
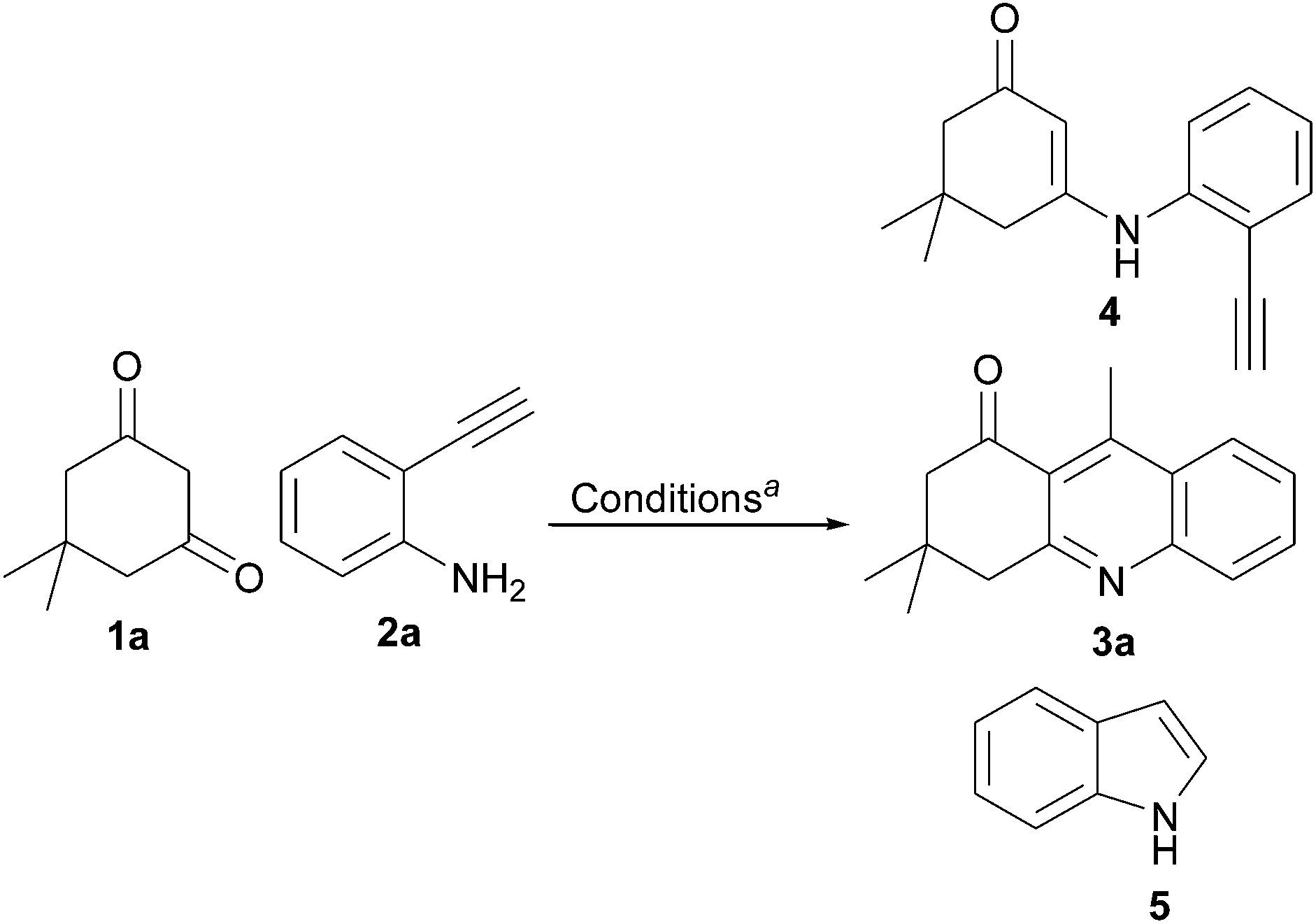
Synthesis of 4-methyl-2,3-disubstituted quinoline scaffolds via environmentally benign Fe(III) catalysed sequential condensation, cyclization and aromatization of 1,3-diketone and 2-ethynylaniline†
Mahalingam Sivaraman and
Paramasivan T. Perumal*
Organic Chemistry Division, CSIR-Central Leather Research Institute, Adyar, Chennai, 600020, Tamilnadu, India. E-mail: ptperumal@gmail.com; Fax: +91-044-24911589
First published on 29th September 2014
Abstract
Environmentally benign Fe(III)-catalysed sequential condensation, cyclization and aromatization of 1,3-diketone and 2-ethynylaniline derivatives to the substituted quinoline scaffolds with good to excellent yield in shorter reaction time is described.
