DOI:
10.1039/C4RA03303E
(Paper)
RSC Adv., 2014,
4, 31084-31093
Microscopic and thermodynamic analysis of PEG–β-lactoglobulin interaction
Received
11th April 2014
, Accepted 26th June 2014
First published on 26th June 2014
Abstract
We report the binding of milk β-lactoglobulin (β-LG) with PEG-3000, PEG-6000 and methoxypoly(ethylene glycol) anthracene (mPEG-anthracene) in aqueous solution at pH 7.4, using multiple spectroscopic methods, thermodynamic analysis, transmission electron microscopy (TEM) and molecular modeling. Thermodynamic and spectroscopic analysis showed that polymers bind β-LG via van der Waals interactions, hydrogen bonding and hydrophobic interactions, with overall binding constants KPEG-3000–β-LG = 9.2 (±0.9) × 103 M−1, KPEG-6000–β-LG = 9.7 (±0.7) × 103 M−1 and KmPEG-anthracene–β-LG = 5.5 (±0.5) × 104 M−1. The binding affinity was mPEG-anthracene > PEG-6000 > PEG-3000. Transmission electron microscopy analysis showed significant changes in protein morphology as polymer–protein complexation occurred, with a major increase in the diameter of the protein aggregate. Modeling showed several hydrogen bonding systems between PEG and the different amino acid stabilized polymer–β-LG complexes. The free binding energy indicated that the interaction process is spontaneous at room temperature. Furthermore, mPEG-anthracene is a stronger protein binder than PEG-3000 and PEG-6000, due to its major hydrophobic characteristics.
Introduction
Polyethylene glycol (PEG) (Scheme 1) is a non-ionic hydrophilic polymer with stealth behavior, which is often used to increase the stability of particles and proteins under physiological conditions. PEG conjugation to therapeutic proteins has emerged as an effective strategy for drug-delivery.1–5 PEGylation of peptides and proteins significantly alters the structure and function of the protein.5
Indeed, the mass and structure of PEG have been shown to play an important role in PEG conjugated protein delivery. For example, small PEGs are more rapidly cleared from blood than larger ones.6–8 It has also been shown that the structure of PEG molecule markedly influences the PEG–protein conjugation.9,10
Despite extensive investigations on PEG conjugated proteins, the effect of mass and PEG structure on protein–polymer complexation is poorly understood. Therefore, the aim of this study was to evaluate the effects of both mass and structure of the PEG polymers on the protein–polymer interactions.
β-Lactoglobulin (β-LG) is an abundant milk protein with the ability to bind to a wide range of hydrophobic and hydrophilic compounds.11–20 Under physiological conditions, β-LG exists as a mixture of monomers and dimers, the equilibrium ratio of which depends on the association constant of the dimer and the protein concentration. Each monomer consists of 162 amino acid residues and has a molecular mass of 18 kDa.21,22 β-LG was used as a simple model protein to provide a better understanding of how the mass and structure of PEG affect polymer–protein interactions at the molecular level. In this study, we report the spectroscopic analysis, thermodynamic parameters, TEM analysis and molecular modeling of the β-LG complexes with PEG-3000, PEG-6000 and mPEG-anthracene (Scheme 1), in aqueous solution at pH 7.4. The structural analysis regarding protein binding sites and the effects of PEG hydrophilic and hydrophobic compositions on the β-LG stability and morphology is also reported here.
 |
| | Scheme 1 Chemical structures of PEG, mPEG and mPEG-anthracene. | |
Experimental section
Materials
β-Lactoglobulin (A variant, purity > 90%), PEG-3000 and PEG-6000 were purchased from Sigma Chemical Company and used as supplied. Methoxypoly(ethylene glycol) anthracene (mPEG-anthracene) was from Polymer Source (Quebec). All other chemicals were of reagent grade and used without further purification.
Preparation of stock solutions
PEG-3000, PEG-6000 and mPEG-anthracene were dissolved in Tris–HCl solution (pH 7.4). The β-lactoglobulin was dissolved in an aqueous solution (4 mg ml−1 to obtain 0.25 mM protein content) containing Tris–HCl (pH 7.4). The protein concentration was determined spectrophotometrically using the extinction coefficient of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 M−1 cm−1 at 280 nm.23
600 M−1 cm−1 at 280 nm.23
FTIR spectroscopic measurements
Infrared spectra were recorded on a FTIR spectrometer (Impact 420 model, Digilab), equipped with a deuterated triglycine sulphate (DTGS) detector and KBr beam splitter, using AgBr windows. The solution of the polymer was added in a dropwise fashion to the protein solution, with constant stirring to ensure the formation of a homogeneous solution and to reach the target polymer concentrations of 15, 30 and 60 μM, with a final protein concentration of 60 μM. Detailed spectroscopic treatments were carried out according to previous reports.14,24
Analysis of protein conformation
Analysis of the secondary structure of β-lactoglobulin and its PEG complexes was carried out using FTIR spectroscopic analysis based on the literature reports.14,25 The secondary structure of the protein was determined from the shape of the amide I band, located at 1660–1650 cm−1. The FTIR spectra were smoothed and their baselines were corrected automatically using the built-in software of the spectrophotometer (OMNIC ver. 7.3). Thus, the root-mean square (rms) noise of every spectrum was calculated. By means of the second derivative in the spectral region of 1700–1600 cm−1, five major peaks for β-lactoglobulin and the complexes were resolved. The above spectral region was deconvoluted by the curve-fitting method with the Levenberg–Marquardt algorithm and the peaks related to the α-helix (1660–1650 cm−1), β-sheet (1640–1610 cm−1), turn structure (1680–1660 cm−1), and β-antiparallel (1692–1680 cm−1) were adjusted and the areas were measured using the Gaussian function. The area of all components of the bands assigned to a given conformation were then summed up and divided by the total area. The curve fitting analysis was performed using the GRAMS/AI Version 7.01 software of the Galactic Industries Corporation.
Fluorescence spectroscopy
Fluorescence spectra were recorded using a Perkin-Elmer LS55 spectrometer. The β-lactoglobulin fluorescence emission was recorded at λex = 295 nm and λem 342 nm. A stock solution of β-lactoglobulin (30 μM) in Tris–HCl buffer was prepared at 24 ± 1 °C. Samples containing 0.06 ml of the above protein solution and various polymer solutions were mixed to obtain the final polymer concentrations, ranging from 15 to 45 μM, with a constant β-LG content (30 μM).
Transmission electron microscopy
The specimens were observed using a Philips EM 208S microscope operating at 180 kV. The morphology of the complexes of β-LG with PEG-3000 and mPEG-anthracene, in aqueous solution at pH 7.4, were observed using transmission electron microscopy (TEM). One drop (5–10 μl) of the freshly-prepared mixture [β-lactoglobulin solution (60 μM) + polymer solution (60 μM)] in Tris–HCl buffer (24 ± 1 °C) was deposited onto a glow-discharged carbon-coated electron microscopy grid. The excess liquid was absorbed by a piece of filter paper and a drop of 2% uranyl acetate negative stain was added before drying at room temperature. The particle diameter was estimated using IMAGEJ® software analysis of the TEM micrographs. It should be emphasized that our determination of the size was based on at least 5 groups (∼1 cm × 1 cm of the TEM image), each containing almost 100 particles and the average size was deduced from these groups.
Molecular modeling
Docking studies were carried out using ArgusLab 4.0.1 software (Mark A. Thompson, Planaria Software LLC, Seattle, Wa, http://www.arguslab.com). The β-LG structures were obtained from the literature report22 and the PEG three-dimensional structures were generated from PM3 semi-empirical calculations using Chem3D Ultra 11.0. The whole protein was selected as a potential binding site, since no prior knowledge of such a site was available in the literature. The docking runs were performed on the ArgusDock docking engine using regular precision with a maximum of 1000 candidate poses.
Results and discussion
Fluorescence spectroscopy and binding parameters for the polymer–β-LG adducts
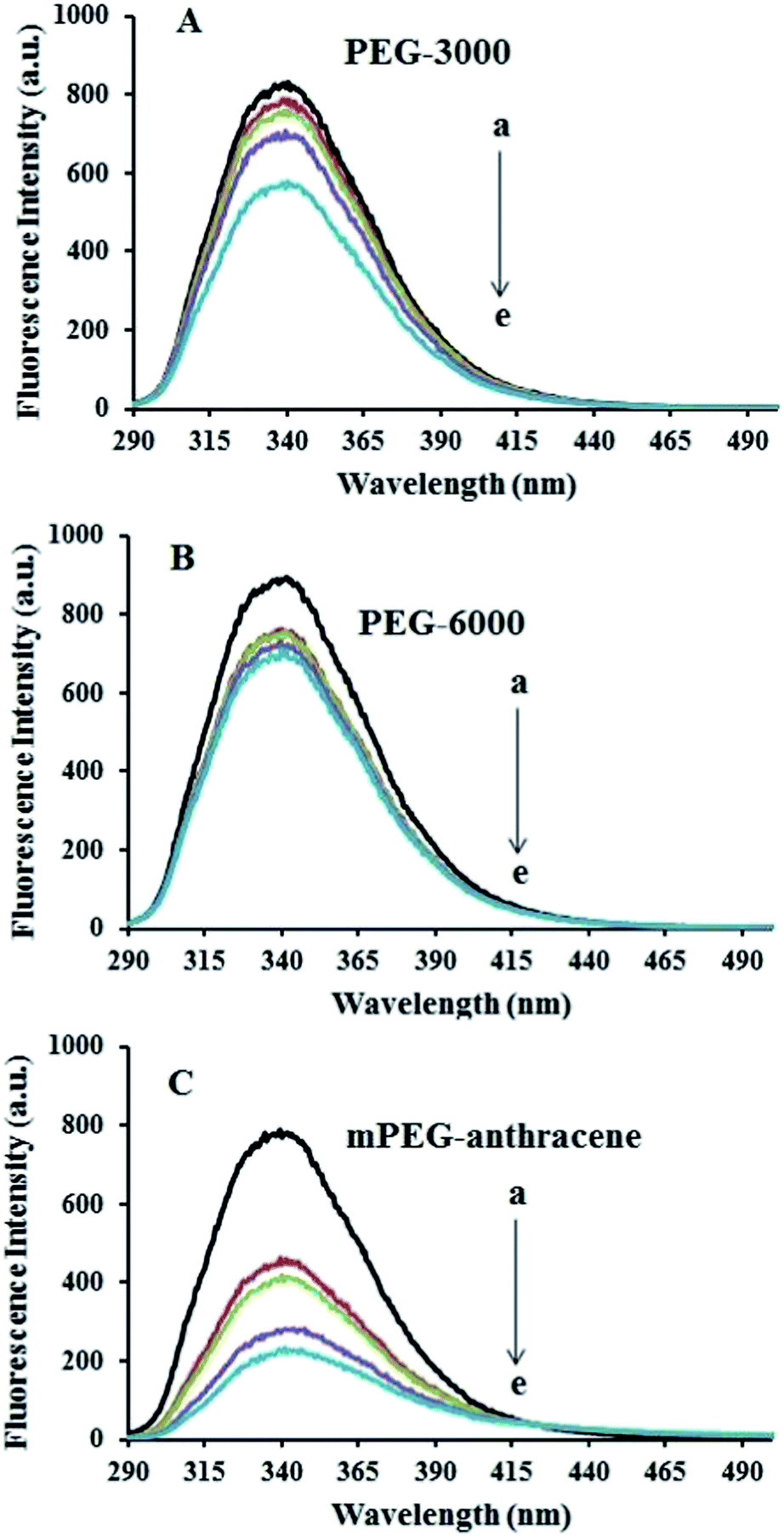
The intrinsic fluorescence of β-LG was studied in the presence of various polymer concentrations, in order to determine the nature of the interaction and the binding parameters between PEG and β-LG. The fluorescence emission spectra of β-LG (10 μM) at pH 7.4, with different concentrations of PEG-3000, PEG-6000 and mPEG-anthracene, are shown in Fig. 1. The maximum emission (λem) of free β-LG was observed at 342 nm. The fluorescence intensity of β-LG gradually decreased as the polymer concentration increased (Fig. 1A–C). These changes indicate that the quenching of the protein intrinsic fluorescence is due to polymer–protein complexation.11,12 Furthermore, a minor blue shift (from 342 to 340 nm) was observed with increasing PEG concentration. This blue shift was more pronounced for mPEG-anthracene (from 342 to 338 nm). The blue shift indicates that the fluorophore (i.e., tryptophan) is experiencing a more hydrophobic environment compared to its native state. This means that proteins in the presence of PEG undergo a conformational change, such that the tryptophan residue (fluorophore) inside β-LG becomes more exposed to the surface after the polymer–protein complex formation. This is also consistent with the FTIR results, which show major protein conformational changes upon polymer interaction (this will be discussed further on).
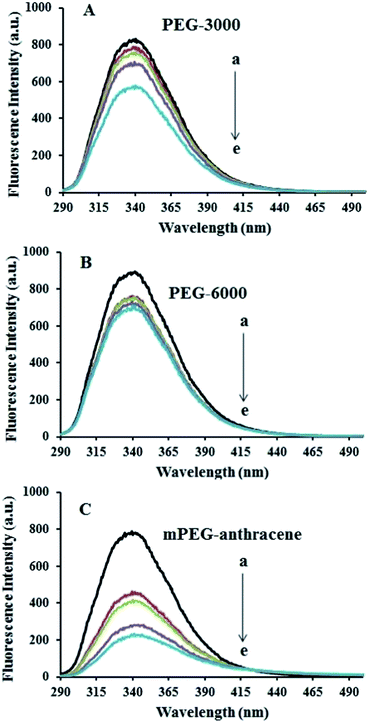 |
| | Fig. 1 Fluorescence emission spectra of β-LG in 10 mM Tris–HCl under varying concentrations of (A) PEG-3000, (B) PEG-6000 and (C) mPEG-anthracene. λex = 295 nm. T= 298.15 K, pH 7.4, [β-LG] = 30 μM. The concentrations of polymers from (a) to (e) are 0, 15, 25, 35 and 45 μM, respectively. | |
The fluorescence quenching of a protein by a ligand can be dynamic, static or both.26,27 To elucidate the nature of the fluorescence quenching mechanism, the fluorescence data were analyzed using the Stern–Volmer equation (eqn (1))
| | |
F0/F = 1 + KSV[Q] = 1 + Kqτ0[Q]
| (1) |
where,
F0 and
F represent the steady-state fluorescence intensities in the absence and presence of the quencher,
Kq is the quenching rate constant,
τ0 is the average lifetime of the protein in the absence of the quencher (1.2 ns) for free β-LG at neutral pH,
12 [
Q] is the molar concentration of the quencher and
KSV is the Stern–Volmer constant.
28 The plots of
F0/
F versus [
Q] show a linear feature for all polymers (
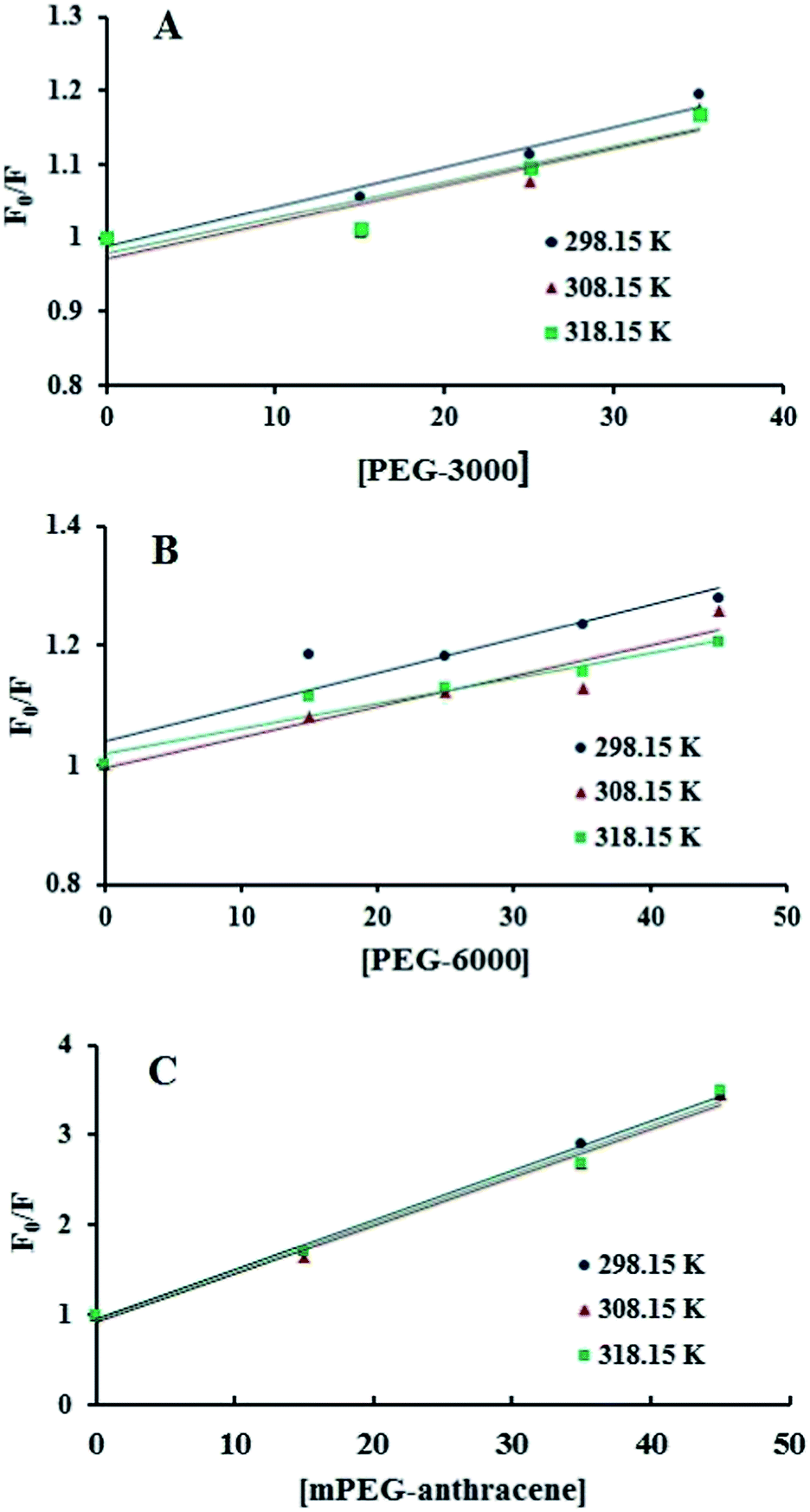
Fig. 2), which means that static or dynamic quenching can occur.
26,27 The values of
KSV were obtained from the slope of the linear regressions of the Stern–Volmer plots (
Fig. 2), and the
Kq values were deduced from
eqn (1) and are listed in
Table 1. The
Kq values for all PEG–β-LG complexes (at 298.15 K) were found to be greater than the maximum value for a diffusion-controlled quenching process (10
10 M
−1 s
−1),
29,30 which shows that the quenching mechanism of β-LG by PEG is static.
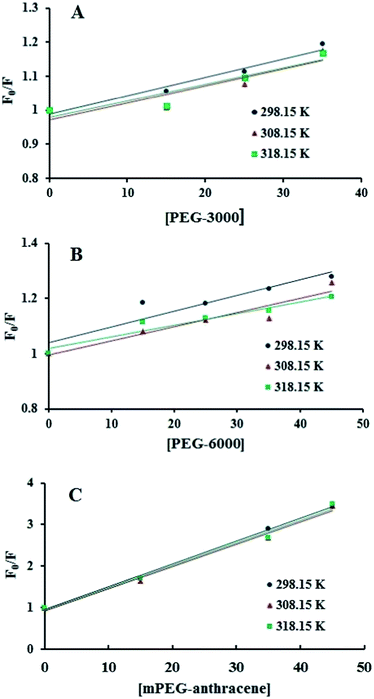 |
| | Fig. 2 Stern–Volmer plots for the fluorescence quenching data of PEG-3000–β-LG (A), PEG-6000–β-LG (B) and mPEG-anthracene–β-LG (C) at three different temperatures, at pH 7.4. | |
Table 1 The quenching constants for the PEG–β-LG complexes at three different temperatures
| Complexes |
Temperature (K) |
Quenching constants |
| Stern–Volmer constant, KSV (M−1) |
Quenching rate constant, Kq (M−1 S−1) |
| PEG-3000–β-LG |
298.15 |
9.2 × 103 |
7.67 × 1012 |
| 308.15 |
3.2 × 103 |
2.67 × 1012 |
| 318.15 |
2.5 × 103 |
2.08 × 1012 |
| PEG-6000–β-LG |
298.15 |
9.7 × 103 |
8.08 × 1012 |
| 308.15 |
5.0 × 103 |
4.17 × 1012 |
| 318.15 |
4.2 × 103 |
3.5 × 1012 |
| mPEG-anthracene–β-LG |
298.15 |
5.5 × 104 |
4.58 × 1013 |
| 308.15 |
4.5 × 104 |
3.75 × 1013 |
| 318.15 |
4.1 × 104 |
3.41 × 1013 |
Additionally, dynamic or static quenching can also be determined by temperature variations. In the case of dynamic quenching, a higher temperature results in faster diffusion and consequently, in a larger quenching rate constant (Kq). In contrast, in static quenching, raising the temperature results in decreasing complex stability and decreasing of the static quenching constant.26,27 As shown in Table 1, the values of KSV and Kq decreased with increasing temperature, which confirms that static quenching is predominant in these PEG–protein complexes.
It should be noted that in the case of static quenching, the Stern–Volmer quenching constant (KSV) can be taken as the binding constant of the quencher to the fluorophore.28–31 The calculated binding constant for PEG shows that PEG-3000 binds to β-LG with binding affinities in the order of 103 M−1 (Table 1). Increasing the PEG chain to PEG-6000 has no major effect on the affinities of PEG–β-LG binding (Table 1). However, substitution of anthracene (a hydrophobic molecule) in PEG leads to a huge increase in the KSV value from 103 M−1 to 104 M−1 for the mPEG-anthracene–β-LG adduct (Table 1). Therefore, it is evident that hydrophobic forces play an important role in PEG–protein binding.
Thermodynamic analysis of the polymer–β-LG adducts
The thermodynamic parameters were analyzed in order to examine the different intermolecular forces involved in the formation of the polymer–protein complexes with PEG-3000, PEG-6000 and mPEG-anthracene. The thermodynamic parameters (standard enthalpy changes, ΔH; standard entropy changes, ΔS and standard Gibbs free energy changes, ΔG) for the PEG–protein interaction were determined at pH 7.4, and conducted at three different temperatures: 298.15, 308.15 and 318.15 K, using eqn (2) and (3).| |
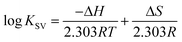 | (2) |
where, T and R are the temperature and gas constant, respectively.
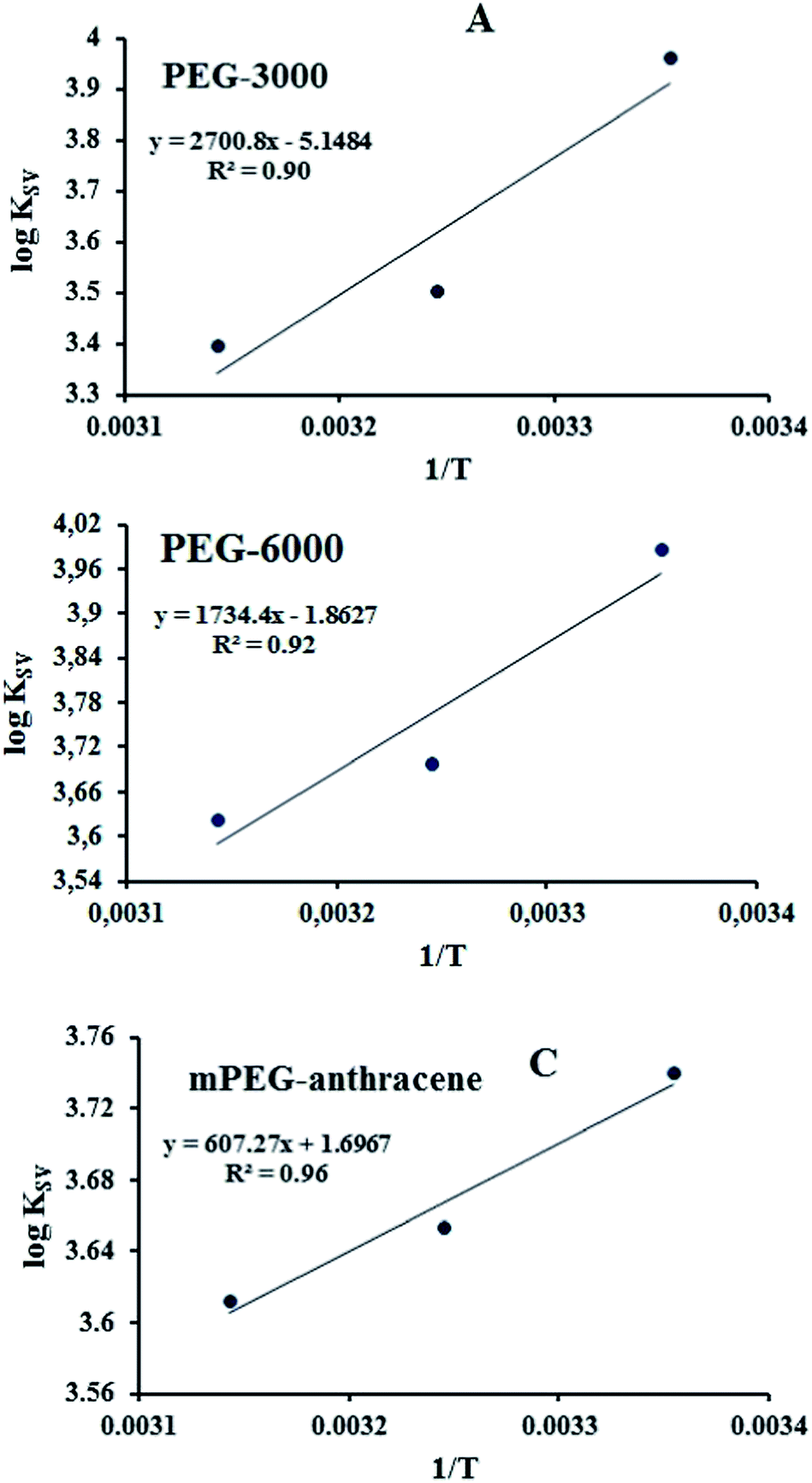
According to the binding constant (KSV) measured at three temperatures (298.15, 308.15 and 318.15 K), the ΔH and ΔS values were estimated from the linear relationship between log KSV and the reciprocal thermodynamic temperature (1/T) (Fig. 3 and Table 2). The ΔG was calculated using eqn (3). As one can see, ΔH and ΔS were found to be −51.71 kJ mol−1 and −98.58 J mol−1 K−1 for the binding between PEG-3000 with β-LG, and −33.21 kJ mol−1 and −35.66 J mol−1 K−1 for the binding between PEG-6000 with β-LG. While for mPEG-anthracene, ΔH and ΔS were found to be 11.63 kJ mol−1 and 32.49 J mol−1 K−1 (Table 2). All three PEG–β-LG complexes have negative ΔH and ΔG, which show that the binding process was exothermic and spontaneous at different temperatures.
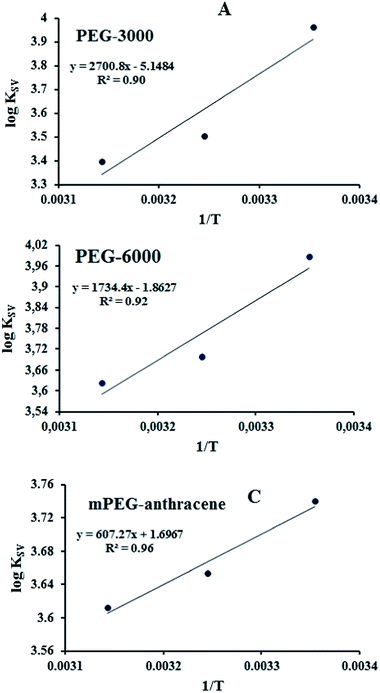 |
| | Fig. 3 van’t Hoff plots of β-LG interaction with the three polymers; PEG-3000 (A), PEG-6000 (B) and mPEG-anthracene (C). | |
Table 2 Thermodynamic parameters for PEG–β-LG complexes and the nature of interaction predicted from these parameters
| Complexes |
Thermodynamic parameters |
Predominant interaction |
| ΔH (kJ mol−1) |
ΔS (J mol−1 K−1) |
ΔG (kJ mol−1) |
| PEG-3000–β-LG |
−51.71 |
−98.58 |
−22.32 (298.15 K) |
Van der Waals forces and hydrogen bonding |
| −21.34 (308.15 K) |
| −20.35 (318.15 K) |
| PEG-6000–β-LG |
−33.21 |
−35.66 |
−22.57 (298.15 K) |
Van der Waals forces and hydrogen bonding |
| −22.21 (308.15 K) |
| −21.86 (318.15 K) |
| mPEG-anthrancene–β-LG |
−11.63 |
32.49 |
−21.31 (298.15 K) |
H-bonding and hydrophobic interactions |
| −21.64 (308.15 K) |
| −22.96 (318.15 K) |
ΔS is a measure of the disorder in a system during the reaction and thus, in polymer–protein complex formation, ΔS involves two main processes with opposite contributions: (i) the approaching of PEG to β-LG, which results in the decrease of freedom, giving a negative ΔS and (ii) the binding of PEG to β-LG, which could lead to protein conformational changes increasing the freedom of the complex, and giving a positive ΔS. Therefore, the positive value of ΔS indicates that entropy provides a contribution to the standard Gibbs free energy changes (ΔG). Furthermore, the positive ΔS is frequently taken as typical evidence of a hydrophobic interaction.32
Interestingly, it is found that in the binding between PEG-3000 and PEG-6000 with β-LG, the major contribution to ΔG arises from the ΔH (negative value), rather than the ΔS (negative value), so the binding process is enthalpy driven (Table 2). Therefore, the negative ΔH and ΔS values (Table 2) suggest the involvement of van der Waals forces and hydrogen bonding in PEG-3000–β-LG and PEG-6000–β-LG complex formation.33,34 However, a decrease in entropy (Table 2) is attributed to the formation of hydrogen bonds between PEG-3000 and PEG-6000 with β-LG, which markedly reduces the degrees of freedom, because the system is more ordered after the complex formation. In contrast, in the binding reaction between mPEG-anthracene and β-LG, both ΔH (negative value) and ΔS (positive value) were favorable to a polymer–protein complexation (Table 2). However, the negative enthalpy (ΔH = −11.63 kJ mol−1) and entropy (TΔS = −9.68 kJ mol−1 at 298.15 K) for mPEG-anthracene–β-LG, both provide about the same contribution to ΔG, which indicates that the binding process is both enthalpy and entropy driven (Table 2). It can therefore be concluded that hydrogen bonding and hydrophobic interactions are the main forces behind the binding of mPEG-anthracene with β-LG.35 These findings also suggest that PEG–protein complexation is more stable when both hydrogen bonding and hydrophobic contacts are involved in the binding process.
FTIR spectroscopic analysis of the polymer–β-LG complexes
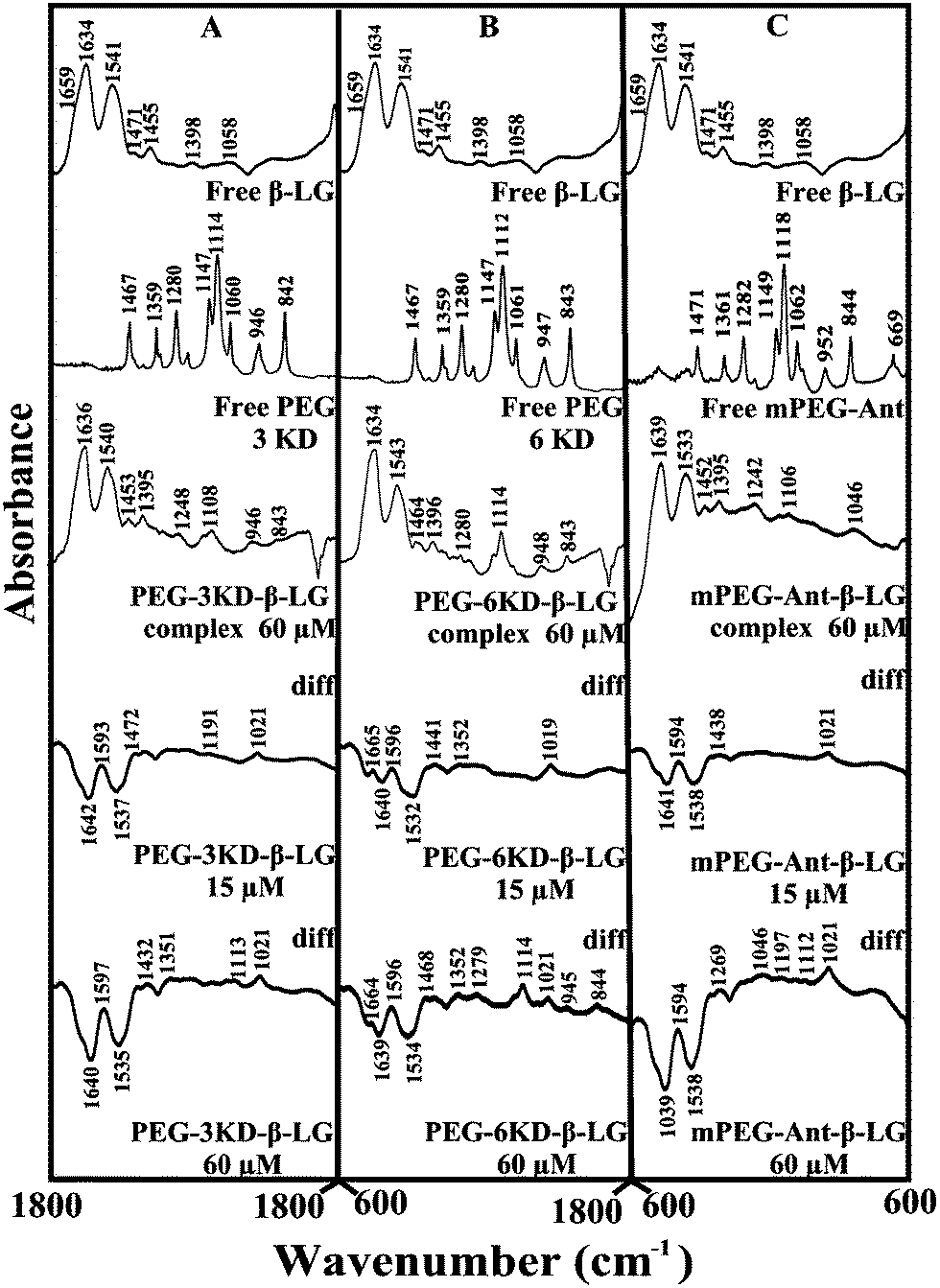
The polymer–protein interaction was characterized by infrared spectroscopy. The protein amide I band at 1660–1650 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations) and amide II band at 1550–1530 cm−1 (C–N stretching coupled with N–H bending modes)36 can be used to assess the polymer–protein complexation. The amide I band is also used to quantify protein conformational changes upon ligand interaction.14,25
O stretching vibrations) and amide II band at 1550–1530 cm−1 (C–N stretching coupled with N–H bending modes)36 can be used to assess the polymer–protein complexation. The amide I band is also used to quantify protein conformational changes upon ligand interaction.14,25
Given that there was no major spectral shifting for the protein amide I and II bands upon polymer interaction, difference spectra [(β-LG solution + polymer solution) − (β-LG solution)] were generated in order to monitor the intensity variations of the amide bands. The results are shown in Fig. 4. At a low polymer concentration of 15 μM, several negative features were observed in the difference spectra for the protein amide I and amide II at 1642 and 1537 cm−1 (PEG-3000–β-LG), at 1640 and 1533 cm−1 (PEG-6000–β-LG) and at 1641 and 1538 cm−1 (mPEG-anthracene–β-LG) (Fig. 4A–C, diff., 15 μM). As the polymer concentration increased to 60 μM, larger negative features were observed for protein amide I and amide II at 1640 and 1535 cm−1 (PEG-3000–β-LG), at 1639 and 1534 cm−1 (PEG-6000–β-LG) and at 1639 and 1538 cm−1 (mPEG-anthracene–β-LG), upon polymer–protein complexation (Fig. 4A–C, diff., 60 μM). These negative features are related to the intensity reduction of the protein amide I and amide II bands upon polymer–protein complexation. These results indicate that the polymer–protein interaction occurs with the protein CO and C–N groups. It is worth mentioning that the observed decrease in intensity of amide I and amide II in the presence of polymers is also due to a change in protein conformation, which is discussed below. This is also consistent with the fluorescence results, which showed major protein conformational changes upon polymer interaction.
