
Charge induced formation of crystalline network polymers†
Arsalan A. Rajaa and
Cafer T. Yavuz*ab
aGraduate School of EEWS, Korea Advanced Institute of Science and Technology (KAIST), Yuseong-gu, Daejeon 305-701, Republic of Korea. E-mail: yavuz@kaist.ac.kr; Fax: +82-42-350-2248; Tel: +82-42-350-1718
bDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Republic of Korea
First published on 4th November 2014
Abstract
Order in purely organic network polymers is hard to achieve, as reversible, dynamic covalent bond formation is required. Strategies have focused on thermodynamically controlled transformations, as kinetics would not seemingly favour reversibility. Herein, we report formation of crystalline network polymers under kinetically favoured conditions by using quaternary ammonium salt linked networks. Charged bulky bridges align, even under fast reaction times (20 minutes) if the rotational freedom is granted. Adding vicinal methyl substituents blocks the ordering, hence forming amorphous networks. Raman experiments and SEM images reveal stacking of 2D layers.
Introduction
Nanoporous organic polymers,1 ordered or not, offer significant promise in the fields of gas capture & storage,2 optoelectronics3 and catalysis.4 Low density, high surface area, tunable pore size and structure, and ease of tailoring functionality make them5 an attractive alternative to inorganic/hybrid porous materials.6The structural regularity in purely organic porous polymers7 is extremely difficult to control, owing to strong covalent bonding that tends to produce disordered materials.8 On the contrary, inorganic porous materials, like zeolites,9 and hybrid porous materials,10 such as metal–organic frameworks (MOFs) are usually crystalline and highly porous. Therefore, a challenge arises for the designed synthesis of organic network polymers, in order to meet the demands for both crystallinity and porosity. Even though a variety of rigid organic building units with different structural configurations are accessible and several synthetic schemes are developed,5 it is yet not straightforward to produce ordered porous organic polymers. While the permanent porosity requires rigid building units, reversible formation of covalent bonds under thermodynamic control is assumed to be required for crystallinity in organic network polymers. In 2005, Yaghi and co-workers introduced crystalline porous organic polymers,11 namely covalent organic frameworks (COFs), by making use of reversible condensation reaction, and in 2007, they reported first synthesis of crystalline 3D COFs.12 Almost similar 2D COFs with a relatively mild synthetic route at ambient pressure was introduced in 2006 by Lavigne and co-workers.13 Whereas, Thomas and co-workers demonstrated14 that the structural regularity in the organic network polymers is not always guaranteed even if prerequisites for crystallinity have been well met; a match in shape and angles in rigid building units shall also be accounted for reversible formation of covalent bonds under controlled thermodynamic conditions.
Recently, new classes of charged porous organic polymers (CPOPs), with quaternary phosphonium cations15 and quaternary borate anions16 as centres for tetrahedral networks, have been introduced with high surface area but unordered network structure. The diamondoid framework topology, that is, imposed by the tetrahedral monomer could be the reason for high surface area, like inorganic zeolites with silica/alumina tetrahedral network. This is also the case of microporous organic polymers17 (MOPs) with tetrahedral networks18 containing carbon or adamantane centres.19 Unlike these CPOPs, we report here a simple design strategy for ordered, charged organic networks that rely on polymerization under kinetic (instead of thermodynamic) control. Surprisingly, quaternary ammonium based CPOPs have never been constructed whereas charged molecules of supramolecular nature that contain ammonium struts have been studied extensively for mechanochemical applications. Stoddart, a pioneer of molecules for nanoelectromechanical systems (NEMS), showed one step preparation of tetracationic receptors20 from 1,4-bis(bromomethyl)benzene and 4,4′-bipyridine. More recently, he introduced a hydrocarbon scavenger ‘ExBox’,21 based on 1,4-bis(bromomethyl)benzene and 4,4′-(1,4-phenylene)-bispyridine, which led to a family of tetracationic cyclophanes.
In this work, we produced purely organic, charged networks, both crystalline and amorphous, termed as ionic covalent organic polymers, i-COPs, based on a cationic backbone with accompanying anions, via simple, catalyst free, room temperature, open air, SN2 (nucleophilic substitution) reaction, using commercially available monomers.
Experimental
Materials and methods
All the chemicals and compounds used were commercially available and used as received without further purification. 1,3,5-tris(bromomethyl)benzene, 4,4′-dipyridyl, 1,2-bis(4-pyridyl)ethane, 4,4′-trimethylenedipyridine, 1,2-bis(4-pyridyl)ethylene, sodium tetrafluoroborate and potassium trifluoromethanesulfonate were purchased from Sigma-Aldrich, USA. Ammonium hexafluorophosphate was bought from Acros Organics, USA. 1,3,5-Tris(bromomethyl)-2,4,6-trimethylbenzene was obtained from TCI, Japan. All the solvents purchased from SAMCHUN, South Korea were dried and stored in anhydrous conditions.The elemental analyses (CHN) were performed on Flash 2000 CHNS analyzer, product of Thermo Scientific, USA. FT-IR spectra were recorded as KBr pellet using a PerkinElmer FT-IR spectrometer. Solid-state CP/MAS 13C SSNMR spectra were recorded on a Agilent 400 MHz, 54 mm NMR DD2 spectrometer. Thermogravimetric analysis (TGA) was performed on a NETZSCH-TG 209 F3 instrument by heating the samples to 800 °C at 10 °C min−1 in N2 and air atmosphere. The graphitic nature of i-COPn was evaluated from Raman spectrum between 1250–1700 cm−1, measured by high resolution dispersive Raman microscope, ARAMIS by Horiba Jobin Yvon, France. N2 physisorption isotherms for i-COPs were obtained with a Micromeritics ASAP 2020 accelerated surface area and porosimetry analyzer at 77 K. Prior to analysis, the samples were degassed at 125 °C for 5 h under vacuum. The adsorption–desorption isotherms were evaluated to give the pore parameters, including BET and Langmuir surface area, pore size, and pore volume. CO2 adsorption and desorption isotherms for i-COPs were measured at 273 K and 298 K by using isochoric system, ASAP 2020, Micrometrics Inc., USA. High power powder X-ray diffraction (PXRD) patterns of the samples were acquired from 2–80° by a Rigaku D/MAX-2500 (18 kW) Micro-area X-ray diffractometer. SEM images for samples, were taken on field emission SEM (Magellan400 by FEI Company, USA), that were prepared by dispersing the material onto a sticky carbon surface attached to a flat-circular aluminium sample holder. Osmium was coated on the samples under pressure of 0.06 mbar for 15 minutes via osmium coater (HPC-1SW, Vacuum Device Inc., Japan) before imaging.
Synthetic procedure
The ionic covalent organic polymers, i-COPs, were synthesized using two different cores, 1,3,5-tris(bromomethyl) benzene and 1,3,5-tris(bromomethyl)-2,4,6-trimethylbenzene to study the effect of vicinal methyl substituent on the crystallinity. The enhanced ordered formation was achieved by increasing the distance between two connecting ends of linkers. The impact of various solvents and different reaction temperatures on the degree of crystallinity, for i-COPn, n = 1, 3, 5, 7 yielded from core, 1,3,5-tris(bromomethyl) benzene, is also studied. The solid-state synthesis also yielded crystalline i-COPn, n = 1, 3, 5, 7 and amorphous i-COPm, m = 2, 4, 6, 8 from core, 1,3,5-tris(bromomethyl) benzene and 1,3,5-tris(bromomethyl)-2,4,6-trimethylbenzene, respectively in 20–40 minutes only. Lastly, ion exchanged was employed to tune the characteristics of charged polymers, in addition to testify the existence of replaceable ions in the charged networks.General procedure for preparation of ionic covalent organic polymers (i-COPs)
1,3,5-tris(bromomethyl) benzene or 1,3,5-tris(bromomethyl)-2,4,6-trimethylbenzene dissolved in solvent was added dropwise to flask containing either 4,4′-dipyridyl, 1,2-bis(4-pyridyl)ethane, 4,4′-trimethylenedipyridine or 1,2-bis(4-pyridyl)ethylene dissolved in solvent with continuous stirring at ambient temperature. The precipitates (i-COP 1–8) formed were stirred for 72 h before washing with THF. Finally, i-COP 1 (light greenish to light yellowish precipitates), i-COP 2 (dark yellow precipitates), i-COP 3 (light pink precipitates), i-COP 4 (light yellow precipitates), i-COP 5 (mint green precipitates), i-COP 6 (off white precipitates), i-COP 7 (light cream precipitates) and i-COP 8 (dark yellow precipitates) were recovered on Whatman® 1001-110 filter paper (using conical glass funnel), washed with THF and dried at room temperature for 8 h and then placed in an oven at 50 °C under vacuum for 12 h. i-COP 1, 2, 3, 4, 5, 6, 7 and 8 were obtained in 75, 82, 83, 86, 71, 93, 72 and 83% yield, respectively (Scheme 1).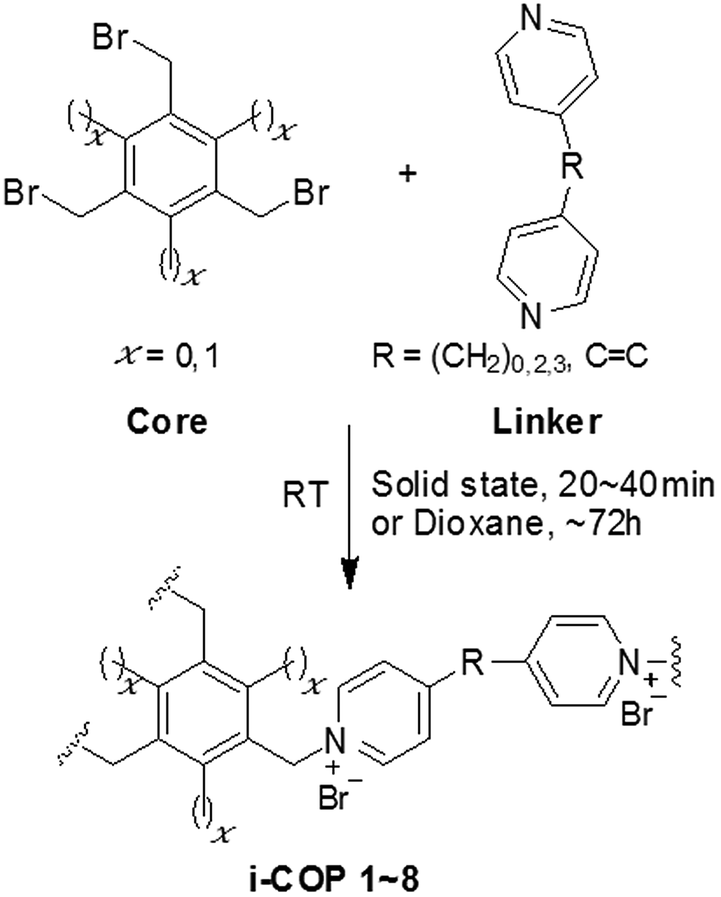 | ||
| Scheme 1 Synthesis of ionic covalent organic polymers (i-COPs), at room temperature and ambient conditions. | ||
Results and discussion
At first, the two nucleophilic nitrogens of all the linkers are distanced apart enough that there is no intramolecular cyclization using three binding nodes (electrophilic carbon). Insoluble powders22,23 precipitating (as early as 20 min) are a clear indication for the network formation. Secondly, only the unsubstituted core, 1,3,5-tris(bromomethyl)benzene, leads to ordered polymer structures, i-COPn, n = 1, 3, 5 and 7 (Fig. 1). On the contrary, the presence of methyl groups in 1,3,5-tris(bromomethyl)-2,4,6-trimethylbenzene rejects planar extension of network structure through highly unfavourable interaction of charged ammonium salts that form spontaneously, and the bulky, hydrophobic methyl substituents (i-COPs 2, 4, 6, 8).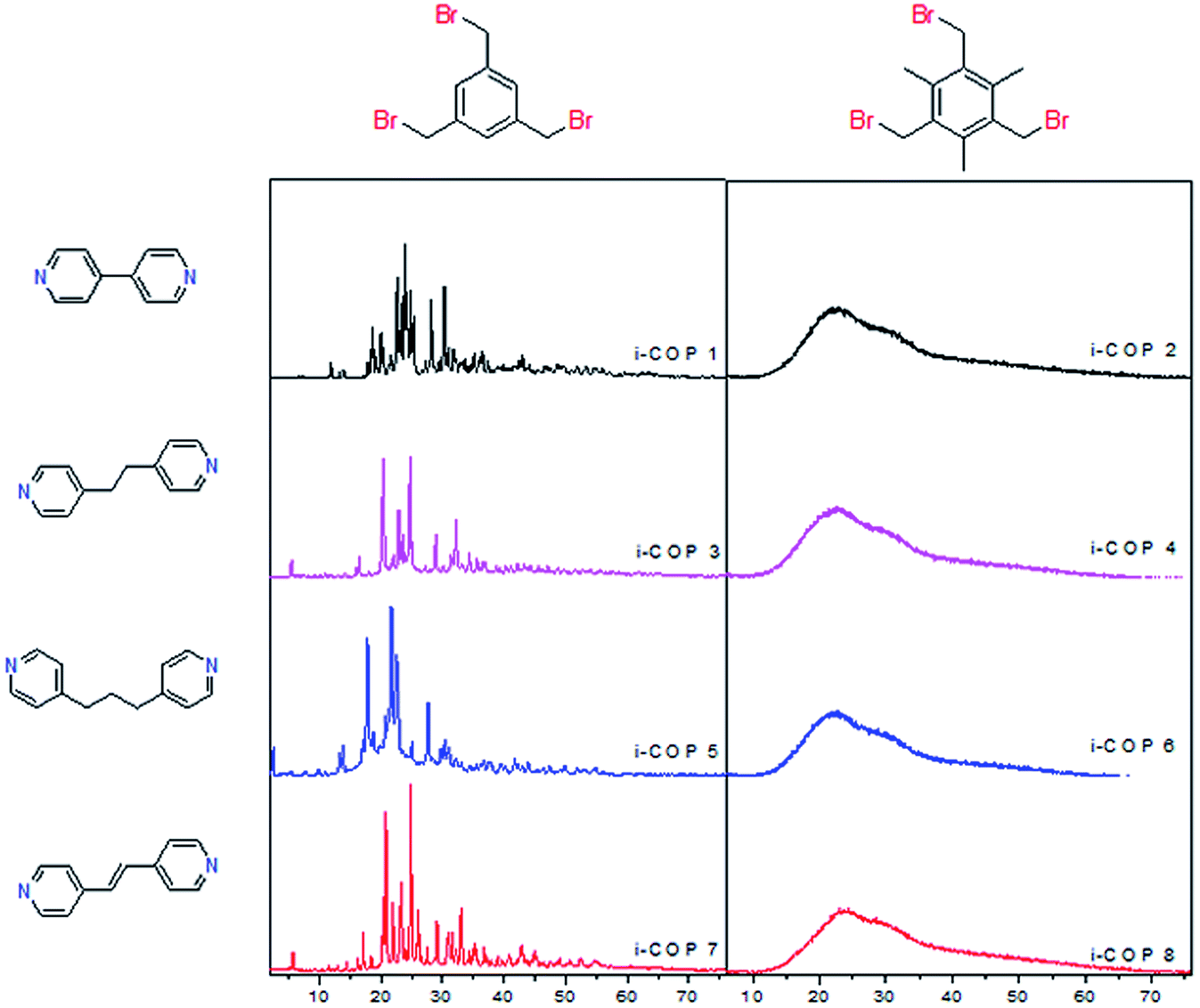 | ||
| Fig. 1 Powder X-ray diffraction patterns of i-COPs with variation of cores and linkers. Vicinal methyl substituents prevent ordering in i-COPs, showing steric effects due to charged spacers as the prime reasons. | ||
Effect of secondary species (e.g. solvents, bases) could become significant in hierarchical ordering.7c,24 Almost identical PXRD patterns are observed on the change of reaction solvents (Fig. S1†) and in the case of a solvent-free synthesis (a.k.a. green chemistry)25 no change in the ordered formation of network polymer structures (i-COPn) is found (Fig. S2†). The ambient open air solid state synthesis,26 at room temperature yielded same i-COP networks in much faster times, 20–40 minutes only. Solvent-less synthesis also show that ordering is unaffected by the use/lack of solvents, and more importantly the concentration of monomers.
There seems to be two possibilities remain in creating order for i-COPs: (1) chemical structures of cores and linkers, (2) temperature of the reaction. In the odd numbered i-COPs, it is evident that the quaternary ammonium centres with a large bromine anion sitting nearby would be able to repel each other to the lowest energy positions, the furthest away from each other (Br-size 3.7–3.9 Å vs. benzene 4–5 Å). And since there are only hydrogens to block their positioning, charged centres enjoy orienting with each other in the most energetically favourable fashion. This anticipation, in fact, drew us to test the substitution effect where bulky methyls are introduced to force the ammonium centres to not have rotational freedom. As expected, this resulted in i-COPm, m = 2, 4, 6 and 8, unordered network polymers (Fig. S3†). The selection of the cores, therefore, verified structural directing effects of substituent, leading to a conviction that lack of rotational space is very unpopular for highly rigid building motif. Although it is highly desired, our attempts to resolve the XRD patterns with respect to finding the preferential crystal orientation have failed. Temperature of the reaction was screened next (Fig. S4†), only to find no observable change in the crystallographic patterns or intensities. Furthermore, the formation of an ordered morphology that is consistent with the crystalline nature of i-COPn is evidenced by SEM, which reveals the formation of blocks that look mostly rectangular like wooden chips of variable sizes. On the other hand, SEM images for the amorphous polymers present agglomerates, which prove non-uniform morphology of i-COPm (Fig. 2).
 | ||
| Fig. 2 Scanning electron micrographs of i-COPs. Crystalline i-COPs (odd numbers) show plate like assemblies, showing a preferred planar orientation. The corresponding amorphous i-COPs (even numbers) are agglomerates of irregular geometry, a preferred alignment to minimize surface energy. | ||
i-COPs, by design, should produce two dimensional (2D) structures, if ordered. The planarity of the 2D systems like graphene are often studied with Raman spectroscopy, a widely used tool to identify sp2 and sp3 hybridized atoms.27 The Raman spectrum depends on the existence of sp2 rings or chains, clustering of sp2 phase, ratio of sp2/sp3 and bond disorder.28 The crystal symmetries for pristine graphene only shows the G peak by Raman spectroscopy; as the disorder appears, D peak(s) appears and may be attributed to sp3 hybridized carbon atoms.29 The polyaromatic materials show G band at ∼1585 (attributed to E2g mode) and D band at ∼1360 (originated from A1g mode) due to stretching of bond of sp2 atoms in both rings and chains, and breathing modes (defect sites) of sp2 atoms in rings, respectively.30 For i-COPn, Raman spectrum indicates the G peaks (1598 cm−1 for i-COP 1, 1607 cm−1 for i-COP 3, 1606 cm−1 for i-COP 5 and 1630 cm−1 for i-COP 7), which is characteristic of crystalline symmetry (Fig. 3).
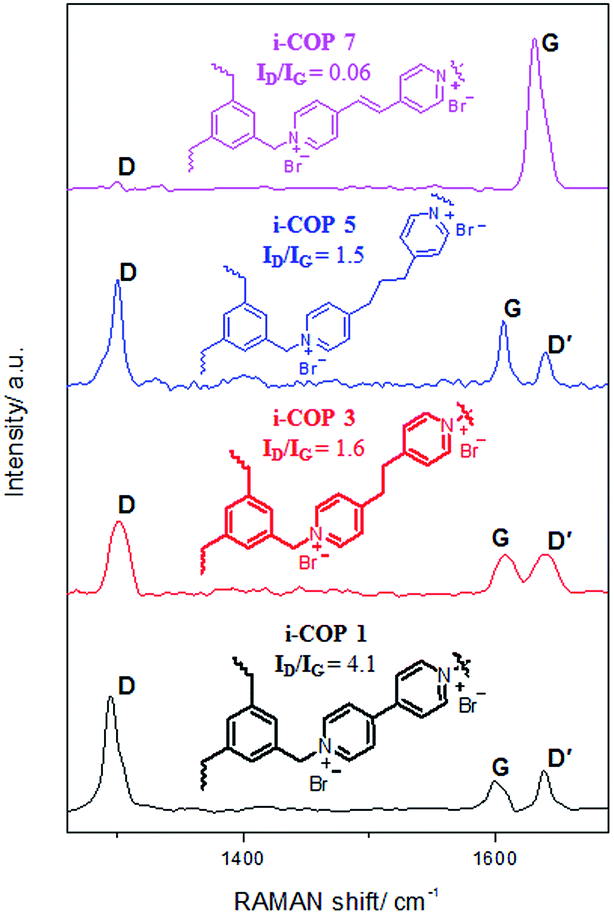 | ||
| Fig. 3 Raman spectrum for crystalline ionic covalent organic polymers (i-COPn, n = 1, 3, 5 and 7), where ID and IG represents the intensity of D and G peak, respectively. | ||
Even though low intensity D and D′ peaks exist in i-COP 1, their intensities reduce as the space between two nucleophilic nitrogens of dipyridyl based linker increase, which is in agreement with PXRD patterns (Fig. 1). This is evident in i-COP 3 and 5, proving the reduction in disorders/defects and in i-COP 7 the π–π stacking prevent the twist in linker structure leading to ordered polymer that is nearly identical in symmetry to pristine graphene. The defects are generally quantified by the ratio of intensity of D to G peaks, ID/IG; high ratio represents more defects.31 It is found that ID/IG ratio for i-COP 1 is 4.1 which goes down to 0.06 for i-COP 7, a value very similar to that of pristine graphene, ID/IG ∼ 0 (Table S1†).
Porosity32 in 2D systems often requires preventing dense packing; a good example is the polymers of intrinsic microporosity (PIM),33 which has 90° kinks in every repeating unit leading to substantial porosity at the expense of crystallinity.34 Weber et al. also synthesized aromatic polyamide and polyimide based on spirobifluorene with a 90° kink in repeating units further demonstrating that bent structures prevent efficient packing for a crystal order.35 Since all the i-COPs are designed to favour dense chain packing, low surface areas and pore sizes were observed from nitrogen physisorption isotherms (Fig. S7 and Table S2†) as well as low CO2 capture capacities (Fig. S8†). Nevertheless, i-COPs showed moderate CO2/N2 selectivities (up to 145 in i-COP 4) mainly due to lack of the preferential binding of nitrogen at the few available pore openings.
Ion exchange
In order to explore the charged nature of the i-COPs we turned to ion exchange, which is usually employed in charged structures to tune their properties according to the field of application. Fischer et al. exchanged lithium of ABNs with sodium and manganese ion, and also reported oxidation of styrene with a higher selectivity of 81% for [Mn(bpy)2]2+–ABN catalyst.16,36 Ship-in-a-bottle synthesis approach37 enabled them to develop [Mn(bpy)2]2+–ABN catalyst. To form porous hybrid gel materials, Coskun and coworkers presented the incorporation of imidazolium cation onto pyrene to interact to ‘reduced graphene oxide’.38 At first, 1-bromopyrenemethanol was synthesized and then functionalized with N-methylimidazole to yield a charged molecule. For ion exchange, the aqueous solutions of NH4PF6, KBF4 and Li(CF3SO2)2N were used to exchange (quantitatively) bromide ion with PF6−, BF4− and (CF3SO2)2N−, respectively. Among all the i-COPs synthesized herein, we succeeded to exchange (quantitatively) bromide counter ions of i-COP 1 with a variety of anions that include PF6−, BF4− and CF3SO3− by treating with aqueous solutions of NH4PF6 (Pf), NaBF4 (Bf) and CF3SO3K (Tf), respectively (Fig. S9†). Because of the aliphatic bridges between rigid aromatic rings of the other i-COPs, the exchanged anion slightly reorganized the order in the symmetric structure leading to minor shifts in PXRD patterns in the case of Pf and Tf and amorphous nature in Bf (Fig. S10†). Such structural flexibility was also observed in porous coordination polymers (PCPs),39 metal–organic frameworks (MOFs)40 and coordination polymer networks (CPNs)41 which was identified by slight differences in their PXRD patterns.Conclusions
In conclusion, purely organic, charged, crystalline network polymers namely, ionic covalent organic polymers (i-COPs) were prepared via rapid nucleophilic substitutions under kinetic control. It was shown that the structural regularity is neither dependent on temperature nor on the solvent. We believe that this is because the limitation of binding motif to only pyridyl-bromomethyl coupling and the steric factors lead to restriction in structural freedom. The solid-state reaction turned to be extremely efficient without compromise on the ordered structures. By controlling and simplifying variation in reaction conditions and composition, we concluded that charged ammonium centres repel the vicinal methyl substituents, and bromines reject each other, leading to a maximal spacing and thus order in the super structure. As the dense chain packing was observed, i-COPs had low surface areas and did not show significant gas capture. The ordered network polymers were also found to have graphitic nature, which was further tuned by selecting non-twisting building unit to yield crystalline network polymer (i-COP 7) like pristine graphene. The synthesized materials contain free replaceable counter anions, which enable us to anticipate their use to wide variety of applications, for instance, ion exchange, catalysis, electrochemical applications, such as, Li+ batteries or Na+ batteries and DSSCs.15,16 We envision that the surface area could be adjusted by tuning the nature of cationic polymer networks to achieve high gas capture.Acknowledgements
We acknowledge the financial support by Korea CCS R&D Centre, Basic Science Research Program through the National Research Foundation of Korea (NRF), ICT & Future Planning (2013R1A1A1012998), and IWT (NRF-2012-C1AAA001-M1A2A2026588).References
- (a) Q. Wei and S. L. James, Chem. Commun., 2005, 1555–1556 RSC; (b) J. Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670–2672 RSC; (c) N. B. McKeown, P. M. Budd and D. Book, Macromol. Rapid Commun., 2007, 28, 995–1002 CrossRef CAS; (d) B. S. Ghanem, K. J. Msayib, N. B. McKeown, K. D. M. Harris, Z. Pan, P. M. Budd, A. Butler, J. Selbie, D. Book and A. Walton, Chem. Commun., 2007, 67–69 RSC; (e) C. D. Wood, B. Tan, A. Trewin, F. Su, M. J. Rosseinsky, D. Bradshaw, Y. Sun, L. Zhou and A. I. Cooper, Adv. Mater., 2008, 20, 1916–1921 CrossRef CAS.
- (a) R. W. Tilford, S. J. Mugavero, P. J. Pellechia and J. J. Lavigne, Adv. Mater., 2008, 20, 2741–2746 CrossRef CAS PubMed; (b) Y. Li and R. T. Yang, AIChE J., 2008, 54, 269–279 CrossRef CAS; (c) H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed; (d) C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed; (e) J. T. Yu, Z. Chen, J. Sun, Z. T. Huang and Q. Y. Zheng, J. Mater. Chem., 2012, 22, 5369–5373 RSC.
- (a) S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed; (b) E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef CAS PubMed.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- (a) P. S. Burgess and W. T. McGeorge, Science, 1926, 64, 652–653 CAS; (b) O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- (a) A. I. Cooper, CrystEngComm, 2013, 15, 1483 RSC; (b) A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed; (c) F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed; (d) F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klock, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570 CrossRef CAS PubMed.
- P. M. Budd, Science, 2007, 316, 210–211 CrossRef CAS PubMed.
- (a) D. W. Breck, W. G. