Enhanced electrocatalytic activities of pulse-mode potentiostatic electrodeposited single-crystal, fern-like Pt nanorods†
Huei-Yu Choua,
Ming-Chi Tsaia,
Sung-Yen Weib,
Yu-Hsuan Weiad,
Tsung-Kuang Yeha,
Fu-Rong Chena,
Chen-Chi M. Mac,
Chuen-Horng Tsai*a and
Chien-Kuo Hsieh*d
aDepartment of Engineering and System Science, National Tsing Hua University, Hsingchu, 30013, Taiwan, ROC
bDepartment of Advanced Optoelectronic Materials and Devices, Industrial Technology Research Institute, Chutung 31040, Taiwan, ROC
cDepartment of Chemical Engineering, National Tsing Hua University, Hsingchu 30013, Taiwan, ROC
dDepartment of Materials Engineering, Ming Chi University of Technology, New Taipei City 24301, Taiwan, ROC. E-mail: jack_hsieh@mail.mcut.edu.tw
First published on 4th July 2014
Abstract
Single-crystal and fern-like Pt nanorods (Pt-NR) were electrodeposited on carbon paper. The electrocatalytic activity of prepared Pt-NR electrodes, as determined by the catalyst mass activity (MA) and specific activity (SA) for the methanol oxidation reaction, are respectively 4.59 times and 10.54 times better than that of a commercial Pt-black catalyst.
Platinum (Pt) is a very important material for many applications, and syntheses of size- and shape-controlled Pt or Pt-embedded nanostructures are among today's most important research topics. Because of their excellent physical and chemical characteristics, they have been widely used as catalysts in many areas such as fine chemical synthesis, hydrogen production, gas sensors and electrocatalysis.1,2
Among the important applications, Pt is the key catalyst in proton exchange membrane (PEM) fuel cells.3 The core component in such a fuel cell is the membrane electrode assembly (MEA), which generally comprises a thin, flat proton exchange membrane with catalyzed electrodes bonded to both sides. Conventionally, the electrodes are made of electronconducting carbon cloth or carbon paper (CP) plated with a mixture of carbon-black-supported catalyst particles and a proton-conducting ionomer. For a PEM fuel cell to operate efficiently, the catalyst must be both highly dispersed and nanoscale in size so as to optimize the rate of hydrogen or methanol oxidation reaction at the anode. Supporting evidence for these requirements is that the activity of a nanocatalyst is known to correlate strongly with its morphology and crystallinity.4,5
For the electrocatalyst in a fuel cell to exhibit high efficiency, three qualities are required: (1) large specific area, (2) three phase contact (including electronic conductivity through the catalyst support), and (3) high durability during and after MEA assembling. In our previous study,6 we demonstrated that carbon nanotubes (CNT) grown directly on carbon cloth could possess high specific surface area and display uniform catalyst (Pt or its alloy) dispersion, and therefore exhibit high catalytic efficiency. However, damage to (and indeed, collapse of) the catalytic CNT layer is likely to occur during the MEA assembly, suggesting the importance of an alternative assembly method.7
It has been found that Pt has distinct electrocatalytic activity on different crystalline planes,8 suggesting the maximum electrocatalytic activity may be achieved by controlling single crystallinity in fabricating the catalysts. To date, most relevant research has focused on syntheses of Pt nanocrystals with specific crystallinities,9 mainly polycrystalline structure.1,10 For example, tetrahexahedral Pt nanocrystals with high-index facets have been synthesized by electrochemical approaches.9 Very recently, however, some single-crystal Pt structures with unique anisotropic structures, such as one-dimensional nanowires and nanorods, have been reported and have attracted considerable attention because of their attractive surface properties and superior catalytic efficiencies.11–13 For example, Xia and coworkers generated single-crystal Pt nanowires by a polyol process combined with the introduction of a trace amount of ionic iron species with PVP (used as a surfactant).11 Song and coworkers produced Pt nanowires networks by chemical reduction using a soft template.12 Sun and coworkers synthesized Pt nanowires and/or nanoflowers by aging hexachloroplatinic acid in a formic acid aqueous solution for several hours to 3 days under ambient conditions.13,14
A challenge to synthesize single-crystal Pt nanowires or nanorods by surfactant- and template-free approaches under mild conditions still remains. Another key challenge is to increase catalyst dispersion and electronic conductivity between the gas diffusion layer and the catalysts themselves. In this communication, we address both these challenges by proposing a novel pulse-mode potentiostatic electrodeposition (PPE) technique using a simple aqueous electrolyte to directly synthesize single-crystal, fern-like Pt nanorods (Pt-NR) on a CP surface. Then, we examine the electrocatalytic activity of such a system for the oxidation of formic acid (CH2O2), formaldehyde (HCHO), and methanol (CH3OH).
