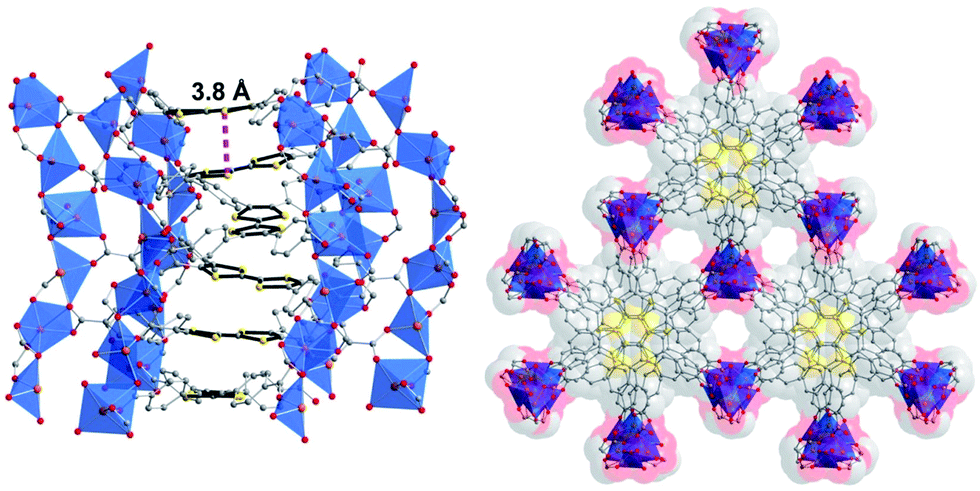 | ||
| Fig. 10 Dincă's TTF-TB MOF showing inter-layer separation and helical arrangement of TTF units. Reprinted (adapted) with permission from ref. 75. Copyright (2012) American Chemical Society. | ||
3.4 BIPO˙−
The development of coordination chemistry with 2,3′-biimidazo[1,2-α]pyridin-2′-one (BIPO˙−, see Fig. 11) has been exclusively pursued by Yong and co-workers. As well as salts76 and 1D polymers,77,78 the team has been successful in synthesising 1D, 2D and 3D structures.79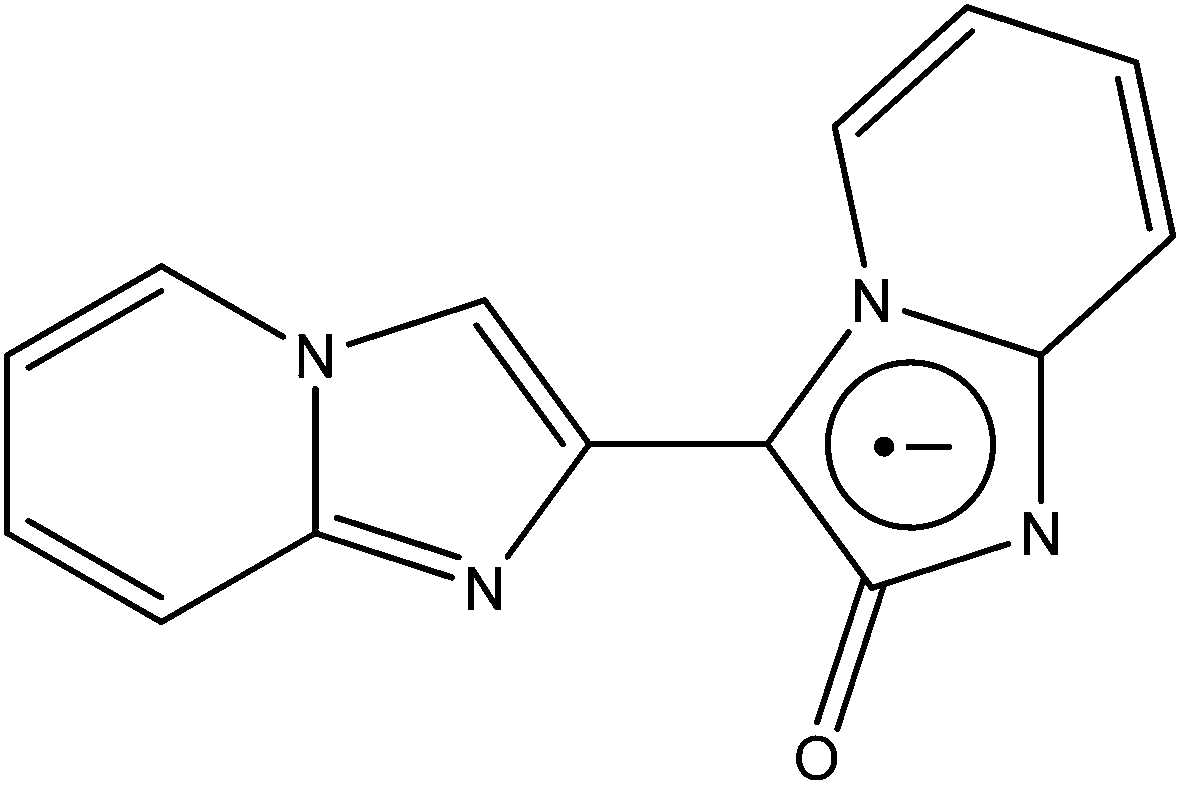 | ||
| Fig. 11 The radical ligand BIPO˙−. | ||
Cd3(BIPO˙−)4(BDC) is a 3D MOF in which a 1D spirocyclic chain of Fe ions is bridged by BIPO radicals extends along the c axis, while BDC ligands chelate the non-spirometals such that they extend in an alternating fashion along the a and b axes (see Fig. 12). The resultant framework is then interpenetrated with a second identical lattice. Since Cd2+ is a d10 diamagnetic ion, magnetic measurements revealed the interaction of the radicals, which proved to be AF.
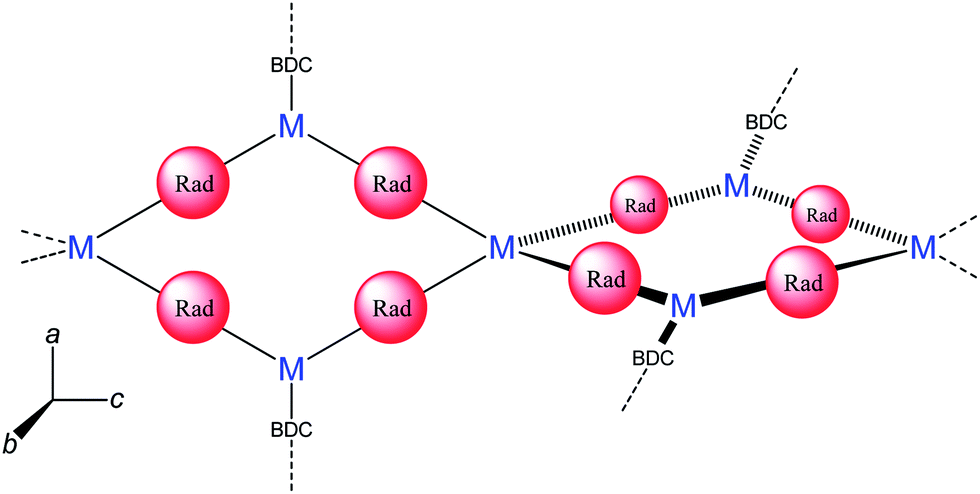 | ||
| Fig. 12 Schematic fragment of Cd3(BIPO−)4(BDC) chain. | ||
Fe3(BIPO˙−)4(BDC) is essentially isostructural with Cd3(BIPO˙−)4(BDC) described above, however the difference in metal radii means that the BDC can no longer chelate to bridge the spirochains (Fe⋯O = 2.535 Å). Strictly speaking this renders the material 1D, but we include it here since its properties appear highly relevant to the discussion. The χMT value at RT is higher than that expected for isolated spins, while χMT displays a continuous decline with decreasing temperature tending towards zero at 0 K. Clearly the ground state is not FM, and it is apparent that intrachain AFM exchange would not lead to a diamagnetic ground state; due to the mismatch in spin amplitude between the Fe(II) (S = 2) and radical (S = 1/2), a ferrimagnetic chain would result. In the absence of more data it would appear that interchain AFM exchange leads to a diamagnetic material.
