DOI:
10.1039/C4RA08940E
(Paper)
RSC Adv., 2014,
4, 50693-50698
Cobalt complexes of BODIPY as precatalyst for the photooxidation of water and DHN†
Received
20th August 2014
, Accepted 26th September 2014
First published on 26th September 2014
Abstract
To develop molecular catalysts for the generation of dioxygen, the photophysical properties of cobalt complexes of boradiazaindacene (BODIPY) derivatives were studied. It is found that the intramolecular energy transfer due to d–π interaction between boradiazaindacene (BODIPY) and the cobalt central in aqua-cobalt complexes, which was confirmed by DFT calculation, leads to the quenched fluorescence emission being recovered completely. The effect of link spacers (benzyl groups) on the intramolecular d–π interaction was discussed. Results show that cobalt complexes of BODIPY derivatives can be photo-sensitizers and a good blue LED light driven precatalyst for the oxidation of water. The dioxygen generation was further confirmed by the photooxidization of 1,5-dihydroxylnaphthalene (DHN) catalyzed by cobalt complexes using water as the oxygen source. This will give a new path to design photo-active complexes to be used as PDT in hypoxic environments.
Introduction
Light-activated water oxidation is the crucial step of artificial photosynthesis and a fundamental reaction of solar-energy conversion in both natural and artificial photosynthetic systems.1 An earth-abundant system for evolving oxygen from water is photosystem II (PSII), which can cause the photoinduced removal of electrons and protons from metal-aqua intermediates via low-energy pathways, eventually powered by visible light. Visible light induced removal of electrons and protons from metal-aqua intermediates via low-energy pathways can carry out by the bioinspired water oxidation catalysts (WOCs).2–4 Since Nocera's report on a Co-based water oxidation catalyst (WOC), catalysts made of cobalt have received increased attention due to their advantages of low cost and good activity.5 Two representatives of homogeneous cobalt catalysts of WOC are the Co(II) pentapyridine complex and the Co(II) porphyrin complex.6,7 Generally, homogeneous water oxidation to form dioxygen proceeding only in the presence of Ru(bpy)32+ (bpy = 2,2′-bipyridine). A common feature for those reported Co(II) complexes, which are capable of water oxidation, is that they need Ru(bpy)32+ (bpy = 2,2′-bipyridine) as the photosensitizer and persulfate (S2O8)2− as the sacrificial electron acceptor.
Non-ruthenium photosensitizer is rarely reported. Boradiazaindacenes (BODIPYs) are very popular fluorophores with high quantum yields, large extinction coefficients and a longer absorption wavelength and fluorescence emission in the visible spectral region.8 The cobalt(II)/(III) redox potential can be tuned by the change of Lewis base interaction and self-exchange electron transfer.9–11 The dinuclear Co(II)Co(III) mixed-valence complex of tridentate ligand N-methyl-N,N-bis(2-pyridylmethyl)amine (L) has been reported as electrocatalytically water oxidation catalysis.12 The redox and kinetic properties of photogenerated intermediates and reactive transients can be changed by the tunable set of ligands. Thus, the conjugation of BODIPY (organic photosensitizers) and Co(II) complexes of di(picolyl)amine through a linker may form a visible light driven catalysts to the oxidization of water. Generally, energy donor and energy acceptor were widely used to construct triplet photosensitizers based on fluorescence resonance energy transfer (FRET). If there is intramolecular fluorescence resonance energy transfer from the photoexcited sensitizer to ground-state triplet oxygen (3O2) in BODIPY-based cobalt(II) complexes, BODIPY-cobalt(II)–water system could be used to generate 1O2 in hypoxic environments. By following this strategy, we report the solution behaviour and photophysical properties of three BODIPY-based fluorescent cobalt(II) complexes of BDA (BDA13 = 8-[di(2-picolyl)amine-n-benzyl]-4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacen; n = meta- (m-BDA), or para- (p-BDA)) (Fig. 1, Scheme 1).