Introduction
Quinoline scaffolds receive much attention due to their pharmacological activity and their presence in the numerous biologically active natural products, particularly in alkaloids.1 Among the numerous methodologies reported for the synthesis of these scaffolds,2 the Friedländer method is an important one; it involves the reaction of 2-aminoacetophenone and a carbonyl compound possessing an active methylene group, in the presence of base or acid.3,4 Recently modified Friedländer methods2,4 and transition metal-catalysed reactions have received considerable attention,5 having the advantages of higher yield and shorter reaction time. In spite of these advantages, the above methods suffer from functionalising substituent at the aryl ‘A’ ring of the quinoline scaffolds (Scheme 1, eqn (1)). Substitution of an aryl ‘A’ ring influences the electronic properties of a molecule, which may affect its biological activity. Metal residues are not allowed to be present in pharmaceutical industries; for this reason, biocompatible iron-catalysed environmentally benign methods are preferred.6 To overcome the above issues we planned the synthesis of 4-methyl-2,3-disubstituted quinolines having an aryl ‘A’ ring with varying substituents indirectly and under environmentally benign conditions. From the previous results, we found that a 2-ethynylaniline in which substitution is possible in the aryl ring is the best choice for an alternative to 2-aminoacetophenone with restricted substitution possibilities (Scheme 1, eqn (2)).7 | ||
| Scheme 1 Concept of synthesis of 4-methyl-2,3-disubstituted quinolines. | ||
To test this hypothesis, we have to choose the correct catalyst that would facilitate condensation of 2-ethynylaniline and 1,3-diketone and further cyclization (Scheme 1, eqn (3), path a), rather than intramolecular cyclization of 2-ethynylaniline followed by coupling of 1,3-diketone, which has already been reported by Antonio Arcadi et. al. (Scheme 1, eqn (3), path b).8 A β-enamino carbonyl compound was formed from the condensation of 2-ethynylaniline and 1,3-diketone having bi-nucleophile character,9,10 which would prefer 6-exo-dig cyclization (C-nucleophile) (Scheme 1, eqn (3), path a1) and aromatization to the expected 4-methyl-2,3-disubstituted quinolines over the 5-endo-dig cyclized product as reported by Hiromichi Fujioka et. al. (N-nucleophile) (Scheme 1, eqn (3), path a2).10a
Results and discussion
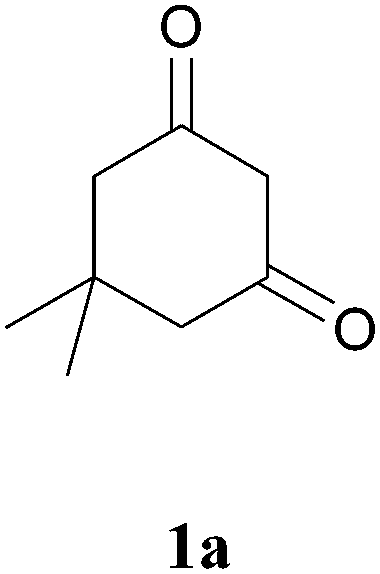
With above-mentioned task to test this methodology, first we chose an iron salt as catalyst from the interesting results of iron-catalysed synthesis of α-carbonyl furan derivatives by Huanfeng Jiang et. al.6d and iron-catalysed construction of oxa- and azacycles through the Prins cyclization by Martín et. al.6g It was observed that 1 equivalent of 1a and 1 equiv. of 2a with Fe(OTs)3·6H2O as catalyst at 80 °C with 1,2-DCE as solvent gave the expected product at a 52% yield with a trace amount of the condensation product 4 and intramolecular cyclized product 5 as a side product (Table 1, entry 1). FeCl3 and FeCl2 gave reduced yields of 41% and 36%, respectively (Table 1, entry 2 and 3 respectively). Changing to toluene as a choice of solvent at 110 °C, the yield increased to 81% (Table 1, entry 4). When the above-mentioned reaction was carried out at room temperature, only traces of 3a was obtained (Table 1 entry 5). Increasing or decreasing the catalyst loading resulted in decreased yield (Table 1, entries 6,7 and 8). When the reaction was carried out in ethanol as solvent at 80 °C, only 11% of 3a with 3-ethoxy-5,5-dimethylcyclohex-2-enone as a major product was obtained (Table 1, entry 9). On further attempts, we found that 1.5 equivalent of 1a and 1 equivalent of 2a with 0.1 equivalent of catalyst Fe(OTs)3·6H2O at 110 °C gave the best yield of 3a at 86% (Table 1, entries 10–12). No trace of product was identified when the reaction was carried out in the absence of catalyst (Table 1, entry 13). Finally, we achieved Fe(OTs)3·6H2O-catalysed sequential condensation, cyclization and aromatization of 1,3-diketone and 2-ethynylaniline in a one-pot manner. To the best of our knowledge this is the first report of the synthesis of 4-methyl-2,3-disubstituted quinolines by an iron(III)-catalysed reaction of 2-ethynylaniline and 1,3-diketones.
|
||||
|---|---|---|---|---|
| Entry | Catalyst (equivalents) | Solvent | Temp (°C) | Yieldb of 3a (%) |
| a 1 equivalent of 1a and 1 equivalent of 2a under nitrogen atmosphere for 2 h.b Isolated yield.c Trace amount of 4 and 5 was observed.d We observed 3-ethoxy-5,5-dimethylcyclohex-2-enone as a major product.e 1.2 equivalents of 1a.f 1.5 equivalents of 1a.g 2.0 equivalents of 1a. | ||||
| 1 | Fe(OTs)3·6H2O (0.1) | 1,2-DCE | 80 | 52c |
| 2 | FeCl3 (0.1) | 1,2-DCE | 80 | 41c |
| 3 | FeCl2 (0.1) | 1,2-DCE | 80 | 36c |
| 4 | Fe(OTs)3·6H2O (0.1) | Toluene | 110 | 81 |
| 5 | Fe(OTs)3·6H2O (0.1) | Toluene | R.T | Trace |
| 6 | Fe(OTs)3·6H2O (0.05) | Toluene | 110 | 69 |
| 7 | Fe(OTs)3·6H2O (0.2) | Toluene | 110 | 77 |
| 8 | Fe(OTs)3·6H2O (0.5) | Toluene | 110 | 70 |
| 9 | Fe(OTs)3·6H2O (0.1) | Ethanol | 80 | 11d |
| 10e | Fe(OTs)3·6H2O (0.1) | Toluene | 110 | 81 |
| 11f | Fe(OTs)3·6H2O (0.1) | Toluene | 110 | 86 |
| 12g | Fe(OTs)3·6H2O (0.1) | Toluene | 110 | 78 |
| 13 | Fe(OTs)3·6H2O (0) | Toluene | 110 | — |
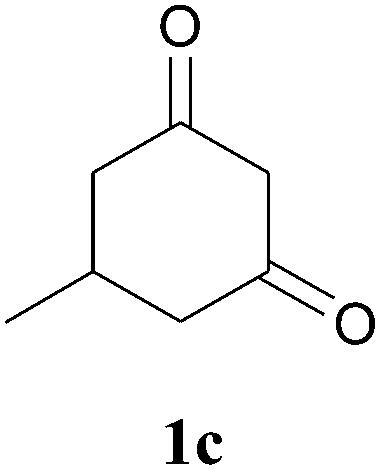
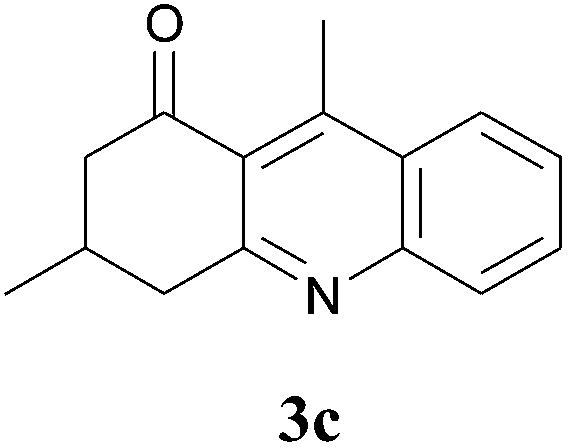
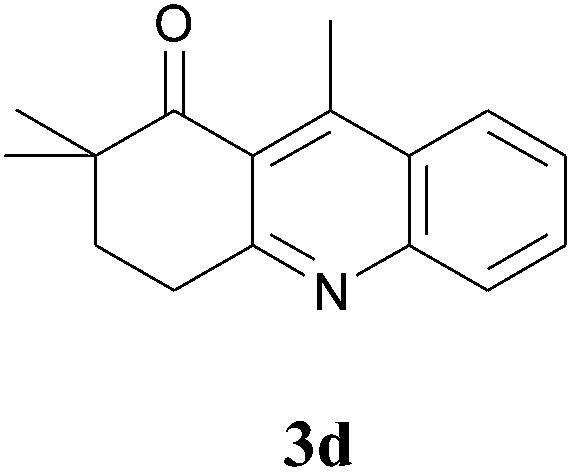
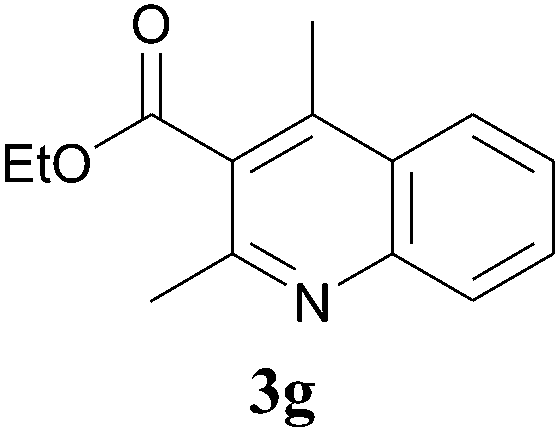
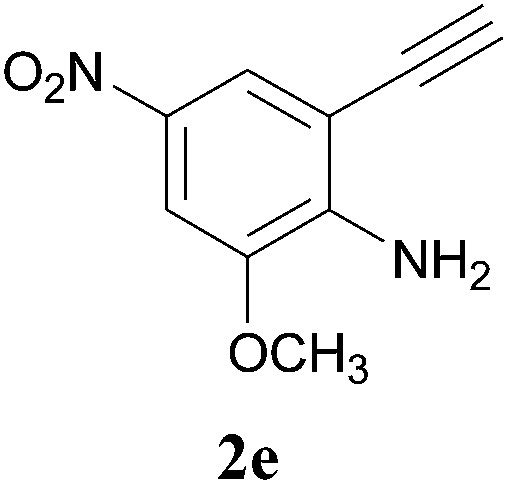
The product structure was confirmed by comparing the NMR of isolated compound 3a with previously reported data.4d After confirming the product structure and with optimal reaction conditions in hand, we tested the scope of the reaction and the results are presented in Table 2. All the six-membered cyclic 1,3-diketone gave a similar yield compared to parent dimedone (Table 2, entries 1–4), whereas five-membered cyclic 1,3-diketone gave a reduced yield of 73% (Table 2, entry 5). The diketone 1d exclusively afforded the single isomer 3d, whose structure was confirmed by 2D NMR experiments. We distinguished between the possible isomeric structures 3d and 3d′ by HMBCs. The aliphatic 2CH3 give a signal at 1.24 ppm with 6 integral values in 1H NMR, which give HSQCs with 24.8 ppm carbon. In HMBCs, two CH3 protons gave a correlation with 205.5 ppm carbonyl carbon, which clearly confirms the structure of 3d and excludes the possibility of 3d′. If the structure were 3d′, then two CH3 protons would not give HMBCs with 205.5 ppm carbonyl carbon (Fig. 1). Diketones like acetyl acetone, ethyl and methyl acetoacetate gave reduced yield when compared with dimedone (Table 2, entries 6–8). Next, we studied the effect of substituents on the 2-ethynylaniline, and the similar yields obtained: the substituents on the 2-ethynylaniline aryl ring had no effect (Table 2, entries 9–12). 2-[(trimethylsilyl)ethynyl]aniline (2f) gave a reduced yield of 77% with trace amounts of condensation product (Table 2, entry 13).
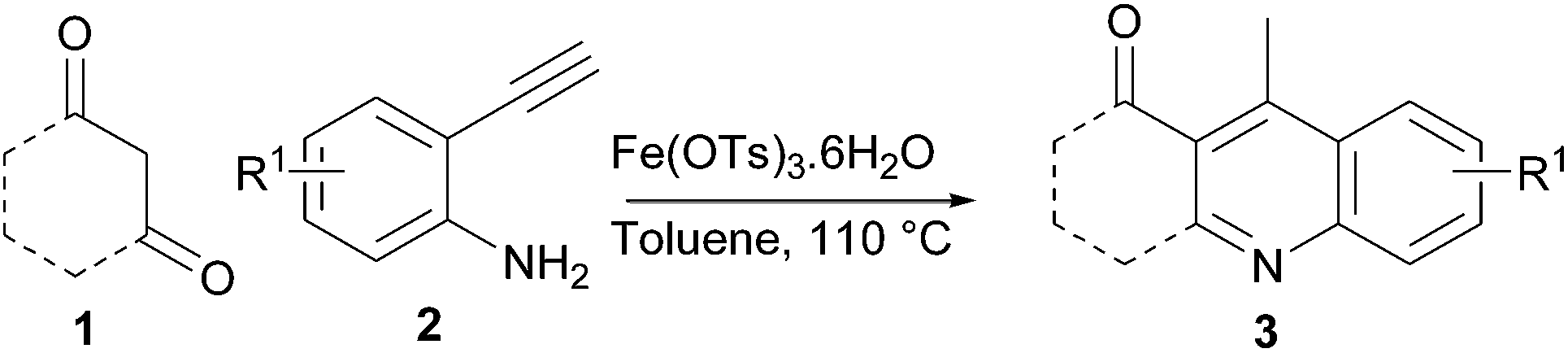











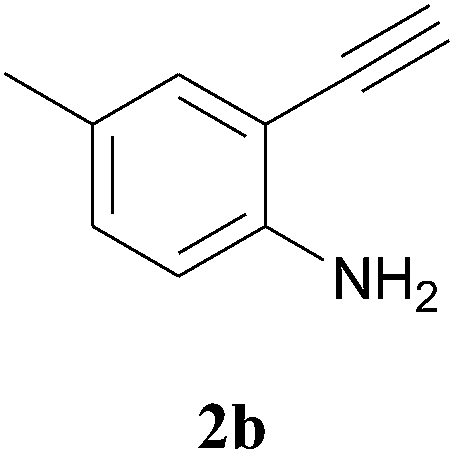


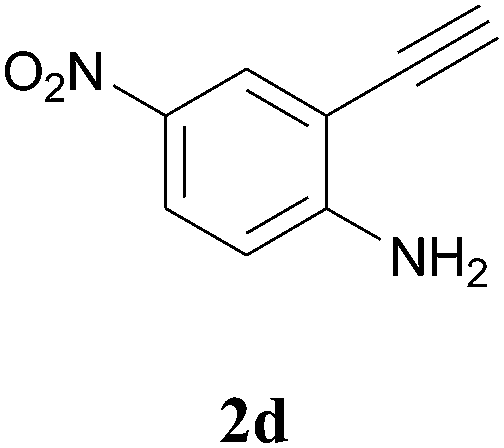


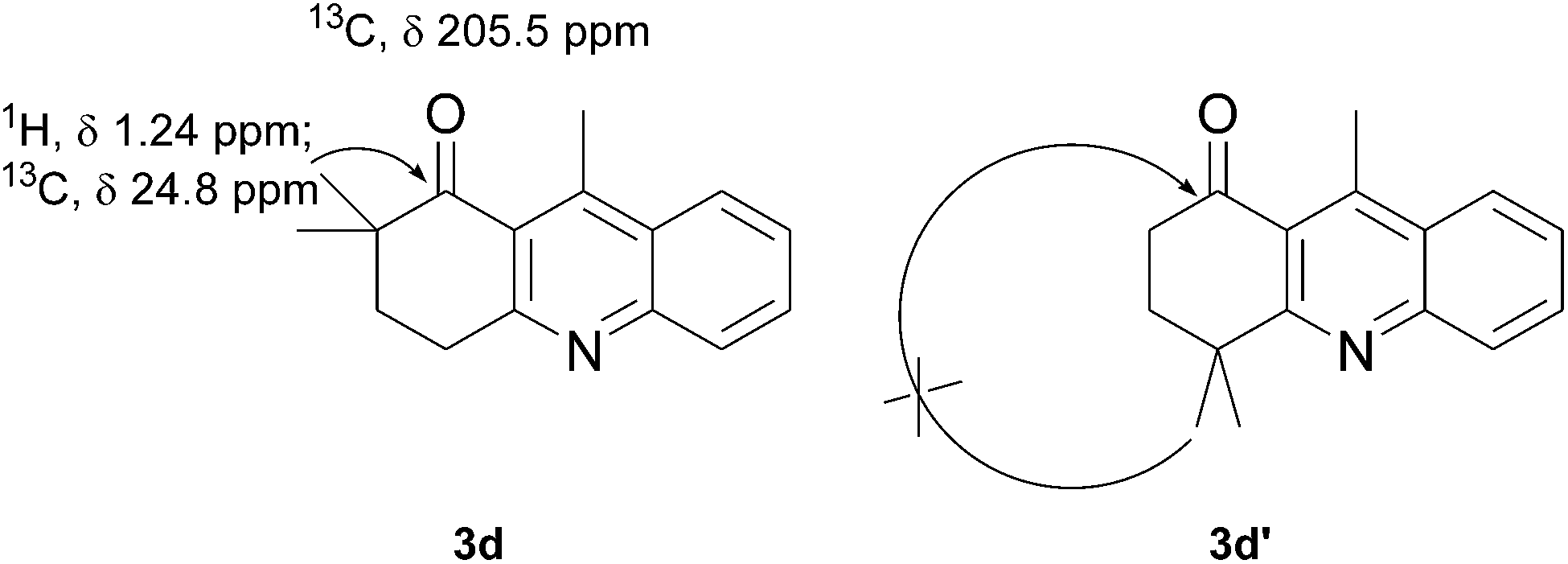 | ||
| Fig. 1 Confirmation of 3d by HMBCs. | ||
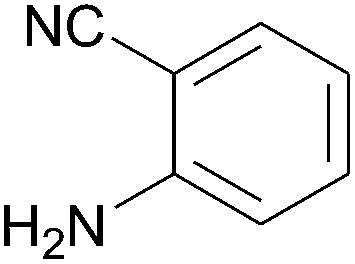
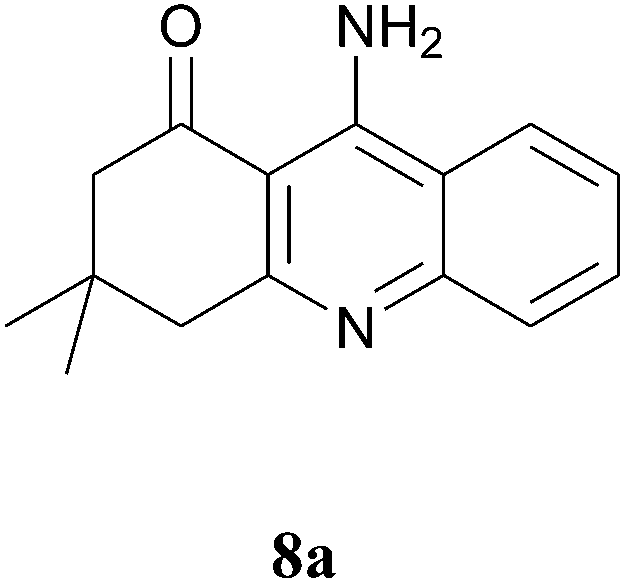
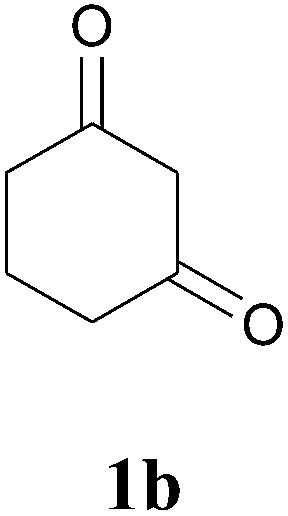
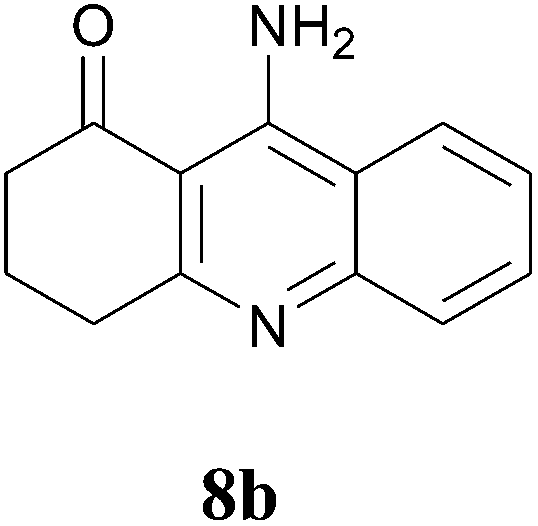
In further attempts to synthesize 4-aminoquinoline derivatives from 1,3-diketone 1 and 2-amino benzonitrile 7, only cyclic 1,3-diketones (1a and 1b) gave the expected product whereas acyclic 1,3-diketone (1g and 1h) failed. When comparing dimedone 1a (Table 3, entry 1) and cyclohexa-1,3-dione 1b (Table 3, entry 2), the former gave comparatively more yield. The acyclic 1,3-diketone reaction proceeded to enamine formation and failed to further cyclize (Table 3, entry 3 and 4).


From the intermediate 4 obtained in Table 1, (entries 1–3) we concluded that the reaction goes through the intermediate 4. Based on the previous report, we proposed a plausible mechanism for this cascade cyclization, in which iron acts as a Lewis acid (Scheme 2).6f,11 Initial activation of carbonyl group of 1,3-diketone 1a and subsequent condensation with 2a leads to the intermediate 4. The triple bond of 4 is activated by the catalyst and 6-exo-dig cyclization with enone carbon C2 to give intermediate II via I. Elimination of a proton and protodemetallation of intermediate II gave intermediate III, which on further aromatisation led to the product 3a.
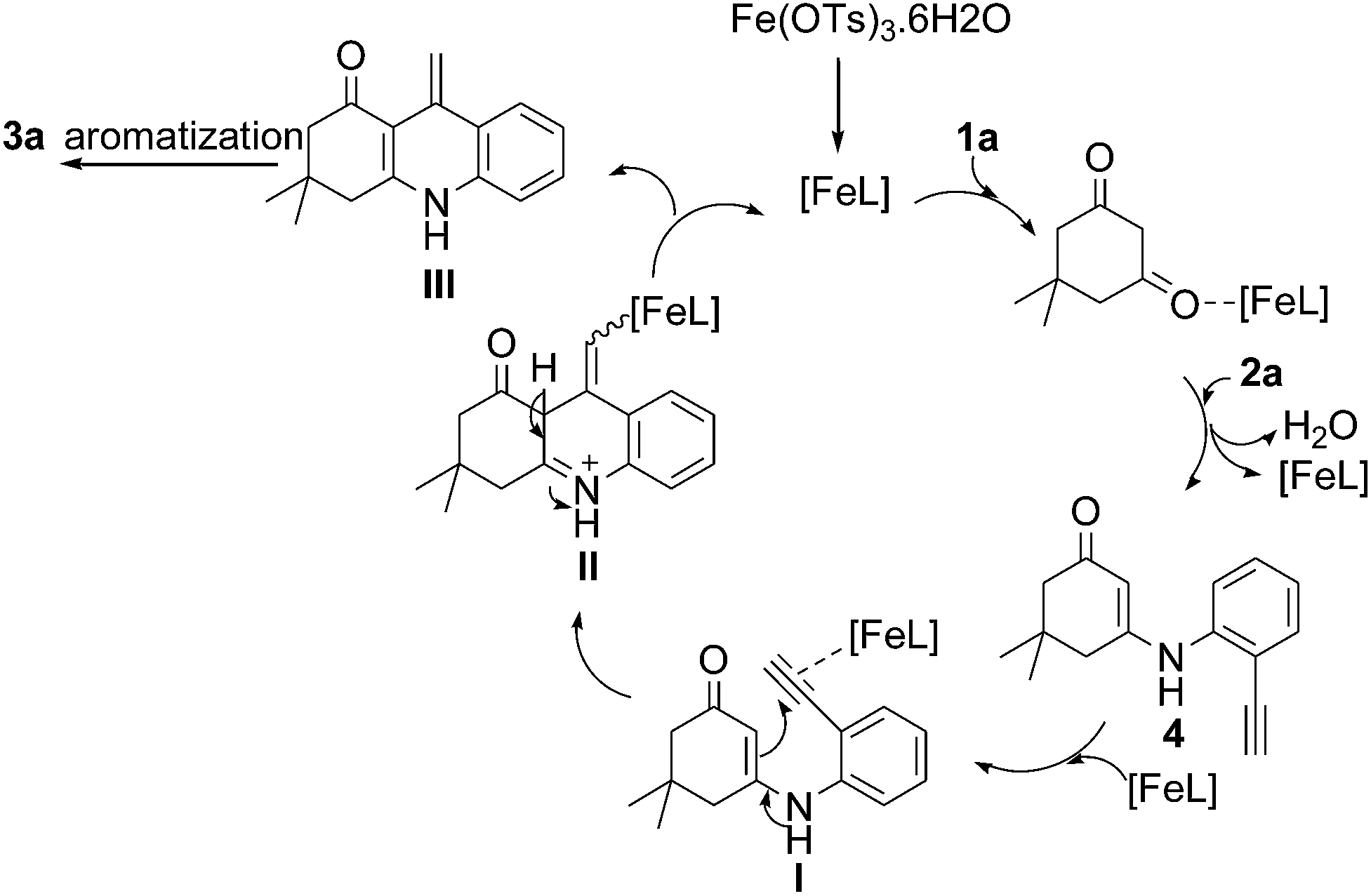 | ||
| Scheme 2 Plausible mechanism for 3a. | ||
The mechanism was further confirmed by control experiments: the reaction of intermediate product 4 under standard conditions (Table 1, entry 11) gave a quantitative yield of 3a (Scheme 3, eqn (1)), which shows that the reaction mechanism goes through the intermediate 4. Also, to check whether the reaction goes through the Lewis acid-catalysed hydration of 2-ethynylaniline and followed (release of water on enamine formation) by the reaction of 1a and 2a under the 0.1 equivalent of catalyst Fe(OTs)3·6H2O at 110 °C of toluene reflux in the presence of 4 Å molecular sieves, we found that it gave the expected product in a similar yield when compared to the Table 1, entry 11 (Scheme 3, eqn (2)). This proves the proposed mechanism, where the initial enamine formation is followed by Fe(III)-catalysed C-cyclization directly onto the alkyne and not through the acid-catalysed hydration of the alkyne (Scheme 3).
 | ||
| Scheme 3 Control experiments for the confirmation of reaction mechanism. | ||
When compared to the report by Zhu et al.,9a our method has the advantages of a shorter reaction time and use of a catalytic amount of biocompatible iron catalyst, whereas in the Zhu et al. report, the reaction required longer time and use of a stoichiometric amount of p-toluenesulfonic acid in the reaction medium. In addition, our report is mechanistically different from the report by Zhu et al.
Conclusions
In conclusion, we have described the synthesis of 4-methyl-2,3-disubstituted quinoline in a cascade manner that features environmentally benign biocompatible and inexpensive iron(III) as a catalyst with only water as the side product, with good to excellent yield in a shorter reaction time using readily available starting materials. In this method, substitutions are possible in the aryl ‘A’ ring of quinoline, which is difficult using the previously reported methods. Further reactions of 2-ethynylanilines and 1,3-diketone are underway in our lab.Experimental section
General methods
Purification of crude compounds was done by column chromatography using silica gel (100–200 mesh, Himedia). FTIR spectra were recorded on a Perkin-Elmer Spectrum Two TM IR spectrometer; absorbances are reported in cm−1. NMR spectra were recorded on a Bruker 400 MHz spectrometer. The chemical shifts are reported in parts per million (ppm) relative to the internal standard TMS (δ = 0 ppm) for 1H NMR (recorded at 400 MHz) and relative to the central CDCl3 resonance (δ = 77.16 ppm) for 13C NMR (recorded at 100 MHz). The coupling constants J are given in Hz. High-resolution mass spectra were recorded on Q-TOF micro mass and JEOL GCMATE II GC-MS spectrometers. All solvents and commercially available chemicals were used as received and 2-alkynylaniline derivatives (2a–f) were prepared according to the literature reports.12General procedure for synthesis of compounds 3a–3l
To a stirred solution of the 1,3-diketone 1 (1.5 equiv.) in toluene, the 2-ethynylaniline 2 (1.0 equiv.) and Fe(OTs)3·6H2O (0.1 equiv.) were added, and the resulting mixture was heated at 110 °C for 2 h. The reaction mixture was diluted with water and extracted with ethyl acetate. The combined organic phases were washed with brine solution and dried over Na2SO4. Removal of the solvent under reduced pressure and purification by column chromatography furnished the desired product. For all new compounds (3d and 3i–3l), spectral data are given below and for the reported compounds (3a–3c, 3e–3h, 8a, and 8b), see ref. 2 and 4.3,4-Dihydro-2,2,9-trimethylacridin-1(2H)-one (3d)
Compound 3d was prepared by general procedure above. After purification by column chromatography (PE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc 6:4) the desired compound was isolated as a brownish orange viscous fluid. IR (Neat) νmax: 475, 757, 797, 861, 960, 1073, 1138, 1115, 1194, 1258, 1385, 1425, 1495, 1563, 1613, 1681, 2864, 2927, 2964, 3070 cm−1; 1H NMR (400 MHz, CDCl3): δ = 8.14 (d, J = 8.4 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 3.26 (t, J = 6.5 Hz, 2H), 2.94 (s, 3H), 2.02 (t, J = 6.5 Hz, 2H), 1.24 (s, 6H); 13C NMR (100 MHz, CDCl3): δ = 205.5, 161.4, 150.1, 147.8, 131.4, 129.0, 127.7, 126.3, 125.4, 125.1, 43.6, 34.9, 30.5, 24.8, 16.2; HRMS (ESI/[M + H]+) calcd for C16H18NO, 240.1388, found 240.1393.
:EtOAc 6:4) the desired compound was isolated as a brownish orange viscous fluid. IR (Neat) νmax: 475, 757, 797, 861, 960, 1073, 1138, 1115, 1194, 1258, 1385, 1425, 1495, 1563, 1613, 1681, 2864, 2927, 2964, 3070 cm−1; 1H NMR (400 MHz, CDCl3): δ = 8.14 (d, J = 8.4 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 3.26 (t, J = 6.5 Hz, 2H), 2.94 (s, 3H), 2.02 (t, J = 6.5 Hz, 2H), 1.24 (s, 6H); 13C NMR (100 MHz, CDCl3): δ = 205.5, 161.4, 150.1, 147.8, 131.4, 129.0, 127.7, 126.3, 125.4, 125.1, 43.6, 34.9, 30.5, 24.8, 16.2; HRMS (ESI/[M + H]+) calcd for C16H18NO, 240.1388, found 240.1393.
3,4-Dihydro-3,3,7,9-tetramethylacridin-1(2H)-one (3i)
Compound 3i was prepared by general procedure above. After purification by column chromatography (PE:EtOAc 6:4) the desired compound was isolated as a yellow viscous liquid. IR (Neat) νmax: 579, 615, 694, 718, 769, 804, 859, 1063, 1102, 1176, 1231, 1287, 1349, 1365, 1447, 1522, 1560, 1595, 1673, 2852, 2924, 3068 cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.76 (s, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.40 (d, J = 8.5 Hz, 1H), 2.97 (s, 2H), 2.85 (s, 3H), 2.46 (s, 2H), 2.37 (s, 3H), 0.94 (s, 6H); 13C NMR (100 MHz, CDCl3): δ = 200.9, 160.2, 149.0, 146.9, 136.2, 133.8, 128.9, 127.7, 124.5, 124.2, 54.9, 48.5, 32.21, 28.4, 22.0, 16.0; HRMS (ESI/[M + H]+) calcd for C17H20NO, 254.1545, found 254.1545.
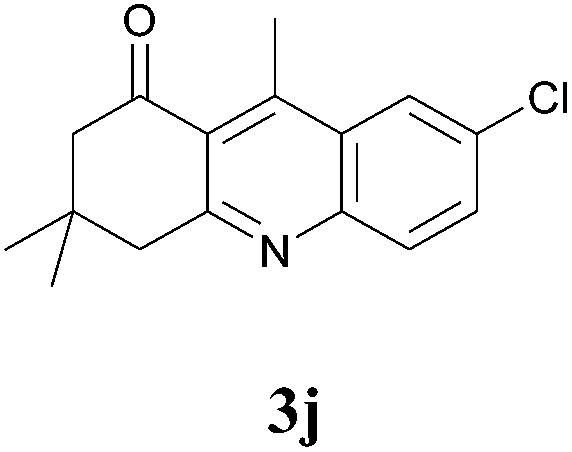
7-Chloro-3,4-dihydro-3,3,9-trimethylacridin-1(2H)-one (3j)
Compound 3j was prepared by general procedure above. After purification by column chromatography (PE:EtOAc 6:4) the desired compound was isolated as a brownish yellow viscous liquid. IR (Neat) νmax: 453, 512, 573, 651, 697, 833, 888, 971, 1090, 1132, 1116, 1214, 1277, 1300, 1337, 1369, 1386, 1468, 1565, 1606, 1682, 2871, 2933, 2956, 3045 cm−1; 1H NMR (400 MHz, CDCl3): δ = 8.14 (s, 1H), 7.91 (d, J = 9.0 Hz, 1H), 7.67 (d, J = 9.0 Hz, 1H), 3.14 (s, 2H), 2.99 (s, 3H), 2.65 (s, 2H), 1.12 (s, 6H); 13C NMR (100 MHz, CDCl3): δ = 200.5, 161.5, 148.7, 146.7, 132.4, 132.3, 130.9, 128.5, 124.8, 124.6, 54.9, 48.5, 32.2, 28.4, 16.1; HRMS (ESI/[M + H]+) calcd for C16H17ClNO, 274.0999, found 274.1002.
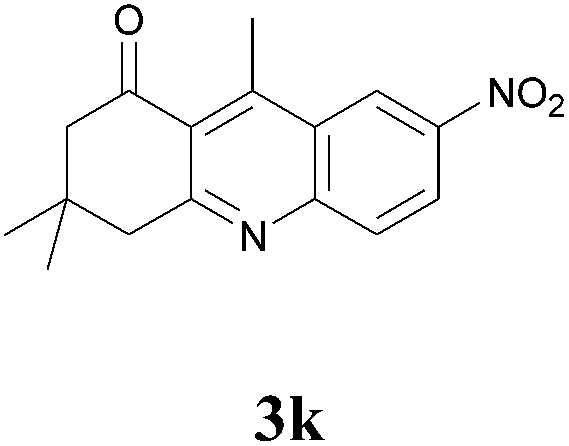
3,4-Dihydro-3,3,9-trimethyl-7-nitroacridin-1(2H)-one (3k)
Compound 3k was prepared by general procedure above. After purification by column chromatography (PE:EtOAc 6:4) the desired compound was isolated as a yellow viscous liquid. IR (Neat) νmax: 435, 495, 580, 646, 691, 741, 856, 895, 912, 971, 1066, 1123, 1214, 1266, 1303, 1371, 1445, 1488, 1521, 1564, 1617, 1687, 2851, 2950, 3106 cm−1; 1H NMR (400 MHz, CDCl3): δ = 9.20 (s, 1H), 8.52 (d, J = 9.2 Hz, 1H), 8.13 (d, J = 9.2 Hz, 1H), 3.23 (s, 2H), 3.15 (s, 3H), 2.72 (s, 2H), 1.16 (s, 6H); 13C NMR (100 MHz, CDCl3): δ = 199.9, 165.2, 151.7, 150.5, 145.5, 131.1, 126.9, 125.5, 124.8, 122.8, 54.7, 48.8, 32.2, 28.4, 16.4; HRMS (ESI/[M + H]+) calcd for C16H17N2O3, 285.1239, found 285.1240.
3,4-Dihydro-5-methoxy-3,3,9-trimethyl-7-nitroacridin-1(2H)-one (3l)
Compound 3l was prepared by general procedure above. After purification by column chromatography (PE:EtOAc 6:4) the desired compound was isolated as a brownish yellow viscous liquid. IR (Neat) νmax: 513, 613, 740, 790, 846, 874, 1058, 1110, 1182, 1247, 1338, 1372, 1395, 1490, 1528, 1564, 1585, 1615, 1688, 2871, 2958, 3107 cm−1; 1H NMR (400 MHz, CDCl3): δ = 8.80 (s, 1H), 7.88 (s, 1H), 4.20 (s, 3H), 3.32 (s, 2H), 3.12 (s, 3H), 2.71 (s, 2H), 1.15 (s, 6H); 13C NMR (100 MHz, CDCl3): δ = 200.1, 164.2, 156.1, 151.7, 145.7, 142.7, 127.6, 126.1, 114.2, 102.9, 57.1, 54.8, 49.0, 32.3, 28.4, 16.7; HRMS (ESI/[M + H]+) calcd for C17H19N2O4, 315.1345, found 315.1345.
Acknowledgements
One of the authors, M.S. thanks the Council of Scientific and Industrial Research, New Delhi, India for the research fellowship.Notes and references
- (a) Y.-L. Chen, K.-C. Fang, J.-Y. Sheu, S.-L. Hsu and C.-C. Tzeng, J. Med. Chem., 2001, 44, 2374 CrossRef CAS PubMed; (b) D. Dubé, M. Blouin, C. Brideau, C.-C. Chan, S. Desmarais, D. Ethier, J.-P. Falgueyret, R. W. Friesen, M. Girard, Y. Girard, J. Guay, D. Riendeau, P. Tagari and R. N. Young, Bioorg. Med. Chem. Lett., 1998, 8, 1255 CrossRef; (c) R. D. Larsen, E. G. Corley, A. O. King, J. D. Carroll, P. Davis, T. R. Verhoeven and P. J. Reider, J. Org. Chem., 1996, 61, 3398 CrossRef CAS; (d) A. A. Joshi and C. L. Viswanathan, Bioorg. Med. Chem. Lett., 2006, 16, 2613 CrossRef CAS PubMed; (e) P. Narender, U. Srinivas, M. Ravinder, B. A. Rao, C. Ramesh, K. Harakishore, B. Gangadasu, U. S. N. Murthy and V. J. Rao, Bioorg. Med. Chem., 2006, 14, 4600 CrossRef CAS PubMed; (f) A. Nayyar, A. Malde, R. Jain and E. Coutinho, Bioorg. Med. Chem., 2006, 14, 847 CrossRef CAS PubMed; (g) X. Franck, A. Fournet, E. Prina, R. Mahieux, R. Hocquemiller and B. Figadère, Bioorg. Med. Chem. Lett., 2004, 14, 3635 CrossRef CAS PubMed; (h) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC; (i) J. P. Michael, Nat. Prod. Rep., 2007, 24, 223 RSC; (j) J. P. Michael, Nat. Prod. Rep., 2005, 22, 627 RSC; (k) J. P. Michael, Nat. Prod. Rep., 2004, 21, 650 RSC; (l) J. P. Michael, Nat. Prod. Rep., 2003, 20, 476 RSC; (m) J. L. McCormick, T. C. McKee, J. H. Cardellina and M. R. Boyd, J. Nat. Prod., 1996, 59, 469 CrossRef CAS PubMed; (n) J. P. Michael, Nat. Prod. Rep., 1999, 16, 697 RSC.
- M. A. Zolfigol, P. Salehi, A. Ghaderi and M. Shiri, Catal. Commun., 2007, 8, 1214 CrossRef CAS PubMed and references therein.