The 2D framework, Fe3(BIPO˙−)2(μ2-OH)2(μ2-H2O)(BDC), consists of a magnetically rich 1D chain which is extended into two dimensions via BDC2− ligands. The 1D chain has a fairly complicated structure; the BIPO˙− ligand coordinates to three metals in a 3.121 mode (Harris notation).80 Fe1 bridges to Fe2 through (i) the backbone of one BIPO˙, (ii) a bridging carboxylate and (iii) a μ2-oxygen from a second BIPO˙. Fe2 bridges to Fe3 though (i) a μ2-oxygen from a BIPO˙, (ii) a μ2-hydroxyl and (iii) a μ2-water. Finally, Fe3 bridges to Fe1 through (i) the backbone of one BIPO˙, (ii) a μ2-hydroxyl and (iii) a bridging carboxylate (see Fig. 13). It is therefore difficult to ascribe exact exchange mechanisms to the empirical observations, which like the previous example exhibits a continuous decrease in χMT with a decrease in temperature (though not to zero) from a RT value higher than that expected for isolated spins. The influence of the radical on the chain is difficult to ascertain, especially since we might expect exchange through the BIPO˙ backbone to be somewhat weaker than the other competing pathways.
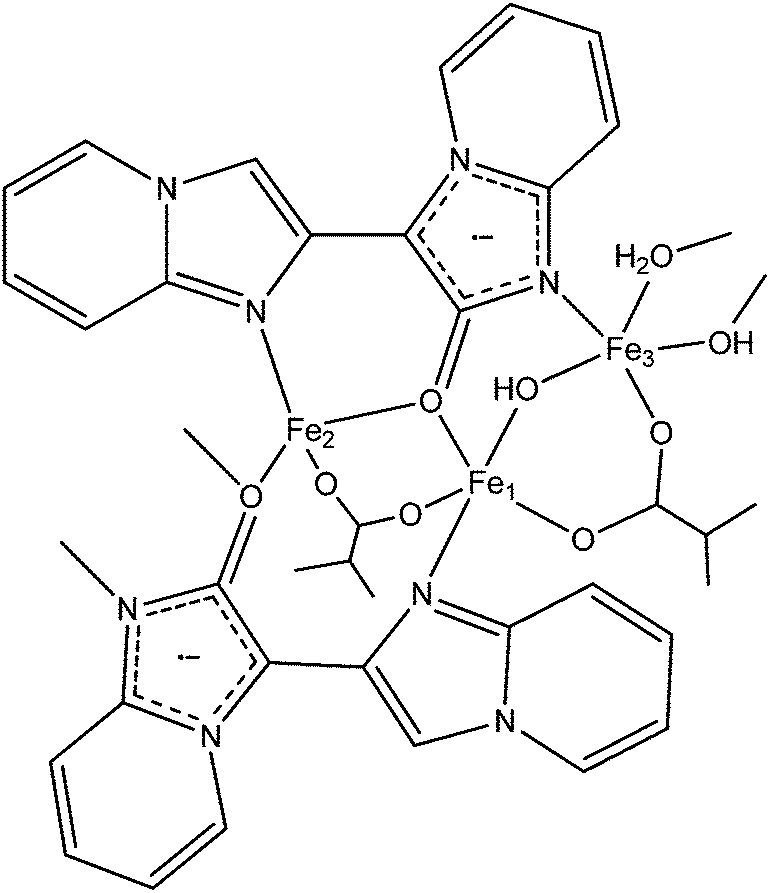 | ||
| Fig. 13 Schematic diagram of Fe3(BIPO˙−)2(μ2-OH)2(μ2-H2O)(BDC) fragment. (Dangling bonds from N/O coordinate to a metal centre on the adjacent fragment, only partial BDC ligands shown.) | ||
While the exchange processes in these last two examples are not well understood, Yong and co-workers have demonstrated a key aspect which has been instrumental in the magnetic ordering previously observed in SMMs, namely the bridging of paramagnetic metal ions through very short bridging units such as carboxylates and μ2-oxygens.
These three frameworks were also studied for their luminescence properties. All three exhibited fluorescence to some degree, despite the presence of iron in the second and third materials which tends the quench such phenomena. In each case the emission was slightly altered from that of the bare radical ligand, which was attributed to ligand–metal interactions.
3.5 Semiquinone radicals
The availability of isolable stable radicals means that the majority of organic centred spin is present prior to MOF synthesis, however spin generation is also possible in concert with framework construction. This has recently been achieved by two groups using solvothermal synthesis techniques involving 1,4-benzoquinone derivatives. Monge and co-workers reported the reaction of the sodium salt of anthraquinone disulfonate (AQDS2−, see Fig. 14) with lanthanoid salts in carefully controlled conditions to yield a MOF with bilayer 63 honeycomb topology and integral AQDS˙3− radical anions.81 The radical anions are π stacked in perfect face-to-face columns with small interlayer distances (alternating ca. 3.4 and 3.7 Å) which are often accompanied by high conductivity. Indeed a single crystal measurement revealed semi-conductor behaviour with very high charge mobility.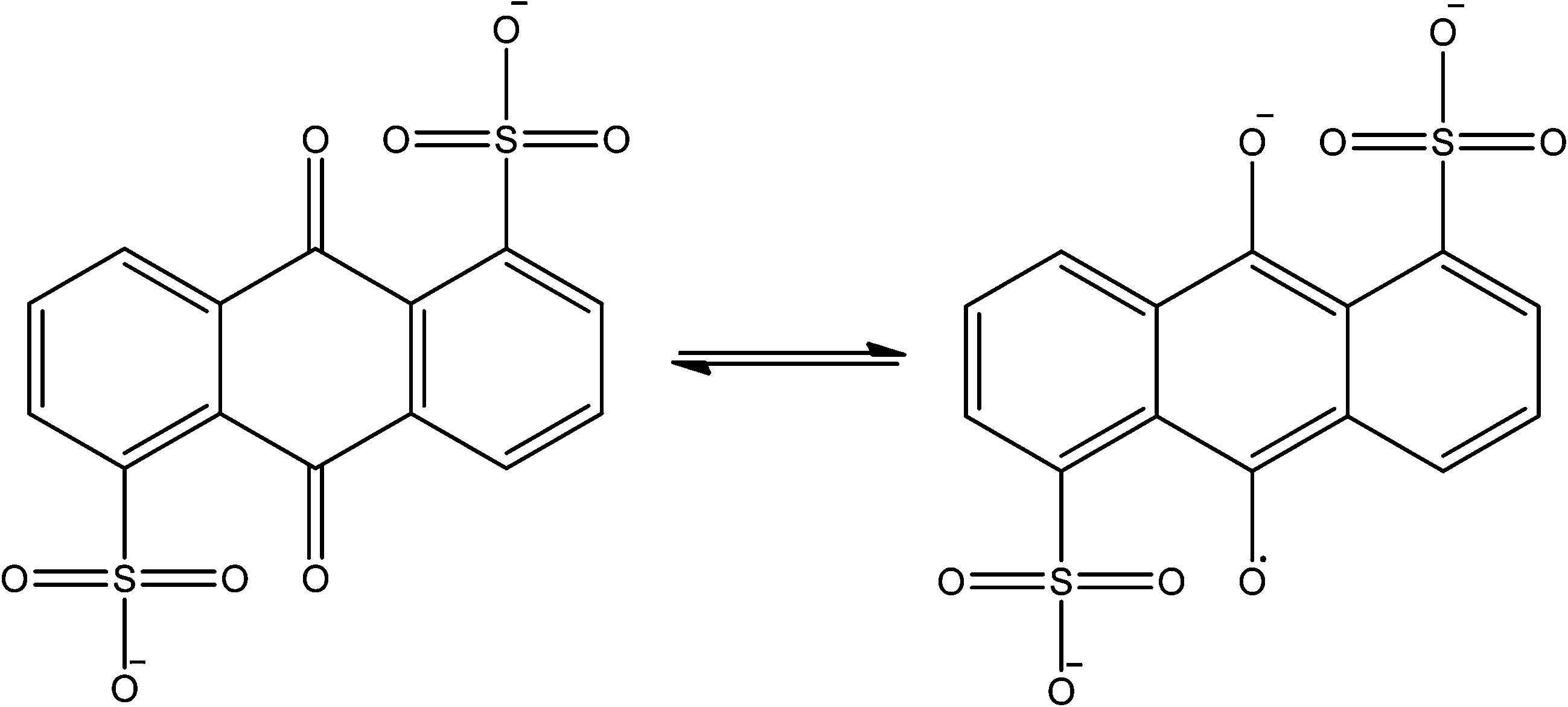 | ||
| Fig. 14 The AQDS2−/AQDS˙3− redox couple. | ||
Yong and co-workers go one step beyond Monge by synthesising the quinone precursor (pyrido[1,2-α]pyrido[2′,1′:2,3]imidazo[4,5-f]benzimidazole-6,13-dione) in situ which was subsequently reduced to the semi-quinone radical (see Fig. 15) before incorporation in the framework.82 In this case the reaction of the radical with copper(II) chloride resulted in a mixed valent copper chloride double-stranded corrugated chain, bridged into two-dimensions by the semi-quinone. The semi-quinone species are π stacked in a slightly offset manner with a consistent interlayer distance of just 3.33 Å. Such a system should deliver very good electrical conductivity, however there are no reports of the necessary measurements to-date.