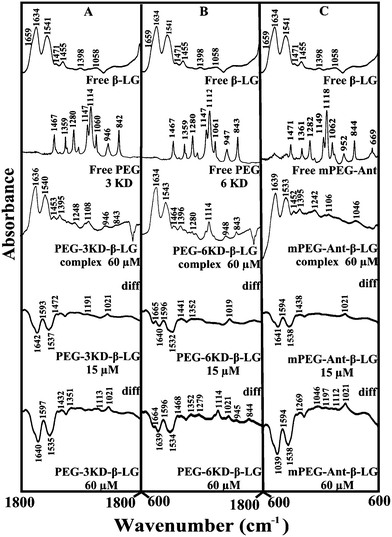 |
| | Fig. 4 FTIR spectra in the region of 1800–600 cm−1 of hydrated films (pH 7.4) for free β-LG (60 μM) and its PEG complexes for (A) PEG-3000, (B) PEG-6000 and (C) mPEG-anthracene, with difference spectra (diff.) (bottom two curves) obtained at different polymer concentrations (indicated on the figure). | |
In order to evaluate the conformational changes of β-LG upon PEG interaction, a quantitative analysis of the protein secondary structure for the free β-LG and its complexes (β-LG–PEG) has been carried out and the results are shown in Fig. 5 and Table 3. The free β-LG has 58% β-sheet (1640, 1623 cm−1), 11% α-helix (1655 cm−1), 14% turn structure (1667 cm−1) and 17% β-antiparallel (1679 cm−1), as previously reported.14,18 Upon PEG interaction, major changes of the α-helix from 11% (free β-LG) to 34% (PEG-3000–β-LG), 28% (PEG-6000–β-LG) and 46% (mPEG-anthracene–β-LG) with a reduction of β-sheet structure from 58% (free β-LG) to 47% (PEG-3000–β-LG), 52% (PEG-6000–β-LG) and 29% (mPEG-anthracene–β-LG), were observed (Table 3). The major alterations to the β-LG conformation (reduction of the β-sheet and increase of the α-helix structure) are due to partial protein destabilization. Our results are consistent with those of recent studies using CD spectroscopy, which showed that PEG–BSA interaction alters protein conformation.37,38
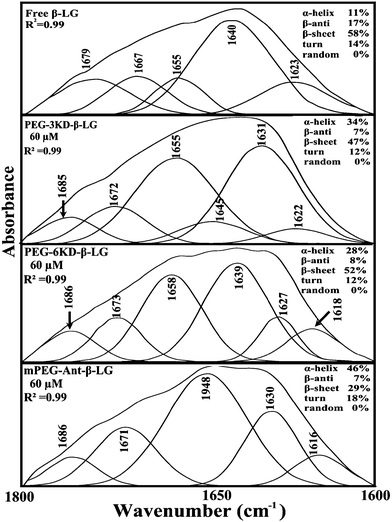 |
| | Fig. 5 Second derivative resolution enhancement and curve-fitting of the amide I region (1700–1600 cm−1) for free β-LG and its PEG complexes with 60 μM polymer (pH 7.4). | |
Table 3 Secondary structure analysis for amide I region (infrared) in free β-LG and its polymer complexes at pH 7.4
| Amide I (cm−1) components |
Free β-LG (%) 60 μM |
PEG-3000 (%) 60 μM |
PEG-6000 (%) 60 μM |
mPEG-anthracene (%) 60 μM |
| α-Helix (±2) 1654–1660 |
11 |
34 |
28 |
46 |
| β-Sheet (±2) 1614–1637 |
58 |
47 |
52 |
29 |
| Turn (±2) 1670–1678 |
14 |
12 |
12 |
18 |
| β-Antiparallel (±1) 1680–1691 |
17 |
7 |
8 |
7 |
Morphological characteristics of the polymer–β-LG aggregates
The changes in the morphological aggregation of β-LG molecules after polymer complexation can be observed visually using transmission electron microscopy (TEM). The TEM images of β-LG in the absence and presence of PEG-3000 and mPEG-anthracene in aqueous solution at pH 7.4 are shown in Fig. 6. As the TEM image of the native β-LG (i.e., without polymers Fig. 6A) shows, it is likely that we have minor spherical aggregates. Aggregates take the form of white masses due to the negative staining procedure employed. The observed changes in the TEM image are consistent with the fact that as a globular protein, native β-LG is almost spherical with a packing density at the interior of the molecule, which shows that the hydrophobic amino acid residues tend to be buried inside the protein, whereas hydrophilic charged groups are located on the surface, in contact with the aqueous phase as reported in the literature.39–41 The particle size analysis of β-LG in the absence of polymers shows that the particle size ranges from 3 to 12 nm with a mean diameter of 5.48 ± 2.29 nm (Fig. 6A).
