New insights into dihydrogenphosphate recognition with dirhenium(I) tricarbonyl complexes bridged by a thiourea moiety†
Anna L. Blackburn,
Naomi C. A. Baker and
Nicholas C. Fletcher*
The School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, UK. E-mail: n.fletcher@qub.ac.uk; Fax: +44 (0)28 9097 6524; Tel: +44 (0)28 9097 5479
First published on 15th April 2014
Abstract
Three thiourea bridged 2,2′-bipyridine ligands bearing either a single thiourea group (L1), or two units separated by either a para (L2) or meta-substituted (L3) aromatic spacer, along with the corresponding bis(fac-tricarbonylrhenium(I)) complexes are reported. The three ligands all show the anticipated binding to acetate. However 1H NMR titrations reveal an unusual cooperative binding to, and selectivity for, two dihydrogenphosphate ions. The rhenium(I) complexes similarly demonstrate unusual sigmoidal titration curves, and in the case of {Re(CO)3Br}2(μ-L1) a surprisingly strong interaction to two anions. These were further exemplified in the emissive behaviour leading to the conclusion that there is an unusual interaction with dihydrogenphosphate, giving an initial increase in the emission, followed by a decrease and a blue shift in wavelength possibly as a result of partial deprotonation. It appears that dihydrogenphosphate binds cooperatively, with the addition of a second anion enhancing the interaction of the first, probably by proton transfer; this could explain the remarkable selectivity for phosphate seen with many reported anion receptors.
Introduction
The selective recognition of anions has developed over the last two decades into a vibrant area of research1 with considerable emphasis placed on the selective recognition of phosphate salts due to their prevalence in biology.2,3 One successful approach is the use of metal-based receptors, where positive electrostatic interactions can be combined with an acidic proton to encourage hydrogen bonding,4 and then exploit a change in the redox, photophysical or magnetic properties to create a sensor.5–7 Anion selective systems able to operate in aqueous media remains a considerable challenge however.8,9 The inclusion of urea groups has proved to be particularly successful in the recognition of fluoride, acetate and dihydrogenphosphate10–12 as shown by the development of systems involving two13–15 or three urea or thiourea moieties,16–18 including tripodal architectures.19–25 These demonstrate considerable selectivity for specific anions, even in protic media.In the creation of metal-complex anion receptors, 2,2′-bipyridine has been widely exploited,26 however the number of situations where it has been combined with a urea function remains surprisingly small. A notable recent example reported by Kitchen et al. highlights a tris-bipyridine complex of ruthenium(II) bearing an appended aryl urea where the emission can be “turned on” by the addition of orthophosphate, but decreased with pyrophosphate.27 Custelcean and co-workers have also highlighted that de novo computer-aided design can be used to design a sulfate receptor via the formation of a M4L6 tetrahedral cage using a simple linear ligand with two bipyridine ligands bridged by a methylene urea group (Fig. 1a) which in the presence of nickel(II) was able to extract tetrahedral oxoanions from aqueous solution.28,29
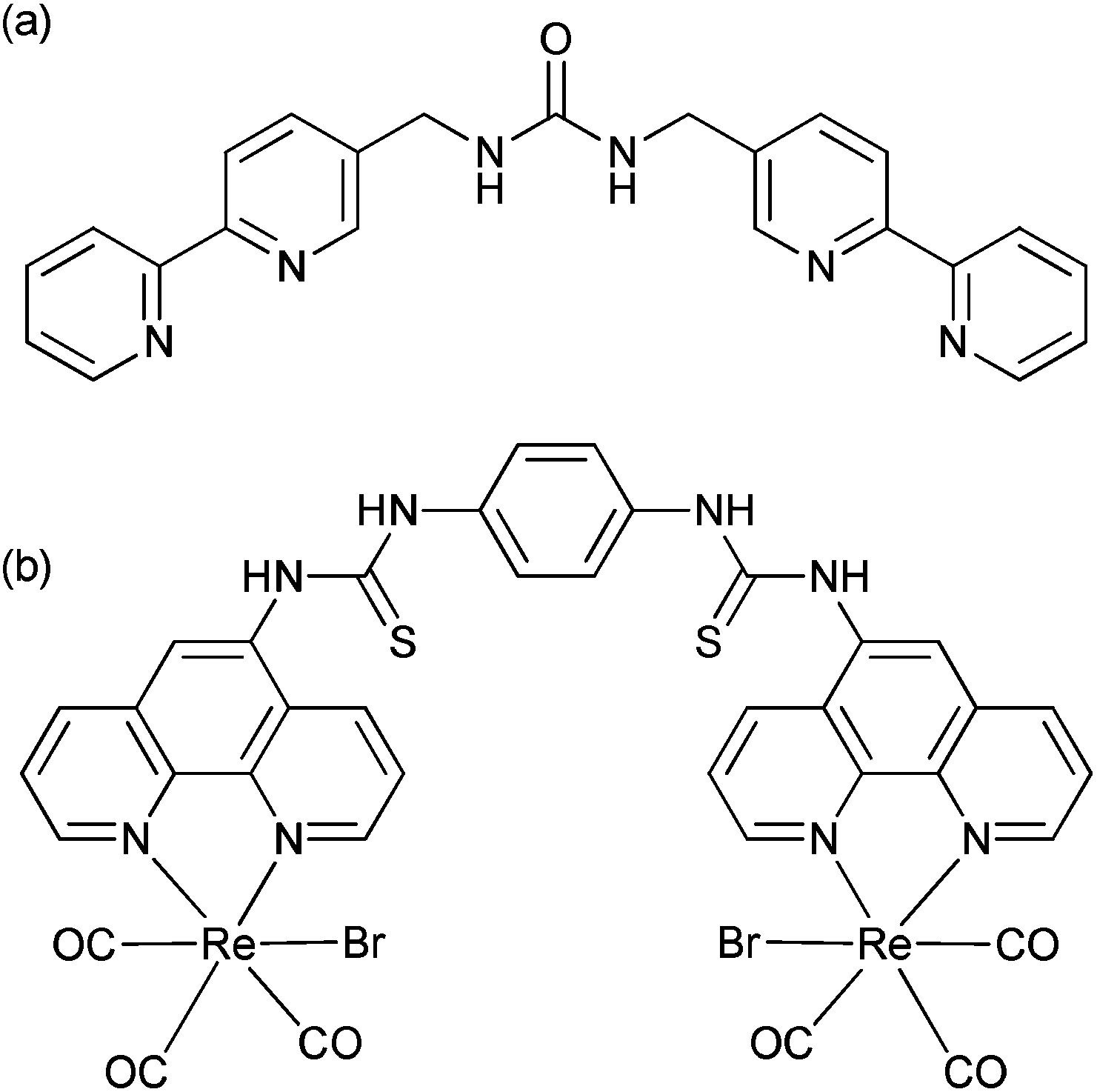 | ||
| Fig. 1 (a) Ligand system previously reported by Custelcean and co-workers28,29 and (b) the dirhenium complex reported by Hanan.30 | ||
Organometallic complexes bearing appropriate hydrogen bond donor groups have been the feature of a number of anion bonding studies.31–33 One family of metal complexes that has shown considerable promise in the design of emissive sensors has been that of the fac-rhenium(I) tricarbonyl diimine fragment, where the sixth coordination site can accommodate either a neutral ligand to give a monovalent cation, or a halide to provide a neutral complex. Anion recognition has been observed in a variety of complexes,34–40 and previously we have shown that large perturbations in the metal-to-ligand excited state luminescence can be achieved on a successful anion recognition event.41,42 In our studies we noted that dihydrogenphosphate can disrupt an unconventional hydrogen bond from an amide proton to the metal carbonyl. The group of Lees has also highlighted that dirhenium(I) carbonyl complexes with polarized N–H groups, including amidic, thioamidic and thiourea bridges, display outstanding sensitivity and selectivity toward a variety of anionic species including acetate and cyanide.43 In another recent example, Hanan30 and co-workers have described a dirhenium species bridged by two thiourea groups (Fig. 1b) which demonstrates a strong affinity for fluoride, acetate and dihydrogenphosphate. Interestingly, this latter anion showed an unusual situation where the binding of the second anion appeared to be counter intuitively greater than the binding of the first anion.
Thiourea offers even greater opportunities for anion recognition than urea itself due to the enhanced acidity of the N–H groups,44 and the observation that it readily permits deprotonation with a number of anionic species. Consequently it offers opportunities to demonstrate remarkably strong anion recognition and possibilities to design sensors capable of operating in aqueous media. To further explore these phenomena, we report here the synthesis of the thiourea analogue of the ligand recently reported by Custelcean et al. and its dirhenium(I) complex.28,29 We then extend this to two systems which include two thiourea groups, with the intention of understanding the remarkable preference for dihydrogenphosphate over other tetrahedral oxo-anions with what superficially look like simple linear luminescent complexes.
