NHC–gold(I) catalysed [4 + 2] cycloaddition–acyclic addition of dialkyl substituted propargylic esters with 1,3-diphenylisobenzofuran: synthesis of novel benzo[c]fluorenols and substituted dienes†
Ramesh Kotikalapudi,
A. Leela Siva Kumari and
K. C. Kumara Swamy*
School of Chemistry, University of Hyderabad, Hyderabad 500 046, A. P., India. E-mail: kckssc@yahoo.com; kckssc@uohyd.ernet.in; Fax: +91-40-23012460; Tel: +91-40-23134856
First published on 28th March 2014
Abstract
A gold carbene complex [IPrAuCl/AgSbF6] catalysed novel cycloaddition of propargylic esters with 1,3-diphenylisobenzofuran is reported. A [4 + 2] cycloaddition followed by sequential aromatic allylation leading to pentannulation with the expulsion of benzoic acid, then 1,2-phenyl migration coupled with ring opening and aromatisation leading to a new class of benzofluorenols, is discovered. This process involves facile multiple C–C bond formation. An accompanying second pathway involving the attack of benzoyloxy anion on the central allenic carbon affording substituted dienes is also observed. Key products are characterised by X-ray structure determination.
Introduction
Gold catalysis has emerged as an important area in modern organic synthesis during the last decade.1 The catalytic activity of gold salts finds many applications in recent methodologies for the construction of new C–C and C–X bonds1c–f,2 leading to a wide range of heterocycles and carbocycles.3 Due to the alkynophilicity of gold catalysts, propargylic esters can undergo inter-/intra-molecular cycloaddition reactions.4 In addition to [4 + 2] cycloaddition,5 [4 + 3] cycloaddition of propargylic esters with α,β-unsaturated imines and furan leading to azepines4a,e and trienes4c,d respectively has been reported. Pioneering studies of Hashmi's group on gold catalysed intramolecular cyclisation of alkynes and furans has led to a new methodology for the synthesis of various phenol derivatives.6 Very recently, formation of phenols via cyclisation of acetylenes and furans has been reported by Echavarren's group.7 In continuation of our work on the reactions of propargylic alcohol/ester/and alkyne chemistry,8 we report herein NHC–gold(I) catalysed [4 + 2] cycloaddition and acyclic addition of dialkyl substituted propargylic esters with 1,3-diphenylisobenzofuran (IBF) leading to benzo[c]fluorenols and substituted dienes. Benzofluorene derivatives are organic electroluminescent compounds and used as key components in organic light emitting diodes.9 They are the core structures for kinamycins which are strongly active natural products against gram positive bacteria.10 They are also potent anticancer and antimicrobial agents.10 Natural products seongomycin,11a cysfluretin,11b shikometabolins11c and fluostatins11d also comprise benzofluorene core structure.Results and discussion
The propargylic esters 1a–l used in the present study were synthesised from the corresponding silyl substituted propargylic esters (see ESI;† species I–V) or propargylic alcohols.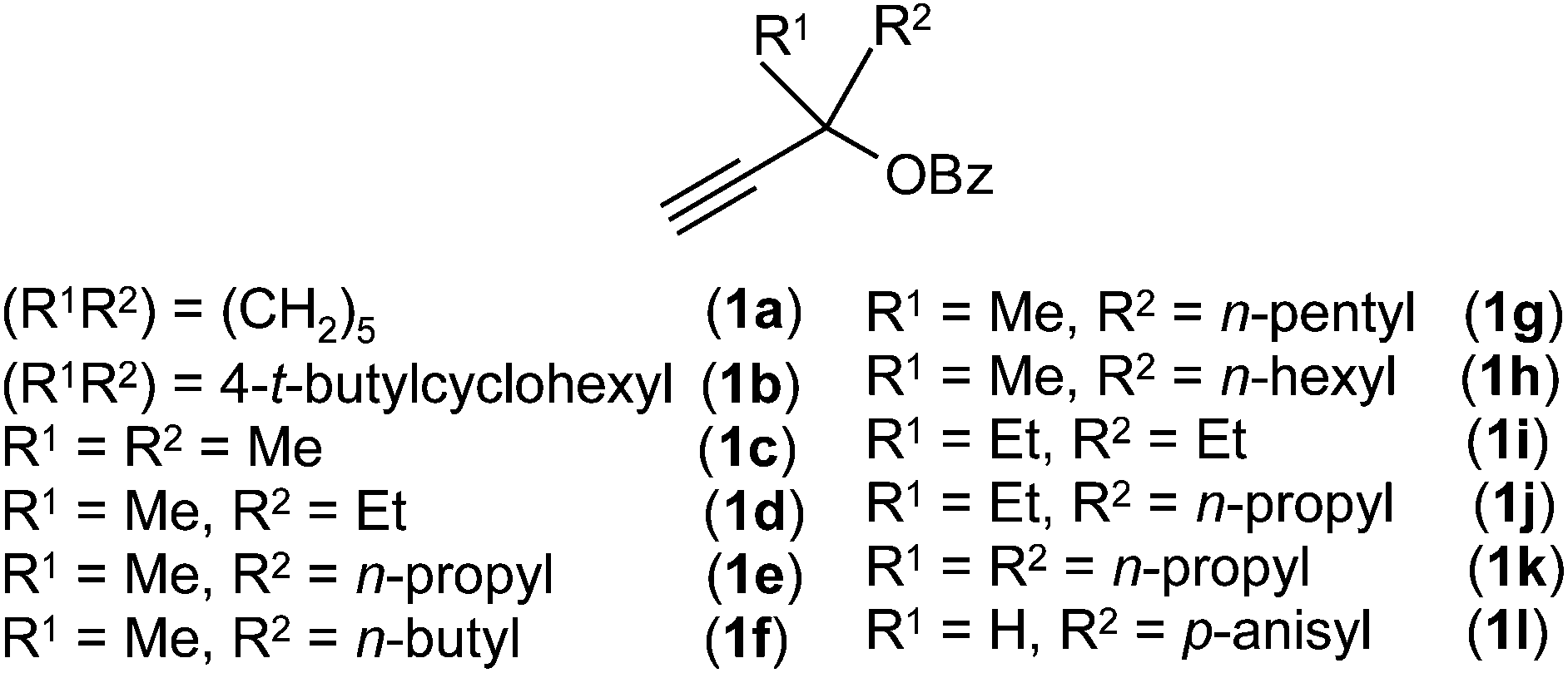
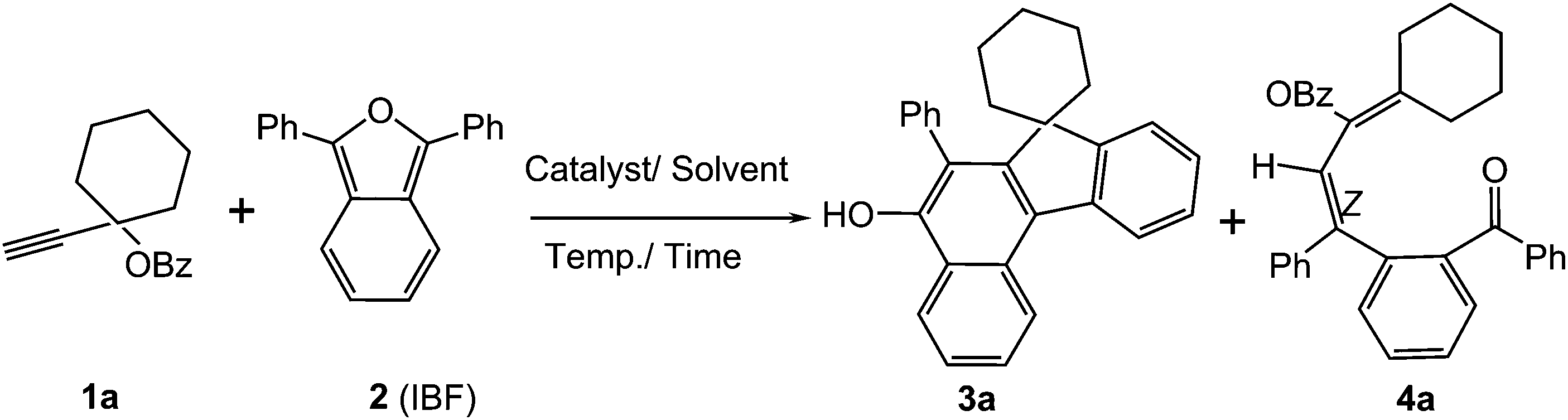
For optimisation, we chose cycloaddition of propargylic ester 1a with IBF 2 (Scheme 1). First it was confirmed that 1a did not react with 1,3-diphenylisobenzofuran (IBF, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 molar ratio) in dichloromethane at rt under catalyst-free condition (Table 1, entry 1). Then, gold carbene complex was employed as the catalyst. To our delight, we found that 2 mol% each of IPrAuCl12 and AgSbF6 in CH2Cl2 worked well to lead to benzofluorenols 3a and substituted diene 4a in 36% and 54% yields respectively (Table 1, entry 2) based on 1a. The combined yield (3a + 4a; after isolation) is excellent (90%). An additional important point is that the Rf values of the two products are significantly different, and hence the benzofluorenols can be very easily isolated. Hence our trials were directed towards improving the yield of benzofluorenol derivatives rather than dienes. Thus when the quantity of IBF was reduced to 1.2 mmol equivalents, the yield of benzofluorenol was increased to 52% whereas yield of diene was 34% [combined yield 86%; Table 1, entry 3]. Equimolar ratio of substrates led to benzofluorenol and diene in 57% and 33% respectively (entry 4) whereas as 1.2:1 ratio of alkyne and IBF afforded improved yield of benzofluorenol (58%) with a combined yield of 91% (entry 5). Use of 2:1 ratio of alkyne and IBF led to decrease in the yield of benzofluorenol to 48% (entry 6). At 55 °C, the reaction afforded 32% of benzofluorenol only (entry 7). Use of 1-ethynylcyclohexyl acetate did not improve the yield of benzofluorenol (53%, entry 8). Silver-free IPrAu(NCMe)SbF6 (ref. 13) led to 42% of benzofluorenol with overall yield of 82% (entry 9). Interestingly, use of 2 mol% IPrAuCl/AgOTf gave only the diene product in 48% yield (Table 1, entry 10). Changing the silver salt to AgNTf2 or AgBF4 led to benzofluorenol in lower yields (entries 11 and 12); 2 mol% IMesAuCl/AgSbF6 could drive the reaction to obtain benzofluorenol in 50% of yield (entry 13). PicAuCl2, IPrAuCl or AgSbF6 individually were not effective to form benzofluorenol (entries 14, 15 and 16). Use of 2 mol% Ph3PAuCl/AgSbF6 led to diene product only (entry 17). Solvents like CH3CN, THF and dioxane (entries 18, 19 and 20) in the presence of 2 mol% IPrAuCl/AgSbF6 did not perform well in forming the benzofluorenol; it should be noted that the catalytic system in entry 18 can be treated as AgCl + [IPrAu(NCMe)SbF6].13 Hence it is concluded that 2 mol% IPrAuCl/AgSbF6 in dichloromethane at rt is the best choice for the formation of the benzofluorenol (entry 5). Details on the optimisation of the products using various screening conditions are presented in Table 1.