 |
| | Fig. 1 (a) Absorption spectra of complexes Co3 (A) and Co2 (B) in MeCN–H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) obtained from experimental observation (in black line) and TD-DFT/PCM calculation (in blue line). For the latter, a Lorentzian function has been adopted with the spectral line width set to be 70 nm. The theoretical spectra are right shifted by 60 nm for the complex Co3 and 40 nm for the complex Co2 from TD-DFT excitation energies. (b) Assignment of the strongest absorption peak (peak 2) of [(m-BDA-e)Co + CH3CN + H2O]+ (Co2). The molecular orbitals are obtained through DFT calculations at the B3LYP/LanL2DZ*+6-31G* level. (c) Assignment of the strongest absorption peak (peak 2) of [(p-BDA-e)Co + CH3CN + H2O]+ (Co3). The molecular orbitals are obtained through DFT calculations at the B3LYP/LanL2DZ*+6-31G* level. :1) obtained from experimental observation (in black line) and TD-DFT/PCM calculation (in blue line). For the latter, a Lorentzian function has been adopted with the spectral line width set to be 70 nm. The theoretical spectra are right shifted by 60 nm for the complex Co3 and 40 nm for the complex Co2 from TD-DFT excitation energies. (b) Assignment of the strongest absorption peak (peak 2) of [(m-BDA-e)Co + CH3CN + H2O]+ (Co2). The molecular orbitals are obtained through DFT calculations at the B3LYP/LanL2DZ*+6-31G* level. (c) Assignment of the strongest absorption peak (peak 2) of [(p-BDA-e)Co + CH3CN + H2O]+ (Co3). The molecular orbitals are obtained through DFT calculations at the B3LYP/LanL2DZ*+6-31G* level. | |
 |
| | Scheme 1 Formation of aqua-cobalt complexes, the intramolecular d–π interaction in Co2 and poor d–π interaction in Co3. | |
Results and discussions
The solution behaviour and photophysical properties
The [(m-BDA)CoX]X (X = Cl (Co1), or NO3− (Co2)) was obtained by mixing BDA with cobalt(II) salt in MeCN solution, the solution behaviour was studied by ES-MS and UV-vis spectra. The main peak at m/z (%) = 297.42 (100) corresponds to species [(m-BDA)Co]2+, which indicates the 1:1 ratio for m-BDA and Co2+ in Co1 (Fig. S1a†). The main peak for Co2 in MeCN at m/z (%) = 656.06 (100) corresponds to the species [(m-BDA)Co + NO3−]+. ES-MS data demonstrate that the [(m-BDA)Co]+ core is stable in ES-MS condition. In MeCN–H2O solution, the coordinated NO3− group in [(m-BDA)Co + NO3−]+ can be replaced by solvent molecules. When p-BDA was used, [(p-BDA)Co + NO3−]+ (Co3) was obtained. The main peaks for Co2 and Co3 in MeCN–H2O are m/z (%) = 656.06 (100) corresponding to the species [(m-BDA-e)Co(CH3CN)(H2O)]+ (or [(p-BDA-e)Co(CH3CN)(H2O)]+ for Co3), demonstrating the existence of aqua-cobalt complexes (Fig. S1,† Scheme 1). The UV spectra of m-BDA had bands at 229, 262, 312 and 499 nm (Fig. 1 and S2†). The band at 499 nm can be attributed to the π–π* transition of the BODIPY structure. The coordination of Co2+ with m-BDA causes the π–π* transition at 230 nm and 260 nm blue shifted to 204 nm and 258 nm, respectively. The peak intensity at 499 nm in Co2 is larger than that in Co3 indicating that metal–ligand interaction among Co1–Co3 may be different.
Based on the similar species for Co2 and Co3 in MeCN–H2O (9:1) solution with different absorption intensity in 498 nm, TD-DFT/PCM calculation was carried out. Fig. 1(b) and (c) make a comparison between the scaled absorption spectra of Co2 and Co3 in MeCN–H2O (9:1) solution with the Lorentzian functions14 and the experimental result, displaying a qualitative agreement in the shape of the absorption spectra. Based on species, the calculated UV-vis absorption bands (π → π*) for peak 1 and peak 2 are located at 260 (peak 1) and 460 nm. The dipole-allowed vertical excitation energies of aqua-metal species [(m-BDA-e)Co(CH3CN)(H2O)]+ (Co2) or [(p-BDA)Co(CH3CN)(H2O)]+ (Co3) are mainly assigned to be π → π* transitions. As schematically illustrated in Fig. 1b and c, the π orbitals of peak 2 in complex Co3 are mainly contributed from p orbitals of C and N atoms in ligands, and 3d orbital of Co atom and p orbital of O atoms has great contribution to the π orbitals of peak 2 in complex Co2. The π* orbitals are dominated by the p orbitals of C and N atoms in ligands for complex Co2 and Co3.
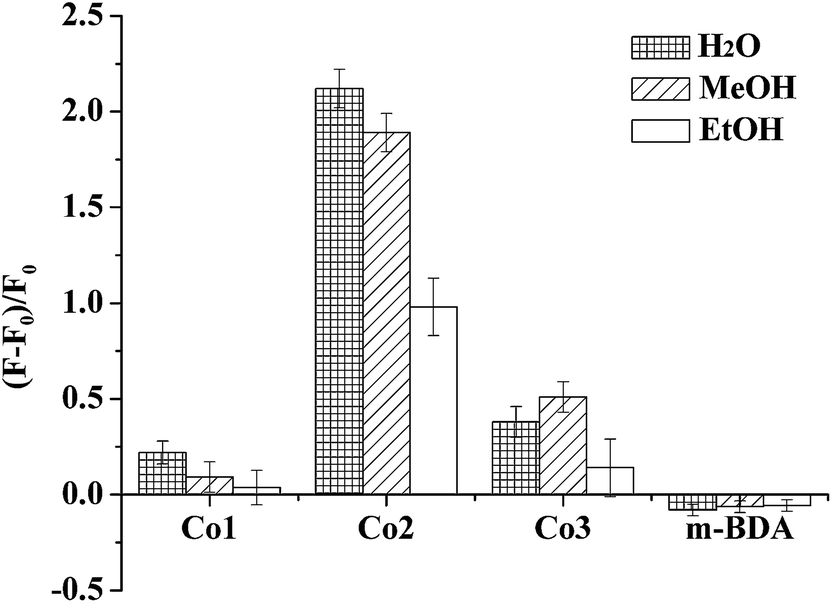
Based on the 3d orbit of Co atom has contribution to the π orbitals of in Co2, solvent effect on complexes has been studied by fluorescence titration. The emission of the complex Co2 was weaker than that of m-BDA. It indicates that the coordination of Co(II) ions results intramolecule non-emission energy transfer. However, the quenched emission can be recovered upon addition of water (Fig. 2 and S3†). Because 3d orbital of Co atom and p orbital of O atoms has great contribution to the π orbitals in Co2, the coordination of water with Co2 leads to the fluorescence emission of m-BDA at 509 nm recovered completed with the quantum yield Φ = 0.615 for Co2 (Φ = 0.610 for m-BDA, Table S1†) when excitated at 460 nm (MeCN:H2O = 6:1, v/v). So, Co2 can be a “turn-on” fluorescence sensor of water. The increase effect for same amount of H2O, MeOH, EtOH is in the order of H2O > MeOH > EtOH. Compared with Co2, Co1 and Co3 is less sensitive to water. As for m-BDA, the H2O can decrease the emission. Experimental results demonstrate that water molecules can coordinate with central Co(II) atom of Co2 with the change of intramolecule energy transfer between m-BDA and metal ions based on the effective d–π interaction (Scheme 1).
 |
| | Fig. 2 Fluorescence intensity changes of cobalt complexes and ligand (1 μM, 3 mL) in the presence of different additives (15 mL). The excitation wavelength was 460 nm. | |
Electrochemical properties
Characterization of Co1–Co3 by cyclic voltammetry in MeCN (or aqueous phosphate buffer at pH 7.2) shows an irreversible peak at 0.76 V (or 0.89 V and 0.89 V), assigned to the Co(II)/Co(III) redox potential (Fig. 3 and S4†). In aqueous phosphate buffer (PB), the intense anodic wave beginning at 1.24–1.32 V (reference electrode, SCE) indicates the oxidation of water.6,7 The catalytic water oxidation was supported by the presence of cathodic waves at −0.680 V and −0.61 mV for Co1 and Co2 in pH 7.2, respectively. There is no obvious cathodic wave in the range of 0 to −0.7 V for Co3 and Co(NO3)2. These show that Co2 may be a photochemical pre-catalyst for the oxidation of water. Scanning cathodically in the phosphate buffer (pH 8.5) a cathodic wave for Co2 appeared in −0.65 V, which shifted to −0.57 V when irradiated with LED light. These indicate Co2 catalyzed water oxidation influenced by pH and light. Moreover, the LED light enhanced catalytic current was observed when Co2 coated FTO was used as working electrode confirming that the water oxidation catalyzed by Co2 is assisted by LED light (Fig. S4d†).
 |
| | Fig. 3 Cyclic voltammograms of Co2 (1 mM) in 0.1 M TBAP in MeCN (black line) and in MeCN:PB (PB = phosphate buffer) v/v = 6:1, red line) and irradiated by blue LED light (4 W) for 5 min (blue line). (a) pH = 7.2; (b) pH 8.5. | |
Water oxidation catalyzed by complexes
The complexes catalyzed water oxidation was further confirmed by measuring the blue LED light (4 W) irradiated oxygen evolution. Consideration on potential application in biological environment, water oxidation was carried out in phosphate buffer (pH 7.2). The obvious oxygen bubble can be observed when the complexes (Co1 and Co2) were added to MeCN–H2O–Na2S2O8 solution. The turnover frequency (TOF) of Co1 and Co2 is 0.09 s−1 and 0.26 s−1 at MeCN–H2O–Na2S2O8 solution (Table 1). This value is comparable to the values reported for many using the RuII(bpy)32+/S2O82− system mononuclear noble metal WOCs, such as cobalt porphyrins (CoTPPS) (TOF ∼ 0.17 s−1),11 In this system, measurements were also carried out in the MeCN–H2O, as controls. The TOF value greatly decreased indicating S2O82− could be an electron acceptor for the complex catalyzed water oxidation. It was found that rate and the total amount of oxygen produced in Co2–H2O system depend on the Co2 concentration, showing saturation-inhibition behaviour (Fig. 4). A number of control experiments were carried out to verify that [(m-BDA)Co(CH3CN)(H2O)]+ (Co2) is the pre-catalysis. The free m-BDA, Co(NO3)2 and complex Co3 featuring the analogous ligand (p-BDA) can't catalyze the oxidation of water to release dioxygen under identical conditions. Moreover, in black, less oxygen can be monitored indicating that Co2 catalyzed oxidation of water is driven by LED light. Experimental results demonstrate that the combination of m-BDA and Co(NO3)3 forming Co2 is essential for the catalytic activity. Although the main components of [(p-BDA)Co(CH3CN)(H2O)]+ (Co3) and [(m-BDA)Co(CH3CN)(H2O)]+ (Co2) in MeCN–H2O are similar, the Co3 with an analogous ligand p-BDA can't oxidize water to release dioxygen. We deduce that the effective electron transfer between m-BDA and central metal ions based on the interaction between 3d orbit to π orbit is a necessary factor for the Co(II) complexes catalyzed water oxidation. Nam found that [CoII(Me6tren)(OH2)]2+ is the precatalyst in light-driven water oxidation, which decompose to give cobalt oxide nanoparticles which are the real catalyst.15 However, dynamic light scattering (DLS) measurement of the solution after photochemical oxidation does not reveal the presence of any dispersion due to nanoparticles formation. All these results suggest that Co2 may be a molecular precatalyst to the oxidation of water, and ES-MS data show the existence of [(m-BDA)Co]2+ core in the after reaction system (Fig. S1c†). The possible mechanism is shown in Scheme 2.
Table 1 Cobalt(II) complexes catalyzed water oxidationa
| |
Additives |
O2 (μM) |
V0 (μM min−1) |
TOF (s−1) |
TON |
| A water oxidation was carried out in MeCN–water (or 0.08 M Na2S2O8 in phosphate buffer (v/v = 2:1) with complexes (1 mM) at 298 K. |
| Co1 |
S2O82− (pH = 7.2) |
145.8 |
15.58 |
0.09 |
48.6 |
| Co2 |
H2O |
343.5 |
35.37 |
0.19 |
114.45 |
| S2O82− (pH = 7.2) |
448.9 |
45.97 |
0.26 |
149.62 |
| S2O82− (pH = 10.1) |
402 |
41.40 |
0.23 |
134 |
| Co3 |
S2O82− (pH = 7.2) |
0 |
— |
— |
— |
| m-BDA |
H2O |
0 |
— |
— |
— |
 |
| | Fig. 4 O2 production from an aqueous solution containing Co2 in MeCN–0.08 M phosphate buffer containing 0.08 M Na2S2O8 (v/v = 2:1), pH 7.2 (Co2 concentration:  , 0.2 mM; , 0.2 mM;  , 0.4 mM; , 0.4 mM;  , 0.6 mM; , 0.6 mM;  , 0.8 mM; , 0.8 mM;  , 1.0 mM; , 1.0 mM;  , 1.7 mM) at 25 °C. , 1.7 mM) at 25 °C. | |
 |
| | Scheme 2 Possible mechanism for Co2 catalyzed water oxidation. | |
Photooxidation of DHN
The light-driven water oxidation catalysed by Co2 was further confirmed by the photoexcited oxidation of dihydroxylnaphthalene (DHN) using water as oxygen source. It is reported that DHN can react with singlet oxygen producing juglone.16 Upon addition of water to the MeCN solution of Co2, the absorption of DHN at 301 nm decreased and the absorption of the product juglone at 427 nm increased gradually (Fig. 5a).