Results and discussion
Synthesis
The preparation of thiourea and urea adducts derived from 2,2′-bipyridine complexes are not common in the literature,28,45 and a new synthetic strategy has been developed via an unreported isothiocyanate derivative from 5-aminomethyl-2,2′-bipyridine.46 This was achieved in reasonable yield via the amination of 5-chloromethyl-2,2′-bipyridine46 rather than the more traditional route through the analogous bromo adduct47 which has proved to be a severe irritant, and in our experience is not a procedure that can be easily scaled-up to give multi-gram quantities. Isolation of 5-isothiocyanatomethyl-2,2′-bipyridine was readily achieved following the procedure reported by Wong et al.48 via the tosyl chloride mediated decomposition of a dithiocarbamic acid salt in 61% yield, avoiding the more traditional route via thiophosgene. Subsequent combination of 5-isothiocyanatomethyl-2,2′-bipyridine and 5-aminomethyl-2,2′-bipyridine, exploiting pyridine as both solvent and base, resulted in the ditopic ligand L1 (Scheme 1) in 73% yield, the identity of which was confirmed by 1H NMR spectroscopy and electrospray mass spectrometry. Subsequent complexation of ligand L1 with Re(CO)5Br was initially attempted in toluene, but the poor solubility of the ligand resulted in the necessity to use dry DMSO, which was subsequently removed by vacuum distillation and recrystallization from hot toluene giving the product as a bright yellow solid in 62% yield. This was characterised by 1H NMR spectroscopy, highlighted by a considerable downfield shift for the bipyridine proton peaks in comparison to the spectrum of L1 (Fig. S1 and S2†), and by electrospray mass spectrometry with a peak at 1110.8530 corresponding to the protonated complex.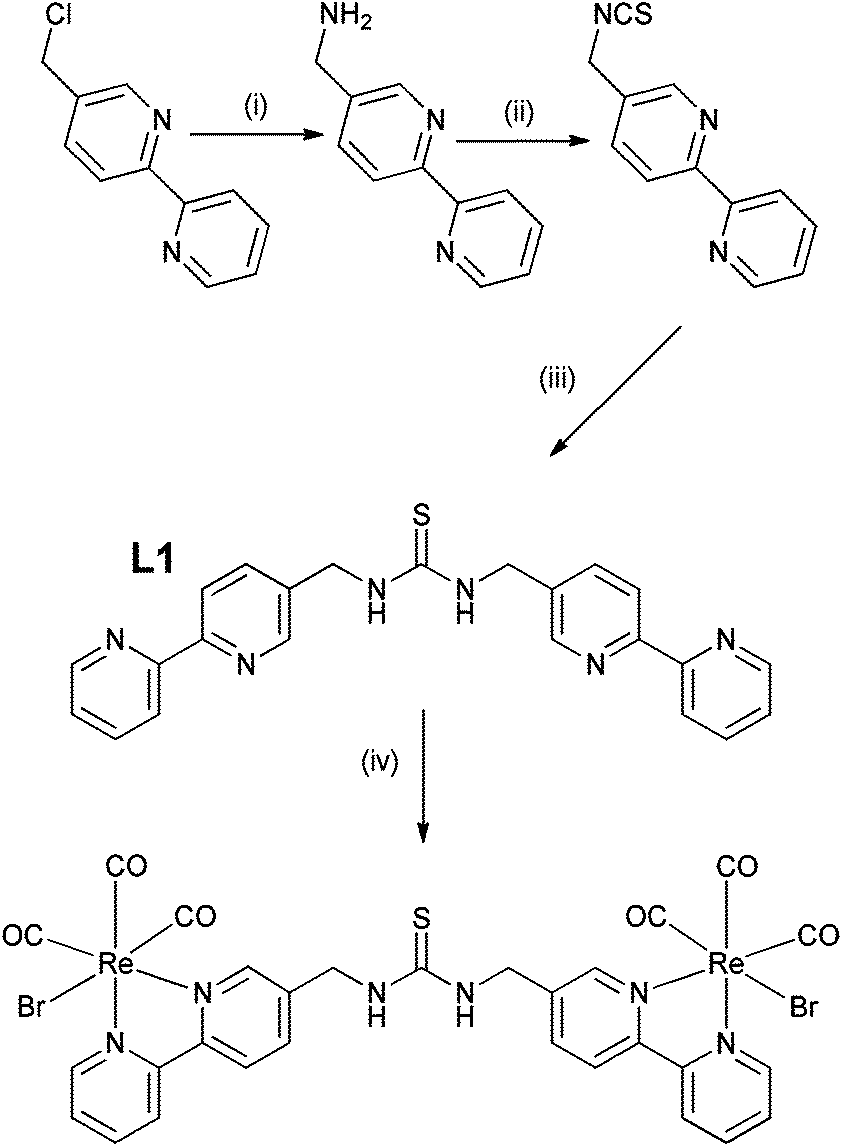 | ||
| Scheme 1 Synthetic pathway to {Re(CO)3Br}2(μ-L1); (i) hexamethylenetetraamine, DCM, reflux 16 h, then 12 M HCl, reflux 16 h, (ii) CS2, NEt3, dry THF stirred for 16 h followed by tosyl chloride and stirred for 16 h, (iii) 5-aminomethyl-2,2′-bipyridine, pyridine, reflux 48 h, and (iv) [Re(CO)5Br], DMSO, 75 °C for 16 h under N2. | ||
To investigate whether the system could be extended, and to introduce two anion binding centres, isothiocyanatomethyl-2,2′-bipyridine was also reacted with a range of aromatic diamines but without success, with only single thiourea adducts being formed. It was decided therefore to react 5-aminomethyl-2,2′-bipyridine with the corresponding diisothiocyanates. 1,4-Phenylenediisothiocyanate was readily prepared following Wong's procedure48 however the 1,3-functionalised analogue could not be isolated, but was obtained using thiophosgene via a published method.49 Ligands L2 and L3 (Scheme 2) were subsequently prepared in the presence of triethylamine in 70 and 86% yield respectively using a similar procedure as described for L1. These two ligands were also successfully bonded to two Re(CO)3Br moieties in DMSO, and characterised by 1H NMR spectroscopy (Fig. S3–S6†) and high resolution electrospray mass spectrometry with the molecular ion less one bromide being observed at 1182.9620 and 1182.9681 respectively (theoretical 1182.9689) with an appropriate isotopic distribution.
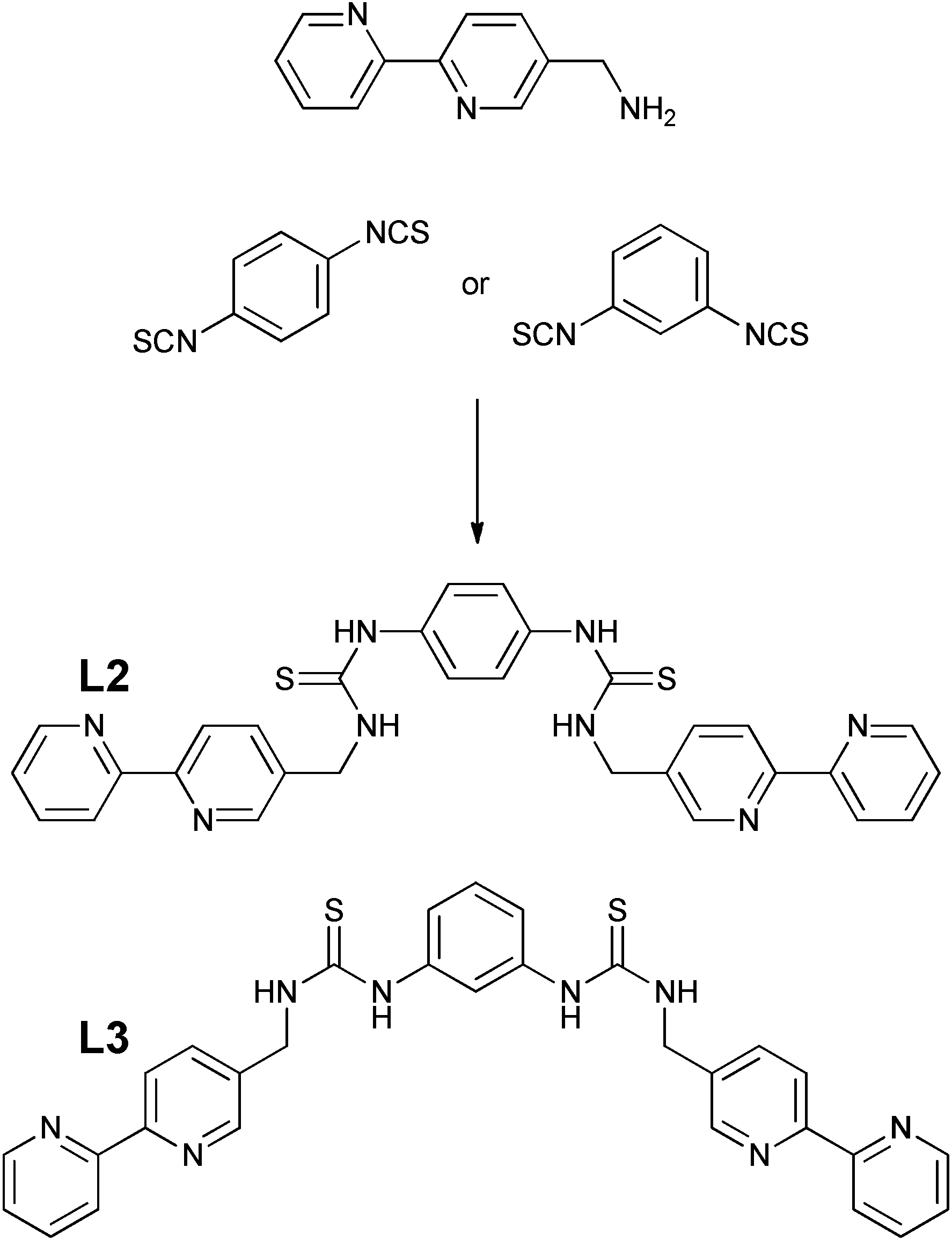 | ||
| Scheme 2 Synthetic pathway to L2 and L3. | ||
The rhenium complexes all proved to be bright yellow in colour and the UV/vis spectrum of {Re(CO)3Br}2(μ-L2) and {Re(CO)3Br}2(μ-L3) revealed the anticipated ligand centred (LC) π–π* transition at approximately 290 nm (Table 1), a shoulder at 320 nm, and a broad metal-to-ligand-charge-transfer (MLCT) absorption in the range of 370 to 390 nm (Fig. S7†). The corresponding emission spectra of all of the complexes show a weak luminescence around 610 nm (excited at 380 nm) and a quantum yield of approximately half that of [Re(CO)3(bpy)Br] (Fig. S8†).50
| Complex | Absorptiona | Emissiona | ||||
|---|---|---|---|---|---|---|
| λmax ± 2 nm | ε × 103 dm−3 mol−1 cm | λmax ± 2 nm | ε × 103 dm−3 mol−1 cm | λmax ± 2 nm | Φem | |
| a Recorded in aerated CH3CN at 298 K, excited at 380 nm, emission quantum yields (Φem) were calculated relative to [Re(CO)3Br(bpy)] (7.8 × 10−3) in acetonitrile.50 | ||||||