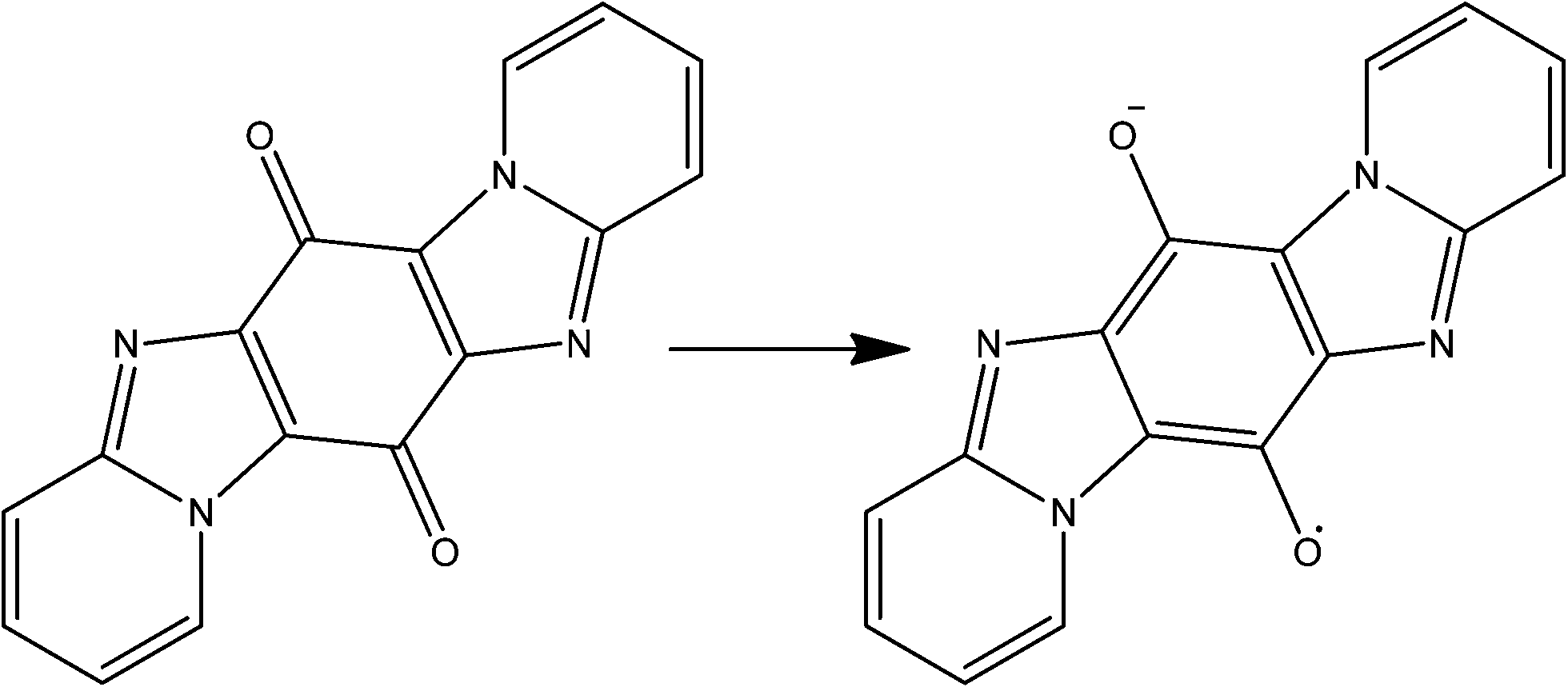 | ||
| Fig. 15 Monge's quinone/semi-quinone. | ||
3.6 Pyridinium radicals
N-Substituted-4,4′-pyridiniums and the better known N,N′-disubstituted-4,4′-bipyridiniums (viologens) whilst not themselves radicals, are often readily photoreduced to highly coloured radical species. The group of Fu has successfully utilised N-(3-carboxyphenyl)-4,4′-bipyridinium (CPBPY, see Fig. 16) to generate MOFs which upon radiation can maintain viologen radicals.83,84 Subsequent exposure to the atmosphere allows a return to the diamagnetic state, a process which can be accelerated thermally. The two 3D MOFs are composed of d10 metals with either meta- or para-benzene dicarboxylate, and are both doubly interpenetrated. The long lived nature of the pyridinium radical can be ascribed to the condensed packing within the crystal which restricts diffusion of quenching oxygen. The interpenetration also brings together the electron donor carboxyl units and pyridyl acceptor units into sufficiently close proximity for intermolecular charge transfer.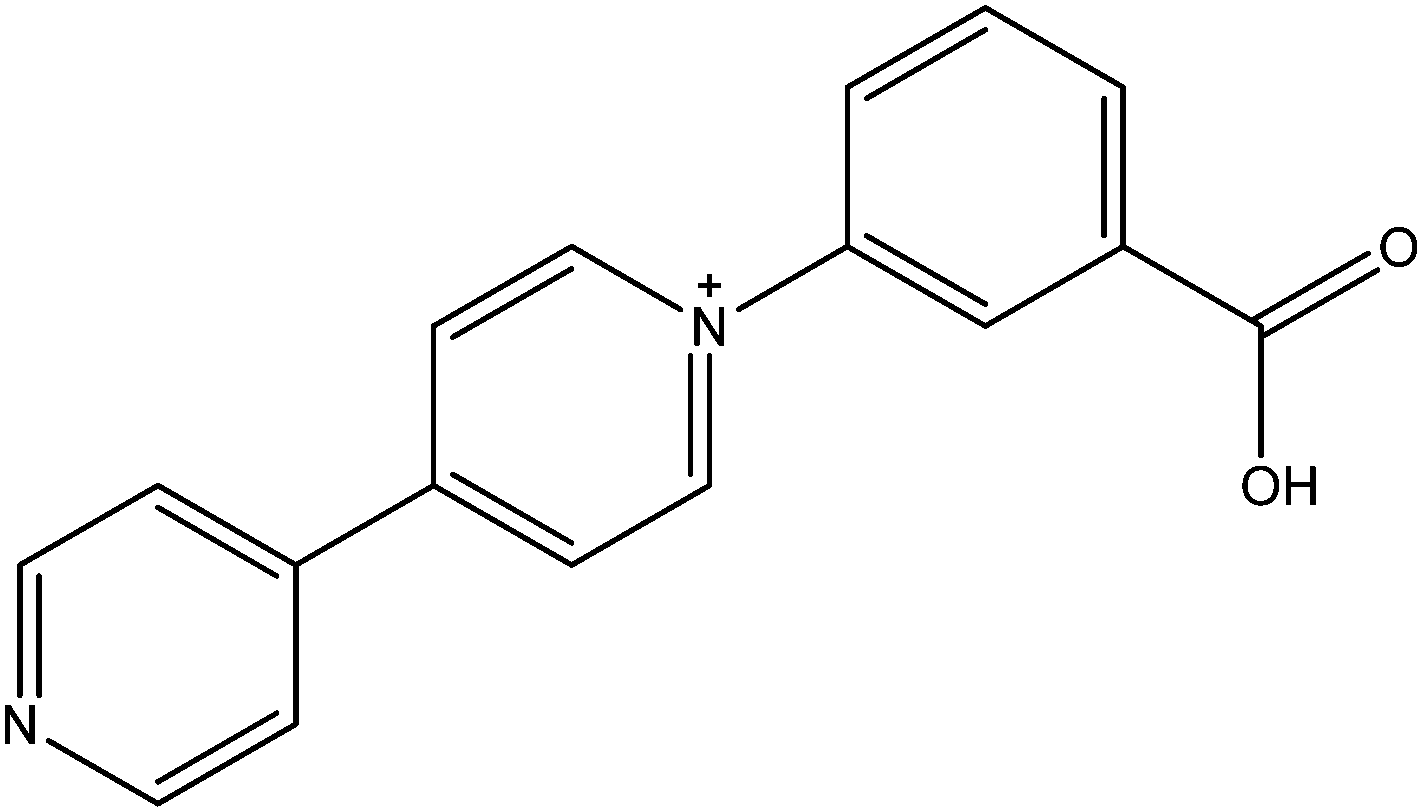 | ||
| Fig. 16 The viologen CPBPY. | ||
Using 1,3,5-benzenetricarboxylate with CPBPY the Fu group has also synthesised a 2D MOF with a three-fold accordion interpenetrating mode. Such is the density of packing that the radical framework is stable under ambient conditions for in excess of one month. Furthermore, the material is switchable between the states, which offer different fluorescent and photochromic behaviours upon application of UV light and thermal energy (see Fig. 17).
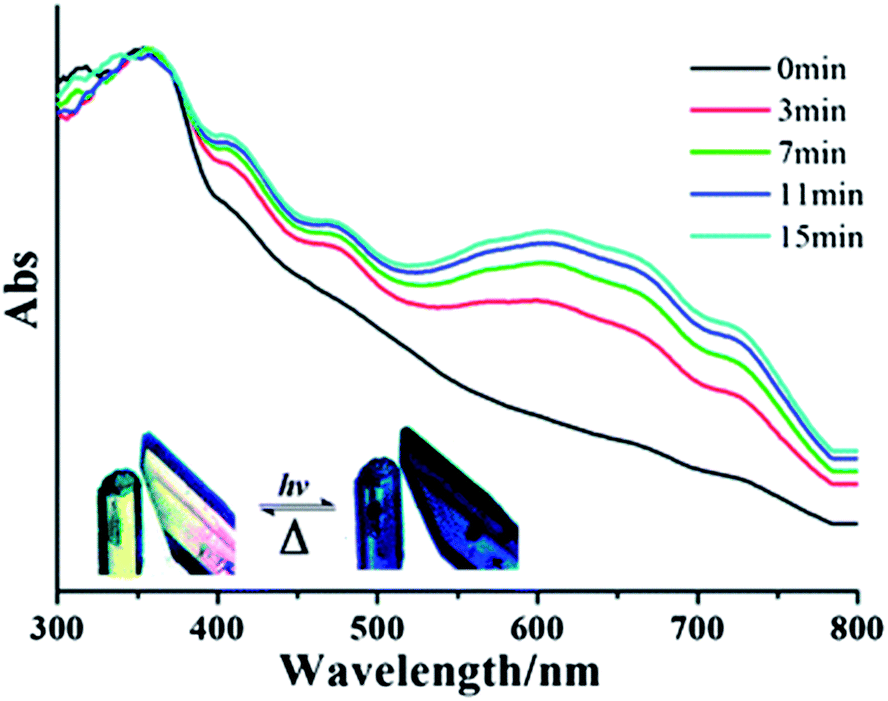 | ||
| Fig. 17 UV-vis spectral changes of Fu's close packed 2D MOF upon photoirradiation. Reprinted with permission from ref. 83. Copyright (2012) Royal Society of Chemistry. | ||
3.7 Triarylmethyl radicals
This class of radical, which are derivatives of the very first radical to be characterised,85 have been mostly pursued by Veciana and co-workers. Since the publication of Cu3(PTMTC˙)2(py)6(CH3CH2OH)2(H2O), (PTMTC˙ = polychlorinated triphenylmethyl tricarboxylate radical, see Fig. 18), MOROF-1 (MOROF = metal–organic radical open-framework) in 2003,86 they have included the same radical ligand in frameworks with cobalt in MOROF-2 (ref. 87) and MOROF-3 (ref. 88) and with terbium.89 The higher denticity polychlorinated triphenylmethyl hexacarboxylate (PTMHC˙) has also been incorporated into a copper framework.90 All of this work has been well summarised by the authors previously.91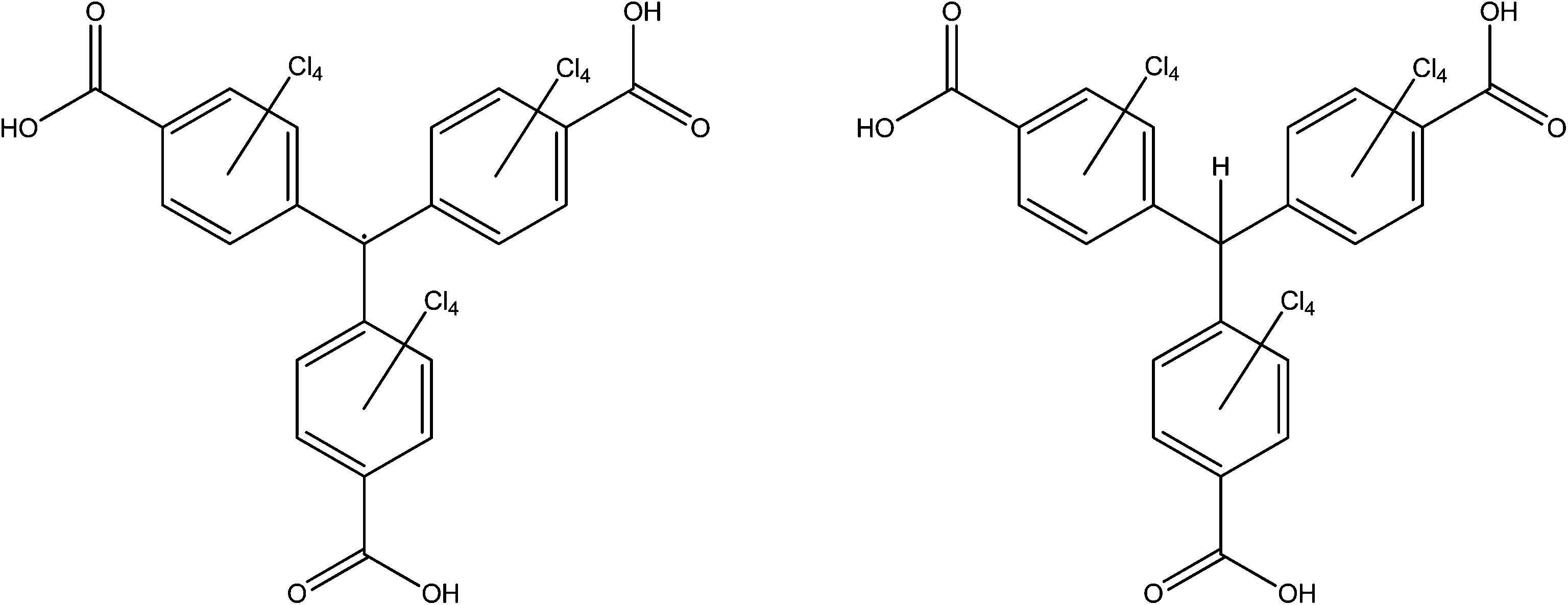 | ||
| Fig. 18 The ligands (PTMTC˙)H3 and (αH-PTMTC)H3. | ||
Since 2011, they have published two further closely related series Ln(DMF)3(PTMTC˙) (Ln = Sm, Eu, Gd, Tb, Dy),92 and Ln(PTMTC˙)(EtOH)2H2O·xH2O,yEtOH (Ln = Tb, Gd, Eu),93 which differ slightly in their formulae and preparation but considerably in their properties.
A principal target for the use of radicals and lanthanoid ions was to create a multifunctional material possessing both magnetic and luminescent properties. The authors elegantly demonstrated the effect of the radical by additionally synthesising isostructural frameworks containing the closed-shell analogue of PTMTC˙, αH-PTMTC (see Fig. 18). In luminescent-active metals, luminescence was observed for all frameworks containing αH-PTMTC but none containing PTMTC˙. This can be attributed to quenching of the luminescence by the radical ligand. Furthermore, the ability to analyse the magnetic properties of both open- and closed-shell analogues allowed a comparison of the magnetic exchange both with and without a radical mediator. Importantly, they collected crystal structures of both to confirm that the structures were adequately similar to allow fair comparison. As expected, room temperature measurements were in accordance with isolated spins. Low temperature measurements revealed that lanthanoid-radical exchanges in PTMTC˙ frameworks were weakly FM, while data from the αH-PTMTC frameworks showed that lanthanoid–lanthanoid interactions were weakly AFM. Analysis of the europium frameworks permitted quantification of the radical–radical exchange since the ground state of europium(III), which is exclusively populated at low temperature, is non-magnetic (f6, 7F0). This suggested that radical–radical exchange was AF.
The weakness of the exchange precludes long range ordering, at least above 2 K, but nonetheless the proof of principal has been established, and there is broad scope for increasing the magnitude of exchange.