 |
| | Fig. 5 (a) UV-vis absorption spectral change for the photooxidation of DHN (1.0 × 10−4M) using Co2 (5 × 10−6 M) as the photo-sensitizer in CH3CN:H2O = 9:1 solution. (a–g = 0, 0.5, 1, 2, 3, 4, 5 h). Irradiation with blue LED light (440–480 nm, 4 W cm−2). (b) UV-vis absorption spectra for the photooxidation of DHN (1.0 × 10−4M) using m-BDA (5 × 10−6 M) as the photo-sensitizer in CH3CN:H2O = 9:1 solution. a–g = 0, 0.5, 1, 2, 3, 4, 5 h). Irradiation with blue LED light (440–480 nm, 4 W cm−2). | |
When m-BDA was used, nearly no absorption at 427 nm appeared. Control experiments carried out in MeCN solution, no juglone band at 427 nm appeared (Fig. 5b). Therefore, experimental results demonstrate that the O2 does come from the photo-irradiated oxidation of water catalyzed by Co2. Moreover, the intramolecular d–π interaction in Co2 increases the intersystem crossing yield and makes possible the photoenergy transfer from photoexcited-BODIPY moiety to ground-state triplet oxygen (3O2) to generate 1O2 (Scheme 3). When the photooxidation of DHN was carried out in CH3CN:D2O (v/v = 9:1) system in the presence of Co2, bleaching of the 498 nm band, belonging to BODIPY core in m-BDA, and indeed obvious formation of juglone band at 427 nm indicate that the 1O2 is stable in D2O and BODIPY core can be destroyed by a fair amount of juglone (Fig. S5a†). The limited life time for singlet oxygen in water makes the BODIPY core is stable. The rate of 1O2 produced in CH3CN/H2O system is 0.113 ΔA h−1, which increases to 0.153 ΔA h−1 in CH3CN/D2O system in the first one hour. While in the subsequent 4 h, the rate of 1O2 produced in CH3CN/D2O system is 0.025 ΔA h−1, which is near twice as much in CH3CN/H2O system (Fig. S5b†). Experimental results demonstrate the LED light-driven production of 1O2 in Co2–H2O (or Co2–D2O) system. A common method to generate 1O2 is by energy transfer from a photoexcited sensitizer to ground-state triplet oxygen (3O2). This strategy is the basis of current PDT, but requires the presence of oxygen at the target site. However, tumor cells are often hypoxic. Here, the LED light-driven production of 1O2 in Co2–H2O system gives a new way to the formation of singlet oxygen without any exogenous of oxygen gas.