| Re(CO)3Br(Bpy) | 294 | 8.55 | 377 | 3.02 | 579 | 7.8 × 10−3 |
| 304 | 8.20 | |||||
| 317 | 7.33 | |||||
| {Re(CO)3Br}2(L1) | 298 | 26.4 | 375 (sh) | 3.38 | 612 | 4.8 × 10−3 |
| 308 | (sh) | |||||
| 321 | (sh) | |||||
| {Re(CO)3Br}2(L2) | 296 | 42.2 | 375 (sh) | 4.85 | 606 | 4.0 × 10−3 |
| 308 | (sh) | |||||
| 321 | (sh) | |||||
| {Re(CO)3Br}2(L3) | 292 | 28.1 | 380 (sh) | 5.26 | 608 | 6.4 × 10−3 |
| 308 | (sh) | |||||
| 321 | (sh) | |||||
1H NMR anion binding studies – ligands
Studies were undertaken to screen compound L1's behaviour in the presence of a variety of common anions by 1H NMR spectroscopy in a 50% mixture of CD3CN and DMSO-D6. This revealed that there is no change in the relative peak position of any of the observed signals, including that of the thiourea NH resonance, with the addition of up to ten equivalents of the tetrabutylammonium (TBA) salts of NO3−, Br− and HSO4−, while the addition of the corresponding chloride salt leads to a small downfield shift of the thiourea proton signal (Δδ = 0.25 ppm with ten equivalents, Fig. 2a and S9†). However a sequential titration with fluoride causes the complete loss of the NH signal, consistent with proton exchange, or even full deprotonation (Fig. S9†).44,51,52 Acetate gives a shift in peak position permitting the determination of a binding constant (pβ1 = 2.8) using a simple one to one host to anion model (WINEQNMR253 Fig. S10a†), which is typical for a simple hydrogen bond pairing in this competitive solvent mixture (Table 2).54 TBA H2PO4 also results in a definite change in the 1H NMR spectrum (Fig. S10b†), however the nature of the observed shift of the thiourea protons is notably different from a typical anion titration, as seen here with acetate (Fig. 2a). At low concentration there is little movement in the NH peak position, but upon the addition of one equivalent there is a much larger shift giving an unusual sigmoidal curve suggestive of a cooperative effect. A model combining two dihydrogenphosphate ions to the one ligand gives a good reproducible fit (pβ1 = 2.6 and pβ2 = 6.1) confirming a surprisingly high degree of cooperativity on the introduction of a second dihydrogenphosphate ion. To further demonstrate this surprising stoichiometry, a 1H NMR Job plot analysis55 was undertaken (Fig. 3, S11 and Table S1†) which is highly suggestive of the proposed two anions to one ligand stoichiometry.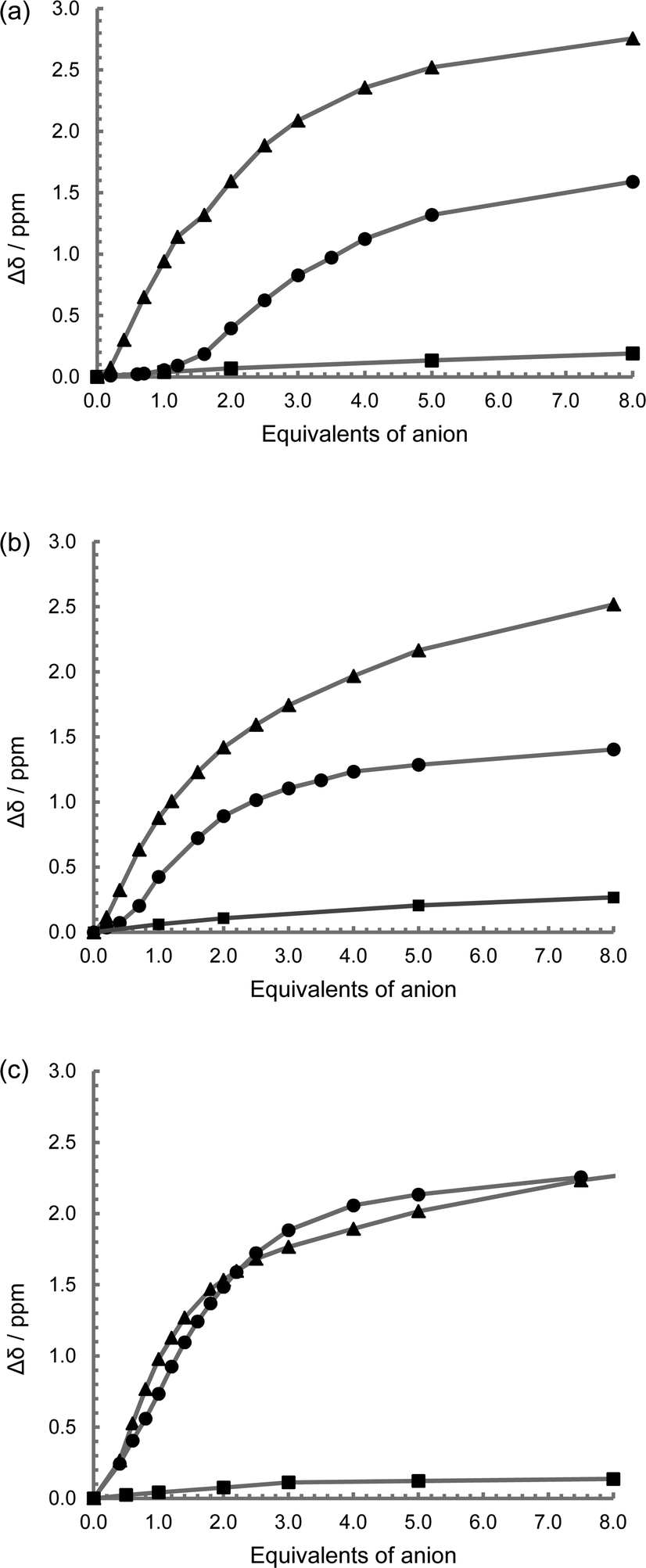 | ||
| Fig. 2 1H NMR spectroscopic change in NH peak position on the introduction of TBA OAc (▲), TBA Cl (■) and TBA H2PO4 (●); (a) L1, (b) L2, and (c) L3 (50% DMSO-D6–CD3CN 400 MHz, 298 K, at approx. 1 × 10−3 mol dm−3). | ||
| Host | OAc− | H2PO4− | ||
|---|---|---|---|---|
| pβ1 | pβ2 | pβ1 | pβ2 | |
| a Errors estimated to be ≤5%; T = 298 K. Data obtained from the movement of the CH2NH signal.b Data unavailable due to loss of the NH signal. | ||||
| L1 | 2.8 | — | 2.6 | 6.1 |
| L2 | 3.8 | 6.0 | 5.1 | 8.3 |
| L3 | 3.5 | 4.4 | 4.7 | 8.0 |
| [{Re(CO)3Br}2L1] | 3.2 | — | 6.9 | >10 |
| [{Re(CO)3Br}2L2] | 4.4 | 6.9 | b | b |
| [{Re(CO)3Br}2L3] | 5.0 | 7.8 | 3.8 | 7.4 |
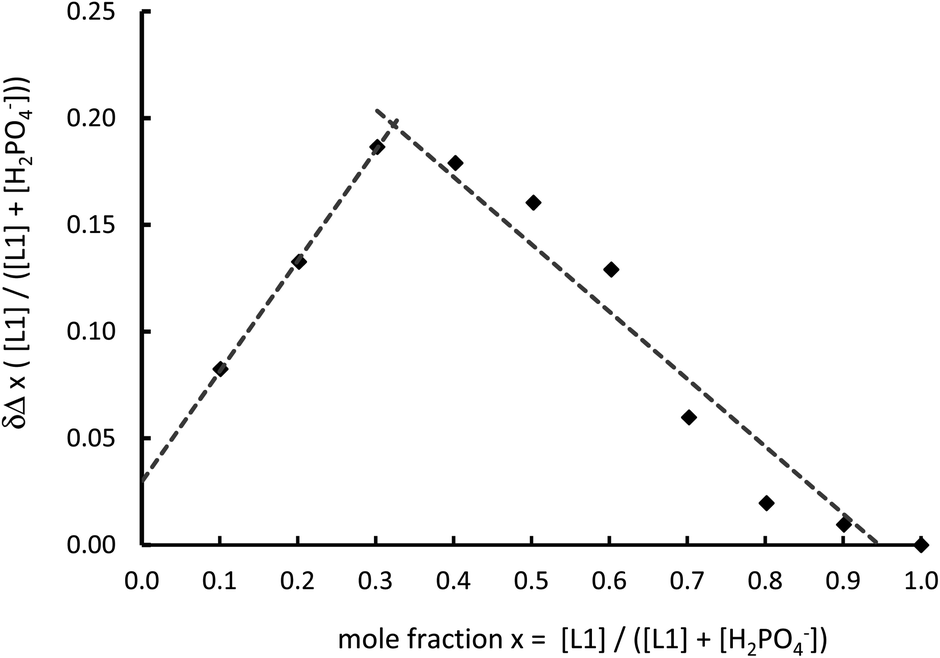 | ||
| Fig. 3 Job plot analysis in the 1H NMR spectroscopic change in NH peak position on the introduction of TBA H2PO4 (total concentration 1.26 mmol dm−3, 50% DMSO-D6–CD3CN 400 MHz, 298 K). | ||
For L2 and L3, the 1H NMR spectroscopic titrations behave in a similar manner to that observed with L1, with no interaction observed with the introduction of up to ten equivalents of TBA NO3, Br and HSO4, while with L2 a sequential titration with fluoride again causes the loss of the NH resonance consistent with a high degree of deprotonation. The addition of the corresponding chloride salt leads to a small downfield shift of the thiourea protons signal (Δδ = 0.25 ppm with ten equivalents) (Fig. 2b, 2c, S12a and S13a†). A significant and predictable interaction is observed with acetate, which fits well to a simple two to one stoichiometry, and it is assumed that one acetate binds to each of the two thiourea groups with reasonable pβ2 values consistent with the literature54 (pβ2 = 6.0 and 4.4 for L2 and L3 respectively; Fig. S12b and 13b†). The observed difference in the behaviour of the two compounds probably arises from the differences in the steric constraint resulting from the para and meta-functionalised spacers (L2 and L3 respectively).