To control the Pt-NR crystal structure during synthesis, we investigated the effect of non-hydrogen-bubble formation during electrodeposition. An applied potential of less than −0.241 VSCE during electrodeposition is known to cause the generation of hydrogen bubbles by the standard hydrogen reduction reaction (SHRR).15 Such hydrogen-bubble formation apparently alters the morphologies of Pt that has been reduced on the CP surface, in our case, causing the reduced Pt to assume a petal-like structure.
We used the PPE technique by applying periodic cycles of high positive potential (1.2 VSCE), followed by the negative potential (−0.24 VSCE), slightly higher than the value known to induce the SHRR. As expected, hydrogen bubbles were not generated during electrodeposition, and Pt-NR was electrodeposited on the CP surface. Obviously, then, as has previously been reported, hydrogen-bubble adsorption on the electrode surface in an aqueous sulphuric electrolyte during deposition can affect the final morphology of the reduced Pt metal.16
Fig. 1(a) shows a typical SEM micrograph of the fern-like Pt-NR so created. Each Pt-NR is approximately 300 nm to 1 μm long. Fig. 1(b) shows a TEM micrograph of the Pt-NR. Their length is approximately 600 nm, consistent with that determined by SEM observation. Fig. 1(c) and (d) show HR-TEM micrographs of the regions indicated by circles in Fig. 1(b). The Pt-NR end and branch both possess a single-crystal structure. The insets in Fig. 1(c) and (d) show respectively the selected-area electron diffraction (SAED) patterns at corresponding region, both these SAED from [001] electron beam zone axis show the typical F.C.C. diffraction, not only the end of stem and branch but also whole Pt-NR show the same diffraction pattern, which indicates that the single crystallinity of the fern-like Pt-NR.
 | ||
| Fig. 1 (a) SEM micrograph of Pt-NR electrodeposited on carbon paper; (b) typical TEM micrograph of prepared Pt-NR; (c) HR-TEM micrograph of the tip region circled in (b) and the related SAED pattern; (d) HR-TEM image of the branch region circled in (b) and the related SAED pattern. | ||
Thus, the PPE technique can be used to synthesize the single-crystal Pt-NR. We herein suggest the associated mechanism is as follows. During the application of −0.24 VSCE, reaction S1 and S2 occur (ESI†). Moreover, during the application of 1.2 VSCE, other reactions may occur, as previously suggested.9 Specifically, reduced Pt may react with H2O to form the adsorption product Pt(O)ad (reaction S4 in ESI†), which in turn, again reacts with H2O to form PtO2 (reaction S5 in ESI†).17–19 During the next application of −0.24 VSCE, these resultants then form pure Pt by reactions (S1), (S2), and (S6)–(S8) in ESI.† In other words, during PPE, the nucleation/growth of Pt atoms and subsequent structural modifications on the Pt surface occur continuously. It has been shown that the PPE technique without the influence of hydrogen bubbles enables the synthesis of fern-like single crystals in an aqueous solution containing sulphuric acid and Pt precursor salt.
To determine the catalytic efficiency of the as-prepared Pt-NR catalyst and compare it with that of a commercial Pt-black catalyst for the reaction of methanol oxidation, we performed cyclic voltammetry (CV) measurements in an Ar-saturated 0.5 M H2SO4 + (a) 1 M CH3OH, (b) 0.1 M CH2O2, and (c) 0.1 M HCHO in aqueous solution, respectively.
Fig. 2(a) shows the voltammograms from fifth cycle for each catalyst. Table 1 lists the following electrochemical characteristics derived for each catalyst: forward peak potential (VP), forward peak current density (if), ratio of if to reverse peak density (if/ib), and mass activity (MA, the peak current density of methanol oxidation obtained from cyclic voltammogram per unit of Pt loading mass). Compared with Pt black, Pt-NR exhibits a distinctly higher if value for the methanol oxidation reaction. The peaks that appeared during the reverse scans signify the desorption of intermediate products generated by methanol oxidation during the forward scans. It has been suggested that these intermediates might be associated with the adsorption of CO (an intermediate during methanol oxidation) on the Pt surfaces, a process that has been termed “CO poisoning”.4,20 Consequently, the if/ib value can be considered to represent the CO tolerance of the catalysts. From Table 1, for Pt-NR, we see that the if/ib value is significantly better (23%) than that for Pt black, suggesting more efficient CO desorption on Pt-NR. From Fig. 2(a) and Table 1, we see that the VP value of Pt-NR (0.63 VSCE) is lower than that of Pt black (0.64 VSCE), indicating that Pt-NR exhibits superior electrocatalytic activity. In addition, at a given potential of 0.5 VSCE, iP of Pt-NR (76.1 mA cm−2) is 3.59 times better than that of Pt black (21.2 mA cm−2) of Pt black catalysts. Similarly, at a given current density of 40 mA cm−2, the potential of Pt-NR (0.45 VSCE) is 120 mV lower than that of Pt black (0.57 VSCE), resulting in lower overpotential during methanol oxidation.
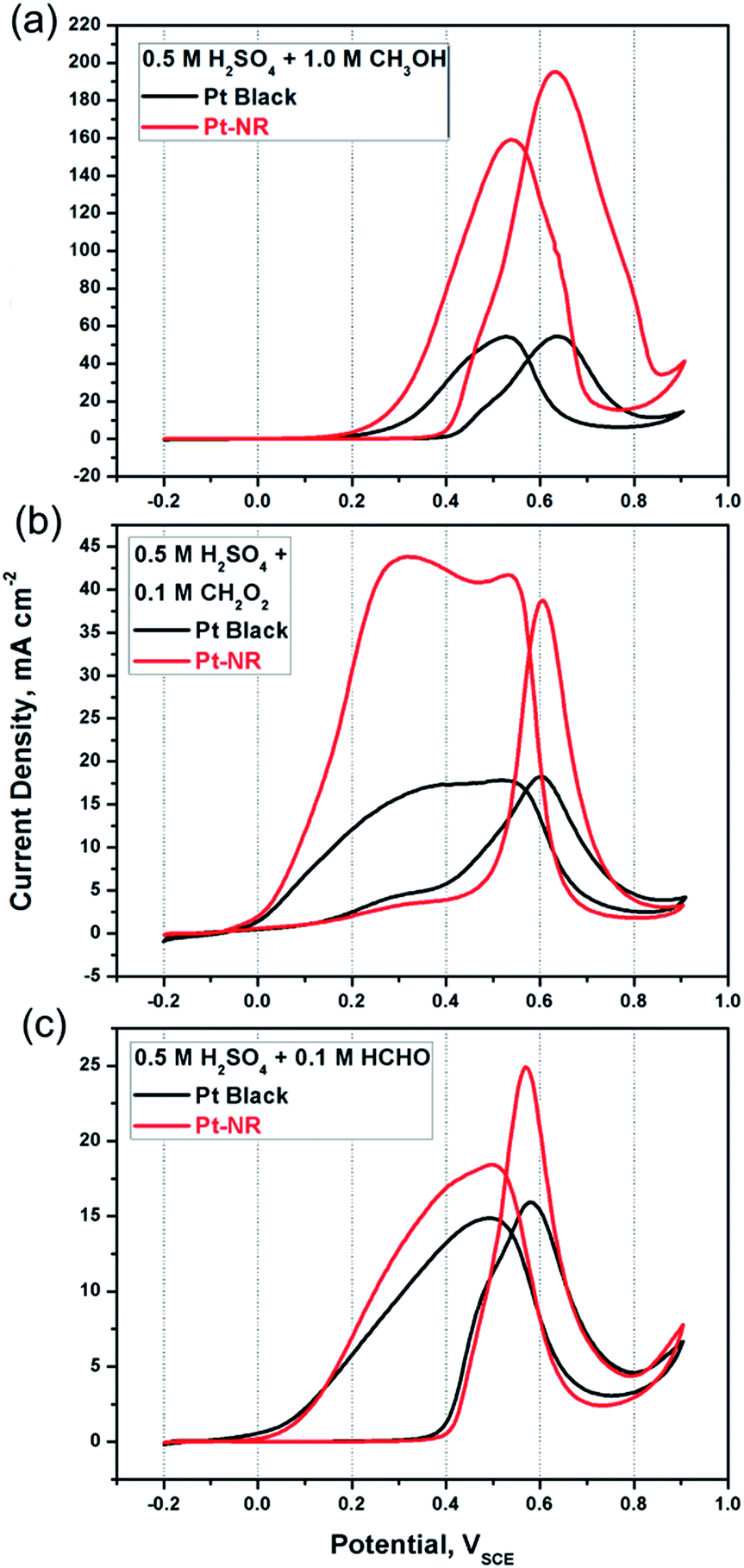 | ||
| Fig. 2 Cyclic voltammograms of the Pt-NR catalyst (red curve) and Pt-black catalyst (black curve) tested in Ar-saturated 0.5 M H2SO4 for the oxidation reactions of (a) 1 M CH3OH, (b) 0.1 M CH2O2, and (c) 0.1 M HCHO in aqueous solutions. The scan rate was 20 mV s−1. The test temperature was controlled at 30 °C. | ||
| Specimen | Loadingsa (mg cm−2) | VPb (VSCE) | ifb (mA cm−2) | if/ibb | MAc (mA mgPt−1) | SAd (mA cmPt−2) |
|---|---|---|---|---|---|---|
| a ICP-MS analysis of Pt catalyst prepared sample.b Forward peak current density obtained from the voltammograms (Fig. 2(a)) per unit Pt loading mass.c The mass and specific activities are obtained by the normalizing the peak current (if) to the Pt loadings and electrochemical surface active area (ECSA), respectively (ESI†).d The mass and specific activities are obtained by the normalizing the peak current (if) to the Pt loadings and electrochemical surface active area (ECSA), respectively (ESI†). | ||||||
| Pt-NR | 0.55 | 0.63 | 195 | 1.23 | 354.5 | 9.036 |
| Pt black | 0.65 | 0.64 | 54 | 1 | 77.1 | 0.857 |
Thus, compared with Pt black, Pt-NR exhibits not only a higher if/ib value, but also a higher if value, indicating its considerably superior catalytic efficiency for methanol oxidation reaction. To verify this superior catalytic efficiency, we also examined the performance of both catalysts for CH2O2 and HCHO oxidation. From Fig. 2(b) for CH2O2, we see the following: (1) at 0.2 VSCE, the oxidation reaction begins, resulting in the decomposition of CH2O2 to CHOO or COOH.4 (2) At 0.6 VSCE, forward peaks appear, caused by the formation of CO2 with if of 40 mA cm−2 on the Pt surface. (3) During the reverse scan from 0.65 to near 0 VSCE, peaks appear, caused by the desorption of CO2 gas from the Pt surfaces (desorption bubbles can be clear seen during the experiments). The if value of Pt-NR is higher than that of Pt black, suggesting a higher rate of catalytic oxidation of formic acid and intermediates.21,22 Similar results have been found that Pt-NR again has a higher if value (decomposition of HCHO forms HCH or HCO and further formation of CO on Pt surfaces at about 0.58 VSCE) and both the reverse peak current densities are caused by CO desorption from the Pt surfaces. For Pt black, a shoulder appears at 0.5 VSCE during the forward scan, caused perhaps by indirect HCHO decomposition and CO formation, which may be a rate-determining-step for methanol oxidation. In contrast, for Pt-NR, no such reaction occurs, suggesting that single-crystal Pt-NR exhibits better electrocatalytic activity for methanol oxidation than a commercial Pt black.
Fig. 3 shows the electrochemical surface area (ECSA) measurements of both Pt-NR and Pt black in 0.5 M H2SO4 at 30 °C. As shown in Fig. 3, the significantly strong peak appeared at −0.15 VSCE indicated that our synthesized Pt-NR showed ECSA features characteristic of Pt(110) surfaces rich (ESI†).23,24 A lower poisoning effect of CO(ad) will rarely occur on the Pt(110) due to the Pt catalyst surface atomic configuration (ESI†).25 By comparison, the current density of Pt black without peak probably because the carbon may wrap around Pt to reduce the effective surface planes.26 Due to the high surface area carbon used as the support for Pt black, a wider double layer (D.L.) region can be seem of Pt black in Fig. 3. The electrochemical characteristics of specific activity (SA) derived from the Had area of ECSA listed in Table 1, are 9.036 mA cm−2 and 0.857 mA cm−2 for Pt-NR and Pt black, respectively. The catalyst specific activity (SA) of Pt-NR is 10.54 times better than that of a commercial Pt black. We herein suggest the Pt(110) rich of Pt-NR enhanced the electrocatalytic activity.
 | ||
| Fig. 3 ECSA of cyclic voltammograms of the Pt-NR catalyst (red curve) and Pt black catalyst (black curve) tested in Ar-saturated 0.5 M H2SO4. The scan rate was 20 mV s−1. The test temperature was controlled at 30 °C. | ||
To investigate the effect of Pt loading on the efficiency of methanol oxidation, we calculated the associated MA and SA values (also listed in Table 1). For Pt-NR, which has less loading mass, the MA value is 4.59 times higher (354.5 mA mg−1) than that for Pt black (77.1 mA mg−1), the SA value is 10.54 times higher (9.036 mA cm−2) than that for Pt black (0.857 mA cm−2), indicating that our prepared single-crystal, fern-like Pt-NR exhibits superior electrocatalytic activity for methanol oxidation. These results suggest the benefit of the PPE technique for the preparation of single-crystal Pt catalysts.
Conclusions
We synthesized single-crystal, fern-like Pt nanorods on the surface of carbon paper by a pulse-mode potentiostatic electrodeposition technique without use of either surfactant or template. Compared with a commercial Pt-black catalyst, the prepared Pt nanorods exhibit definitely improved electrocatalytic activity (4.59 times better mass activity, and 10.54 times better specific activity) for methanol oxidation. More detailed electrochemical and physical characterizations of the prepared nanorods are in progress, with the purpose of optimizing their use as electrodes in PEM-based fuel cells and other electrochemical applications.Acknowledgements
This study was supported financially by the National Science Council of Taiwan under grant NSC 100-2623-E-007-010-ET.Notes and references
- Z. Y. Zhou, N. Tian, Z. Z. Huang, D. J. Chen and S. G. Sun, Faraday Discuss., 2008, 140, 81–92 RSC.
- C. K. Hsieh, M. C. Tsai, C. Y. Su, S. Y. Wei, M. Y. Yen, C. C. M. Ma, F. R. Chen and C. H. Tsai, Chem. Commun., 2011, 47, 11528–11530 RSC.
- C. Koenigsmann and S. S. Wong, Energy Environ. Sci., 2011, 4, 1161–1176 CAS.
- P. Ferrin and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 14381–14389 CrossRef CAS PubMed.
- K. Yamamoto, T. Imaoka, W. J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397–402 CrossRef CAS PubMed.
- M. C. Tsai, T. K. Yeh and C. H. Tsai, Int. J. Hydrogen Energy, 2011, 36, 8261–8266 CrossRef CAS PubMed.
- M. C. Tsai, T. K. Yeh, C. Y. Chen and C. H. Tsai, Electrochem. Commun., 2007, 9, 2299–2303 CrossRef CAS PubMed.
- E. Herrero, K. Franaszczuk and A. Wieckowski, J. Phys. Chem., 1994, 98, 5074–5083 CrossRef CAS.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- Y. J. Song, S. B. Han and K. W. Park, Mater. Lett., 2010, 64, 1981–1984 CrossRef CAS PubMed.
- J. Y. Chen, T. Herricks, M. Geissler and Y. N. Xia, J. Am. Chem. Soc., 2004, 126, 10854–10855 CrossRef CAS PubMed.
- Y. Song, R. M. Garcia, R. M. Dorin, H. R. Wang, Y. Qiu, E. N. Coker, W. A. Steen, J. E. Miller and J. A. Shelnutt, Nano Lett., 2007, 7, 3650–3655 CrossRef CAS PubMed.
- S. Sun, D. Yang, G. Zhang, E. Sacher and J. P. Dodelet, Chem. Mater., 2007, 19, 6376–6378 CrossRef CAS.
- S. H. Sun, G. X. Zhang, D. S. Geng, Y. G. Chen, M. N. Banis, R. Y. Li, M. Cai and X. L. Sun, Chem.–Eur. J., 2010, 16, 829–835 CrossRef CAS PubMed.
- D. Dobos, Electrochemical Data: A Handbook for Electrochemists in Industry and Universities, Elsevier, 1975 Search PubMed.
- M. C. Tsai, T. K. Yeh, Z. Y. Juang and C. H. Tsai, Carbon, 2007, 45, 383–389 CrossRef CAS PubMed.
- V. I. Birss, M. Chang and J. Segal, J. Electroanal. Chem., 1993, 355, 181–191 CrossRef CAS.
- G. Jerkiewicz, G. Vatankhah, J. Lessard, M. P. Soriaga and Y. S. Park, Electrochim. Acta, 2004, 49, 1451–1459 CAS.
- K. A. Friedrich, K. P. Geyzers, A. J. Dickinson and U. Stimming, J. Electroanal. Chem., 2002, 524, 261–272 CrossRef.
- H. S. Liu, C. J. Song, L. Zhang, J. J. Zhang, H. J. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95–110 CrossRef CAS PubMed.
- B. E. Conway, J. Electroanal. Chem., 2002, 524, 4–19 CrossRef.
- G. Q. Lu, A. Crown and A. Wieckowski, J. Phys. Chem. B, 1999, 103, 9700–9711 CrossRef CAS.
- N. Furuya and S. Koide, Surf. Sci., 1989, 220, 18–28 CrossRef CAS.
- J. Solla-Gullon, F. J. Vidal-Iglesias, A. Lopez-Cudero, E. Garnier, J. M. Feliu and A. Aldaza, Phys. Chem. Chem. Phys., 2008, 10, 3689–3698 RSC.
- M. Hepel, I. Dela, T. Hepel, J. Luo and C. J. Zhong, Electrochim. Acta, 2007, 52, 5529–5547 CrossRef CAS PubMed.
- J. Xie, X. G. Yang, B. H. Han, Y. Shao-Horn and D. W. Wang, ACS Nano, 2013, 7, 6337–6345 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary reactions. See DOI: 10.1039/c4ra04153d |
| This journal is © The Royal Society of Chemistry 2014 |