
Fabrication of highly-stable Ag/CA@GTA hydrogel beads and their catalytic application†
Shuxian Huang,
Zuoyi Xiao*,
Shangru Zhai,
Bin Zhai,
Feng Zhang and
Qingda An*
Dalian Polytechnic University, Faculty of Light Industry and Chemical Engineering, No. 1 Qing Gongyuan, Gan Jingzi District, Dalian City, Liaoning Province, China. E-mail: zyxiao@dlpu.edu.cn; anqingda@dlpu.edu.cn; Tel: +86-411-86324887
First published on 6th November 2014
Abstract
To enhance the stability of a catalyst that uses alginate as a carrier, Ag nanoparticle-loaded glutaraldehyde-crosslinked calcium alginate (Ag/CA@GTA) hydrogel beads were prepared. The Ag/CA@GTA hydrogel beads were characterized by UV-visible spectroscopy, X-ray diffraction, Fourier-transform infrared spectroscopy, transmission electron microscopy and X-ray photoelectron spectroscopy analyses. Given that the mechanical strength of the support was remarkably enhanced by crosslinking alginate with GTA, the resulting Ag/CA@GTA beads exhibited high stability and durable activity, with more than 97% conversion within 10 successive cycles, for the catalytic reduction of 4-nitrophenol to 4-aminophenol by NaBH4 in aqueous solution. These Ag/CA@GTA hydrogel beads were expected to have potential as a highly efficient, cost-effective and eco-friendly heterogeneous catalyst for industrial applications.
Introduction
Noble metal nanoparticles (NPs) are receiving considerable attention because of their unique chemical properties and significant potential applications in sensors, optical devices, electronics, catalysts, and biology.1–4 When metal NPs are directly used as the catalyst in chemical synthesis reactions, their high surface energy usually causes serious aggregation problems and results in remarkably decreased intrinsic catalytic activity.5–7 To avoid the aggregation of metal NPs, considerable efforts have been devoted to immobilizing NPs onto different supports, including the use of solid beads, hollow spheres, as well as inorganic mesoporous materials and hydrogels with a three-dimensional (3D) network structure.8–13 Among them, grafting or growing metal NPs on spheres usually results in inhomogeneous distribution, low density, and poor stability.14,15 However, the advantages are enhanced stability and controllable particle sizes that aid in embedding metal NPs into supports with interconnected pores.16Polysaccharide hydrogels are attracting considerable interest from bioengineering and nanoengineering perspectives. Given their non-toxicity and biocompatibility, natural hydrogels are widely applied in the fields of bioscience and biotechnology. As a hydrogel, alginate acid is a linear polysaccharide containing 1,4-linked-β-D-mannuronic acid (M) and α-L-guluronic acid (G) in an irregular orderthat is produced from brown algae.17 After adding a divalent cation such as Ca2+, Cu2+, Fe2+, Zn2+ and Ba2+, sodium alginate can easily transform into a stable hydrogel, forming an egg-box-like structure.18 Hence, alginate hydrogel is usually used as a support for the immobilization of cells, enzymes, and biological medicines. Alginate hydrogel is stable in most organic solvents and possesses a large amount of functional groups. These properties allow alginate hydrogel to act as a scaffold for loading metal NPs, making it suitable for catalytic applications. Several groups have reported the fabrication of NP-embedded alginate hydrogels for various applications. Otari et al. reported the synthesis of Ag–Alg biohydrogels using alginate-immobilized microorganisms at ambient condition. This material exhibited efficient heterogeneous catalytic activity in the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP).4 Chtchigrovsky et al. described the preparation and characterization of a library of alginate-supported palladium NPs, which was used as a catalyst for Suzuki coupling reaction.19 Ai et al. successfully fabricated silver NPs in situ grown on magnetically separable alginate-based biohydrogels by an environment-friendly light-driven approach.20 Based on the aforementioned work, the preparation procedure of these materials is quite simple, and the resulting catalysts display high catalytic efficiency toward the corresponding reactions. However, the circulation usability, which is important for industrial application, is unsatisfactory.
Divalent cation-solidified alginate hydrogels are known to able to exchange with Na+ upon addition to a basic solution.21 The “egg-box” structure is easily damaged and alginate hydrogel redissolves in the solution because of the ion-exchange effect. To solve this problem, different chemicals including methanal, glutaraldehyde (GTA), and epichlorohydrin have been chosen as crosslinking agents that could enhance the mechanical strength of alginate hydrogel. Among them, GTA is widely used to crosslink alginate to improve its chemical and mechanical properties. For example, Liu et al. prepared composite TiO2-impregnated GTA-crosslinked alginate beads and used them for the photoreduction of Cr(VI) in acid aqueous solution.22 Chen et al. reported that GTA-crosslinked humic acid-immobilized sodium alginate is an effective absorbent for the removal of Cu(II) ions from wastewater.23 However, no study has explored GTA-crosslinked alginate beads and their catalytic activity for use as catalyst support.