The addition of TBA dihydrogenphosphate to L2 and L3 both result in a very similar sigmoidal titration curves for both of the two thiourea NH resonances, similar in shape to that exhibited by L1 (Fig. S12c and 13c†). Using a two to one model (similar to that used for acetate), binding constants of approximately pβ1 ≈ 5 and pβ2 ≈ 8 were obtained for both complexes (WINEQNMR253). While this does not indicate cooperativity in itself, there is definitely something unexpected occurring giving rise to the sigmoidal shape to the titration curve. There is a very small difference in the NH resonances associated with the first anion binding, typically Δδ = 0.3 ppm on both the NH signals, while the second anion causes a more dramatic change of the order of Δδ ≈ 1.5 ppm (depending on the sample and the NH resonance considered). Further, after one equivalent of the dihydrogenphosphate with ligand L3, there was considerable perturbation on the bridging aromatic group with a dramatic down-field shift for the signal attributed to H4 and H6, while H2 and H3 demonstrated movement in an up-field direction (Fig. S13c†). For all these samples, dilution of the compound did not result in a change in the peak position of any of the 1H resonances indicating that this surprising phenomenon is not as a result of an aggregation. However considering the behaviour observed with L1, and the fact that this sigmoidal shape in the titration curve is not seen with the addition of acetate, along with the rigid separation between the two thiourea groups in L2 and L3, the situation is probably a little more complicated and will be discussed subsequently.
1H NMR anion binding studies – complexes
With the inclusion of the metal fragment, the rhenium complexes were similarly investigated using 1H NMR spectroscopic anion titrations under the same conditions used with L1 to L3. {Re(CO)3Br}2(μ-L1) with NO3−, HSO4−, Br− and ClO4− salts resulted in no perturbation to the spectra (in 50% CD3CN–DMSO-D6; Fig. S14†), as seen with the free ligand L1, while chloride exhibits a slight deviation in the thiourea peak position (Δδ = 0.2 ppm with ten equivalents; Fig. 4 and S14†). Surprisingly, fluoride shows no immediate change in the spectrum, but once one equivalent had been added, a small downfield shift is observed before the peaks broaden and become uninterpretable consistent with proton exchange.44,51,52 With acetate, a similar behaviour is found to that of the free ligand (Fig. 4a and S15a†), with a binding constant, pβ1 = 3.2, assuming a one to one stoichiometry, suggesting that the presence of the two Re(CO)3 groups has a marginal affect on the affinity for acetate.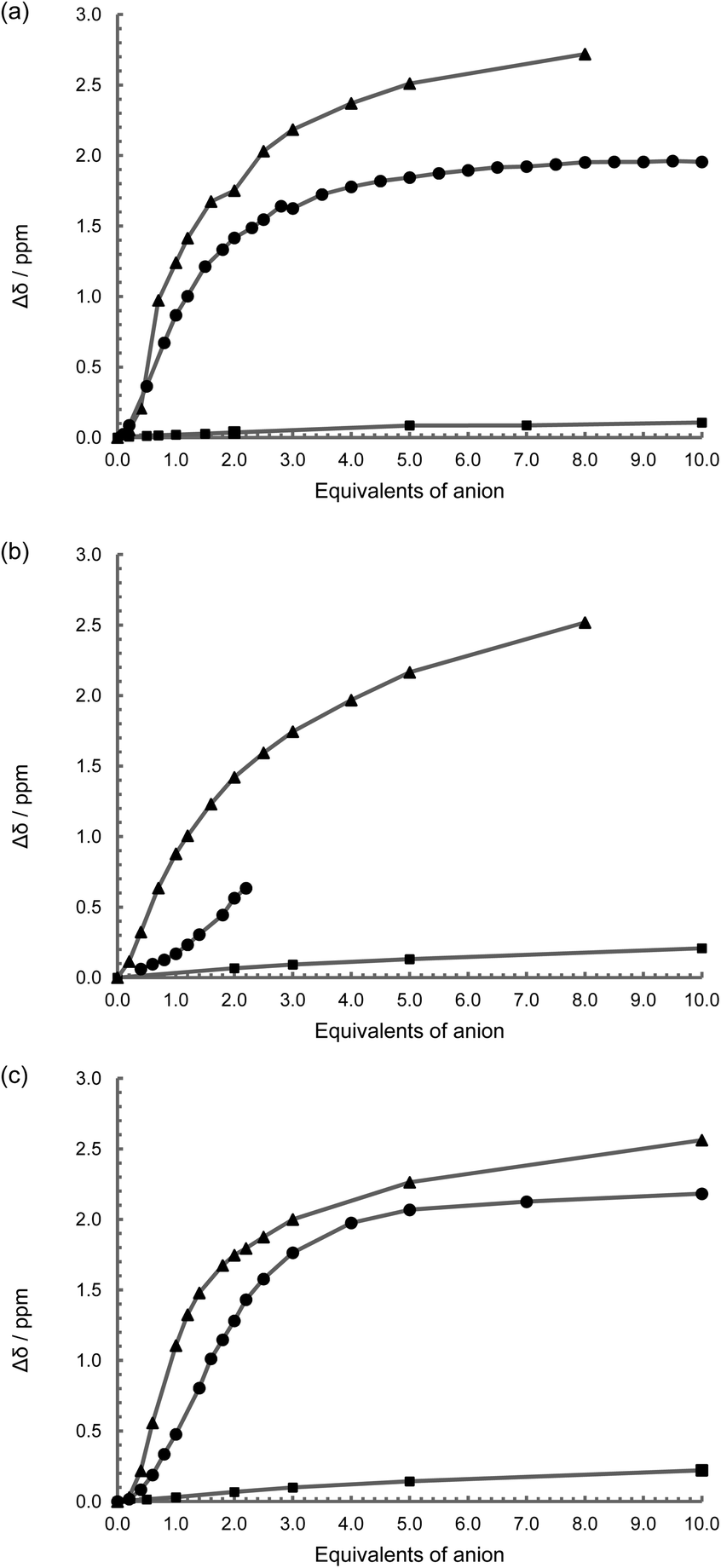 | ||
| Fig. 4 1H NMR spectroscopic change in CH2NH peak position on the introduction of TBA OAc (▲), TBA Cl (■) and TBA H2PO4 (●); (a) {Re(CO)3Br}2(μ-L1), (b) {Re(CO)3Br}2(μ-L2), and (c) {Re(CO)3Br}2(μ-L3); (50% DMSO-D6–CD3CN) 400 MHz, 298 K, at approx. 1 × 10−3 mol dm−3. | ||
With the introduction of TBA H2PO4 to {Re(CO)3Br}2(μ-L1) in a 50% DMSO-D6–CD3CN mixture, there is a significant down-field shift of the NH proton signal (Fig. S15b†), and a change in the methylene peak position, with the singlet broadening and eventually becoming a multiplet implying a conformational change and a more rigid arrangement around the flexible methylene groups – i.e. they become conformationally frozen making the two protons diastereotopic. A slight downfield shift in the peaks attributed to the 3 and 4 positions on the bipyridine is also noted. A value of pβ2 of over 10 is obtained by modelling the system to a two to one stoichiometry confirming a surprisingly strong interaction with the dihydrogenphosphate (Table 2). Given that these values are too high to be accurate, the titration was repeated in DMSO-D6 with 0.5% H2O, however in the more protic environment there is a rapid loss of the N–H proton signal consistent with proton exchange. Under the same conditions, the presence of the fac-tricarbonyl rhenium(I) centre results in an enhanced interaction to this anion when compared to the free ligand (L1).