3.8 Triarylamminum radicals
Isoelectronic with triarylmethyl radicals are triarylamminum radicals which are readily accessed from triarylamines by intermolecular charge transfer with a suitable electron acceptor. Whilst employed extensively in a range of mixed valent,94 donor–acceptor,95 and organic–polymeric systems,96 their incorporation into any kind of 3D inorganic system appears extremely limited. Two recent publications report the tris(4-(pyridin-4-yl)phenyl)amine ligand incorporated in 3D frameworks with copper97 and manganese,98 though both are highly interpenetrated with limited, if any, pore space. Secondly, generation of the radical state was not trivial; in the former case the radical cation was unstable and in the latter, generation of the ammonium was generally concomitant with oxidation of Mn(II) to Mn(III) which likely led to ligand dissociation and framework degradation.3.9 Thiazyl radicals
Whilst thiazyl radicals are included in a great deal of inorganic literature such as salts99,100 and 1D coordination polymers101,102 there exist no MOFs with a thiazyl unit integrated within the framework scaffold. There are reports of thiazyl guests within a MOFs such as that given by Rawson and co-workers in 2011.103 The system consisted of benzodithiazolyl (BDTA˙) or methylbenzodithiazolyl (MBDTA˙) radicals (see Fig. 19) within the well-known MOF, MIL-53(Al).104 By virtue of π*–π* dimerisation, these radicals tend to be EPR silent when arranged in their lowest energy head-to-head stacking arrangement, though the energy jump to head-to-tail is very small. X-ray diffraction studies show that confinement of BDTA˙ pairs within the MIL-53 cavity is relatively facile due to favourable size matching. Conversely, the extra methyl in MBDTA˙ causes distortion of the framework and disorder in the orientation of the radical (see Fig. 20), the latter presenting as a sharper, more intense EPR signal. It is therefore evident that confinement of species within a MOF can alter the behaviour of its guests.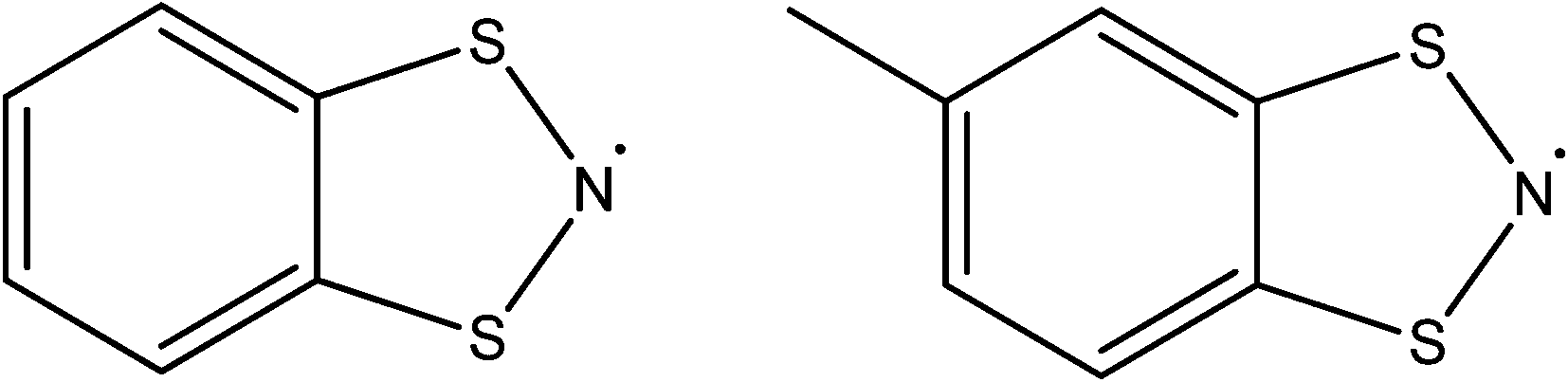 | ||
| Fig. 19 The radical guests BDTA˙ and MBDTA˙. | ||
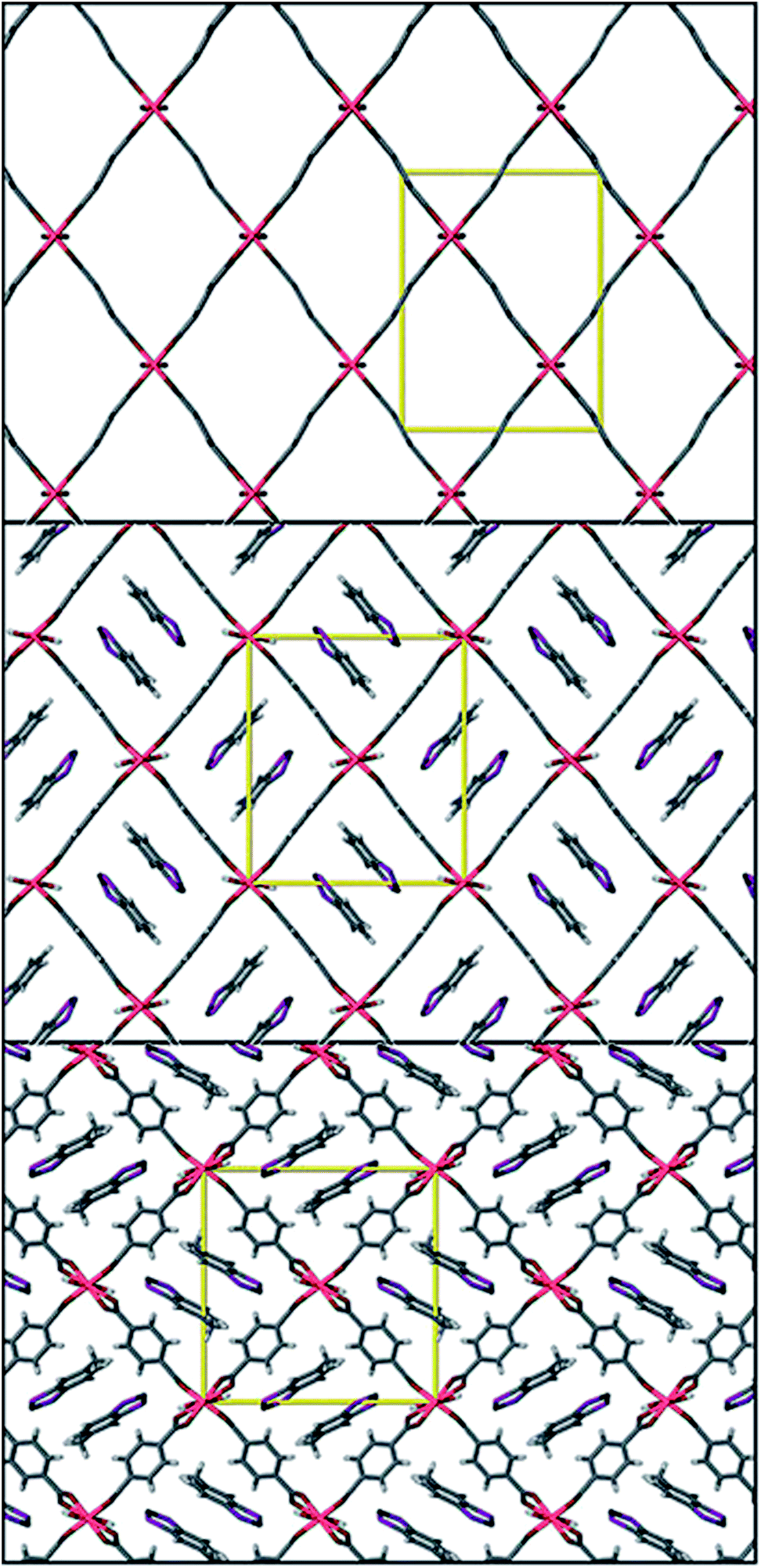 | ||
| Fig. 20 Top to bottom; MIL-53(Al) viewed down the a axis, MIL-53(Al)@ BDTA˙ and MIL-53(Al)@MBDTA˙ both viewed down the c axis. Reprinted with permission from ref. 103. Copyright (2012) American Chemical Society. | ||
3.10 Aromatic diimides
The chemistry and properties of aromatic diimides, particularly naphthalenediimides (NDIs, see Fig. 21)105 and their inclusion in MOFs106 have been reviewed previously. In brief, they are typically electron deficient ligands whose reduction is reversible and achievable at modest potentials. A large number of papers report MOFs including NDIs (particularly as bispyridyl-NDI) for their structural characteristics (length, rigidity, planarity) and ease of synthesis rather than for their electrochemical properties. Whilst it has been established that the monoradical anion and dianion forms of NDI can be accessed via in situ electrochemistry,107 most reports discuss the chemochromic,108 photochromic109 and electrochromic110 properties arising from the electron deficiency of the NDI core. The redox activity of the related pyromellitic diimide unit has been exploited for its variable affinity for the quadrupole moment of CO2, and such observations suggest exciting potential for MOFs incorporating such ligands in electrical swing adsorption.111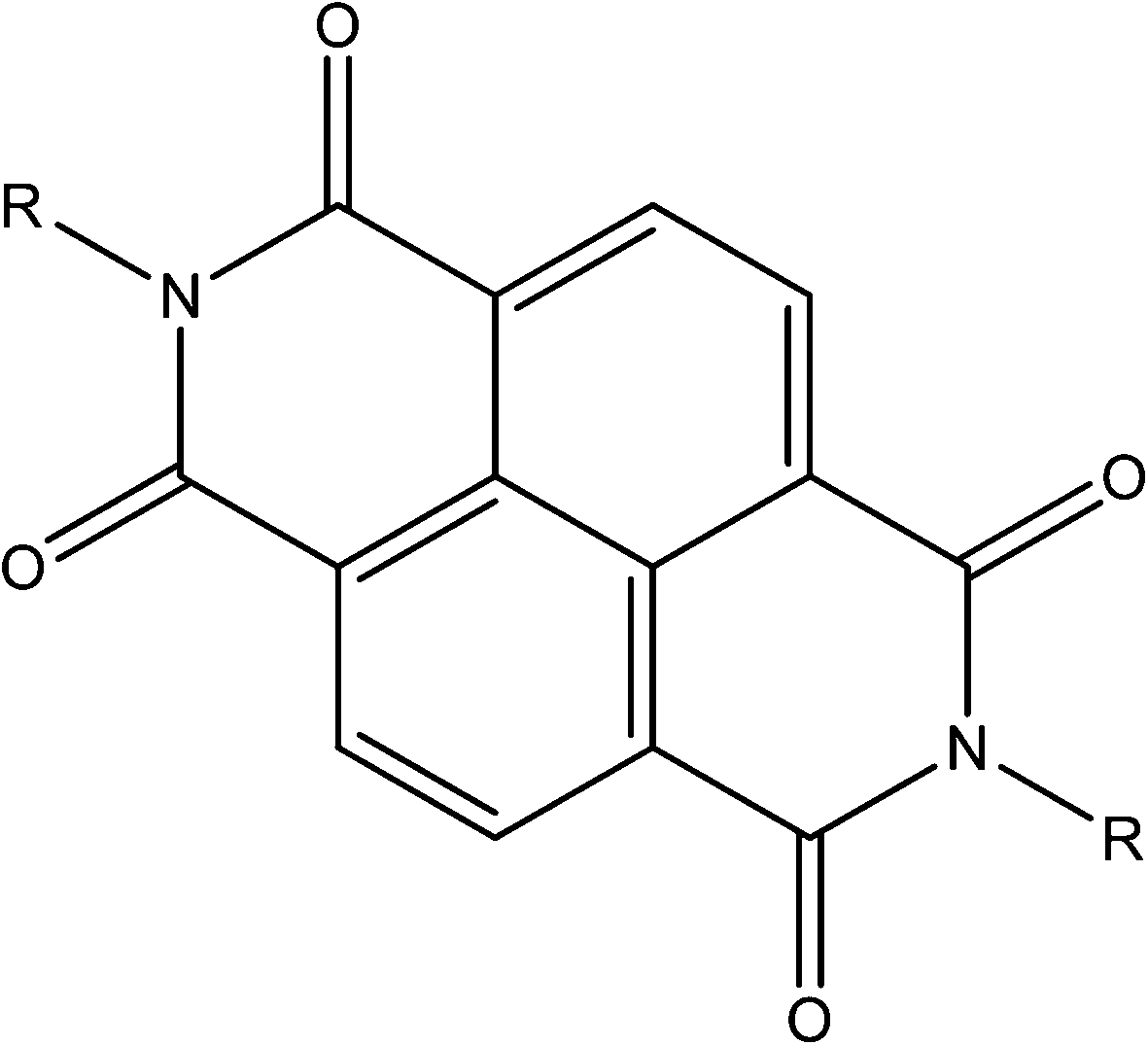 | ||
| Fig. 21 The generic NDI core. | ||
4. Conclusions and future outlook
In this review, we have sought to highlight the most recent developments (since 2011) in the area of radical inclusion into MOFs, whilst drawing attention to critical aspects of prior work in the field. In view of the rapid rate of advancements, the preceding three years have been marked by a number of key results that highlight the tremendous prospects for future developments in the field.At a fundamental level, measurements of magnetic exchange have enabled the elucidation of the mechanisms of long range magnetism, as well as the potential for technologically-useful switchability due to the presence of bistable states that can be modulated with external stimuli such as pressure, temperature and light. Emerging research has also demonstrated interesting semiconducting properties of MOFs incorporating radical ligands. These measurements have typically been achieved using pressed pellet and single crystal conductivity experiments, along with microwave conductivity and time of flight techniques for determination of electron/hole mobilities. While these quantities are often used to characterise the degree of conductivity in the materials, elucidation of the mechanisms of charge transport are less well characterised, underscoring the enormous scope that exists for further advances in the field.