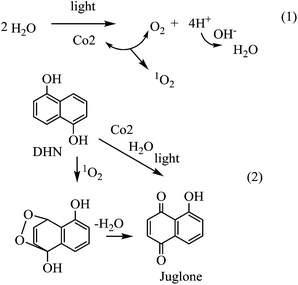 |
| | Scheme 3 The photooxidation of DHN catalyzed by Co2. | |
Conclusions
The different metal–ligand interaction in aqua-metal species [(n-BDA)Co(CH3CN)(H2O)]+ (n = meta- or para-) were studied. DFT calculation confirmed that the 3d orbits of Co atom have great contribution to the π orbitals of UV-vis absorption bands in Co2. Fluorescence titration results demonstrate that the coordination of water with the central cobalt ion in Co2 leads to the recovered fluorescence emission of BDA due to the possible intramolecular electrostatic interaction. Moreover, effective LED light driven intramoleculer electron transfer makes Co2 can be a pre-catalyst to the oxidation of water to release dioxygen. Thus, Co2 can be a photochemical and good white LED light driven pre-catalyst to the oxidation of water. Photooxidation of DHN shows generation of 1O2 during the process of the LED-light driven the oxidation of water in Co2–water system. This gives new path to generate 1O2 using water as oxygen source. Our results demonstrate that the conjugation of BODIPY (organic photosensitizers) and Co(II) complexes of di(picolyl)amine through a proper spacer may form a light driven catalysts for the oxidization of water If the effective intramolecular d–π interaction exists. This will give new path to design photo-active complexes to be used as PDT in hypoxic environments.
Experimental section
Materials and measurements
8-[Di(2-picolyl)amine-n-benzyl]-4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacen; n = meta- (m-BDA), or para- (p-BDA).13 All the fluorescence spectra and absorption spectra were measured in the mixture solution of tris(hydroxymethyl)amino-methane (Tris) solution and DMSO (Tris–DMSO) (Tris:DMSO = 9:1, v/v) at pH 7.4. Mass spectra were measured on a Finnigan LCQ mass spectrograph. Fluorescence spectra were determined on a Varian Cary Eclipse fluorescence spectrophotometer. Absorption spectra were determined on a Shimadzu UV-Visible 2450 spectrophotometer. Cyclic voltammograms were recorded using a CHI-730 chemical workstation and a platinum working electrode (PE), a platinum-wire auxiliary electrode, and a saturated calomel reference electrode (SCE). Bu4N+ClO4− (TBAP) from Sigma-Aldrich was used as the supporting electrolyte at a concentration of 0.1 M. Potentials are reported relative to Fc+/Fc in TBAP/MeCN; the potential of ferrocene is 0.415 V against SCE. The C, H and N microanalyses were performed on Vario EL elemental analyzer. The molar electrical conductivities in DMF solution containing 10−4 mol L−1 complex were measured at 25 ± 0.1 °C using a BSA-A conductometer. Thermogravimetric analysis was carried out in nitrogen atmosphere with a heating rate of 10 °C min−1 using a Shimadzu DT-40 thermal analyzer.
Computational details
Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were performed to rationalize the experimental absorption spectra by using the Gaussian 09 program.17 The complexes were optimized at the B3LYP/LanL2DZ*+6-31G* level. Here, the basis set 6-31G*+LanL2DZ* (using 6-31G* basis set for C, N, O, B, F, and H atoms, adding the f-type polarization functions to Co atom at the basis set LanL2DZ) was employed in view of the influence of d and f functions on the absorption spectra. The exponent, 2.78 of f orbital for Co atom was selected as those in previous work.18 The vertical electronic excitation energies of complexes were then obtained through TD-DFT calculation at the same level.
Synthesis of complexes
Synthesis of [(m-BDA)CoCl2]·H2O (Co1). m-BDA (104 mg, 0.19 mmol) were mixed with CoCl2·6H2O (46 mg, 0.19 mmol) in 10 mL CH3CN and then stirred for 1 h to give dark red solution. After cooling to room temperature, ethyl ether was added and dark red powders (102 mg) were obtained. Yield 81%. Anal. found: C, 56.65; H, 5.03; N, 10.32. Calcd. (%) for C32H34BCl2CoF2N5O: C, 56.19; H, 4.98; N, 10.24. UV-vis (MeCN/nm) (ε × 10−4, dm3 mol−1 cm−1): 230 (4.64), 314 (0.91), 498 (5.42). IR (KBr, ν/cm−1): IR (KBr, ν/cm−1): 3418 m, 3065–2846 m, 1604 m, 1528 s, 1420 s, 1280 s, 1183 s, 755 m, 964 s. ∧m (S cm2 mol−1): 15. The thermal analysis (TG) of Co1 was shown in Fig. S6a.†
Synthesis of [(m-BDA)Co(NO3)2](CH3CN)·(H2O)2 (Co2). Co2 was synthesized using same process as Co1 except Co(NO3)2·6H2O (112 mg, 0.21 mmol) was used. Yield 81%. 102 mg, 83%. Anal. found: C, 51.42; H, 5.01; N, 14.15. Calcd. (%) for C34H39BCoF2N8O8: C, 51.29; H, 4.90; N, 14.08. UV-vis (MeCN/nm) (ε × 10−4, dm3 mol−1 cm−1): 204 (10.31), 263 (2.67), 314 (1.81), 498 (7.86). IR (KBr, ν/cm−1): 3405 m, 3002–2846 w, 1621 m, 1530 s, 1393 s, 1295 s, 1196 s, 741 m, 969 s. ∧m (S cm2 mol−1): 19. The thermal analysis (TG) of Co2 was shown in Fig. S6b† (left).