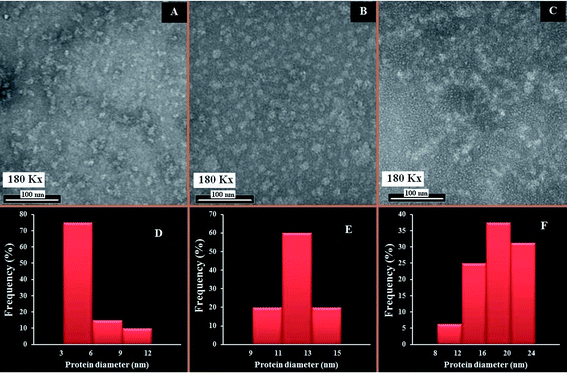 |
| | Fig. 6 TEM images showing the morphology of β-LG at pH 7.4 and 24 °C: in the absence of the polymers (A); and in the presence of PEG-3000 (B) and mPEG-anthracene (C). Histograms of β-LG particle size distribution: in the absence of the PEG (D); and in the presence of PEG-3000 (E), and mPEG-anthracene (F). The concentrations of β-LG and polymers were 60 μM in all samples. | |
Upon mixing β-LG with PEG-3000 and mPEG-anthracene, the stable dispersions of the β-LG aggregates become more evident in the TEM images (Fig. 6B and C). These results suggest that PEG–protein complexation causes a major change in protein morphology. As a consequence, segments of different β-LG molecules may interact through hydrophobic contacts or by forming hydrogen bonds, leading to aggregation. The TEM images show that the number and size of the spherical aggregates are increased after the PEG–β-LG complexes are formed (Fig. 6B, C, E and F), compared to the small aggregates observed in the native β-LG molecules (Fig. 6A and D). The size aggregate analysis reveals that the mean diameter of the aggregation of β-LG with PEG-3000 and mPEG-anthracene is 12.09 and 17.57 nm, respectively (Fig. 6E). It is worth mentioning that the increases in the diameters of the β-LG aggregates are consistent with those of the binding constants estimated by the fluorescence data, which shows that more stable complexes are formed with mPEG-anthracene (Table 1).
These results can be explained by the nature of the interactions between β-LG and the PEG-polymers. As we mentioned earlier, in the section related to the FTIR and fluorescence spectroscopic studies, the polymer induced major perturbations of the secondary protein structure. The secondary structure of a protein is the specific geometric shape caused by intramolecular hydrogen bonding of the amide groups. Therefore, in the case of the PEG-3000–β-LG complex, the increases in the diameter of the β-LG aggregate (110%) is due to the intermolecular hydrogen bonding between PEG-3000 and the protein, which constitutes the predominant interaction force of protein–polymer complexation, as shown by the thermodynamic analysis. Thus, the intramolecular hydrogen bonds of the amide groups are lost at the expense of the intermolecular hydrogen bonds between PEG and β-LG, which causes the changes in the secondary structure of the protein and thereby its morphology. In the case of the mPEG-anthracene–β-LG complex, the 300% increase in the β-LG aggregate diameter is due to the fact that hydrogen bonding and hydrophobic interactions are the main forces behind protein–polymer complexation. In the native protein, this larger morphological perturbation comes from the fact that the hydrophobic amino acid residues are buried inside the protein core. Thus, to promote the hydrophobic interactions between mPEG-anthracene and β-LG, the protein must expose its hydrophobic residues to the surface and adopt a conformation which facilitates complex formation, thereby causing a larger perturbation in the protein secondary structure than that of PEG-3000 (Table 3).
Docking studies
A docking study was used to locate the preferred polymer binding sites with the protein. The models of the docking for the polymer are shown in Fig. 7 and Table 4. Several amino acids are located in the vicinity of PEG and PEG-anthracene (Fig. 7 and Table 4). The presence of several H-bonding systems stabilizes the PEG–β-LG and PEG–anthracene complexes (Table 4). As one can see, different amino acids are involved in PEG and PEG–anthracene complexation due to the mainly hydrophilic character of PEG and the very hydrophobic nature of PEG–anthracene. This is consistent with the more stable complexation of mPEG-anthracene with β-LG, rather than with PEG-3000 and PEG-6000 (Table 1). These findings are also in agreement with the TEM results, which show a larger increase in protein diameter with mPEG-anthracene than those of the PEG-3000 and PEG-6000 (Fig. 6). Since β-LG contains a large hydrophobic domain it seems that the hydrophobicity of the anthracene residue results in a stronger mPEG-anthracene–protein complex formation.
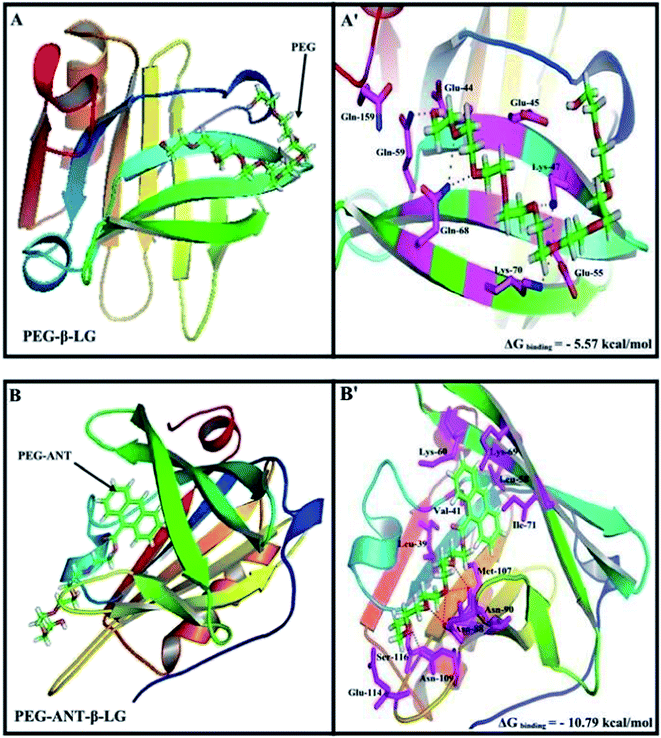 |
| | Fig. 7 Docking results of the PEG-β-LG and PEG-anthracene–β-LG complexes. View of the nearest amino acids surrounding PEG with H-bonding network and free binding energy. | |
Table 4 Amino acid residues involved in the PEG–β-LG complexesa
| Complex |
Amino acids in the vicinity of PEG |
ΔG binding (kcal mol−1) |
| *Hydrogen bonding involved with these amino acids. |
| PEG–β-LG |
*Glu-44, Glu-45, **Lys-47, Glu-55, *Gln-59, **Gln-68, *Lys-70, Gln-159 |
−5.57 |
| PEG-Ant–β-LG |
Leu-39, Val-41, Leu-58, Lys-60, Lys-69, Ile-71, **Asn-88, **Asn-90, Met-107, *Asn-109, Glu-114, Ser-116 |
−10.79 |
Conclusions
Spectroscopic results showed that PEG mass and composition have a major effect on polymer–β-LG interactions. mPEG-anthracene forms more stable complexes than PEG-3000 and PEG-6000. Thermodynamic analysis showed that the polymer–protein binding process is enthalpy and entropy driven with hydrogen bonding, van der Waals interactions and hydrophobic contacts. Polymer–protein interactions alter the β-LG conformation, with mPEG-anthracene causing larger perturbations of the protein conformation than PEG-3000 and PEG-6000, due to its major hydrophobic characteristics. Major alterations of protein morphology were observed as protein–PEG complexation occurred, with a drastic increase in the diameter of the protein aggregates. Modeling showed the presence of several hydrogen bonding systems between PEG and different amino acids that stabilized the polymer–β-LG complexes. These results provide an important insight into the binding mechanism of proteins with PEG, and its derivatives, and increase our understanding about the effects of PEG on polymer–protein interactions.