For the complexes {Re(CO)3Br}2(μ-L2) and {Re(CO)3Br}2(μ-L3), they again show no interaction with NO3−, HSO4−, Br− and ClO4− salts in 50% CD3CN–DMSO-D6, while similarly chloride exhibits only a slight deviation in the thiourea peak position (Fig. 4b, 4c, S16a and S17a†). Tentative calculations of pβ2 for chloride are of the order of 2, but given the size of the change in peak position in both damp DMSO-D6, and 50% CD3CN–DMSO-D6, precise values cannot be accurately determined (Δδ = 0.2 ppm with ten equivalents). With acetate the complexes show similar behaviour in binding to the first anion to that displayed by the free ligands L2 and L3 respectively. However the association to the second anion is considerably greater with the complex, presumably due to the influence of the metal centre and associated carbonyl groups (Fig. S16b and S17b†).
The 1H NMR spectroscopic titrations with {Re(CO)3Br}2(μ-L2) and TBA dihydrogenphosphate were not successful. While it can be seen from the change in position of the visible NH protons signals that an interaction is occurring (Fig. 4b and S16c†), with the addition of over one equivalent of the anion, the peak broadens to such an extent that it effectively disappears, although the start of a sigmoidal curve similar to that seen with the free ligand, and in keeping with the other dihydrogenphosphate titrations, is in evidence. Similar behaviour is also observed in DMSO-D6 with 0.5% H2O. With {Re(CO)3Br}2(μ-L3) on the other hand, while the signal broadens indicative of a degree of proton exchange in the solvent system used, it can be followed sufficiently to again highlight a sigmoidal curve (Fig. 4c, S17c†) and gives similar stability constants (pβ1 = 3.8 and pβ2 = 7.4) to that of the free ligand L3 suggestive of a degree of cooperativity in the binding of the second anion. And again, there are similar perturbations to the aromatic linkage as seen with ligand L3 itself.
UV/vis and emission anion binding studies
The complexes {Re(CO)3Br}2(μ-L1), {Re(CO)3Br}2(μ-L2) and {Re(CO)3Br}2(μ-L3) UV/vis absorption behaviour with a range of TBA salts in acetonitrile prove again that NO3−, HSO4−, Br−, Cl− and ClO4− salts result in little or no perturbation to the spectra, while the addition of TBA fluoride, shows a decrease in the LC transition (Fig. S18†). Similar behaviour is seen by UV spectroscopy with acetate (Fig. S19†) and to a lesser extent with dihydrogenphosphate indicative of a degree of selectivity. However the observed change in the spectra is very marginal and is probably best related to the capacity of the anion to affect deprotonation of the thiourea, rather than a measure of a “recognition” event with F− > OAc− > H2PO4− > Cl−.44,51 An attempt was made to complete a Job plot analysis of {Re(CO)3Br}2(μ-L1) in the presence of TBA dihydrogenphosphate, while examining the concentration corrected change in the absorption at both 235 nm and 300 nm. Given the very marginal perturbations in the UV absorption, the data is rather tentative but would appear to illustrate the proposed two to one stoichiometry observed with the ligand, and proposed for the complexes (Fig. S20†).There are no observed perturbations in the emission spectra of {Re(CO)3Br}2(μ-L1), {Re(CO)3Br}2(μ-L2) and {Re(CO)3Br}2(μ-L3) (excited at 380 nm) on the addition of NO3−, Br−, and ClO4−. Chloride salts result in a very slight drop in the emissive behaviour in the order of 2% quenching for example with {Re(CO)3Br}2(μ-L1) (Fig. S21†). HSO4− gives a slight increase followed by a consistent decrease in the order of 8% quenching for example with {Re(CO)3Br}2(μ-L3) (Fig. S22†). The introduction of fluoride anions however results in a gradual decrease (20% and 50% drop with ten equivalents in {Re(CO)3Br}2(μ-L1) and {Re(CO)3Br}2(μ-L2) respectively) consistent with deprotonation.
The addition of acetate results in a different behaviour between the various complexes under investigation; the simpler complex, {Re(CO)3Br}2(μ-L1), initially gives a slight increase in the luminescence, before a gradual decrease. This is understood to be binding, followed by a degree of deprotonation (Fig. S23a†). However for {Re(CO)3Br}2(μ-L2) a significant quenching in the emission is observed in a similar fashion to that seen with fluoride. It is therefore assumed that the presence of the aryl group makes the NH group more acidic in agreement with previous studies.30,44
With the addition of TBA dihydrogenphosphate, the emission titrations result in interesting behaviour suggestive of two competing processes. With {Re(CO)3Br}2(μ-L1), an increase in luminescence (Fig. 5a) is observed alongside a slight blue shift of the order of 10 nm. There is a subsequent gradual decrease; a plot of the integrated spectra (Fig. 5b) reveals an unusual sigmoidal titration curve, with a two to one ratio confirming the 1H NMR data. With the more extended system {Re(CO)3Br}2(μ-L2), the titration results in a reproducibly small initial increase in luminescence (up to 0.7 equivalents) followed by a significant decrease along with a 20 nm blue shift in the maximum peak position (Fig. 6a). Complex {Re(CO)3Br}2(μ-L3) shows an intermediate situation with initially an increase along with a subsequent and considerable hypsochromic shift before a quenching mechanism is invoked (Fig. 6b).
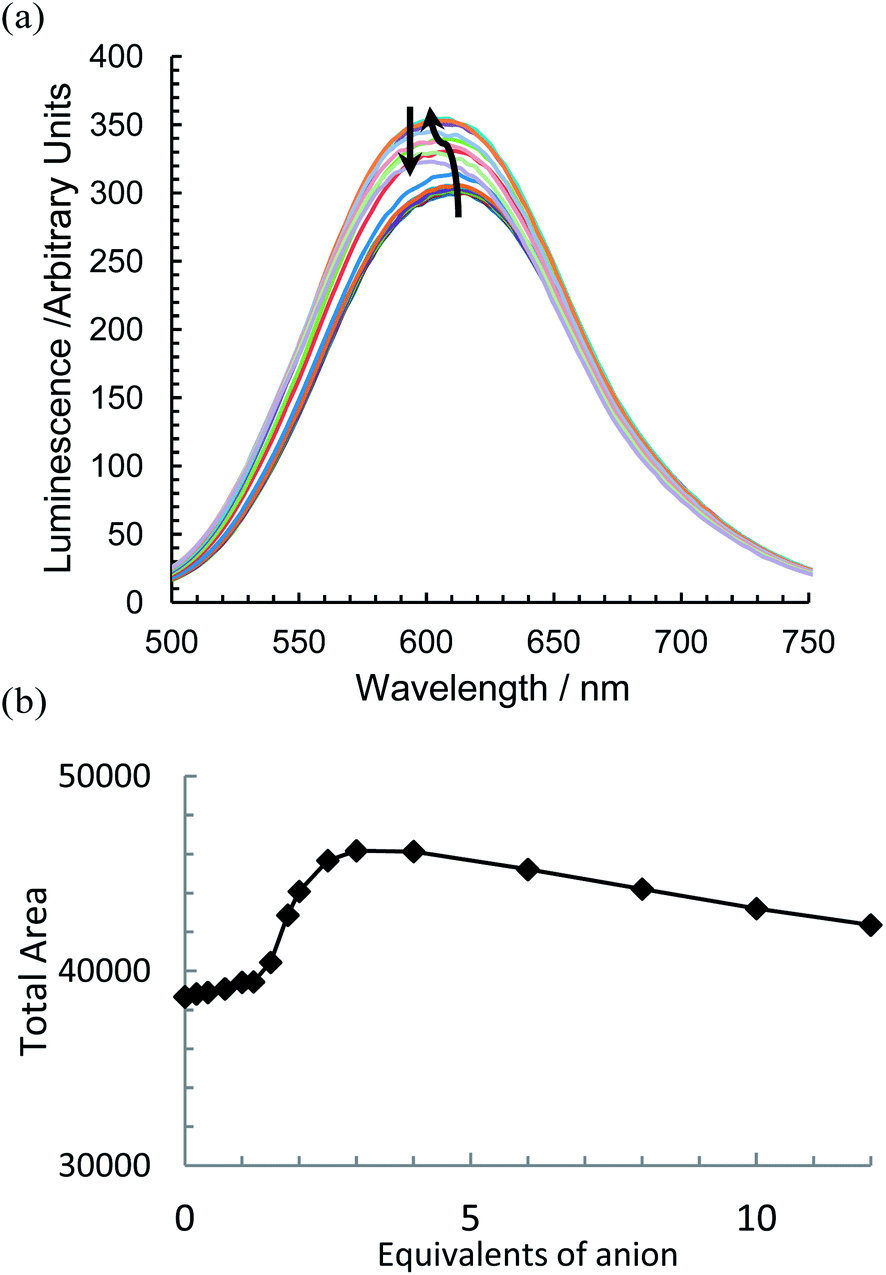 | ||
| Fig. 5 The change in emissive behaviour of (a) {Re(CO)3Br}2(μ-L1) on the introduction of TBA H2PO4 and (b) the change in integrated area (CD3CN, 298 K, at approx. 1 × 10−5 mol dm−3). | ||
Although attempts to get stability constants from the emission data were considered, given the complexity in the system, it proved to be difficult to obtain meaningful data. The luminescence in such systems is attributed to a long lived meta stable excited triplet state predominantly located on the bipyridine ligand, which decays to the HOMO possessing mainly metal-carbonyl character.56 In the systems investigated here there appear to be two emissive states at approximately 580 nm and 610 nm. Initially the transition from the lower energy state is dominant, but this state appears to be more sensitive to the anions interacting with the thiourea groups, leading to the observed blue shift. However this is complicated by there being two competing processes occurring in the three systems considered. While further spectroscopic investigations are required to verify this, it would appear that binding the anion initially enhances the fluorescence, presumably by either blocking a solvent, or oxygen induced radiationless decay processes. However the act of deprotonation provides an alternative non-emissive decay pathway as exemplified by the more basic anions fluoride and acetate, and the more acidic aryl thioureas in L2 and L3.