:1.5 molar ratio) in dichloromethane at rt under catalyst-free condition (Table 1, entry 1). Then, gold carbene complex was employed as the catalyst. To our delight, we found that 2 mol% each of IPrAuCl12 and AgSbF6 in CH2Cl2 worked well to lead to benzofluorenols 3a and substituted diene 4a in 36% and 54% yields respectively (Table 1, entry 2) based on 1a. The combined yield (3a + 4a; after isolation) is excellent (90%). An additional important point is that the Rf values of the two products are significantly different, and hence the benzofluorenols can be very easily isolated. Hence our trials were directed towards improving the yield of benzofluorenol derivatives rather than dienes. Thus when the quantity of IBF was reduced to 1.2 mmol equivalents, the yield of benzofluorenol was increased to 52% whereas yield of diene was 34% [combined yield 86%; Table 1, entry 3]. Equimolar ratio of substrates led to benzofluorenol and diene in 57% and 33% respectively (entry 4) whereas as 1.2:1 ratio of alkyne and IBF afforded improved yield of benzofluorenol (58%) with a combined yield of 91% (entry 5). Use of 2:1 ratio of alkyne and IBF led to decrease in the yield of benzofluorenol to 48% (entry 6). At 55 °C, the reaction afforded 32% of benzofluorenol only (entry 7). Use of 1-ethynylcyclohexyl acetate did not improve the yield of benzofluorenol (53%, entry 8). Silver-free IPrAu(NCMe)SbF6 (ref. 13) led to 42% of benzofluorenol with overall yield of 82% (entry 9). Interestingly, use of 2 mol% IPrAuCl/AgOTf gave only the diene product in 48% yield (Table 1, entry 10). Changing the silver salt to AgNTf2 or AgBF4 led to benzofluorenol in lower yields (entries 11 and 12); 2 mol% IMesAuCl/AgSbF6 could drive the reaction to obtain benzofluorenol in 50% of yield (entry 13). PicAuCl2, IPrAuCl or AgSbF6 individually were not effective to form benzofluorenol (entries 14, 15 and 16). Use of 2 mol% Ph3PAuCl/AgSbF6 led to diene product only (entry 17). Solvents like CH3CN, THF and dioxane (entries 18, 19 and 20) in the presence of 2 mol% IPrAuCl/AgSbF6 did not perform well in forming the benzofluorenol; it should be noted that the catalytic system in entry 18 can be treated as AgCl + [IPrAu(NCMe)SbF6].13 Hence it is concluded that 2 mol% IPrAuCl/AgSbF6 in dichloromethane at rt is the best choice for the formation of the benzofluorenol (entry 5). Details on the optimisation of the products using various screening conditions are presented in Table 1.
 | ||
| Scheme 1 Reaction of propargylic ester 1a with 1,3-diphenylisobenzofuran 2. | ||
| Entrya | Alkyne/IBF | Catalyst | Solvent | Time (h) | Combined yieldb (%) (3a + 4a) |
|---|---|---|---|---|---|
| a All reactions were performed at room temperature.b Isolated yield.c Starting material remained.d A diketone14 which is a dimeric form of IBF was also formed.e Oil bath temperature.f 1-Ethynylcyclohexyl acetate was used. The diene product, although present (ca. 20%; tlc), could not be isolated. | |||||
| 1 | 1/1.5 | No catalyst | DCM | 12 | 0c |
| 2 | 1/1.5 | 2% IPrAuCl/AgSbF6 | DCM | 4 | 90 (36 + 54)d |
| 3 | 1/1.2 | 2% IPrAuCl/AgSbF6 | DCM | 4 | 86 (52 + 34) |
| 4 | 1/1 | 2% IPrAuCl/AgSbF6 | DCM | 4 | 90 (57 + 33) |
| 5 | 1.2/1 | 2% IPrAuCl/AgSbF6 | DCM | 4 | 91 (58 + 33) |
| 6 | 2/1 | 2% IPrAuCl/AgSbF6 | DCM | 4 | 84 (48 + 36) |
| 7 | 1.2/1 | 2% IPrAuCl/AgSbF6 | DCM | 4 (at 55 °C)e | 74 (32 + 42) |
| 8 | 1.2/1 | 2% IPrAuCl/AgSbF6 | DCM | 4 | 80 (60 + 20)f |
| 9 | 1.2/1 | 2% IPrAu(NCMe)SbF6 | DCM | 4 | 82 (42 + 40) |
| 10 | 1.2/1 | 2% IPrAuCl/AgOTf | DCM | 12 | 48 (0 + 48)c |
| 11 | 1.2/1 | 2% IPrAuCl/AgNTf2 | DCM | 12 | 62 (26 + 36)c |
| 12 | 1.2/1 | 2% IPrAuCl/AgBF4 | DCM | 4 | 85 (38 + 47) |
| 13 | 1.2/1 | 2% IMesAuCl/AgSbF6 | DCM | 4 | 88 (50 + 38) |
| 14 | 1.2/1 | 3% PicAuCl2 | DCM | 12 | 54 (0 + 54)c |
| 15 | 1.2/1 | 3% IPrAuCl | DCM | 12 | 15 (0 + 15)c |
| 16 | 1.2/1 | 3% AgSbF6 | DCM | 12 | 68 (0 + 68)c |
| 17 | 1.2/1 | 2% Ph3PAuCl/AgSbF6 | DCM | 12 | 65 (0 + 65)c |
| 18 | 1.2/1 | 2% IPrAuCl/AgSbF6 | CH3CN | 12 | ∼32 (trace + 32) |
| 19 | 1.2/1 | 2% IPrAuCl/AgSbF6 | THF | 12 | 38 (10 + 28)c |
| 20 | 1.2/1 | 2% IPrAuCl/AgSbF6 | Dioxane | 12 | 36 (6 + 30)c |
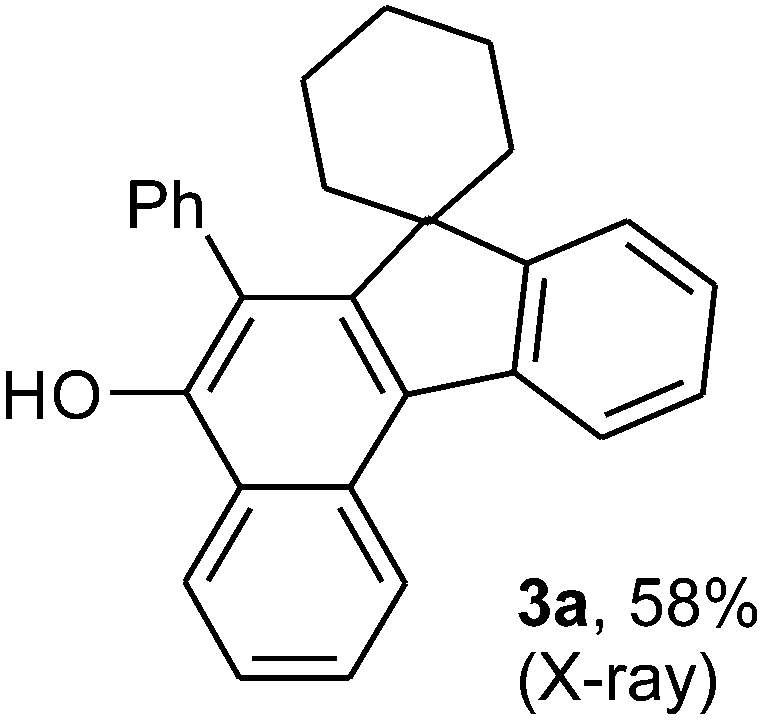
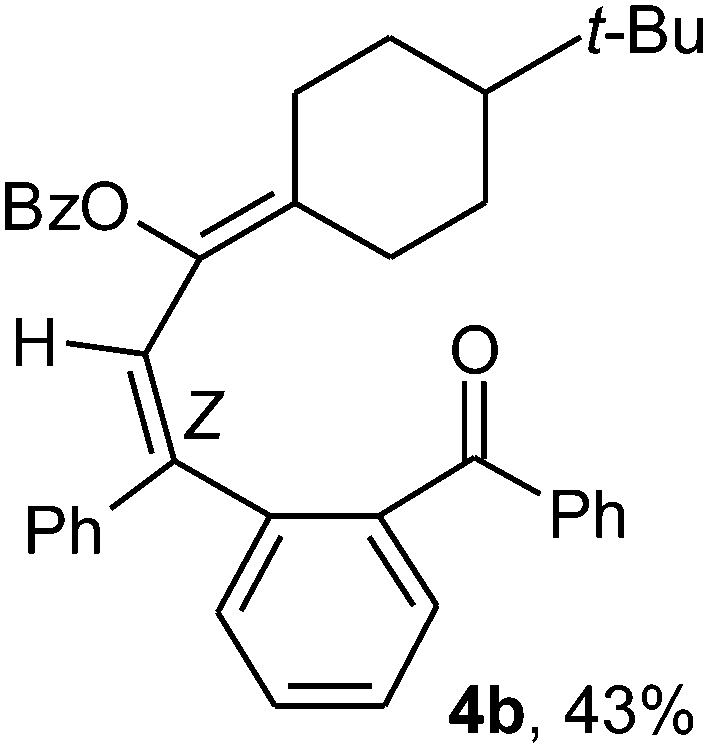
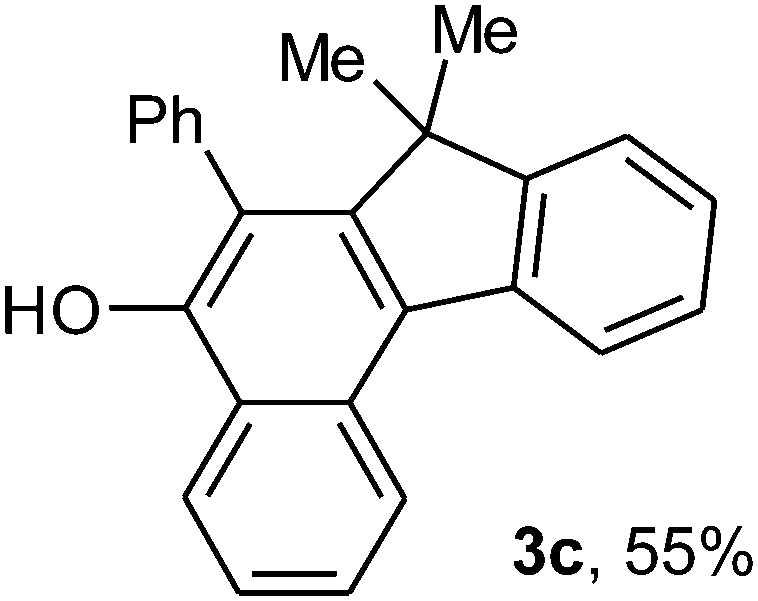
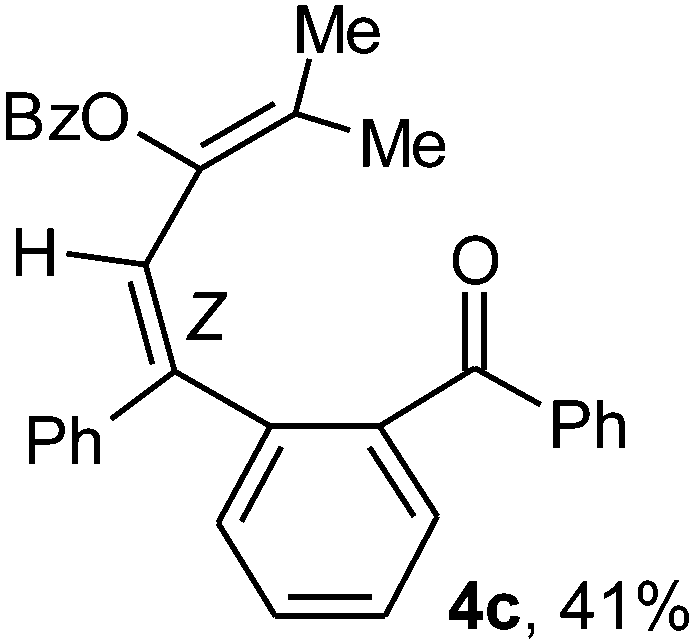
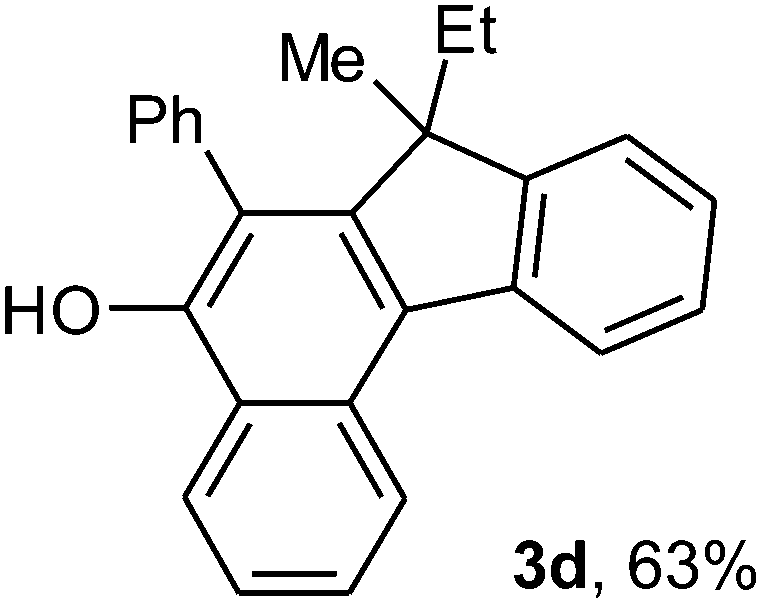
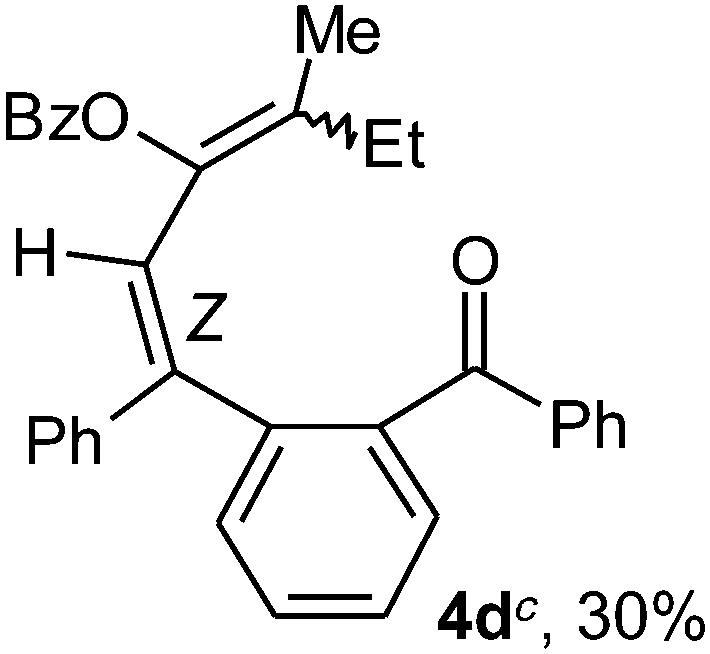
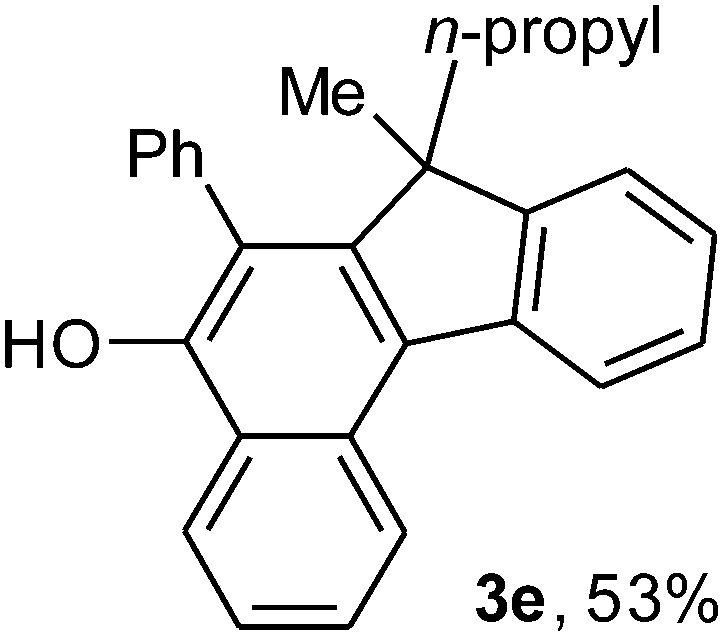
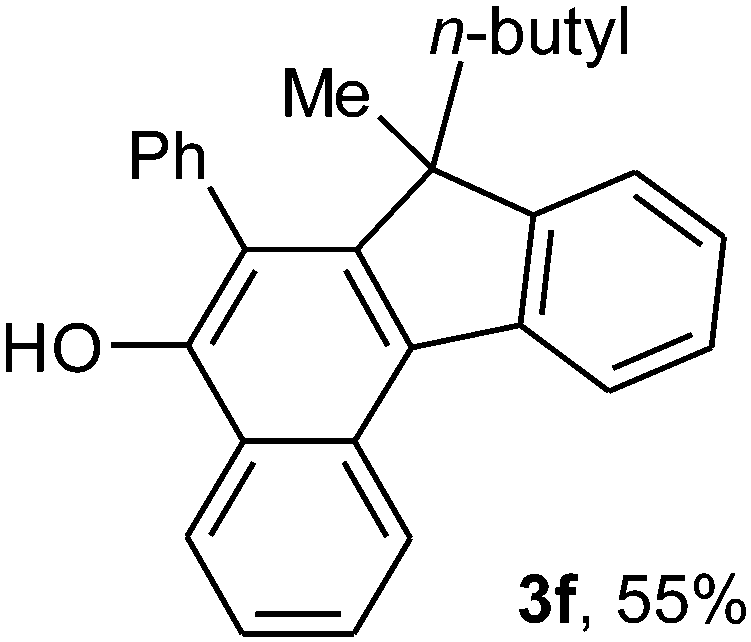
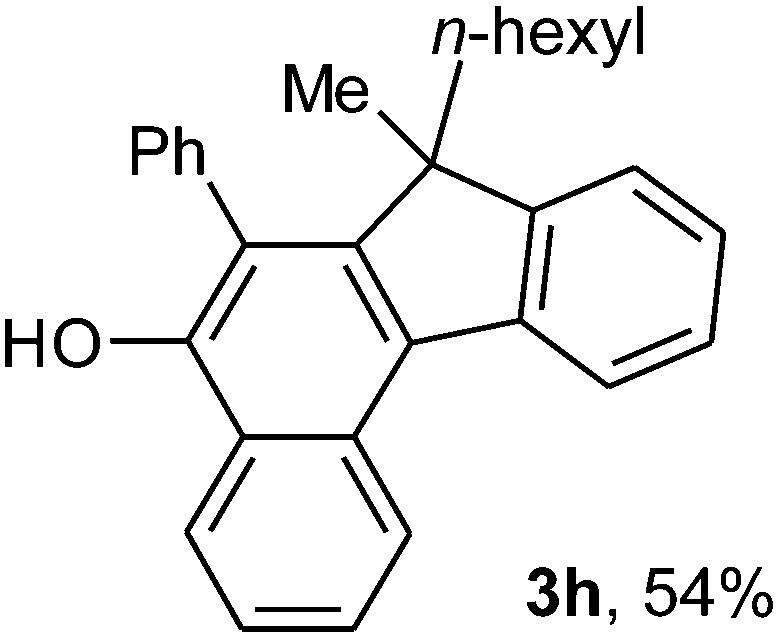
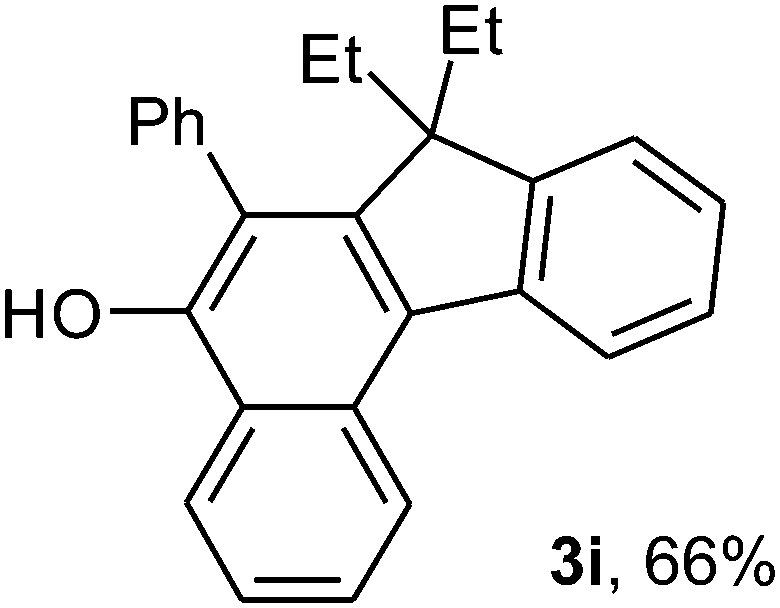
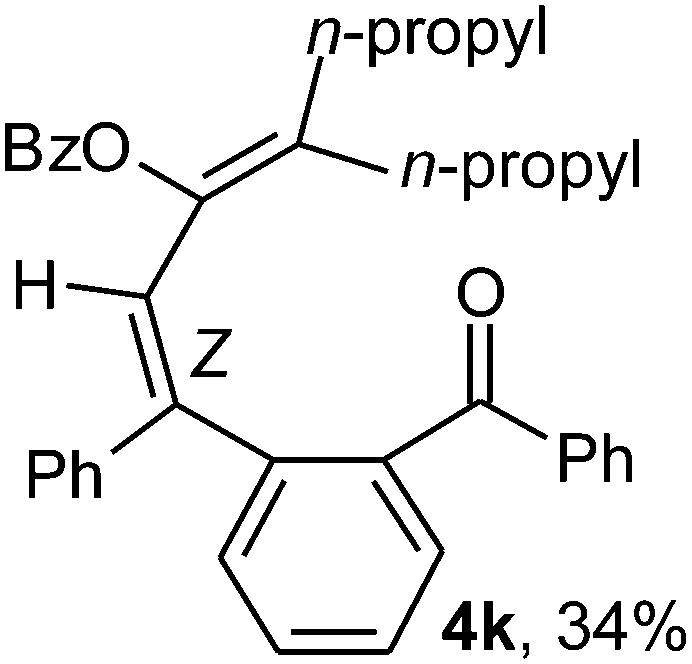
To ascertain the efficacy and generality of the above catalytic system, various propargylic benzoates 1b–1k were treated with IBF (Scheme 1). These reactions afforded products 3(b–k)–4(b–k) (benzofluorenols and dienes) in combined yields of 90–96% with benzofluorenols as predominant and readily isolatable products (Table 2). The structure of benzofluorenol 3a was confirmed by X-ray crystallography (Fig. 1). The substituents were varied in terms of alkyl groups. The configuration at one of the double bonds in the diene product is Z and the other double bond exhibits (E + Z) isomeric mixture (∼1:1) if the alkyl groups are unsymmetrically substituted (Table 2, entries 4–8 and 10). The Rf values of the two diene isomers were very close to each other and hence they were not separated. However, in the case of mono-substituted propargyl benzoate 1l under similar conditions, isomeric dienes 4l and 4l′ were formed (via acyclic addition; Scheme 2). These were separated and the structure of 4l was confirmed by X-ray crystallography (Fig. 2). Use of the terminally substituted ester 1-(phenylethynyl)cyclohexyl benzoate led to a mixture with much of the starting material unreacted.
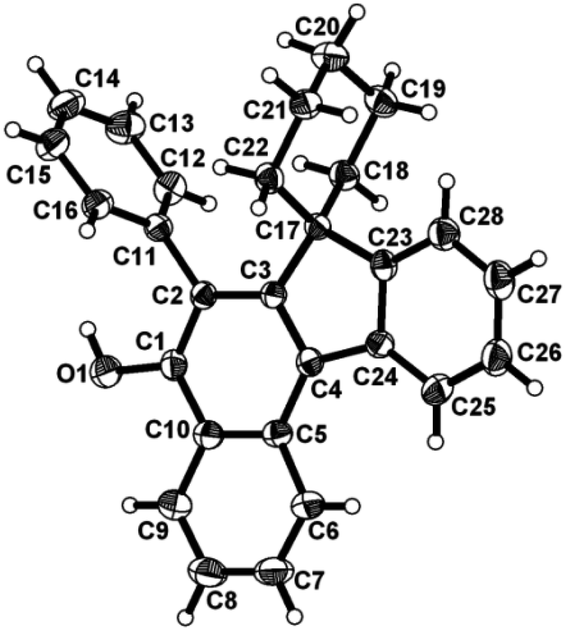 | ||
| Fig. 1 ORTEP diagram for compound 3a. Selected bond lengths with esd's in parentheses: C2–C1 1.377(4), C3–C2 1.422(4), C4–C3 1.389(3), C17–C23 1.518(4), C11–C2 1.485(3). | ||











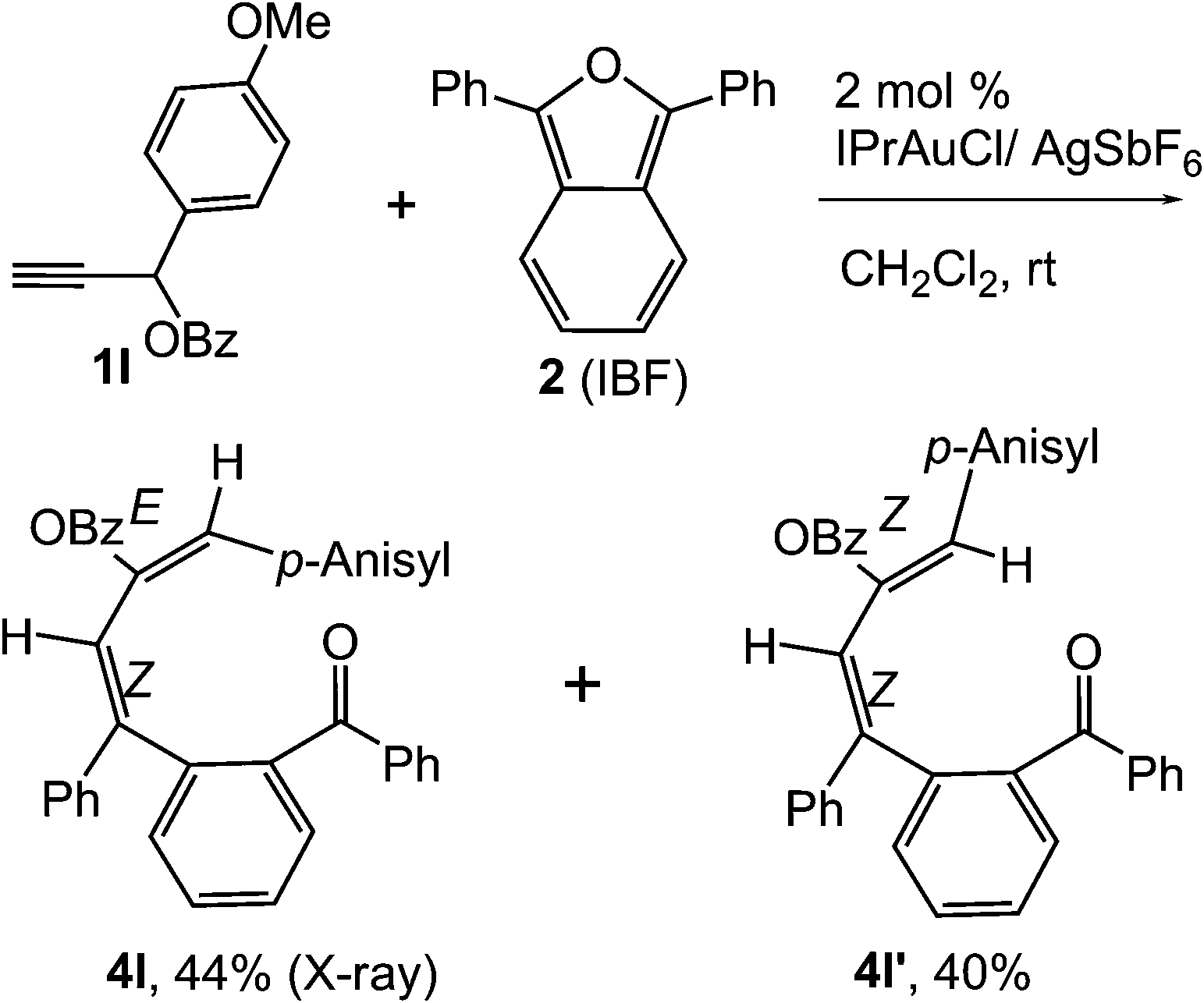 | ||
| Scheme 2 Formation of diene products 4l and 4l′. | ||
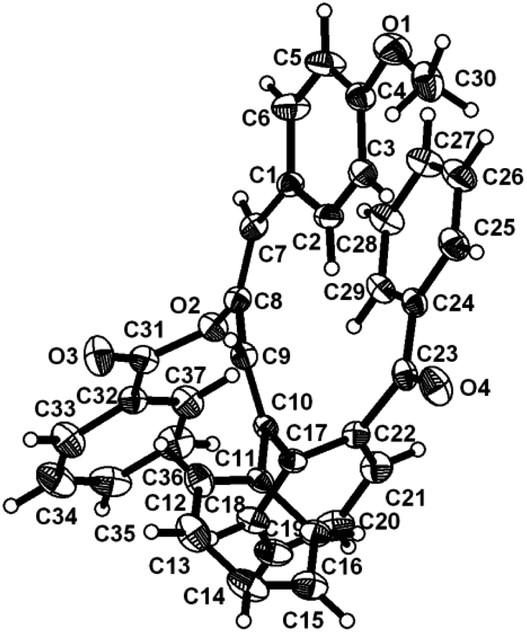 | ||
| Fig. 2 ORTEP diagram for compound 4l. Selected bond lengths with esd's in parentheses: C7–C8 1.320(3), C8–C9 1.445(3), C9–C10 1.328(3), O4–C23 1.218(2). | ||
We have also performed the above reaction by using 4-nitrobenzoate (1a′) and 4-methoxybenzoate (1a′′)15 in place of unsubstituted benzoate 1a (Scheme 3). It was found that the yield of the isolated fluorenol product 3a was increased to 66% in the former while it decreased to 23% in the case of latter. The yield of the diene 5 (29%) or 6 (26%) was low in both the cases.
 | ||
| Scheme 3 Reaction of 4-nitro- and 4-methoxy-benzoate propargylic esters with 2. | ||
The variation in the ester moiety having electron withdrawing –NO2 and electron donating –OMe substrates in the cycloaddition favours a gold–alkyne pathway. The propargylic ester having the –NO2 group resulted in higher yield. This suggests that 1,2- or 1,3-benzyloxy group migration16,17 is less favourable. Based on these results, a plausible mechanism for the above reaction is shown in Scheme 4. Initially, alkyne coordinates to [Au], and undergoes [4 + 2] cycloaddition with 1,3-diphenylisobenzofuran to form VI. This intermediate undergoes elimination of benzoyl group to generate a five membered ring forming the polycycle VII with bridged oxygen between two phenyl rings. A new C–C bond is formed and benzoic acid is eliminated at this stage. Then the ring containing bridged oxygen is opened by the attack of the double bond to form allylic cationic intermediate VIII. The other phenyl group migrates to the adjacent carbocation followed by ketone formation to lead to the intermediate IX. Species IX aromatises to the benzofluorenol product. In the case of monoaryl substituted propargyl ester 1l, 1,2-benzoyloxy group migration is only observed which can be in line with the literature.17d,18
 | ||
| Scheme 4 Proposed reaction pathway for the formation of benzofluorenols and substituted dienes. | ||
For the diene product as shown on the right of the Scheme 4, the allenic intermediate X is formed first; attack of the benzoyloxy anion on the central allenic carbon of followed by reorganization of bonds leads to the diene product.
Conclusions
In summary, a new type of cycloaddition involving propargylic esters with the aid of N-heterocyclic carbene–gold complex under very mild conditions is discovered. The products are novel benzo[c]fluorenols and substituted dienes that are very conveniently isolated. While the former product involves sequential cycloaddition, carbocyclisation, 1,2-phenyl migration, ring opening and aromatisation, the latter involves attack of benzoyloxy anion on central allenic carbon followed by rearrangement. The structures of two such products have been unambiguously established by X-ray structure determination.Experimental section
General procedure for the synthesis of benzofluorenols 3a–k, dienes 4a–l, 4l′ and 5–6
To a mixture of IPrAuCl [IPr = 1,3-bis(diisopropylphenyl)imidazol-2-ylidene] (0.006 g, 0.01 mmol) and AgSbF6 (0.003 g, 0.01) in DCM (2 mL) was added the corresponding propargyl benzoate 1a–l (0.6 mmol) and 1,3-diphenylisobenzofuran 2 (0.5 mmol). The contents were stirred at rt (25 °C) for 4 h. The solvent was removed under vacuum. Products 3a–k were separated from the reaction mixture by column chromatography by using acetone–hexane (1:100) mixture whereas 4a–l, 4l′ and 5–6 (1a′ and 1a′′ were used for these) were isolated by using acetone–hexane (1:50) mixture.
:50) mixture]; yield 0.109 g (58%) using 1a, 0.124 g (66%) using 1a′ and 0.056 g (23%) using 1a′′; Mp 230–232 °C; IR νmax(KBr): 3534, 3052, 2953, 2932, 2843, 1622, 1563, 1439, 1391, 1343, 1219, 1065, 1024, 752, 710, 666 cm−1; 1H NMR (400 MHz, CDCl3): δ 0.75–0.79, 1.32–1.54, 1.76–2.00 and 2.21–2.28 (m, 10H, cyclohexyl-H), 5.10 (s, 1H, Ar-OH), 7.23–7.96, 8.36–8.42 and 8.86–8.88 (m, 13H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 22.3, 25.1, 32.6 and 52.4 (cyclohexyl-CH2), 120.3, 122.2, 123.6, 123.7, 124.3, 124.8, 125.2, 126.7, 126.8, 127.4, 129.0, 129.2, 129.6, 130.5, 132.8, 134.2, 140.8, 149.4, 149.8 and 153.6 (Ar-C); HRMS (ESI): calcd for C28H25O [M+ + H]: m/z 377.1906. Found: 377.1904. X-ray structure has been determined for this compound after crystallisation from CH2Cl2–hexane mixture. CCDC no. 952109.:50) mixture]; yield 0.82 g (33%); IR νmax(neat) 3063, 2926, 2854, 1726, 1671, 1599, 1452, 1276, 1068, 701 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.31–1.53 and 1.87–2.10 (m, 10H, cyclohexyl-H), 6.73 (s, 1H, PhC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.10–7.76 (m, 19H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 26.2, 26.6, 27.0, 28.1, 29.4 (cyclohexyl-C), 121.1, 126.3, 127.5, 128.00, 128.02, 129.1, 129.3, 129.4, 129.7, 130.1, 130.2, 131.3, 132.6, 132.9, 134.1, 136.4, 137.2, 139.2, 140.4, 141.0, 142.6 (alkenyl-C + Ar-C), 164.3 (OCOPh), 196.3 (ArCOPh); HRMS (ESI): calcd for C35H30O3 [M+ + H]: m/z 499.2274. Found: 499.2274.CH), 6.94–7.75 (m, 19H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 27.6, 28.1, 29.3, 32.4 and 47.9 (cyclohexyl-C + t-Bu-C), 121.2, 126.3, 127.4, 127.4, 128.0, 129.3, 129.5, 129.8, 130.1, 130.2, 131.3, 132.5, 132.8, 133.8, 136.4, 137.3, 139.3, 140.4, 141.5 and 142.6 (alkenyl-C + Ar-C), 164.3 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C39H39O3 [M+ + H]: m/z 555.2900. Found: 555.2903.C(CH3)2), 6.69 (s, 1H, PhCCH), 7.10–7.74 (m, 19H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 18.5 and 19.2 (C(CH3)2), 121.6, 126.4, 127.0, 127.5, 127.9, 128.5, 129.2, 129.3, 129.8, 130.0, 130.2, 130.6, 131.3, 132.6, 132.9, 137.1, 139.1, 139.3, 140.2, 140.7, 142.4 (alkenyl-C + Ar-C), 164.1 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C32H26O3 [M+ + H]: m/z 459.1961. Found: 459.1964.C), 1.65 and 2.04 (2 br s, 4H, CH2CH3), 6.67 and 6.73 (2 s, 2H, PhCCH), 7.07–7.77 (m, 38H, Ar-H). In the assignment, the proton numbers are doubled to show the presence of both the isomers; 13C NMR (100 MHz, CDCl3): δ 11.7 and 12.0 (2 CH3CH2), 15.8, 16.5, 25.2 and 26.2 (2 CH3CH2 and 2 CH3CC), 120.6, 121.8, 126.3, 126.5, 127.5, 127.9, 128.1, 129.1, 129.4, 129.7, 129.8, 