In this work, Ag/CA@GTA hydrogel beads with controllable size and dosage of Ag NPs were successfully prepared. Detailed characterizations of the as-prepared material were performed. Analysis results of the catalytic reduction of 4-NP to 4-AP revealed that Ag/CA@GTA hydrogel beads exhibited remarkable and reproducible activities such that they could be recycled for 10 successive reaction cycles with >97% conversion rate. Moreover, different experimental parameters on catalytic performance were also studied.
Experimental
Chemicals
Sodium alginate and CaCl2 were purchased from Sinopharm Chemical Reagent Corporation, China. Ethanol was purchased from Tianjin Kermel Chemical Reagent Factory, China. GTA, 4-NP and NaBH4 were purchased from Aladdin Chemistry Corporation, China. AgNO3 was obtained from Tianjin Chemical Corporation, China. All chemicals were analytical grade and used as received without any further purification.Preparation of Ag/CA hydrogel beads
Sodium alginate (1.2 g) was dissolved in 56 mL deionized water and stirred until evenly dispersed. Different amounts of AgNO3 (1, 3, 5 and 10 wt% of AgNO3 based on the total mass of sodium alginate) were dissolved in 10 mL of deionized water, and then added into the solution containing sodium alginate. 8 mL of NaBH4 with the concentration of 2.3 × 10−4 M was slowly added into the above solution until Ag+ ions were reduced totally. Ag/CA hydrogel beads with a mean diameter of 4 mm were obtained by drop-by-drop addition of the prepared sodium alginate into 10 wt% CaCl2 aqueous solution under continual magnetic stirring. The obtained beads were immersed in the CaCl2 solution for another 6 h for complete gelling. The hydrogel beads were filtered and washed with deionized water to remove extra Cl− and Ca2+. Finally, these Ag/CA hydrogel beads were stored in deionized water for further use.Preparation of GTA Ag/CA@GTA hydrogel beads
A acetone solution was achieved by mixing water and acetone (Vwater:Vacetone = 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The mixtures with different volume ratio of GTA to the acetone solution (0.1, 0.5, 1, 2%) were prepared. Hydrochloric acid was added into the system until pH value was 5. The as-prepared Ag/CA hydrogel beads were crosslinked with the GTA solution at 30 °C under magnetic stirring for 3 h. Finally, the beads were filtered and repeatedly washed with ethanol and water for further use. The preparation procedure was presented in Scheme 1.
:1). The mixtures with different volume ratio of GTA to the acetone solution (0.1, 0.5, 1, 2%) were prepared. Hydrochloric acid was added into the system until pH value was 5. The as-prepared Ag/CA hydrogel beads were crosslinked with the GTA solution at 30 °C under magnetic stirring for 3 h. Finally, the beads were filtered and repeatedly washed with ethanol and water for further use. The preparation procedure was presented in Scheme 1.
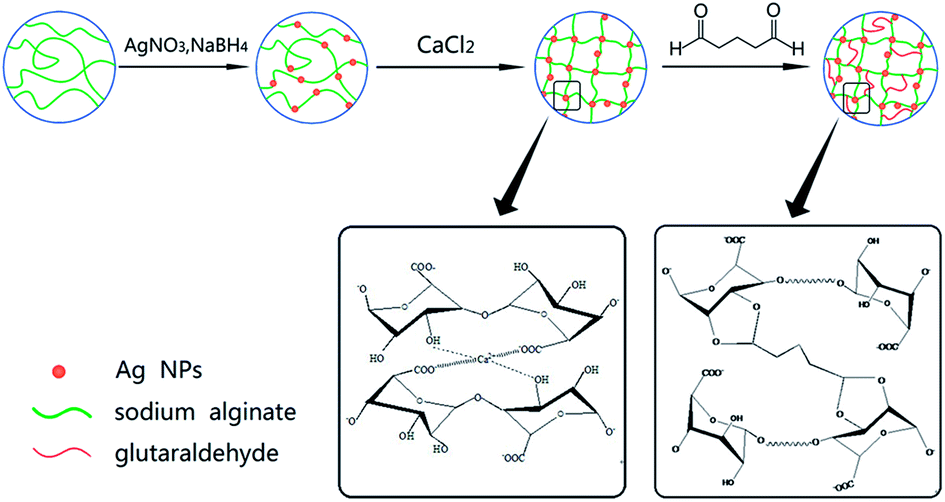 | ||
| Scheme 1 The procedure for preparation of the Ag/CA@GTA hydrogel beads. | ||
Catalytic reaction of 4-NP to 4-AP in the presence of Ag/CA and Ag/CA@GTA hydrogel beads