- (a) P. Friedlander, Chem. Ber., 1883, 15, 2572 CrossRef; (b) J. Marco-Contelles, E. Pérez-Mayoral, A. Samadi, M. Carreiras and E. Soriano, Chem. Rev., 2009, 109, 2652 CrossRef CAS PubMed; (c) M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Adv. Heterocycl. Chem., 2011, 102, 139 CrossRef CAS; (d) B. Reddy, A. Venkateswarlu, G. Reddy and Y. Reddy, Tetrahedron Lett., 2013, 54, 5767 CrossRef CAS PubMed; (e) L. Li and D. Seidel, Org. Lett., 2010, 12, 5064 CrossRef CAS PubMed; (f) A. Bañón-Caballero, G. Guillena and C. Nájera, J. Org. Chem., 2013, 78, 5349 CrossRef PubMed; (g) J. Akbari, A. Heydari, H. Kalhor and S. Kohan, J. Comb. Chem., 2010, 12, 137 CrossRef CAS PubMed.
- (a) A. Hasaninejad, A. Zare, M. Shekouhy and J. Ameri-Rad, Green Chem., 2011, 13, 958 RSC; (b) D. S. Bose, M. Idrees, N. M. Jakka and J. V. Rao, J. Comb. Chem., 2010, 12, 100 CrossRef CAS PubMed; (c) X.-L. Zhang, Q.-Y. Wang, S.-R. Sheng, Q. Wang and X.-L. Liu, Synth. Commun., 2009, 39, 3293 CrossRef CAS; (d) G.-W. Wang, C.-S. Jia and Y.-W. Dong, Tetrahedron Lett., 2006, 47, 1059 CrossRef CAS PubMed; (e) Y. Venkateswarlu, S. R. Kumar and P. Leelavathi, Org. Commun., 2012, 5, 120 CAS; (f) E. Rafiee, F. K. Nejad and M. Joshaghani, Chin. Chem. Lett., 2011, 22, 288 CrossRef CAS PubMed; (g) S. Kumar, A. Saini and J. S. Sandhu, Synth. Commun., 2007, 37, 4071 CrossRef CAS; (h) C.-S. Jia, Z. Zhang, S.-J. Tu and G.-W. Wang, Org. Biomol. Chem., 2006, 4, 104 RSC; (i) M. M. Heravi, N. M. Haj, B. Baghernejad, Y. Beheshtia and F. F. Bamoharram, E–J. Chem., 2010, 7, 875 CrossRef CAS PubMed; (j) J. Yang, J.-N. Tan and Y. Gu, Green Chem., 2012, 14, 3304 RSC; (k) L. Fang, J. Yu, Y. Liu, A. Wang and L. Wang, Tetrahedron, 2013, 69, 11004 CrossRef CAS PubMed.
- (a) C. Gronnier, G. Boissonnat and F. Gagosz, Org. Lett., 2013, 15, 4234 CrossRef CAS PubMed; (b) S. Zhu, L. Wu and X. Huang, J. Org. Chem., 2013, 78, 9120 CrossRef CAS PubMed; (c) S. Sarkar, K. Bera, S. Jalal and U. Jana, Eur. J. Org. Chem., 2013, 6055 CrossRef CAS; (d) T. Xu and G. Liu, Org. Lett., 2012, 14, 5416 CrossRef CAS PubMed; (e) M. Zhang, T. Roisnel and P. H. Dixneuf, Adv. Synth. Catal., 2010, 352, 1896 CrossRef CAS; (f) A. Arcadi, M. Aschi, F. Marinelli and M. Verdecchia, Tetrahedron, 2008, 64, 5354 CrossRef CAS PubMed.
- (a) K. Bera, S. Sarkar, S. Jalal and U. Jana, J. Org. Chem., 2012, 77, 8780 CrossRef CAS PubMed; (b) K. Bera, S. Sarkar, S. Biswas, S. Maiti and U. Jana, J. Org. Chem., 2011, 76, 3539 CrossRef CAS PubMed; (c) Z.-Q. Wang, Y. Lei, M.-B. Zhou, G.-X. Chen, R.-J. Song, Y.-X. Xie and J.-H. Li, Org. Lett., 2011, 13, 14 CrossRef CAS PubMed; (d) H. Jiang, W. Yao, H. Cao, H. Huang and D. Cao, J. Org. Chem., 2010, 75, 5347 CrossRef CAS PubMed; (e) R. M. Carballo, M. Purino, M. A. Ramírez, V. S. Martín and J. I. Padrón, Org. Lett., 2010, 12, 5334 CrossRef CAS PubMed; (f) S. Maiti, S. Biswas and U. Jana, J. Org. Chem., 2010, 75, 1674 CrossRef CAS PubMed; (g) P. O. Miranda, R. M. Carballo, V. S. Martín and J. I. Padrón, Org. Lett., 2009, 11, 357 CrossRef CAS PubMed.
- E. Bosch and L. Jeffries, Tetrahedron Lett., 2001, 42, 8141 CrossRef CAS.
- A. Arcadi, M. Alfonsi, G. Bianchi, G. D'Anniballe and F. Marinelli, Adv. Synth. Catal., 2006, 348, 331 CrossRef CAS.
- (a) C. Peng, Y. Wang, L. Liu, H. Wang, J. Zhao and Q. Zhu, Eur. J. Org. Chem., 2010, 818 CrossRef CAS; (b) S. Cacchi, G. Fabrizi and E. Filisti, Org. Lett., 2008, 10, 2629 CrossRef CAS PubMed.
- (a) K. Murai, S. Hayashi, N. Takaichi, Y. Kita and H. Fujioka, J. Org. Chem., 2009, 74, 1418 CrossRef CAS PubMed; (b) A. Arcadi, S. D. Giuseppe, F. Marinelli and E. Rossi, Adv. Synth. Catal., 2001, 343, 443 CrossRef CAS.
- (a) G. C. Senadi, W.-P. Hu, J.-S. Hsiao, J. Kishore Vandavasi, C.-Y. Chen and J.-J. Wang, Org. Lett., 2012, 14, 4478 CrossRef CAS PubMed; (b) H. Hebbache, Z. Hank, S. Boutamine, M. Meklati, C. Bruneau and J.-L. Renaud, C. R. Chim., 2008, 11, 612 CrossRef CAS PubMed; (c) R. M. Carballo, M. A. Ramírez, M. L. Rodríguez, V. S. Martín and J. I. Padrón, Org. Lett., 2006, 8, 3837 CrossRef CAS PubMed; (d) P. O. Miranda, D. D. Díaz, J. I. Padrón, J. Bermejo and V. S. Martín, Org. Lett., 2003, 5, 1979 CrossRef CAS PubMed.
- C. Praveen, S. Jegatheesan and P. T. Perumal, Synlett, 2009, 2795 CAS and references therein.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra for all new compounds. See DOI: 10.1039/c4ra03666b |
| This journal is © The Royal Society of Chemistry 2014 |