Discussion
The strong interactions observed with fluoride and acetate are consistent with similar thiourea complexes. Fluoride enables deprotonation and the formation of H2F+, while acetate forms a strong double hydrogen bond with the thiourea (Fig. 7a) with binding constants consistent with literature values for similar systems.22–24 However it is evident that there is also a degree of deprotonation occurring with there being a considerable loss in the 1H NMR signal for the NH signal on the introduction of this anion and the UV/vis titration not presenting a clear isobestic point (Fig. S19†). In fact moving to a solution of DMSO-D6 contain 0.5% water, the 1H NMR signal, for all of the complexes disappeared on the addition of less than one equivalent of acetate suggestive of a high degree of proton exchange particularly with the para-substituted aryl bridged complex {Re(CO)3Br}2(μ-L2).51 However where the NH peak could be followed, the peak position gave a good indication of an association process, rather than the competing deprotonation, which reveals an insight into the nature of how dihydrogenphosphate interacts with thiourea groups.The binding of monovalent dihydrogenphosphate is far stronger than we would have anticipated in these systems, and it is anomalously high in comparison to other tetrahedral oxo-anions. Evidently it behaves more like acetate than the majority of the other anions considered, including chloride implying that the presence of the two OH groups is important. However it is the surprising observation of sigmoidal titration curves and the implication of cooperative binding that needs to be considered in more depth. Initially we considered a syn/anti conformational change in the thiourea group,57,58 however a review of the literature reveals that similar anomalies have been reported with dihydrogenphosphate, and not just with thioureas and urea based systems.25,30,59,60 It appears that the binding of a second H2PO4− strengthens the interaction with the first unit, and that acetate can show similar behaviour in certain instances. It has been hypothesised to be as a result of the formation of a hydrogen bond with the conjugate base.51
Lazarides et al. highlighted an unusual situation with a widely spaced diamide with a Re(I) tricarbonyl complex,59 where similar behaviour is attributed to the formation of a phosphate dimer and they propose two possible configurational explanations. Recently both Blažek et al.14 and Amendola et al.61 have also shown that the binding of phosphate to urea provides additional stability though the formation of secondary hydrogen bonds between the two phosphates groups. In addition a study by Gale and co-workers with a diindolylurea complex has also shown an anomalous interaction with dihydrogen phosphate, where neither a simple 1 to 1, nor a 2 to 1, binding model adequately accounted for the observed behaviour in the 1H NMR spectroscopic titrations.62 They concluded that following binding that the pKa for the H2PO4− dropped to permit proton transfer to a second dihydrogen phosphate, which was a slow process on the 1H NMR timescale. It is conceivable that a similar process is occurring here. A review of the Cambridge Crystallographic Database highlighted several hundred examples where two or more phosphates are associated together through hydrogen bonding, including several “anion receptors” bearing amidic NH groups, which also show remarkable selectivity for dihydrogenphosphate.63–65
In the case of ligand L1, where only solution phase behaviour is available to us, and basing this on the previous observations, it is proposed that the second phosphate “piggy backs” on the first formally bonded anion (Fig. 7b). This interaction, with a partial transfer of a proton from the thiourea to the anions then encourages one of the protons from the dihydrogenphosphate from the first anion onto the second, enhancing the anionic character of the formally bound anion thereby strengthening its interaction explaining the observed cooperativity, and the high binding constants. In an attempt to confirm this, a titration was attempted to add both ligand L1 and the complex {Re(CO)3Br}2(μ-L1) into dihydrogenphosphate in DMSO-D6, and monitor the change in the 31P resonance, but unfortunately no change is observed.
With two rhenium centres in the complex {Re(CO)3Br}2(μ-L1), there is a significant enhancement in the binding of the two dihydrogenphosphate groups over that seen with L1 alone, which is not seen with acetate. In light of our previous findings, it is proposed that this corresponds to an involvement of the carbonyl groups, which are available to form additional chelating hydrogen bonds with the phosphates (Fig. 7c).41,42 Unfortunately, the poor solubility of the materials involved in this study prevented further elucidation by a solution IR spectroscopy.
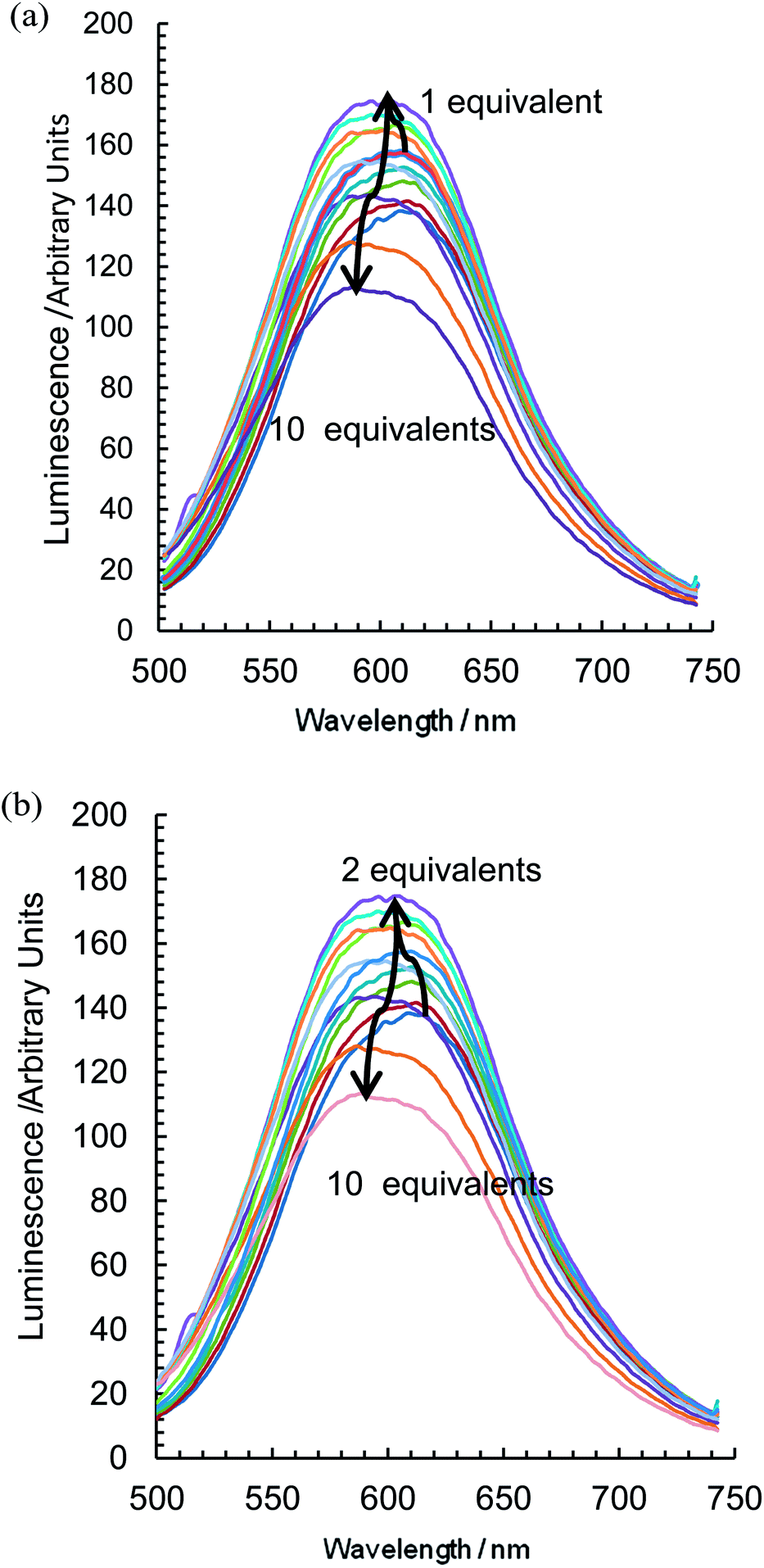 | ||
| Fig. 6 The change in emissive behaviour of (a) {Re(CO)3Br}2(μ-L2) and (b) {Re(CO)3Br}2(μ-L3) on the introduction of TBA H2PO4 (CD3CN, 298 K, at approx. 1 × 10−5 mol dm−3). | ||