The catalytic reaction was carried out in a well-stoppered three flask under the condition of Ar bubbling using Ag/CA and Ag/CA@GTA hydrogel beads as catalyst, respectively. 0.04 g of hydrogel beads were added to 0.4 mL of aqueous 4-NP solution (1.0 × 10−4 M). Subsequently, the above suspension was immediately mixed with 0.64 mL of fresh NaBH4 solution (0.1 M) and 19 mL of deionized water. As the catalytic reaction proceeded, the bright yellow color of the solution faded gradually. The catalytic activity was monitored by a UV-vis spectrophotometer at a time interval of 3 min. After the reaction was completed, the catalysts were separated from the mixture with filtration and washed with ethanol, and then reused in the next cycle. The recycling experiments for the Ag/CA and Ag/CA@GTA hydrogel beads were performed for 5 and 10 times, respectively.Characterizations
Absorption spectra were recorded in a MAPADA UV-1600PC spectrometer equipped with a 1 cm quartz cuvette. FT-IR spectra were recorded on one-B FT-IR spectrometer over KBr pressed pellets. The morphologies of the as-prepared samples with Ag NPs were observed by JEM-2100 TEM (JEOL, Japan). XRD patterns were conducted by Shimadzu XRD-6100 diffractometer using Cu Kα radiation (λ = 1.54060 Å) for a 2θ range of 10° to 80° at a scanning speed of 8° per min. XPS measurements were performed by applying Thermo Scientific ESCALAB250 spectrometer (Thermo VG, USA) with monochromatic AI Kα radiation (1486.6 eV).Results and discussion
Synthesis and characterization of Ag/CA@GTA hydrogel beads
Ag NPs were prepared through a simple and low-energy pathway using natural biopolymer alginate as stabilizer and complexing agent at room temperature. UV-visible (UV-vis) DRS was performed to characterize the formation of Ag NPs in sodium alginate solutions prepared with different amounts of AgNO3. As shown in Fig. 1a, without adding Ag+ precursor, no distinct absorbance peak is observed within the studied range. Upon addition of Ag+ ions, an absorption band at 405 nm is clearly observed, which indicates the presence of Ag NPs. The intensity of the peaks increases with increased AgNO3 concentration, which indicates complete reduction of the introduced Ag+ ions in the sodium alginate solution. However, the resulting samples exhibit various adsorption characteristics within the 250–800 nm range, which can be attributed to the change in size of Ag NPs in the products. Thus, the size of Ag NPs depends on the amount of AgNO3 added during the preparation process. As shown in Fig. 1a, the characteristic band widens with increased concentration of AgNO3. The wide peak indicates the presence of larger Ag NPs. Among all samples, those prepared with 1 wt% and 3 wt% AgNO3 obviously show narrow peaks, indicating that the Ag NPs in these two samples are relatively uniform.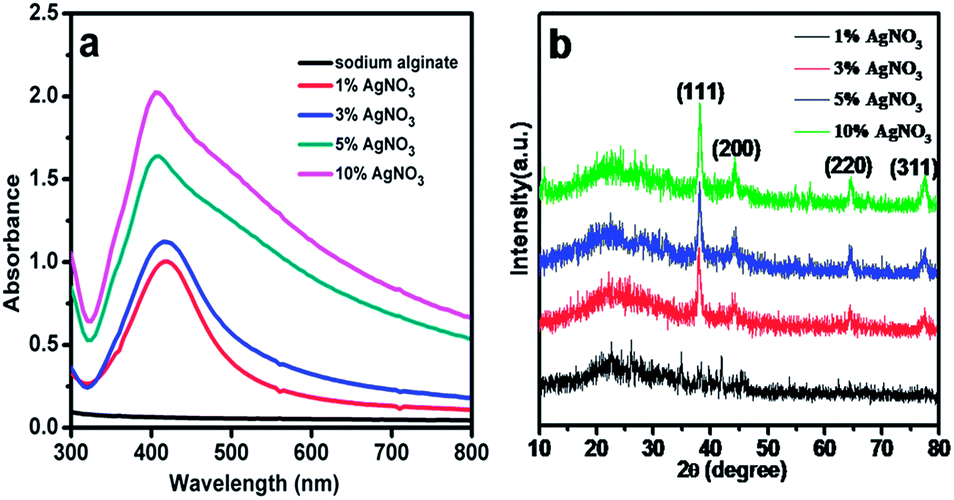 | ||
| Fig. 1 (a) UV-vis absorption spectra of Ag NPs in sodium alginate with different AgNO3 concentration and (b) XRD patterns of Ag/CA@GTA hydrogel beads with different AgNO3 concentration. | ||
The crystallinity of the NPs in Ag/CA@GTA with different amounts of Ag NPs hydrogel beads were characterized by XRD. The corresponding XRD pattern is shown in Fig. 1b. The broad peak centered at 2θ = 25.5° is assigned to the characteristic peak of alginate, indicating its amorphous nature. The well-defined characteristic diffraction peaks at 38.2°, 44.2°, 64.5° and 77.4° correspond to the (111), (200), (220) and (311) planes of the face-centered cubic crystal structure of Ag NPs, respectively.24 The intensity of the characteristic peaks is enhanced with increased the AgNO3 content during the synthesis. It indicates that the amount and size of the Ag NPs in Ag/CA@GTA hydrogel beads are increased. Moreover, the narrow peaks in XRD patterns reveal that the Ag NPs in the prepared samples have a good crystal structure.
TEM analysis provides more information about the size and shape of Ag NPs immobilized in the beads. Fig. 2 shows the TEM images of the Ag NPs in Ag/CA@GTA hydrogel beads at different AgNO3 concentrations. As shown in Fig. 2a, the Ag NPs are well dispersed and spherical, with sizes ranging from 5 nm to 20 nm. With increased AgNO3 weight ratio from 1% to 3%, the Ag NPs in the beads have similar size distributions, except that the dosage of Ag NPs is higher. With more AgNO3 added to the synthetic system, the Ag NPs in the beads further enlarged possibly because of the few complexing positions in the alginate. With increased AgNO3 content to 10%, much more and larger Ag NPs are observed, suggesting that Ag NP aggregation took place during the preparation process. This observation may be due partly to the fact that the sodium alginate solution is insufficiently dense to prevent Ag NP aggregation.25 Analysis results of TEM images are consistent with UV-vis DRS data. What's more, Fig. S1† shows the morphology of the as-prepared Ag/CA@GTA hydrogel beads. A large number of macropores exist on the surface of the beads, which demonstrates that the alginate hydrogel beads are porous and interconnected.
 | ||
| Fig. 2 TEM images of Ag/CA@GTA hydrogel beads obtained with various AgNO3 concentrations: (a) 1%, (b) 3%, (c) 5% and (d) 10%. | ||
Fourier-transform infrared spectroscopy (FT-IR) is widely used for structural investigation. Fig. 3a shows the FT-IR spectra of Ag/CA and Ag/CA@GTA hydrogel beads. By comparison, the FT-IR spectrum of Ag/CA@GTA hydrogel beads exhibits strong and narrow bands at around 1021, 1094 and 1265 cm−1. The weak absorption peak at 2927 cm−1 is associated with the stretching vibration of C–H groups in the six-member ring, which shows a little change. The intensity of the peaks at 1094, 1021 cm−1 and 1265 cm−1, which are attributed to the stretching vibrations of C–O–C and C–O groups, are also improved with increased the amount of C–O–C and C–O groups, suggesting that crosslinking between alginate and GTA occurs.26 The aforementioned results confirm that Ca2+ ions in the “egg-box” structure are substituted by GTA and that crosslinking between GTA and alginate occurs.
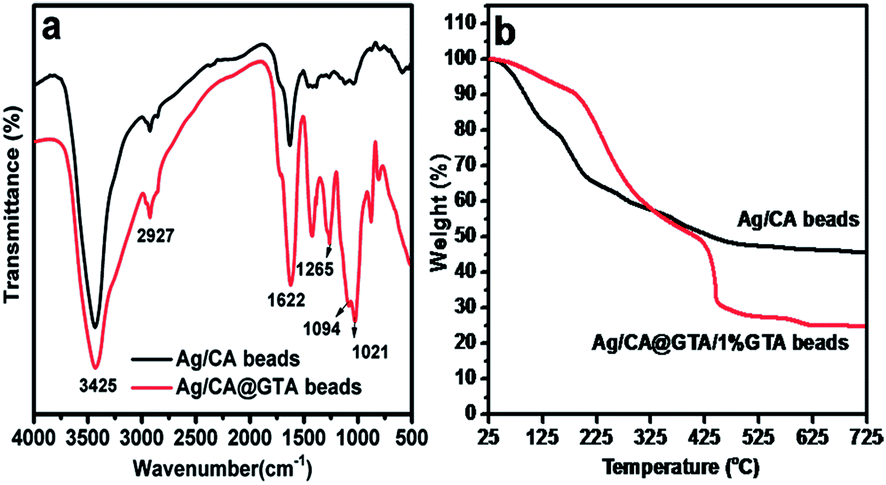 | ||
| Fig. 3 (a) FT-IR spectra and (b) TGA curves of Ag/CA hydrogel beads and Ag/CA@GTA hydrogel beads. | ||
To support the FT-IR results, TGA measurements of the Ag/CA and Ag/CA@GTA hydrogel beads in air were carried out (Fig. 3b). The products remaining after heating at 600 °C in air are mainly from CaO. The remaining weights are reduced from 47% (Ag/CA beads) to 25% (Ag/CA@GTA beads), which can be explained by the substitution of Ca2+ by GTA during the crosslinking process. The TGA results further demonstrate that acetal reaction between GTA and alginate is successfully achieved.
To investigate the surface chemical composition of the Ag/CA@GTA hydrogel beads, the wide scan survey XPS spectrum (Fig. 4) of the Ag/CA@GTA hydrogel beads and high-resolution spectrum of Ag element were analyzed. Fig. 4a presents the wide scan survey XPS spectrum of Ag/CA@GTA hydrogel beads. C 1s, Ca 2p, Ag 3d and O 1s peaks are observed in the Ag/CA@GTA hydrogel beads. The high-resolution spectrum of Ag 3d in Fig. 4b is composed of doublet peaks at 367.7 and 373.7 eV, which are assigned to the 3d5/2 and 3d3/2 peaks of metallic Ag,27,28 respectively. The 6.0 eV gap between the two states also indicates the formation of metallic Ag. However, the Ag 3d5/2 peak appears at a low value (367.7 eV), which further indicates bindings between Ag NPs and O atoms of alginate.29
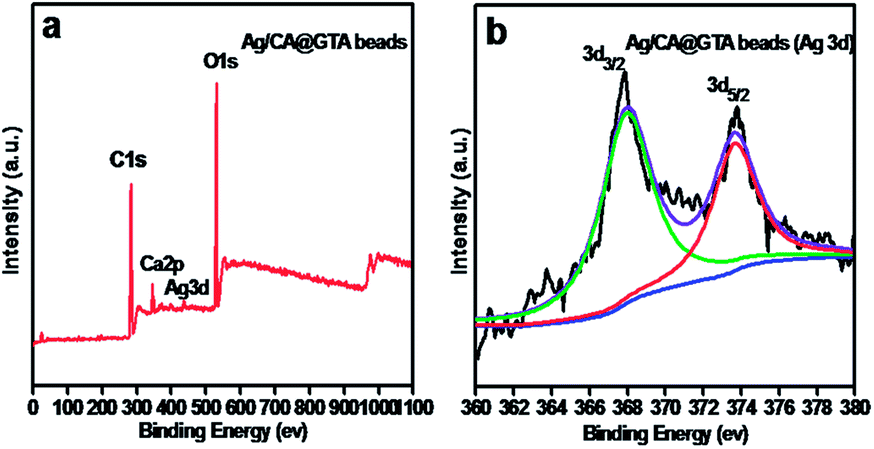 | ||
| Fig. 4 (a) XPS survey spectrum of Ag/CA@GTA hydrogel beads and (b) high resolution core level spectrum of Ag 3d. | ||
Comparison of the catalytic activity using Ag/CA and Ag/CA@GTA hydrogel beads as catalysts
To study the effect of crosslinking on catalyst performance, the catalytic properties of Ag/CA and Ag/CA@GTA hydrogel beads were compared. Herein, the reduction of 4-NP to 4-AP by NaBH4 was chosen as a model reaction to evaluate the catalytic activity of the as-fabricated hydrogel beads. The solution containing 4-NP shows the maximum absorption at 317 nm in the UV-vis spectrum, as shown in Fig. 5a. Upon addition of NaBH4, the characteristic peak assigned to 4-NP shifts from 317 nm to 400 nm because of the formation of 4-NP ions in alkaline conditions.30,31 Upon initiation of the reaction from 4-NP to 4-AP, a new absorption peak at 300 nm is observed. After adding Ag/CA hydrogel beads to the system, the absorption intensity of 4-NP at 400 nm rapidly decreases with prolonged time, accompanied by the appearance of the peak of 4-AP at 300 nm (Fig. 5b). This reduction reaction is completed within 15 min. Given that the amount of NaBH4 is in a large excess in the reaction, the reduction can be regarded as a pseudo-first-order reaction based on the evaluation of the rate constant with regard to 4-NP. Therefore, the reaction kinetics can be described as ln(C/C0) = −kt, where k is the apparent first-order rate constant (min−1), t is the reaction time, and C and C0 are the concentrations of 4-NP at time t and 0, respectively. Fig. 5c shows a linear relationship between ln(C/C0) and t in the reduction catalyzed by Ag/CA hydrogel beads. In our catalytic system, such catalysts could be easily recovered by filtration. Therefore, the catalytic reaction was carried out for five cycles. As shown in Fig. 5d, conversion clearly decreases when the Ag/CA beads are reused for the first cycle. Moreover, the conversion drastically decreases from the initial 95.5% (1st cycle), to 40% (3rd cycle) and final 27.5% (5th cycle).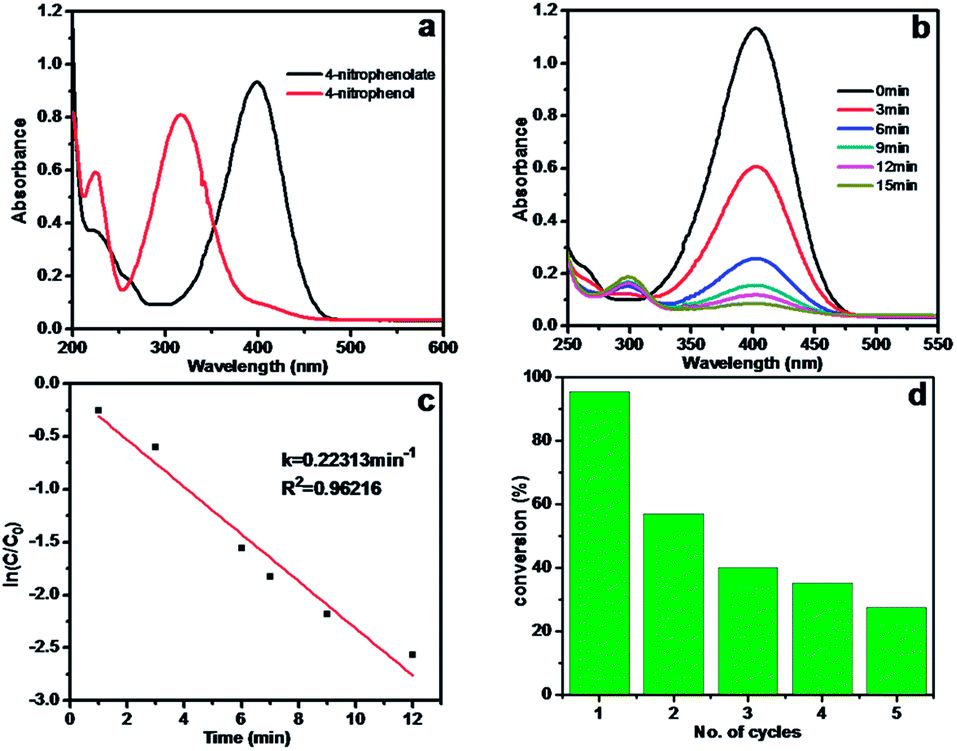 | ||
| Fig. 5 (a) UV-vis absorption spectra of 4-NP in water and in NaBH4 solution, (b) time dependant UV-vis absorption spectra of the reduction of 4-NP by NaBH4 in presence of Ag/CA hydrogel beads, (c) plot of ln(C/C0) against the reaction time, (d) recyclability of the Ag/CA hydrogel beads. | ||
The stability and recyclability of heterogeneous catalysts are quite significant for industrial applications. Therefore, the reason for the rapid loss of catalytic activity should be understood. One possible reason is that the 4-AP product is tightly anchored onto the active site because of the interaction between amine groups and Ag NPs, which may have prevented the substance from diffusing onto the surface of Ag NPs for further reaction. In our opinion, the main reason is that the Ca2+ ions that chelated with the O atoms in CA beads exchange with Na+ ions in the reaction system. Thus, a part of alginate may have redissolved in the solution. This phenomenon could be reflected through the photos taken during the course of recycling (Fig. S2†). After three cycle runs, the Ag/CA hydrogel beads swell. When catalyst use is extended to five times, the beads are seriously damaged, as exhibited in Fig. S2c.† The phenomenon is attributed to the destruction of the 3D network structure caused by ion exchanging. The Ag NPs in the 3D network undoubtedly enter the solution. The calculated mechanism is also evidenced by the XPS result shown in Table 1. After reusing Ag/CA hydrogel beads for five times, the atomic ratio of Ca decreases from 5.84% to 3.98%. The atomic ratio of Ag also decreases from 0.32% to 0.24%. These findings mean that the reduction in the conversion of catalytic reaction is due to the loss of Ag NPs during recycling experiments.
| XPS (At%) | ||||
| Element | Ag/CA beads | Ag/CA beads (after 5 recycles) | Ag/CA@GTA beads | Ag/CA@GTA beads (after 10 recycles) |
| Ag | 0.32 | 0.24 | 0.24 | 0.23 |
| Ca | 5.84 | 3.98 | 2.42 | 2.47 |
The catalytic performance of Ag/CA@GTA hydrogel beads was also examined. Fig. 6a shows the UV-vis spectra of 4-NP in the presence of NaBH4 and CA@GTA hydrogel beads without Ag NPs. Only a slight decrease in the absorbance at 400 nm is observed after mixing for 30 min. This result means that the CA@GTA hydrogel beads without Ag NPs do not catalyze the reaction. When Ag/CA@GTA hydrogel beads are added to the reaction system, the reaction mixture begins to fade and ultimately bleaches from the original yellow color. As shown in Fig. 6b, the time-dependent UV-vis spectra of the reaction display the disappearance of the peak at 400 nm and the gradual development of a new peak at 300 nm, indicating 4-AP formation. The reaction is completed within 30 min, which is longer than that using Ag/CA hydrogel beads as catalyst. Fig. 6c shows that the plots of ln(C/C0) vs. t for the reaction are linear. The corresponding rate constant is found to be 0.07422 min−1, which is less than that using Ag/CA beads as catalyst. One reason is that the pore size is smaller than that of Ag/CA hydrogel beads because of shrinkage after crosslinking, which affects the diffusion rate of the reactant. Another explanation is that the loss of Ag NPs occurs during crosslinking between alginate and GTA, as evidenced by the data in Table 1. By comparison, the atomic ratios of Ag and Ca decrease from the initial 0.32% and 5.84% to the final 0.24% and 2.42%. Alginate has many functional groups.
 | ||
| Fig. 6 Time dependant UV-vis absorption spectra of the reduction of 4-NP by NaBH4 in presence of (a) CA@GTA hydrogel beads and (b) Ag/CA@GTA hydrogel beads, (c) plot of ln(C/C0) against the reaction time, (d) recyclability of the Ag/CA@GTA hydrogel beads. | ||
Ag NPs in the beads definitely have strong affinity for O atoms in –OH and –COOH groups. During the crosslinking between GTA and alginate, acetal formation affects the interaction between Ag NPs and O atoms and induces a decrease in the amount of Ag NPs to some degree.
The reusability of Ag/CA@GTA hydrogel beads as a heterogeneous catalyst was further investigated. The catalytic reaction without the obvious decrease in conversion is performed for 10 times, as exhibited in Fig. 6d. Results demonstrate that the catalyst possesses high stability and excellent reusability because the effect among alginate molecules is adjusted from chelation with Ca2+ to chemical bonding through crosslinking between alginate and GTA. A more stable 3D network structure is formed, as evidenced by the photos of Ag/CA@GTA hydrogel beads after 1, 5 and 10 cycles (Fig. S3†). After the recycling experiment, the morphology of the hydrogel beads remains intact. Therefore, CA hydrogel beads crosslinked by GTA are an ideal support for metallic NP loading toward various catalytic reactions.
In this reaction, the amount of Ag NPs plays a key role in catalytic performance. We thus investigated the effect of Ag NP amount in the hydrogel beads on catalytic performance. The catalysts were fabricated using different amounts of AgNO3 and at the same GTA concentration. As shown in Fig. 7a, the values of the kinetic reaction rate constant k calculated from the slope of the straight line increase from 0.01995 min−1 to 0.06022, 0.07422 and 0.07783 min−1 with increased AgNO3 amount from 1% to 3%, 5% and 10%. When the AgNO3 amount reaches 5%, further AgNO3 addition almost has no effect on the reaction rate, which well agrees with the TEM result in Fig. 2. The TEM image shows that the dosage and size of Ag NPs are similar between the Ag/CA@GTA hydrogel beads prepared with 5% and 10% AgNO3. When the catalytic-activated sites are sufficient, the reaction rate is mainly controlled by the diffusion rate of the substance.
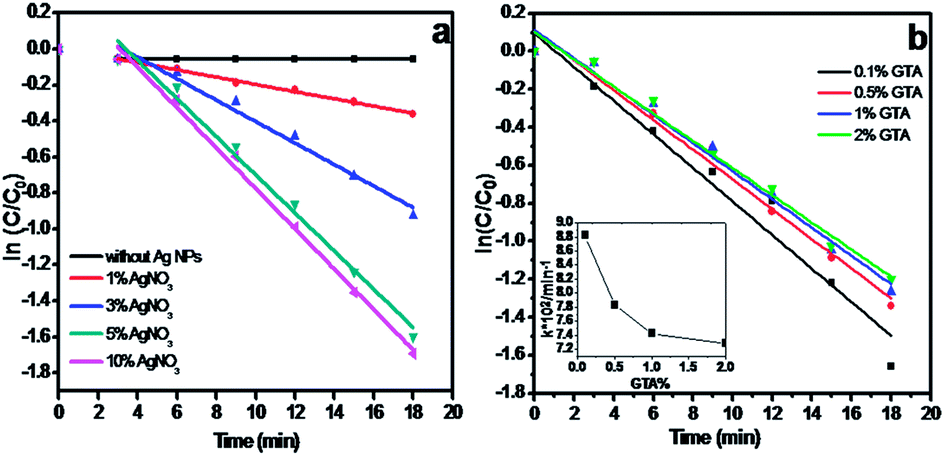 | ||
| Fig. 7 Plot of ln(C/C0) against the reaction time in presence of Ag/CA@GTA hydrogel beads prepared under (a) different amounts of AgNO3 and (b) different concentration of GTA solution. | ||
Apparently, these lines display a good linear relationship between ln(C/C0) and t, which is highly consistent with first-order kinetics. An initial delay time is observed before the onset of rapid catalytic reaction in all lines. This phenomenon has been observed by a number of research groups.32 Generally, a delay time is required for the reactants to diffuse onto the surface of Ag NPs to initiate the reaction, where the size of hydrogel beads is ∼4 mm.20,33 In addition, the reduction of dissolved oxygen in solution usually occurs in such a reaction.34 As shown in Fig. 6a, the addition of hydrogel beads without Ag NPs reduces the reactant concentration within the first 3 min, which demonstrates the above view.
To investigate the effect of GTA on catalytic activity, the Ag/CA hydrogel beads were crosslinked with different amounts of GTA. The dynamics of the resulting beads were then studied. Fig. 7b shows that the reaction rate decreases with increased GTA volume fraction. As shown in Fig. 7b (inset), with increased volume fraction of GTA in solution from 0.1% to 0.5% and 1%, the reaction rate sharply decreases from 8.8 × 10−2 to 7.8 × 10−2 and 7.4 × 10−2. Further addition of the crosslinking agent (2%) does not affect the reaction rate. These results demonstrate that our synthetic system needs about 100 mL of 1% GTA to achieve complete crosslinking with CA hydrogel beads.
Conclusions
We demonstrated a simple and economic route to the synthesis of a stabilized and segregative heterogeneous catalyst using Ag NPs as the active component, alginate as the stabilizer, and GTA as the crosslinking agent. The catalytic activity of the as-prepared GTA-crosslinked Ag/CA hydrogel beads was examined through the catalytic reduction of 4-NP to 4-AP. Compared with the catalytic performance of Ag/CA hydrogel beads as catalyst, the as-formed Ag/CA@GTA hydrogel beads are found to possess excellent activity and high stability because Ca2+ ions are substituted by GTA to form a chemical bond between GTA and alginate through acetal reaction. Moreover, the successful in situ growth of Ag NPs within the alginate network stabilizes the Ag NPs and maintains catalytic performance. Finally, the proposed approach is an ideal platform for fabricating highly efficient catalysts for various catalytic reactions.Acknowledgements
Financial supports from the Educational Commission of Liaoning Province of China (no. L2013218), the Science and Technology Program of Dalian (2013A16GX117) and the Program for Liaoning Innovative Research Team in University (LT2013012) are highly appreciated.Notes and references
- C. M. Cobley, J. Chen, E. C. Cho, L. V. Wang and Y. Xia, Chem. Soc. Rev., 2011, 40, 44 RSC.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302 CrossRef CAS PubMed.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. V. Duyne, Nat. Mater., 2008, 7, 442 CrossRef CAS PubMed.
- S. V. Otari, R. M. Patil, S. R. Waghmare, S. J. Ghosh and S. H. Pawar, Dalton Trans., 2013, 42, 9966 RSC.
- A. Panáček, L. Kvítek, R. Prucek, M. Kolář, R. Večeřová, N. Pizúrová, V. K. Sharma, T. Nevěčná and R. Zbořil, J. Phys. Chem. B, 2006, 110, 16248 CrossRef PubMed.
- J. R. Morones, J. L. Elechiguerra, A. Camacho, K. Holt, J. B. Kouri, J. T. Ramírez and M. J. Yacaman, Nanotechnology, 2005, 16, 2346 CrossRef CAS PubMed.
- C. Baker, A. Pradhan, L. Pakstis, D. J. Pochan and S. I. Shah, J. Nanosci. Nanotechnol., 2005, 5, 244 CrossRef CAS PubMed.
- K. Wang, X. Zhang, C. Niu and Y. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 1272 CAS.
- S. Yang, C. Song, T. Qiu, L. Guo and X. Li, Langmuir, 2013, 29, 92 CrossRef PubMed.
- S. Tang, S. Vongehr and X. Meng, J. Mater. Chem., 2010, 20, 5436 RSC.
- B. Guan, X. Wang, Y. Xiao, Y. Liu and Q. Huo, Nanoscale, 2013, 5, 2469 RSC.
- B. You, J. Yang, Y. Sun and Q. Su, Chem. Commun., 2011, 47, 12364 RSC.
- S. Sharma, P. Sanpui, A. Chattopadhyay and S. S. Ghosh, RSC Adv., 2012, 2, 5837 RSC.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 3827 CrossRef CAS PubMed.
- S. K. Das, C. Dickinson, F. Lafir, D. F. Brougham and E. Marsili, Green Chem., 2012, 14, 1322 RSC.
- S. Kundu, Phys. Chem. Chem. Phys., 2013, 15, 14107 RSC.
- S. Saha, A. Pal, S. Pande, S. Sarkar, S. Panigrahi and T. Pal, J. Phys. Chem. C, 2009, 113, 7553 CAS.
- S. K. Papageorgiou, F. K. Katsaros, E. P. Favvas, G. E. Romanos, C. P. Athanasekou, K. G. Beltsios, O. I. Tzialla and P. Falaras, Water Res., 2012, 46, 1858 CrossRef CAS PubMed.
- M. Chtchigrovsky, Y. Lin, K. Ouchaou, M. Chaumontet, M. Robitzer, F. Quignard and F. Taran, Chem. Mater., 2012, 24, 1505 CrossRef CAS.
- L. Ai, H. Yue and J. Jiang, J. Mater. Chem., 2012, 22, 23447 RSC.
- S. K. Bajpai, S. K. Saxena and S. Sharma, React. Funct. Polym., 2006, 66, 659 CrossRef CAS PubMed.
- Y. Liu, X. Hu, H. Wang, A. Chen, S. Liu, Y. Guo, Y. He, X. Hu, J. Li, S. Liu, Y. Wang and L. Zhou, J. Chem. Eng., 2013, 226, 131 CrossRef CAS PubMed.
- J. Chen, Q. Liu, S. Hu, J. Ni and Y. He, J. Chem. Eng., 2011, 173, 511 CrossRef CAS PubMed.
- R. Xiong, C. Lu, Y. Wang, Z. Zhou and X. Zhang, J. Mater. Chem. A, 2013, 1, 14910 CAS.
- D. Karthiga and S. P. Anthony, RSC Adv., 2013, 3, 16765 RSC.
- A. Saarai, V. Kasparkova, T. Sedlacek and P. Saha, J. Mech. Behav. Biomed. Mater., 2013, i8, i52 Search PubMed.
- Y. Long, J. Wu, H. Wang, X. Zhang, N. Zhao and J. Xu, J. Mater. Chem., 2011, 21, 4875 RSC.
- W. Ren, C. Zhu and E. Wang, Nanoscale, 2012, 4, 5902 RSC.
- Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. B, 2006, 110, 3930 CrossRef CAS PubMed.
- P. Zhang, C. Shao, Z. Zhang, M. Zhang, J. Mu, Z. Guo and Y. Liu, Nanoscale, 2011, 3, 3357 RSC.
- Z. Zhang, C. Shao, Y. Sun, J. Mu, M. Zhang, P. Zhang, Z. Guo, P. Liang, C. Wang and Y. Liu, J. Mater. Chem., 2012, 22, 1387 RSC.
- T. Wu, L. Zhang, J. Gao, Y. Liu, C. Gao and J. Yan, J. Mater. Chem. A, 2013, 1, 7384 CAS.
- Y. Wang and Y. Ni, Analyst, 2014, 139, 416 RSC.
- S. Panigrahi, S. Basu, S. Praharaj, S. Pande, S. Jana, A. Pal, S. K. Ghosh and T. Pal, J. Phys. Chem. C, 2007, 111, 4596 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08801h |
| This journal is © The Royal Society of Chemistry 2014 |