Abbreviations
| PEG | poly(ethylene glycol) |
| mPEG-anthracene | methoxypoly(ethylene glycol) anthracene |
| β-LG | beta-lactoglobulin |
| FTIR | Fourier transform infrared |
| CD | circular dichroism |
| TEM | transmission electron microscopy |
Acknowledgements
The financial support of the Natural Sciences and Engineering Research Council of Canada (NSERC) is highly appreciated.
Notes and references
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed
 .
. - Z. Cao and S. Jiang, Nano Today, 2012, 7, 404–413 CrossRef CAS PubMed .
- A. Li, H. P. Lehmann, G. Sun, S. Samarajeewa, J. S. Zou, F. Zhang, M. J. Welch, Y. Liu and K. L. Wooley, ACS Nano, 2012, 10, 8970–8982 CrossRef PubMed .
- K. Kavitha and G. L. BhalaMurugan, Int. J. Res. Pharm. Biomed. Sci., 2013, 4, 296–304 CAS .
- S. Jevševar, M. Kunstelj and V. G. Porekar, Biotechnol. J., 2010, 5, 113–128 CrossRef PubMed .
- T. Yamaoka, Y. Tabata and Y. Ikada, J. Pharm. Sci., 1994, 83, 601–606 CrossRef CAS .
- S. J. DeNardo, Z. S. Yao, K. S. Lam, A. M. Song, P. A. Burke, G. R. Mirick, K. R. Lamborn, R. T. Donnell and G. L. DeNardo, Clin. Cancer Res., 2003, 9, 3854S–3864S CAS .
- L. E. van Vlerken, T. K. Vyas and M. M. Amiji, Pharm. Res., 2007, 24, 1405–1414 CrossRef CAS PubMed .
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS .
- H. Rachmawati, P. L. Febrina, R. A. Ningrum and D. S. Retnoningrum, Int. J. Res. Pharm. Sci., 2012, 3, 228–233 Search PubMed .
- L. Liang and M. Subirade, J. Phys. Chem. B, 2010, 114, 6707–6712 CrossRef CAS PubMed .
- L. Liang, H. A. Tajmir-Riahi and M. Subirade, Biomacromolecules, 2008, 9, 50–55 CrossRef CAS PubMed .
- X. Liu, L. Shang, S. Jiang, E. Dong and E. Wang, Biophys. Chem., 2006, 121, 218–223 CrossRef CAS PubMed .
- J. Essemine, I. Hasni, R. Caprpentier, T. J. Thomas and H. A. Tajmir-Riahi, Int. J. Biol. Macromol., 2011, 49, 201–211 CrossRef CAS PubMed .
- G. Kontopidis, C. Holt and L. Sawyer, J. Dairy Sci., 2004, 87, 785 CrossRef CAS .
- G. Kontopidis, C. Holt and L. Sawyer, J. Mol. Biol., 2002, 318, 1043–1055 CrossRef CAS .
- D. C. Lange, R. Kothari, R. C. Patel and S. C. Patel, Biophys. Chem., 1998, 74, 45–51 CrossRef CAS .
- D. C. Kanakis, I. Hasni, P. Bourassa, P. Tarantilis, M. G. Polissiou and H. A. Tajmir-Riahi, Food Chem., 2011, 127, 1046–1055 CrossRef PubMed .
- S. Brownlow, J. H. M. Cabral, R. Cooper, D. R. Flower, S. J. Yewdall, A. C. T. Polikarpov, L. Nort and L. Sawyer, Structure, 1997, 5, 481–495 CrossRef CAS .
- F. Zsila, Z. Bikadi and M. Simonyi, Biochem. Pharmacol., 2002, 64, 1651–1660 CrossRef CAS .
- H. A. McKenzie and W. H. Sawyer, Nature, 1967, 214, 1101–1104 CrossRef CAS .
- B. Y. Qin, M. C. Bewley, L. K. Creamer, H. M. Baker, E. N. Baker and G. B. Jameson, Biochemistry, 1998, 37, 14014–14023 CrossRef CAS PubMed .
- M. D. Collini, L. Alfonso and G. Baldini, Protein Sci., 2000, 9, 1968–1974 CrossRef CAS PubMed .
- F. Dousseau, M. Therrien and M. Pezolet, Appl. Spectrosc., 1989, 43, 538–542 CrossRef CAS .
- D. M. Byler and H. Susi, Biopolymers, 1986, 25, 469–486 CrossRef CAS PubMed .
- S. Roufik, S. F. Gauthier, X. J. Leng and S. L. Turgeon, Biomacromolecules, 2006, 7, 419–426 CrossRef CAS PubMed .
- A. Uttam, G. Subhadip, D. N. Kumar, G. Narayani and M. Ramakanta, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2013, 52, 1031–1040 Search PubMed .
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 3nd edn, 2006 Search PubMed .
- G. Zhang, Q. Que, J. Pan and J. Guo, J. Mol. Struct., 2008, 881, 132–138 CrossRef CAS PubMed .
- J. B. Xiao, X. Q. Chen, X. Y. Jiang, X. Y. M. Hilczer and M. Tachiya, J. Fluoresc., 2008, 18, 671–678 CrossRef CAS PubMed .
- G. B. Behera, B. K. Mishra, P. K. Behera and M. Panda, Adv. Colloid Interface Sci., 1999, 82, 1–42 CrossRef CAS .
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS .
- O. K. Abou-Zied and O. I. K. Al-Shihi, J. Am. Chem. Soc., 2008, 130, 10793–10801 CrossRef CAS PubMed .
- Q. Zhou and T. M. Swager, J. Am. Chem. Soc., 1995, 117, 12593–12602 CrossRef CAS .
- J. Tian, S. Wei, Y. Zhao, R. Liu and S. Zhao, J. Chem. Sci., 2010, 132, 391–400 CrossRef PubMed .
- S. Krimm and J. Bandekar, Adv. Protein Chem., 1986, 38, 181–346 CrossRef CAS .
- S. Rawat, R. Suri and D. K. Sahoo, Biochem. Biophys. Res. Commun., 2010, 392, 561–566 CrossRef CAS PubMed .
- V. Kumar, V. K. Sharma and D. S. Kalonia, Int. J. Pharm., 2009, 366, 38–43 CrossRef CAS PubMed .
- Food Colloids and Polymers: Stability and Mechanical Properties, ed. E. Dickinson and P. Walstra, Royal Society of Chemistry, Cambridge, UK, 1993, pp. 332–340 Search PubMed .
- R. N. Zúñniga, A. Tolkach, U. Kulozik and J. M. Aguilera, J. Food Sci., 2010, 75, E261–E268 CrossRef PubMed .
- L. Bateman, A. Ye and H. Singh, J. Agric. Food Chem., 2010, 58, 9800–9808 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.