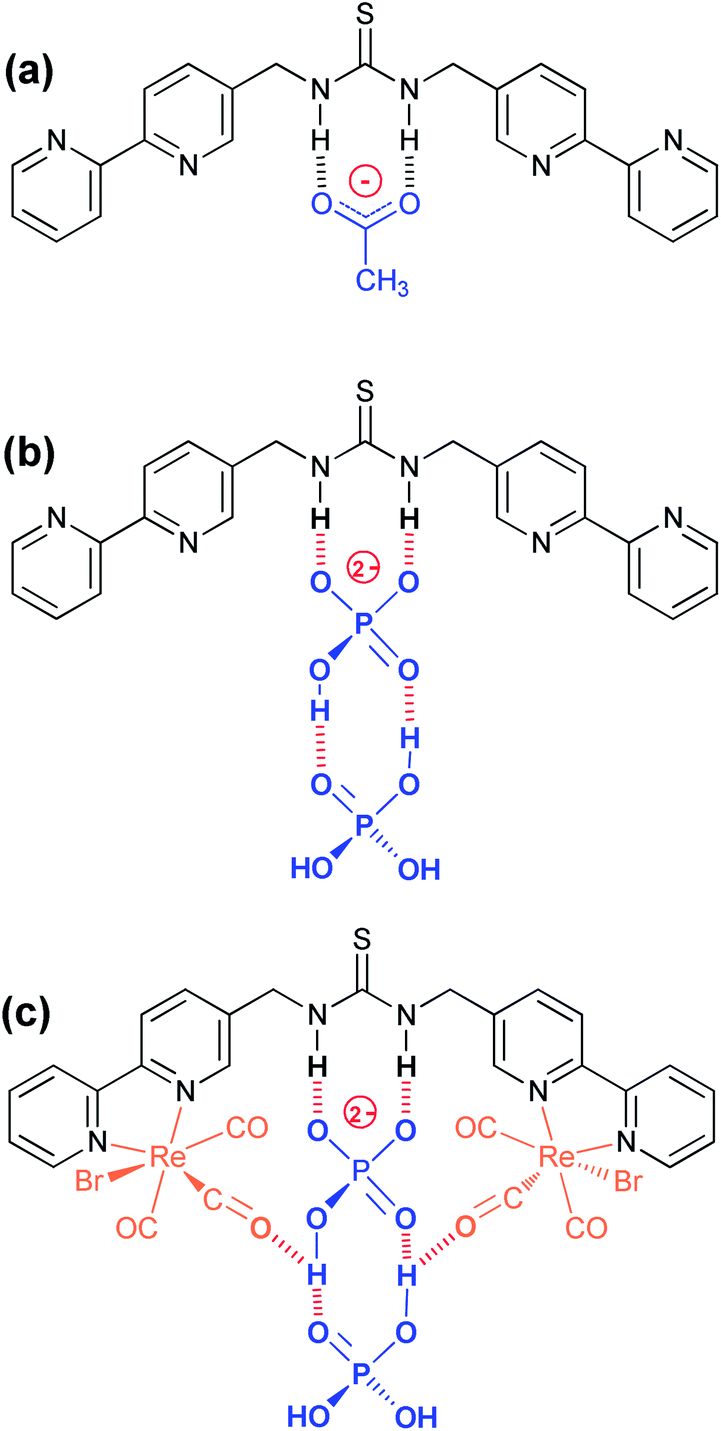 | ||
| Fig. 7 Proposed modes of binding of (a) OAc− and H2PO4− to (b) L and (c) {Re(CO)3Br}2(μ-L1). | ||
In adding a second urea to the ligand system (L2 and L3), both the free ligand and the complexes see a marginal enhancement in the binding consistent with two sites being available to bind anions. In a CD3CN–DMSO-D6 mixture, these fit well to a two to one stoichiometry. However dihydrogenphosphate again shows remarkably strong binding to the second anion that cannot be adequately explained using this simple model. Given the sigmoidal titration curve, the data did fit surprisingly well to the two to one model with goodness-of-fit of less than 2% in each titration curve examined. But the situation is evidently far more complex than this simple model justifies and is consistent with the anomalous results observed by Hanan and co-workers in the related system.30 In DMSO there was some evidence of a degree of proton exchange with the loss of the N–H proton signals on the addition of both acetate and dihydrogenphosphate, but discounting this dynamic effect, the observed cooperativity is not adequately explained given the rigid separation of the two binding sites by an aromatic spacer. In light of the result proposed for the simpler system {Re(CO)3Br}2(μ-L1), it is conceivable that once the two anions are bound to the ligands, a secondary process occurs such as dimerisation, or the formation of larger aggregations. This is also consistent with the observed broadening of the observed aromatic signals in the 1H NMR titrations. However larger assemblies have not yet been evident by electrospray mass spectrometry and remain a hypothesis.
Conclusions
Our results suggest that the binding of acetate and dihydrogenphosphate to thioureas is not a simple binding event, with several competing processes in evidence. As has been previously described, proton exchange, or even deprotonation, of the NH group readily occurs with fluoride and acetate, while there is some evidence that this can also occur with dihydrogenphosphate. However it is also apparent that the latter anion also binds cooperatively, with two units potentially interacting with one thiourea group, and the second anion enhancing the interaction of the first possibly by proton exchange. This accounts for the remarkable selectivity for dihydrogenphosphate in many of the reported protic anion receptors. While this knowledge will enable understanding to assist in designing systems to both encourage selectivity for this intriguing anion, the ramifications of these results extend into understanding many of the commonly observed supramolecular processes in biology involving this ubiquitous anion. For example, the facile addition of a dihydrogenphosphate to ADP to form ATP in ATP synthase could be explained by the cooperativity observed in this study, where the initial stage of simply bringing two negatively charged groups together cooperatively is justified, permitting the subsequent enzyme catalysed dehydration.Experimental
Physical measurements
NMR spectra were recorded using Bruker AV300 (ligands), AV400 (titrations) and DRX500 (complexes) spectrometers. Electronic absorption studies were performed with a Perkin-Elmer Lambda 800 spectrophotometer and emission spectra were recorded on a Perkin-Elmer LS55 spectrofluorimeter, having adjusted the adsorption at the excitation wavelength to be 0.1. Microanalysis, and E.S. and E.I. mass spectrometry were performed by A.S.E.P., The School of Chemistry and Chemical Engineering, Queen's University Belfast.Materials
All reagents were purchased from Sigma Aldrich and used as supplied unless otherwise stated. 5-Chloromethyl-2,2′-bipyridine46 was prepared from 2-acetylpyridine via a Kröhnke synthesis to 5-methyl-2,2′-bipyridine,66 lithiation and subsequent reaction to 5-(trimethylsilyl)methyl-2,2′-bipyridine followed by conversion to the chlorinated derivative by reaction with CsF and Cl3C2Cl3.46 1,3-Phenylene diisothiocyanate was prepared using thiophosgene following a literature procedure.49Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch), 1604 (br, thiourea bend).O stretch), 1551 (br, thiourea bend).O stretch), 1540 (br, thiourea).
O stretch), 1604 (br, thiourea bend).O stretch), 1551 (br, thiourea bend).O stretch), 1540 (br, thiourea).Acknowledgements
This work was supported by a DELNI studentship (ALB) and QUESTOR and ChemVite Ltd. for the studentship (NCAB). Prof. M. D. Ward is thanked for his additional insight.Notes and references
- M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC.
- A. E. Hargrove, S. Nieto, T. Z. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS.
- A. K. H. Hirsch, F. R. Fischer and F. Diederich, Angew. Chem., Int. Ed., 2007, 46, 338–352 CrossRef CAS.
- V. Amendola, D. Esteban-Gomez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343–353 CrossRef CAS.
- V. Amendola and L. Fabbrizzi, Chem. Commun., 2009, 513–531 RSC.
- D. J. Mercer and S. J. Loeb, Chem. Soc. Rev., 2010, 39, 3612–3620 RSC.