Synthesis of [(p-BDA)Co(NO3)2] (Co3). Co3 was synthesized using same process as Co2 except m-PDA (108 mg, 0.20 mmol) was used. 96 mg, 75%. Anal. found: C, 53.32; H, 4.66; N, 13.25. Calcd. (%) for C32H32BCoF2N7O6: C, 53.50; H, 4.49; N, 13.65. UV-vis (MeCN/nm) (ε × 10−4, dm3 mol−1 cm−1): 230 (3.32), 263 (1.12), 312 (0.56), 498 (7.12). IR (KBr, ν/cm−1): 2969–2931 w, 1608 m, 1535 s, 1470 s, 1366 s, 1300 s, 1198 s, 800 m, 977 s. ∧m (S cm2 mol−1): 19. The thermal analysis (TG) of Co3 was shown in Fig. S6b† (right).
Acknowledgements
Financial support of National Science Foundation of China (21271090, 21103073), Youth Science Foundation of Jiangsu Province (BK20130485) and Coordination Chemistry State key Laboratory Foundation of Nanjing University.
Notes and references
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed
 .
. - C. J. Gagliardi, A. K. Vannucci, J. J. Concepcion, Z. F. Chen and T. J. Meyer, Energy Environ. Sci., 2012, 5, 7704–7717 CAS .
- V. S. Thoi, Y. J. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2399 RSC .
- J. L. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudving, Science, 1999, 283, 1524–1527 CrossRef CAS .
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed .
- S. Tanaka, M. Annaka and K. Sakai, Chem. Commun., 2012, 48, 1653–1655 RSC .
- Z. F. Chen, J. J. Concepcion, H. L. Luo, J. F. Hull, A. Paul and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 17670–17673 CrossRef CAS PubMed .
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC .
- A. Han, H. T. Wu, Z. J. Sun, H. X. Jia and P. W. Du, Phys. Chem. Chem. Phys., 2013, 15, 12534–12538 RSC .
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2011, 47, 4249–4251 RSC .
- T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC .
- J. Luo, N. P. Rath and L. M. Mirica, Inorg. Chem., 2011, 50, 6152–6157 CrossRef CAS PubMed .
- Z. Li, Q. Y. Chen, P. D. Wang and Y. Wu, RSC Adv., 2013, 3, 5524–5528 RSC .
- D. C. Harri and M. D. Bertolucci, Symmetry and Spectroscopy: An Introduction to Vibrational and Electronic Spectroscopy, Oxford University Press, New York, 1978, pp. 307–419 Search PubMed .
- D. Hong, J. Jung, J. Park, Y. Yamada, T. Suenobu, Y. M. Lee, W. Nam and F. Fukuzumi, Energy Environ. Sci., 2012, 5, 7606–7616 CAS .
- L. Huang, X. N. Cui, B. Therrien and J. J. Zhao, Chem.–Eur. J., 2013, 19, 17472–17482 CrossRef CAS PubMed .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision A. 02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed .
- A. Höllwarth, M. Böhme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 237–240 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available: It contains the ES-MS, UV, Fluorescence spectra and CV are shown in Fig. S1–S4. Photoxidation of DHN, Fig. S5 in D2O. Synthesis and DSC-TG, Fig. S6 data. See DOI: 10.1039/c4ra08940e |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 




 , 0.2 mM;
, 0.2 mM;  , 0.4 mM;
, 0.4 mM;  , 0.6 mM;
, 0.6 mM;  , 0.8 mM;
, 0.8 mM;  , 1.0 mM;
, 1.0 mM;  , 1.7 mM) at 25 °C.
, 1.7 mM) at 25 °C.