Tantalising prospects are also evident for the realisation of fundamental physical phenomena that arise from the interplay between the magnetic and electronic properties. While this interplay has been extensively probed in organic solids such as the archetypal charge-transfer salts, experimental and theoretical work in MOFs is in its infancy. While an intrinsic interplay between the magnetic and electronic properties can be realised via incorporation of pendant and structurally integrated radicals (see Fig. 1), the presence of void space within the scaffold offers the potential for guest radical incorporation. In the latter case, emerging research has demonstrated the modification of framework properties with guest radicals, highlighting the prospect for developing sensing capabilities of the materials.
The different modes of radical inclusion also provide numerous possibilities for generating structurally- and physically-diverse materials. The majority of radical species discussed here have been incorporated into MOFs as components that are structurally integrated into the backbone of a ligand. The geometry of the resulting assembly is thus dictated by the identity and relative disposition of the ligating groups which are typically carboxylate or pyridyl-based. The incorporation of radicals as pendant or guest species enables the synthesis of materials that are isoreticular with known frameworks. Interestingly, confinement of radical species as guests within of frameworks can significantly alter their behaviour due to the geometric constraints that are imposed by the pore. As the number of reports of MOFs incorporating radical species increase, it is hoped that some general design parameters can be elucidated that offer a guide to structure–property relationships in these systems.
A final key issue we seek to emphasise is the importance of further developments in the theoretical and computational understanding of spin and charge in radical-containing MOFs. Whilst these systems consist of molecular components such as metal clusters (commonly referred to as secondary building units or SBUs) and centres linked by organic bridges, their physical properties are often influenced by long range magnetic and electronic interactions. An all-encompassing theoretical and computational model is therefore required which accounts for properties arising from both short and long range interactions.
Clearly, the scope for future advances in the experimental, theoretical and computational understanding of MOFs incorporating radicals is vast. Developments in these fundamental aspects will almost certainly inspire the integration of radical MOFs into technologically-important device architectures.
Abbreviations
| AFM | Antiferromagnetic |
| AQDS | Anthraquinone disulfonate |
| BDC | 4,4′-Benzene dicarboxylate |
| BDTA˙ | Benzodithiazolyl radical |
| BIPO˙ | 2,3′-Biimidazo[1,2-α]pyridin-2′-one radical |
| BIPY | 2,2′-Bipyridine |
| BPTC | 3,3′,5,5′-Biphenyltetracarboxylate |
| bTbK˙˙ | Bis-TEMPO-bisketal biradical |
| BTC | 1,3,5-Benzene tricarboxylate |
| CPBPY | N-(3-Carboxyphenyl)-4,4′-bipyridinium |
| DCA | Dicyanamide |
| DMF | Dimethyl formamide |
| DTBN˙ | Di-tert-butyl nitroxide radical |
| EPR | Electron paramagnetic resonance |
| FM | Ferromagnetic |
| HKUST | Hong Kong University of Science and Technology |
| MBDTA˙ | Methylbenzodithiazolyl radical |
| MIL | Materials Institute Lavoisier |
| MOF | Metal–organic framework |
| MOROF | Metal–organic radical open-framework |
| NDI | Naphthalene diimide |
| NIT4PY˙ | 2-(4′-Pyridyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide radical |
| NIT-TZ˙ | 2-(2-Thiazole)-4,4,5,5-tetramethylimidazoline-1-oxy-3-oxide radical |
| NMR | Nuclear magnetic resonance |
| PHEN | 1,10-Phenanthroline |
| PTMHC˙ | Triphenylmethyl hexacarboxylate radical |
| PTMTC˙ | Triphenylmethyl tricarboxylate radical |
| SMM | Single molecule magnet |
| ssNMR | Solid state NMR |
| TCE-TTF | 2,3,6,7-Tetrakis(2-cyanoethylthio)-TTF |
| TCM | Tricyanomethanide |
| TCNE | Tetracyanoethylene |
| TCNQ | 7,7,8,8-Tetracyano-p-quinodimethane |
| TEMPO | 2,2,6,6-Tetramethylpiperidine-1-oxy radical |
| TPT | 2,4,6-Tris(4-pyridyl)-1,3,5-triazine |
| TTF | Tetrathiafulvalene |
| TTF-TB | TTF-tetrabenzoate |
| TTF-TC | TTF-tetracarboxylate |
Acknowledgements
The authors gratefully acknowledge support from the Australian Research Council.References
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- H. W. Langmi, J. Ren, B. North, M. Mathe and D. Bessarabov, Electrochim. Acta, 2014 DOI:10.1016/j.electacta.2013.1010.1190.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- M. C. Das, Q. Guo, Y. He, J. Kim, C.-G. Zhao, K. Hong, S. Xiang, Z. Zhang, K. M. Thomas, R. Krishna and B. Chen, J. Am. Chem. Soc., 2012, 134, 8703–8710 CrossRef CAS.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2011, 112, 1105–1125 CrossRef.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, G. F. El, H. P. Yoon, F. Leonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS.
- G. Lorusso, J. W. Sharples, E. Palacios, O. Roubeau, E. K. Brechin, R. Sessoli, A. Rossin, F. Tuna, E. J. L. McInnes, D. Collison and M. Evangelisti, Adv. Mater., 2013, 25, 4653–4656 CrossRef CAS.
- H. Yang, R. L. Sang, X. Xu and L. Xu, Chem. Commun., 2013, 49, 2909–2911 RSC.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS.
- T. Uemura, Bull. Chem. Soc. Jpn., 2011, 84, 1169–1177 CrossRef CAS.
- S.-L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 CAS.
- W. L. Hubbell, C. J. Lopez, C. Altenbach and Z. Yang, Curr. Opin. Struct. Biol., 2013, 23, 725–733 CrossRef CAS.
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS.
- V. V. Khramtsov, In vivo Spectroscopy and Imaging of Nitroxide Probes, Nitroxides – Theory, Experiment and Applications, InTech, 2012 Search PubMed.
- C. L. Hawkins and M. J. Davies, Biochim. Biophys. Acta, 2014, 1840, 708–721 CrossRef CAS.
- T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS.
- S. J. Blundell and F. L. Pratt, J. Phys.: Condens. Matter, 2004, 16, R771–R828 CrossRef CAS.
- A. Rajca, J. Wongsriratanakul and S. Rajca, Science, 2001, 294, 1503–1505 CrossRef CAS.
- N. A. Zaidi, S. R. Giblin, I. Terry and A. P. Monkman, Polymer, 2004, 45, 5683–5689 CrossRef CAS.
- A. Rajca, Chem. – Eur. J., 2002, 8, 4834–4841 CrossRef CAS.
- H. Iwamura, Polyhedron, 2013, 66, 3–14 CrossRef CAS.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS.
- P. Hu, M. Zhu, X. Mei, H. Tian, Y. Ma, L. Li and D. Liao, Dalton Trans., 2012, 41, 14651–14656 RSC.
- I.-R. Jeon, J. G. Park, D. J. Xiao and T. D. Harris, J. Am. Chem. Soc., 2013, 135, 16845–16848 CrossRef CAS.
- T. I. Kawakami, S. I. Takamizawa, Y. Kitagawa, F. Matsuoka, T. Maruta, W. Mori and K. Yamaguchi, Mol. Cryst. Liq. Cryst., 2000, 343, 215–220 CrossRef CAS.
- T. Kawakami, S. Takamizawa, Y. Kitagawa, T. Maruta, W. Mori and K. Yamaguchi, Polyhedron, 2001, 20, 1197–1206 CrossRef CAS.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Ohrstrom, M. O'Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- A. D. Jenkins, P. Kratochvíl, R. F. T. Stepto and U. W. Suter, Pure Appl. Chem., 1996, 68, 2287–2311 CAS.
- B. Jee, K. Koch, L. Moschkowitz, D. Himsl, M. Hartman and A. Pöppl, J. Phys. Chem. Lett., 2011, 2, 357–361 CrossRef CAS.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS.
- P. Kuesgens, M. Rose, I. Senkovska, H. Froede, A. Henschel, S. Siegle and S. Kaskel, Microporous Mesoporous Mater., 2009, 120, 325–330 CrossRef CAS.
- G. W. Peterson, G. W. Wagner, A. Balboa, J. Mahle, T. Sewell and C. J. Karwacki, J. Phys. Chem. C, 2009, 113, 13906–13917 CAS.
- B. J. Burnett, P. M. Barron, C. Hu and W. Choe, J. Am. Chem. Soc., 2011, 133, 9984–9987 CrossRef CAS.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Chem. Sci., 2012, 3, 126–130 RSC.
- O. Karagiaridi, W. Bury, E. Tylianakis, A. A. Sarjeant, J. T. Hupp and O. K. Farha, Chem. Mater., 2013, 25, 3499–3503 CrossRef CAS.
- H. C. Hoffmann, M. Debowski, P. Mueller, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Materials, 2012, 5, 2537–2572 CrossRef CAS.
- Y. Matsuki, T. Maly, O. Ouari, H. Karoui, M. F. Le, E. Rizzato, S. Lyubenova, J. Herzfeld, T. Prisner, P. Tordo and R. G. Griffin, Angew. Chem., Int. Ed., 2009, 48, 4996–5000 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, J. Canivet, S. Aguado, O. Ouari, P. Tordo, M. Rosay, W. E. Maas, C. Coperet, D. Farrusseng, L. Emsley and A. Lesage, Angew. Chem., Int. Ed., 2012, 51, 123–127 CrossRef CAS PubMed.
- M. N. Timofeeva, V. N. Panchenko, A. A. Abel, N. A. Khan, I. Ahmed, A. B. Ayupov, K. P. Volcho and S. H. Jhung, J. Catal., 2014, 311, 114–120 CrossRef CAS.
- A. M. Sheveleva, D. I. Kolokolov, A. A. Gabrienko, A. G. Stepanov, S. A. Gromilov, I. K. Shundrina, R. Z. Sagdeev, M. V. Fedin and E. G. Bagryanskaya, J. Phys. Chem. Lett., 2014, 5, 20–24 CrossRef CAS PubMed.
- H. H. Lin, S. Mohanta, C.-J. Lee and H.-H. Wei, Inorg. Chem., 2003, 42, 1584–1589 CrossRef CAS PubMed.
- T. Kitazawa, H. Sato, C. Kachi-Terajima, K. Yoshida, H. Takagaki, C. Kanadani and T. Saito, Polyhedron, 2011, 30, 3054–3057 CrossRef CAS.
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination polymers: design, analysis and application, Royal Society of Chemistry, 2008 Search PubMed.
- W. Kaim and M. Moscherosch, Coord. Chem. Rev., 1994, 129, 157–193 CrossRef CAS.
- J. S. Miller, Dalton Trans., 2006, 2742–2749 RSC.
- J. S. Miller, Chem. Soc. Rev., 2011, 40, 3266–3296 RSC.
- A. C. McConnell, E. Shurdha, J. D. Bell and J. S. Miller, J. Phys. Chem. C, 2012, 116, 18952–18957 CAS.
- A. C. McConnell, J. D. Bell and J. S. Miller, Inorg. Chem., 2012, 51, 9978–9982 CrossRef CAS PubMed.
- J. G. DaSilva, A. C. McConnell and J. S. Miller, Inorg. Chem., 2013, 52, 4629–4634 CrossRef CAS PubMed.
- J. H. Her, P. W. Stephens, R. A. Davidson, K. S. Min, J. D. Bagnato, K. van Schooten, C. Boehme and J. S. Miller, J. Am. Chem. Soc., 2013, 135, 18060–18603 CrossRef CAS PubMed.
- D. S. Acker, R. J. Harder, W. R. Hertler, W. Mahler, L. R. Melby, R. E. Benson and W. E. Mochel, J. Am. Chem. Soc., 1960, 82, 6408–6409 CrossRef CAS.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- S. Shimomura and S. Kitagawa, J. Mater. Chem., 2011, 21, 5537–5546 RSC.
- H. Miyasaka, Acc. Chem. Res., 2013, 46, 248–257 CrossRef CAS PubMed.
- Z. Zhang, H. Zhao, H. Kojima, T. Mori and K. R. Dunbar, Chem. – Eur. J., 2013, 19, 3348–3357 CrossRef CAS PubMed.
- C. Avendano, Z. Y. Zhang, A. Ota, H. H. Zhao and K. R. Dunbar, Angew. Chem., Int. Ed., 2011, 50, 6543–6547 CrossRef CAS PubMed.
- A. Pearson, A. P. O'Mullane, S. K. Bhargava and V. Bansal, Inorg. Chem., 2012, 51, 8791–8801 CrossRef CAS PubMed.
- T. H. Le, A. Nafady, J. Z. Lu, G. Peleckis, A. M. Bond and L. L. Martin, Eur. J. Inorg. Chem., 2012, 2012, 2889–2897 CrossRef CAS.
- A. Pearson, A. P. O'Mullane, V. Bansal and S. K. Bhargava, Inorg. Chem., 2011, 50, 1705–1712 CrossRef CAS PubMed.
- E. Coronado and P. Day, Chem. Rev., 2004, 104, 5419–5448 CrossRef CAS PubMed.
- N. Martín, Chem. Commun., 2013, 49, 7025–7027 RSC.
- Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780–783 CrossRef CAS PubMed.
- A. Gorgues, P. Hudhomme and M. Salle, Chem. Rev., 2004, 104, 5151–5184 CrossRef CAS PubMed.
- D. Lorcy, N. Bellec, M. Fourmigue and N. Avarvari, Coord. Chem. Rev., 2009, 253, 1398–1438 CrossRef CAS.
- B. Chen, N. W. Ockwig, F. R. Fronczek, D. S. Contreras and O. M. Yaghi, Inorg. Chem., 2004, 44, 181–183 CrossRef PubMed.
- T. L. A. Nguyen, R. Demir-Cakan, T. Devic, M. Morcrette, T. Ahnfeldt, P. Auban-Senzier, N. Stock, A.-M. Goncalves, Y. Filinchuk, J.-M. Tarascon and G. Ferey, Inorg. Chem., 2010, 49, 7135–7143 CrossRef CAS PubMed.
- Y. R. Qin, Q. Y. Zhu, L. B. Huo, Z. Shi, G. Q. Bian and J. Dai, Inorg. Chem., 2010, 49, 7372–7381 CrossRef CAS PubMed.
- L. P. Wu, J. Dai, M. Munakata, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga and Y. Ohno, J. Chem. Soc., Dalton Trans., 1998, 3255–3262 RSC.
- Q. Y. Zhu, J. P. Wang, Y. R. Qin, Z. Shi, Q. H. Han, G. Q. Bian and J. Dai, Dalton Trans., 2011, 40, 1977–1983 RSC.
- J. Olivier, S. Golhen, R. Swietlik, O. Cador, F. Pointillart and L. Ouahab, Eur. J. Inorg. Chem., 2009, 3282–3290 CrossRef CAS.
- T. C. Narayan, T. Miyakai, S. Seki and M. Dincă, J. Am. Chem. Soc., 2012, 134, 12932–12935 CrossRef CAS PubMed.
- G. P. Yong, C.-F. Li, Y. Z. Li and S. W. Luo, Chem. Commun., 2010, 46, 3194–3196 RSC.
- G. P. Yong, S. Qiao and Z. Y. Wang, Cryst. Growth Des., 2008, 8, 1465–1467 CAS.
- G. P. Yong, Y. Li, W. She and Y. Zhang, Chem. – Eur. J., 2011, 17, 12495–12501 CrossRef CAS PubMed.
- G. P. Yong, Y. Z. Li, Y. M. Zhang and W. L. She, CrystEngComm, 2012, 14, 1439–1448 RSC.
- The binding mode of the ligand is referred to according to the notation [X.Y1Y2Y3…Yn] whereby X is the total number of metals bound by the ligand and Y is the number of metals bound by each donor of the ligand. Y is ordered by the Cahn–Ingold–Prelog priority rules such that the oxygen is accounted for before the nitrogens. See: R. A. Coxall, S. G. Harris, D. K. Henderson, S. Parsons, P. A. Tasker and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2000, 2349–2356 RSC.
- F. Gandara, N. Snejko, A. A. de, J. R. Fernandez, J. C. Gomez-Sal, E. Gutierrez-Puebla and A. Monge, RSC Adv., 2012, 2, 949–955 RSC.
- G. P. Yong, Y.-M. Zhang and B. Zhang, CrystEngComm, 2012, 14, 8620–8625 RSC.
- Y. Tan, Z. Fu, Y. Zeng, H. Chen, S. Liao, J. Zhang and J. Dai, J. Mater. Chem., 2012, 22, 17452–17455 RSC.
- Y. Tan, H. Chen, J. Zhang, S. Liao, J. Dai and Z. Fu, CrystEngComm, 2012, 14, 5137–5139 RSC.
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- D. Maspoch, D. Ruiz-Molina, K. Wurst, N. Domingo, M. Cavallini, F. Biscarini, J. Tejada, C. Rovira and J. Veciana, Nat. Mater., 2003, 2, 190–195 CrossRef CAS PubMed.
- D. Maspoch, D. Ruiz-Molina, K. Wurst, C. Rovira and J. Veciana, Chem. Commun., 2004, 1164–1165 RSC.
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, J.-M. Hernandez, G. Vaughan, C. Rovira, F. Lloret, J. Tejada and J. Veciana, Chem. Commun., 2005, 5035–5037 RSC.
- N. Roques, D. Maspoch, I. Imaz, A. Datcu, J. P. Sutter, C. Rovira and J. Veciana, Chem. Commun., 2008, 3160–3162 RSC.
- N. Roques, D. Maspoch, F. Luis, A. Camon, K. Wurst, A. Datcu, C. Rovira, D. Ruiz-Molina and J. Veciana, J. Mater. Chem., 2008, 18, 98–108 RSC.
- V. Mugnaini, M. Mas-Torrent, I. Ratera, C. Rovira and J. Veciana, Supramolecular Soft Matter: Applications in Materials and Organic Electronics, John Wiley & Sons, Inc., 2011 Search PubMed.
- A. Datcu, N. Roques, V. Jubera, I. Imaz, D. Maspoch, J.-P. Sutter, C. Rovira and J. Veciana, Chem. – Eur. J., 2011, 17, 3644–3656 CrossRef CAS PubMed.
- A. Datcu, N. Roques, V. Jubera, D. Maspoch, X. Fontrodona, K. Wurst, I. Imaz, G. Mouchaham, J.-P. Sutter, C. Rovira and J. Veciana, Chem. – Eur. J., 2012, 18, 152–162 CrossRef CAS PubMed.
- C. Lambert and G. Noell, J. Am. Chem. Soc., 1999, 121, 8434–8442 CrossRef CAS.
- B. Wang, Y. Wang, J. Hua, Y. Jiang, J. Huang, S. Qian and H. Tian, Chem. – Eur. J., 2011, 17, 2647–2655 CrossRef CAS PubMed.
- M. Thelakkat, Macromol. Mater. Eng., 2002, 287, 442–461 CrossRef CAS.
- I. Y. Drozdyuk, S. E. Tolstikov, E. V. Tretyakov, S. L. Veber, V. I. Ovcharenko, R. Z. Sagdeev, E. G. Bagryanskaya and M. V. Fedin, J. Phys. Chem. A, 2013, 117, 6483–6488 CrossRef CAS PubMed.
- C. Hua and D. M. D'Alessandro, CrystEngComm, 2014 10.1039/c1033ce42603c.
- K. Awaga, T. Tanaka, T. Shirai, M. Fujimori, Y. Suzuki, H. Yoshikawa and W. Fujita, Bull. Chem. Soc. Jpn., 2006, 79, 25–34 CrossRef CAS.
- W. Fujita and K. Kikuchi, Eur. J. Inorg. Chem., 2014, 2014, 93–100 CrossRef CAS.
- E. M. Fatila, R. Clerac, M. Rouzieres, D. V. Soldatov, M. Jennings and K. E. Preuss, Chem. Commun., 2013, 49, 6271–6273 RSC.
- W. Fujita, K. Kikuchi and K. Awaga, Angew. Chem., Int. Ed., 2008, 47, 9480–9483 CrossRef CAS PubMed.
- S. V. Potts, L. J. Barbour, D. A. Haynes, J. M. Rawson and G. O. Lloyd, J. Am. Chem. Soc., 2011, 133, 12948–12951 CrossRef CAS PubMed.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Ferey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC.
- M. Pan, X. M. Lin, G. B. Li and C. Y. Su, Coord. Chem. Rev., 2011, 255, 1921–1936 CrossRef CAS.
- C. F. Leong, B. Chan, T. B. Faust, P. Turner and D. M. D'Alessandro, Inorg. Chem., 2013, 52, 14246–14252 CrossRef CAS PubMed.
- Y. Takashima, V. M. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- L. Han, L. Qin, L. Xu, Y. Zhou, J. Sun and X. Zou, Chem. Commun., 2013, 49, 406–408 RSC.
- C. R. Wade, M. Li and M. Dincă, Angew. Chem., Int. Ed., 2013, 52, 13377–13381 CrossRef CAS PubMed.
- C. F. Leong, T. B. Faust, P. Turner, P. M. Usov, C. J. Kepert, R. Babarao, A. W. Thornton and D. M. D'Alessandro, Dalton Trans., 2013, 42, 9831–9839 RSC.
| This journal is © The Royal Society of Chemistry 2014 |