- J. W. Steed, Chem. Soc. Rev., 2009, 38, 506–519 RSC.
- D. Jana, G. Mani and C. Schulzke, Inorg. Chem., 2013, 52, 6427–6439 CrossRef CAS.
- S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- C. Caltagirone, C. Bazzicalupi, F. Isaia, M. E. Light, V. Lippolis, R. Montis, S. Murgia, M. Olivari and G. Picci, Org. Biomol. Chem., 2013, 11, 2445–2451 CAS.
- V. Blažek, K. Molcanov, K. Mlinaric-Majerski, B. Kojic-Prodic and N. Basaric, Tetrahedron, 2013, 69, 517–526 CrossRef.
- R. M. Duke and T. Gunnlaugsson, Tetrahedron Lett., 2010, 51, 5402–5405 CrossRef CAS.
- Y. L. Zhang, R. Zhang, Y. X. Zhao, L. G. Ji, C. D. Jia and B. Wu, New J. Chem., 2013, 37, 2266–2270 RSC.
- C. Jia, B. Wu, S. Li, X. Huang, Q. Zhao, Q.-S. Li and X.-J. Yang, Angew. Chem., Int. Ed., 2011, 50, 486–490 CrossRef CAS.
- C. Jia, B. Wu, S. Li, Z. Yang, Q. Zhao, J. Liang, Q.-S. Li and X.-J. Yang, Chem. Commun., 2010, 46, 5376–5378 RSC.
- R. Custelcean, P. Remy, P. V. Bonnesen, D.-E. Jiang and B. A. Moyer, Angew. Chem., Int. Ed., 2008, 47, 1866–1870 CrossRef CAS.
- A. Rajbanshi and R. Custelcean, Supramol. Chem., 2012, 24, 65–71 CrossRef CAS.
- R. Custelcean, A. Bock and B. A. Moyer, J. Am. Chem. Soc., 2010, 132, 7177–7185 CrossRef CAS.
- A. Aldrey, A. Macias, R. Bastida, G. Zaragoza, G. Rama and M. Vazquez Lopez, Org. Biomol. Chem., 2012, 10, 5379–5384 CAS.
- A. R. Jennings and D. Y. Son, Tetrahedron Lett., 2012, 53, 2181–2184 CrossRef CAS.
- P. G. Young, J. K. Clegg, M. Bhadbhade and K. A. Jolliffe, Chem. Commun., 2011, 47, 463–465 RSC.
- P. S. Lakshminarayanan, I. Ravikumar, E. Suresh and P. Ghosh, Chem. Commun., 2007, 5214–5216 RSC.
- M. H. Keefe, K. D. Benkstein and J. T. Hupp, Coord. Chem. Rev., 2000, 205, 201–228 CrossRef CAS.
- J. A. Kitchen, E. M. Boyle and T. Gunnlaugsson, Inorg. Chim. Acta, 2012, 381, 236–242 CrossRef CAS.
- R. Custelcean, J. Bosano, P. V. Bonnesen, V. Kertesz and B. P. Hay, Angew. Chem., Int. Ed., 2009, 48, 4025–4029 CrossRef CAS.
- R. Custelcean, P. V. Bonnesen, N. C. Duncan, X. Zhang, L. A. Watson, G. Van Berkel, W. B. Parson and B. P. Hay, J. Am. Chem. Soc., 2012, 134, 8525–8534 CrossRef CAS.
- A. Bessette, S. Nag, A. K. Pal, S. Derossi and G. S. Hanan, Supramol. Chem., 2012, 24, 595–603 CrossRef CAS.
- N. C. Fletcher and M. C. Lagunas, Top. Organomet. Chem., 2009, 28, 143–170 CrossRef.
- J. Perez and L. Riera, Chem. Commun., 2008, 533–543 RSC.
- J. Perez and L. Riera, Chem. Soc. Rev., 2008, 37, 2658–2667 RSC.
- P. D. Beer, S. W. Dent, G. S. Hobbs and T. J. Wear, Chem. Commun., 1997, 99–100 RSC.
- P. D. Beer, M. G. B. Drew, D. Hesek, M. Shade and F. Szemes, Chem. Commun., 1996, 2161–2162 RSC.
- D. Curiel and P. D. Beer, Chem. Commun., 2005, 1909–1911 RSC.
- C.-O. Ng, S.-W. Lai, H. Feng, S.-M. Yiu and C.-C. Ko, Dalton Trans., 2011, 40, 10020–10028 RSC.
- R. V. Slone, D. I. Yoon, R. M. Calhoun and J. T. Hupp, J. Am. Chem. Soc., 1995, 117, 11813–11814 CrossRef CAS.
- S. S. Sun and A. J. Lees, Chem. Commun., 2000, 1687–1688 RSC.
- B. C. Tzeng, Y. F. Chen, C. C. Wu, C. C. Hu, Y. T. Chang and C. K. Chen, New J. Chem., 2007, 31, 202–209 RSC.
- D. Pelleteret and N. C. Fletcher, Eur. J. Inorg. Chem., 2008, 3597–3605 CrossRef CAS.
- D. Pelleteret, N. C. Fletcher and A. P. Doherty, Inorg. Chem., 2007, 46, 4386–4388 CrossRef CAS.
- K.-C. Chang, S.-S. Sun and A. J. Lees, Inorg. Chim. Acta, 2012, 389, 16–28 CrossRef CAS.
- D. E. Gomez, L. Fabbrizzi, M. Licchelli and E. Monzani, Org. Biomol. Chem., 2005, 3, 1495–1500 CAS.
- C. Janiak, S. Deblon and H. P. Wu, Synth. Commun., 1999, 29, 3341–3352 CrossRef CAS.
- S. A. Savage, A. P. Smith and C. L. Fraser, J. Org. Chem., 1998, 63, 10048–10051 CrossRef CAS.
- J. Eaves, H. Munro and D. Parker, Inorg. Chem., 1987, 26, 644–650 CrossRef CAS.
- R. Wong and S. J. Dolman, J. Org. Chem., 2007, 72, 3969–3971 CrossRef CAS.
- K. A. Jacobson, S. Barone, U. Kammula and G. L. Stiles, J. Med. Chem., 1989, 32, 1043–1051 CrossRef CAS.
- E. Wolcan, G. Torchia, J. Tocho, O. E. Piro, P. Juliarena, G. Ruiz and M. R. Feliz, J. Chem. Soc., Dalton Trans., 2002, 2194–2202 RSC.
- M. Boiocchi, L. Del Boca, D. Esteban-Gomez, L. Fabbrizzi, M. Licchelli and E. Monzani, Chem.–Eur. J., 2005, 11, 3097–3104 CrossRef CAS.
- G. W. Bates, P. A. Gale and M. E. Light, Chem. Commun., 2007, 2121–2123 RSC.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- P. A. Gale, S. E. Garcia-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC.
- M. A. Saeed, F. R. Fronczek and M. A. Hossain, Chem. Commun., 2009, 6409–6411 RSC.
- A. Vlček Jr and S. Zalis, Coord. Chem. Rev., 2007, 251, 258–287 CrossRef.
- M. Olivari, C. Caltagirone, A. Garau, F. Isaia, M. E. Light, V. Lippolis, R. Montis and M. A. Scorciapino, New J. Chem., 2013, 37, 663–669 RSC.
- R. Custelcean, Chem. Commun., 2013, 49, 2173–2182 RSC.
- T. Lazarides, T. A. Miller, J. C. Jeffery, T. K. Ronson, H. Adams and M. D. Ward, Dalton Trans., 2005, 528–536 RSC.
- P. Dydio, T. Zielinski and J. Jurczak, J. Org. Chem., 2009, 74, 1525–1530 CrossRef CAS.
- V. Amendola, M. Boiocchi, D. Esteban-Gomez, L. Fabbrizzi and E. Monzani, Org. Biomol. Chem., 2005, 3, 2632–2639 CAS.
- P. A. Gale, J. R. Hiscock, S. J. Moore, C. Caltagirone, M. B. Hursthouse and M. E. Light, Chem.–Asian J., 2010, 5, 555–561 CrossRef CAS.
- N. C. A. Baker, N. McGaughey, N. C. Fletcher, A. V. Chernikov, P. N. Horton and M. B. Hursthouse, Dalton Trans., 2009, 965–972 RSC.
- C. Caltagirone, P. A. Gale, J. R. Hiscock, M. B. Hursthouse, M. E. Light and G. J. Tizzard, Supramol. Chem., 2009, 21, 125–130 CrossRef CAS.
- F. Szemes, D. Hesek, Z. Chen, S. W. Dent, M. G. B. Drew, A. J. Goulden, A. R. Graydon, A. Grieve, R. J. Mortimer, T. Wear, J. S. Weightman and P. D. Beer, Inorg. Chem., 1996, 35, 5868–5879 CrossRef CAS.
- T. L. J. Huang and D. G. Brewer, Can. J. Chem., 1981, 59, 1689–1700 CrossRef CAS.
- R. Ziessel and J.-M. Lehn, Helv. Chim. Acta, 1990, 73, 1149–1162 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic characterization and titration data. See DOI: 10.1039/c4ra00912f |
| This journal is © The Royal Society of Chemistry 2014 |