129.9, 130.1, 130.2, 131.1, 131.3, 132.5, 132.6, 132.9, 137.2, 137.3, 138.7, 139.0, 139.4, 140.0, 140.6, 140.9, 142.4, 142.5 (Ar-C), 163.9, 164.3 (OCOPh), 196.5 (ArCOPh); HRMS (ESI): calcd for C33H28NaO3 [M+ + Na]: m/z 495.1936. Found: 495.1938.CH), 6.81–7.74 (m, 38H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 14.0, 14.2, 16.4, 17.1, 20.5, 21.0, 34.3 and 35.1 (propyl-C + CH3), 121.1, 121.9, 126.3, 126.5, 127.5, 127.9, 128.1, 129.1, 129.5, 129.8, 129.9, 130.2, 130.3, 130.6, 131.2, 131.3, 132.4, 132.6, 132.8, 137.30, 137.34, 139.3, 139.7, 139.9, 140.1, 140.6, 140.9, 142.4 and 142.6 (Ar-C), 163.9, 164.3 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C34H31O3 [M+ + H]: m/z 487.2274. Found: 487.2273.CH), 7.05–7.75 (m, 38H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 13.9, 14.9, 16.4, 17.0, 22.60, 22.62, 29.3, 29.9, 31.8 and 32.9 (CH3 + butyl-C), 121.0, 121.8, 126.3, 126.4, 127.4, 127.9, 128.00, 128.02, 129.0, 129.3, 129.5, 129.7, 129.9, 130.1, 130.2, 130.7, 131.1, 131.3, 131.4, 132.4, 132.5, 132.8, 137.3, 139.1, 139.2, 140.0, 140.9, 142.4, 142.6, (alkenyl-C + Ar-C), 163.9 and 164.3 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C35H33 O3 [M+ + H]: m/z 501.2430. Found: 501.2432.CH), 6.74–7.79 (m, 38H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 14.0, 16.4, 17.1, 22.4, 22.6, 26.8, 27.0, 27.4, 31.8, 32.1 and 33.2 (n-pentyl-C + CH3), 121.0, 121.9, 126.3, 126.5, 126.7, 127.5, 127.9, 128.1, 128.4, 129.1, 129.4, 129.8, 130.2, 130.8, 131.2, 131.4, 132.4, 132.6, 132.8, 137.4, 139.2, 139.3, 139.5, 139.8, 140.1, 140.7, 142.5 and 142.6 (alkenyl-C + Ar-C), 164.3 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C36H35O3 [M+ + H]: m/z 515.2587. Found: 515.2589.CH), 7.05–7.20, 7.27–7.37, 7.44–7.49, 7.54–7.76 (m, 38H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 14.1, 14.2, 16.4, 17.1, 22.6, 24.7, 27.1, 27.7, 29.2, 31.6, 31.8, 32.1, 33.2, 36.7 (n-C6H13 + CH3), 121.0, 121.8, 126.3, 126.5, 127.1, 127.5, 127.9, 128.00, 128.04, 129.0, 129.4, 129.7, 129.9, 130.2, 130.9, 131.1, 131.3, 131.5, 132.5, 132.6, 132.8, 132.9, 137.2, 137.3, 139.1, 139.2, 139.4, 139.8, 140.0, 140.6, 140.9, 142.4, 142.6 (alkenyl-C + Ar-C), 163.9 and 164.2 (OCOPh), 196.5 (ArCOPh); HRMS (ESI): calcd for C37H37O3 [M+ + H]: m/z 529.2743. Found: 529.2742.CH), 7.00–7.72 (m, 19H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 12.1 and 12.6 (CH2CH3), 22.8 and 23.6 (CH2CH3), 121.2, 126.4, 127.5, 127.9, 128.1, 128.4, 129.1, 129.4, 129.7, 129.9, 130.1, 130.2, 131.1, 132.6, 132.9, 137.3, 137.8, 139.0, 139.5, 139.8, 140.8, 142.6 (alkenyl-C + Ar-C), 164.1 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C34H31O3 [M+ + H]: m/z 487.2274. Found: 487.2272.CH), 7.02–7.72 (m, 38H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 12.1, 12.7, 14.2, 14.4, 20.9, 21.5, 23.4, 24.1, 32.0 and 32.7 (ethyl-C + propyl-C), 121.2, 121.3, 126.4, 127.4, 127.90, 127.96, 128.1, 128.6, 129.2, 129.6, 129.7, 130.0, 130.1, 130.4, 131.1, 132.5, 132.8, 136.5, 136.6, 137.4, 139.5, 139.7, 139.8, 140.8, 142.6 and 142.7 (alkenyl-C + Ar-C), 164.2 (OCOPh), 196.5 (ArCOPh); HRMS (ESI): calcd for C35H33 O3 [M+ + H]: m/z 501.2430. Found: 501.2431.CH), 6.81–6.83, 6.98–7.54 and 7.60–7.71 (m, 19H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 14.2, 14.4, 20.9, 21.5, 32.4 and 33.0 (propyl-C), 126.3, 127.1, 127.4, 127.6, 127.8, 127.9, 128.0, 129.2, 129.5, 129.6, 130.0, 130.1, 131.1, 131.6, 131.8, 132.5, 132.7, 135.2, 137.3, 139.0, 139.3, 139.5, 140.0, 140.7, 142.6 and 144.7 (alkenyl-C + Ar-C), 164.1 (OCOPh), 196.5 (ArCOPh); HRMS (ESI): calcd for C36H35O3 [M+ + H]: m/z 515.2587. Found: 515.2585.CH), 6.76–6.83 and 7.12–7.73 (m, 24H, PhCCH + Ar-H); 13C NMR (100 MHz, CDCl3): δ 55.3 (OCH3), 113.8, 121.6, 124.1, 126.8, 127.3, 127.9, 128.10, 128.14, 129.3, 129.5, 129.8, 130.1, 130.3, 130.5, 131.2, 132.7, 133.0, 136.9, 139.3, 140.5, 142.0, 142.9, 143.9, 159.2 (alkenyl-C + Ar-C), 164.5 (OCOPh), 196.6 (ArCOPh); HRMS (ESI): calcd for C37H28NaO4 [M+ + Na]: m/z 559.1886. Found: 559.1886. X-ray structure was determined for this compound after crystallisation from CH2Cl2–hexane mixture. CCDC no. 952110.CH), 6.59–6.73 and 7.06–7.73 (m, 24H, PhCCH + Ar-H); 13C NMR (100 MHz, CDCl3): δ 55.2 (OCH3), 114.0, 123.1, 124.8, 126.7, 127.1, 127.6, 127.7, 127.8, 128.0, 129.1, 129.3, 129.7, 130.1, 130.2, 131.4, 132.5, 133.2, 137.5, 139.4, 139.6, 140.0, 141.8, 143.9, 159.1 (alkenyl-C + Ar-C), 163.5 (OCOPh), 197.2 (ArCOPh); HRMS (ESI): calcd for C37H28NaO4 [M+ + Na]: m/z 559.1886. Found: 559.1886.CH), 7.13–7.18 (m, 8H, Ar-H), 7.29–7.32 (m, 3H, Ar-H), 7.46 (t, J = 7.2 Hz, 1H, Ar-H), 7.64 (d, J = 7.6 Hz, 2H, Ar-H), 7.70 (d, J = 8.4 Hz, 2H, Ar-H), 8.10 (d, J = 8.4 Hz, 2H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 26.1, 26.7, 27.1, 28.2, 29.5 (cyclohexyl-C), 120.6, 123.0, 126.5, 127.6, 128.0, 129.2, 129.9, 130.1, 130.8, 131.3, 132.7, 134.6, 134.8, 136.3, 136.9, 139.4, 140.5, 141.0, 142.2, 150.3 (alkenyl-C + Ar-C), 162.3 (OCOPh), 196.2 (ArCOPh); HRMS (ESI): calcd for C35H29NO5Na [M+ + Na]: m/z 566.1944. Found: 566.1947.CH), 6.75–6.78 (m, J ∼ 8.8 Hz, 2H, Ar-H), 7.13–7.19 (m, 8H, Ar-H), 7.33–7.39 (m, 3H, Ar-H), 7.47–7.51 (m, J ∼ 8.8 Hz, 3H, Ar-H), 7.75 (d, J = 8.4 Hz, 2H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 26.2, 26.6, 27.0, 28.1, 29.4 (cyclohexyl-C), 55.4 (OCH3), 113.1, 121.4, 121.9, 126.3, 127.4, 127.9, 128.0, 129.2, 130.1, 130.2, 131.3, 131.8, 132.5, 134.0, 136.3, 137.2, 139.2, 140.2, 141.0, 142.6, 163.3 (alkenyl-C + Ar-C) 164.0 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C36H32O4Na [M+ + Na]: m/z 551.2199. Found: 551.2199.
CH), 7.10–7.76 (m, 19H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 26.2, 26.6, 27.0, 28.1, 29.4 (cyclohexyl-C), 121.1, 126.3, 127.5, 128.00, 128.02, 129.1, 129.3, 129.4, 129.7, 130.1, 130.2, 131.3, 132.6, 132.9, 134.1, 136.4, 137.2, 139.2, 140.4, 141.0, 142.6 (alkenyl-C + Ar-C), 164.3 (OCOPh), 196.3 (ArCOPh); HRMS (ESI): calcd for C35H30O3 [M+ + H]: m/z 499.2274. Found: 499.2274.CH), 6.94–7.75 (m, 19H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 27.6, 28.1, 29.3, 32.4 and 47.9 (cyclohexyl-C + t-Bu-C), 121.2, 126.3, 127.4, 127.4, 128.0, 129.3, 129.5, 129.8, 130.1, 130.2, 131.3, 132.5, 132.8, 133.8, 136.4, 137.3, 139.3, 140.4, 141.5 and 142.6 (alkenyl-C + Ar-C), 164.3 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C39H39O3 [M+ + H]: m/z 555.2900. Found: 555.2903.C(CH3)2), 6.69 (s, 1H, PhCCH), 7.10–7.74 (m, 19H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 18.5 and 19.2 (C(CH3)2), 121.6, 126.4, 127.0, 127.5, 127.9, 128.5, 129.2, 129.3, 129.8, 130.0, 130.2, 130.6, 131.3, 132.6, 132.9, 137.1, 139.1, 139.3, 140.2, 140.7, 142.4 (alkenyl-C + Ar-C), 164.1 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C32H26O3 [M+ + H]: m/z 459.1961. Found: 459.1964.C), 1.65 and 2.04 (2 br s, 4H, CH2CH3), 6.67 and 6.73 (2 s, 2H, PhCCH), 7.07–7.77 (m, 38H, Ar-H). In the assignment, the proton numbers are doubled to show the presence of both the isomers; 13C NMR (100 MHz, CDCl3): δ 11.7 and 12.0 (2 CH3CH2), 15.8, 16.5, 25.2 and 26.2 (2 CH3CH2 and 2 CH3CC), 120.6, 121.8, 126.3, 126.5, 127.5, 127.9, 128.1, 129.1, 129.4, 129.7, 129.8, 129.9, 130.1, 130.2, 131.1, 131.3, 132.5, 132.6, 132.9, 137.2, 137.3, 138.7, 139.0, 139.4, 140.0, 140.6, 140.9, 142.4, 142.5 (Ar-C), 163.9, 164.3 (OCOPh), 196.5 (ArCOPh); HRMS (ESI): calcd for C33H28NaO3 [M+ + Na]: m/z 495.1936. Found: 495.1938.CH), 6.81–7.74 (m, 38H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 14.0, 14.2, 16.4, 17.1, 20.5, 21.0, 34.3 and 35.1 (propyl-C + CH3), 121.1, 121.9, 126.3, 126.5, 127.5, 127.9, 128.1, 129.1, 129.5, 129.8, 129.9, 130.2, 130.3, 130.6, 131.2, 131.3, 132.4, 132.6, 132.8, 137.30, 137.34, 139.3, 139.7, 139.9, 140.1, 140.6, 140.9, 142.4 and 142.6 (Ar-C), 163.9, 164.3 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C34H31O3 [M+ + H]: m/z 487.2274. Found: 487.2273.CH), 7.05–7.75 (m, 38H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 13.9, 14.9, 16.4, 17.0, 22.60, 22.62, 29.3, 29.9, 31.8 and 32.9 (CH3 + butyl-C), 121.0, 121.8, 126.3, 126.4, 127.4, 127.9, 128.00, 128.02, 129.0, 129.3, 129.5, 129.7, 129.9, 130.1, 130.2, 130.7, 131.1, 131.3, 131.4, 132.4, 132.5, 132.8, 137.3, 139.1, 139.2, 140.0, 140.9, 142.4, 142.6, (alkenyl-C + Ar-C), 163.9 and 164.3 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C35H33 O3 [M+ + H]: m/z 501.2430. Found: 501.2432.CH), 6.74–7.79 (m, 38H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 14.0, 16.4, 17.1, 22.4, 22.6, 26.8, 27.0, 27.4, 31.8, 32.1 and 33.2 (n-pentyl-C + CH3), 121.0, 121.9, 126.3, 126.5, 126.7, 127.5, 127.9, 128.1, 128.4, 129.1, 129.4, 129.8, 130.2, 130.8, 131.2, 131.4, 132.4, 132.6, 132.8, 137.4, 139.2, 139.3, 139.5, 139.8, 140.1, 140.7, 142.5 and 142.6 (alkenyl-C + Ar-C), 164.3 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C36H35O3 [M+ + H]: m/z 515.2587. Found: 515.2589.CH), 7.05–7.20, 7.27–7.37, 7.44–7.49, 7.54–7.76 (m, 38H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 14.1, 14.2, 16.4, 17.1, 22.6, 24.7, 27.1, 27.7, 29.2, 31.6, 31.8, 32.1, 33.2, 36.7 (n-C6H13 + CH3), 121.0, 121.8, 126.3, 126.5, 127.1, 127.5, 127.9, 128.00, 128.04, 129.0, 129.4, 129.7, 129.9, 130.2, 130.9, 131.1, 131.3, 131.5, 132.5, 132.6, 132.8, 132.9, 137.2, 137.3, 139.1, 139.2, 139.4, 139.8, 140.0, 140.6, 140.9, 142.4, 142.6 (alkenyl-C + Ar-C), 163.9 and 164.2 (OCOPh), 196.5 (ArCOPh); HRMS (ESI): calcd for C37H37O3 [M+ + H]: m/z 529.2743. Found: 529.2742.CH), 7.00–7.72 (m, 19H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 12.1 and 12.6 (CH2CH3), 22.8 and 23.6 (CH2CH3), 121.2, 126.4, 127.5, 127.9, 128.1, 128.4, 129.1, 129.4, 129.7, 129.9, 130.1, 130.2, 131.1, 132.6, 132.9, 137.3, 137.8, 139.0, 139.5, 139.8, 140.8, 142.6 (alkenyl-C + Ar-C), 164.1 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C34H31O3 [M+ + H]: m/z 487.2274. Found: 487.2272.CH), 7.02–7.72 (m, 38H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 12.1, 12.7, 14.2, 14.4, 20.9, 21.5, 23.4, 24.1, 32.0 and 32.7 (ethyl-C + propyl-C), 121.2, 121.3, 126.4, 127.4, 127.90, 127.96, 128.1, 128.6, 129.2, 129.6, 129.7, 130.0, 130.1, 130.4, 131.1, 132.5, 132.8, 136.5, 136.6, 137.4, 139.5, 139.7, 139.8, 140.8, 142.6 and 142.7 (alkenyl-C + Ar-C), 164.2 (OCOPh), 196.5 (ArCOPh); HRMS (ESI): calcd for C35H33 O3 [M+ + H]: m/z 501.2430. Found: 501.2431.CH), 6.81–6.83, 6.98–7.54 and 7.60–7.71 (m, 19H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 14.2, 14.4, 20.9, 21.5, 32.4 and 33.0 (propyl-C), 126.3, 127.1, 127.4, 127.6, 127.8, 127.9, 128.0, 129.2, 129.5, 129.6, 130.0, 130.1, 131.1, 131.6, 131.8, 132.5, 132.7, 135.2, 137.3, 139.0, 139.3, 139.5, 140.0, 140.7, 142.6 and 144.7 (alkenyl-C + Ar-C), 164.1 (OCOPh), 196.5 (ArCOPh); HRMS (ESI): calcd for C36H35O3 [M+ + H]: m/z 515.2587. Found: 515.2585.CH), 6.76–6.83 and 7.12–7.73 (m, 24H, PhCCH + Ar-H); 13C NMR (100 MHz, CDCl3): δ 55.3 (OCH3), 113.8, 121.6, 124.1, 126.8, 127.3, 127.9, 128.10, 128.14, 129.3, 129.5, 129.8, 130.1, 130.3, 130.5, 131.2, 132.7, 133.0, 136.9, 139.3, 140.5, 142.0, 142.9, 143.9, 159.2 (alkenyl-C + Ar-C), 164.5 (OCOPh), 196.6 (ArCOPh); HRMS (ESI): calcd for C37H28NaO4 [M+ + Na]: m/z 559.1886. Found: 559.1886. X-ray structure was determined for this compound after crystallisation from CH2Cl2–hexane mixture. CCDC no. 952110.CH), 6.59–6.73 and 7.06–7.73 (m, 24H, PhCCH + Ar-H); 13C NMR (100 MHz, CDCl3): δ 55.2 (OCH3), 114.0, 123.1, 124.8, 126.7, 127.1, 127.6, 127.7, 127.8, 128.0, 129.1, 129.3, 129.7, 130.1, 130.2, 131.4, 132.5, 133.2, 137.5, 139.4, 139.6, 140.0, 141.8, 143.9, 159.1 (alkenyl-C + Ar-C), 163.5 (OCOPh), 197.2 (ArCOPh); HRMS (ESI): calcd for C37H28NaO4 [M+ + Na]: m/z 559.1886. Found: 559.1886.CH), 7.13–7.18 (m, 8H, Ar-H), 7.29–7.32 (m, 3H, Ar-H), 7.46 (t, J = 7.2 Hz, 1H, Ar-H), 7.64 (d, J = 7.6 Hz, 2H, Ar-H), 7.70 (d, J = 8.4 Hz, 2H, Ar-H), 8.10 (d, J = 8.4 Hz, 2H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 26.1, 26.7, 27.1, 28.2, 29.5 (cyclohexyl-C), 120.6, 123.0, 126.5, 127.6, 128.0, 129.2, 129.9, 130.1, 130.8, 131.3, 132.7, 134.6, 134.8, 136.3, 136.9, 139.4, 140.5, 141.0, 142.2, 150.3 (alkenyl-C + Ar-C), 162.3 (OCOPh), 196.2 (ArCOPh); HRMS (ESI): calcd for C35H29NO5Na [M+ + Na]: m/z 566.1944. Found: 566.1947.CH), 6.75–6.78 (m, J ∼ 8.8 Hz, 2H, Ar-H), 7.13–7.19 (m, 8H, Ar-H), 7.33–7.39 (m, 3H, Ar-H), 7.47–7.51 (m, J ∼ 8.8 Hz, 3H, Ar-H), 7.75 (d, J = 8.4 Hz, 2H, Ar-H); 13C NMR (100 MHz, CDCl3): δ 26.2, 26.6, 27.0, 28.1, 29.4 (cyclohexyl-C), 55.4 (OCH3), 113.1, 121.4, 121.9, 126.3, 127.4, 127.9, 128.0, 129.2, 130.1, 130.2, 131.3, 131.8, 132.5, 134.0, 136.3, 137.2, 139.2, 140.2, 141.0, 142.6, 163.3 (alkenyl-C + Ar-C) 164.0 (OCOPh), 196.4 (ArCOPh); HRMS (ESI): calcd for C36H32O4Na [M+ + Na]: m/z 551.2199. Found: 551.2199.Single crystal X-ray data for compounds 3a, and 4l were collected on an OXFORD diffractometer using Mo-Kα (λ = 0.71073 Å) radiation. The structures were solved by direct methods and refined by full-matrix least squares method using standard procedures.19 Absorption corrections were done using SADABS program, where applicable. In general, all non-hydrogen atoms were refined anisotropically; hydrogen atoms were fixed by geometry or located by a Difference Fourier map and refined isotropically.
Acknowledgements
We thank Department of Science & Technology (DST, New Delhi) for financial support, single crystal X-ray diffractometer and HRMS facility. KCK thanks DST for a J. C. Bose fellowship. RK thanks CSIR (New Delhi) and ALSK thanks UGC for fellowships.Notes and references
- (a) G. J. Hutchings, Gold Bull., 2004, 37, 3–11 CrossRef CAS; (b) A. S. K. Hashmi, Gold Bull., 2004, 37, 51–65 CrossRef CAS; (c) A. Arcadi, Chem. Rev., 2008, 108, 3266–3325 CrossRef CAS PubMed; (d) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC; (e) C. J. Lima and L. Rodríguez, Chem. Soc. Rev., 2011, 40, 5442–5456 RSC; (f) Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467–2505 CrossRef CAS PubMed; (g) E. Feng, Y. Zhou, F. Zhao, X. Chen, L. Zhang, H. Jiang and H. Liu, Green Chem., 2012, 14, 1888–1895 RSC.
- Selected references on gold catalysed C–C and C–X bond formations: (a) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed; (b) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS PubMed; (c) B. H. Lipshutz and Y. Yamamoto, Chem. Rev., 2008, 108, 2793–2796 CrossRef CAS PubMed; (d) C. Praveen, P. Kiruthiga and P. T. Perumal, Synlett, 2009, 1990–1996 CAS; (e) S. M. A. Sohel and R. S. Liu, Chem. Soc. Rev., 2009, 38, 2269–2281 RSC; (f) M. Egi, K. Azechi and S. Akai, Org. Lett., 2009, 11, 5002–5005 CrossRef CAS PubMed; (g) C. Praveen, A. Kalyanasundaram and P. T. Perumal, Synlett, 2010, 0777–0781 CAS; (h) B. Alcaide, P. Almendros and J. M. Alonso, Org. Biomol. Chem., 2011, 9, 4405–4416 RSC; (i) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657–1712 CrossRef CAS PubMed.
- (a) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994–2009 CrossRef CAS PubMed; (b) X. Zeng, Chem. Rev., 2013, 113, 6864 CrossRef CAS PubMed.
- (a) D. J. Gorin, P. Dube and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 14480–14481 CrossRef CAS PubMed; (b) N. D. Shapiro and F. D. Toste, J. Am. Chem. Soc., 2008, 130, 9244–9245 CrossRef CAS PubMed; (c) B. W. Gung, D. T. Craft, L. N. Bailey and K. Kirschbaum, Chem.–Eur. J, 2010, 16, 639–644 CrossRef CAS PubMed; (d) B. W. Gung, L. N. Bailey and J. Wonser, Tetrahedron Lett., 2010, 51, 2251–2253 CrossRef CAS PubMed; (e) H. Liu, X. Li, Z. Chen and W. X. Hu, J. Org. Chem., 2012, 77, 5184–5190 CrossRef CAS PubMed.
- Selected references on gold catalysed [4 + 2] cycloaddition reactions: (a) N. Asao, T. Nogami, S. Lee and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 10921–10925 CrossRef CAS PubMed; (b) N. Asao, H. Aikawa and Y. Yamamoto, J. Am. Chem. Soc., 2004, 126, 7458–7459 CrossRef CAS PubMed; (c) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed; (d) F. Liu, D. Qian, L. Li, X. Zhao and J. Zhang, Angew. Chem., Int. Ed, 2010, 49, 6669–6672 CrossRef CAS PubMed; (e) H. Gao, X. Wua and J. Zhang, Chem. Commun., 2010, 46, 8764–8766 RSC; (f) H. Gao, X. Zhao, Y. Yu and J. Zhang, Chem.–Eur. J, 2010, 16, 456–459 CrossRef CAS PubMed; (g) J. Barluenga, J. Calleja, A. Mendoza, F. Rodriguez and F. J. Fananas, Chem.–Eur. J, 2010, 16, 7110–7112 CrossRef CAS PubMed; (h) H. Gao, X. Wu and J. Zhang, Chem.–Eur. J., 2011, 17, 2838–2841 CrossRef CAS PubMed.
- Selected references on Hashmi's phenol synthesis: (a) A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef CAS; (b) A. S. K. Hashmi, J. P. Weyrauch, M. Rudolph and E. Kurpejović, Angew. Chem., Int. Ed., 2004, 43, 6545–6547 CrossRef CAS PubMed; (c) A. S. K. Hashmi, M. Wçlfle, F. Ata, M. Hamzic, R. Salath and W. Frey, Adv. Synth. Catal., 2006, 348, 2501–2508 CrossRef CAS; (d) A. S. K. Hashmi, M. Rudolph, H.-U. Siehl, M. Tanaka, J. W. Bats and W. Frey, Chem.–Eur. J., 2008, 14, 3703–3708 CrossRef CAS PubMed; (e) A. S. K. Hashmi, J. Hofmann, S. Shi, A. Schütz, M. Rudolph, C. Lothschütz, M. Wieteck, M. Bührle, M. Wçlfle and F. Rominger, Chem.–Eur. J, 2013, 19, 382–389 CrossRef CAS PubMed.
- N. Huguet, D. Leboeuf and A. M. Echavarren, Chem.–Eur. J, 2013, 19, 6581–6585 CrossRef CAS PubMed.
- (a) K. V. Sajna, V. Srinivas and K. C. Kumara Swamy, Adv. Synth. Catal., 2010, 352, 3069–3081 CrossRef CAS; (b) V. Srinivas, K. V. Sajna and K. C. Kumara Swamy, Chem. Commun., 2011, 47, 5629–5631 RSC; (c) R. Kotikalapudi and K. C. Kumara Swamy, Tetrahedron Lett., 2012, 53, 3831–3834 CrossRef CAS PubMed; (d) M. Nagarjuna Reddy and K. C. Kumara Swamy, Eur. J. Org. Chem., 2012, 2013–2022 CrossRef CAS; (e) K. V. Sajna and K. C. Kumara Swamy, J. Org. Chem., 2012, 77, 5345–5356 CrossRef CAS PubMed; (f) R. Kotikalapudi and K. C. Kumara Swamy, Tetrahedron, 2013, 69, 8002–8012 CrossRef CAS PubMed; (g) M. Nagarjuna Reddy and K. C. Kumara Swamy, Org. Biomol. Chem., 2013, 11, 7350–7360 RSC.
- (a) F. Hide, M. A. Diáz-Garciá, B. J. Schwartz and A. J. Heeger, Acc. Chem. Res., 1997, 30, 430–436 CrossRef CAS; (b) C. D. Müller, A. Falcou, N. Reckefuss, M. Rojahn, V. Wiederhirn, P. Rudati, H. Frohne, O. Nuyken, H. Becker and K. Meerholz, Nature, 2003, 421, 829–833 CrossRef PubMed; (c) Y. Wu, J. Li, Y. Fu and Z. Bo, Org. Lett., 2004, 6, 3485–3487 CrossRef CAS PubMed; (d) N. Basari, N. Cindro, D. Bobinac, K. Mlinarić-Majerski, L. Uzelac, M. Kralj and P. Wan, Photochem. Photobiol. Sci., 2011, 10, 1910–1925 RSC; (e) Z. Chen, M. Zeng, J. Yuan, Q. Yang and Y. Peng, Org. Lett., 2012, 14, 3588–3591 CrossRef CAS PubMed; (f) P. J. Coelho, M. A. Salvador, M. M. Oliveira and L. M. Carvalho, Tetrahedron, 2004, 60, 2593–2599 CrossRef CAS PubMed; (g) A. Picot, C. Feuvrie, C. Barsu, F. Malvolti, B. Le Guennic, H. Le Bozec, C. Andraud, L. Toupet and O. Maury, Tetrahedron, 2008, 64, 399–411 CrossRef CAS PubMed.
- (a) H. He, W. Ding, V. S. Bernan, A. D. Richardson, C. M. Ireland, M. Greenstein, G. A. Ellestad and G. T. Carter, J. Am. Chem. Soc., 2001, 123, 5362–5363 CrossRef CAS; (b) S. B. Herzon and C. M. Woo, Nat. Prod. Rep., 2012, 29, 87–118 RSC.
- (a) J. R. Carney, S. Hong and S. J. Gould, Tetrahedron Lett., 1997, 38, 3139–3142 CrossRef CAS; (b) T. Aoyama, W. Zhao, F. Kojima, Y. Muraoka, H. Naganawa and T. Takeuchi, J. Antibiot., 1993, 46, 1471–1474 CrossRef CAS; (c) M. R. Meselhy, S. Kadota, K. Tsubono, M. Hattori and T. Nambaa, Tetrahedron, 1994, 50, 3081–3098 CrossRef CAS; (d) T. Akiyama, S. Harada, F. Kojima, Y. Takahashi, C. Imada, Y. Okami, Y. Muraoka, T. Aoyagi and T. Takeuchi, J. Antibiot., 1998, 51, 553–559 CrossRef CAS.
- A. J. Arduengo III, R. Krafczyk and R. Schmutzler, Tetrahedron, 1999, 55, 14523 CrossRef.
- P. De Frémont, N. Marion and S. P. Nolan, J. Organomet. Chem., 2009, 694, 551–560 CrossRef PubMed.
- L. Berre and L. Georges, Bull. Soc. Chim. Fr., 1967, 11, 4328 Search PubMed.
- K. Kato, H. Nouchi, K. Ishikura, S. Takaishi, S. Motodate, H. Tanaka, K. Okudaira, T. Mochida, R. Nishigaki, K. Shigenobu and H. Akita, Tetrahedron, 2006, 62, 2545 CrossRef CAS PubMed.
- M. B. Smith and J. March, Advanced Organic Chemistry: Reaction, Mechanism and Structure, Wiley, Hoboken, NJ, 6th edn, 2007 Search PubMed.
- (a) X. Huang, T. De Haro and C. Nevado, Chem.–Eur. J., 2009, 15, 5904–5908 CrossRef CAS PubMed; (b) N. Ghosh, S. Nayak and A. K. Sahoo, J. Org. Chem., 2011, 76, 500–511 CrossRef CAS PubMed; (c) T. De Haro, E. Gómez-Bengoa, R. Cribiffl, X. Huang and C. Nevado, Chem.–Eur. J., 2012, 18, 6811–6824 CrossRef CAS PubMed; (d) R. K. Shiroodi and V. Gevorgyan, Chem. Soc. Rev., 2013, 42, 4991 RSC and references cited therein.
- Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and F. D. Toste, Wiley-VCH, Weinheim, 2012 Search PubMed.
- (a) G. M. Sheldrick, SADABS, Siemens Area Detector Absorption Correction, University of Göttingen, Göttingen, Germany, 1996 Search PubMed; (b) G. M. Sheldrick, SHELX-97, A Program for Crystal Structure Solution and Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (c) G. M. Sheldrick, SHELXTL NT Crystal Structure Analysis Package, Bruker AXS, Analytical X-ray System, Madison, WI, 1999; version 5.10 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the synthesis of precursors 1b, 1f and 1h–j, 1H and 13C NMR spectra and CIF files. CCDC 952109 and 952110. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra01105h |
| This journal is © The Royal Society of Chemistry 2014 |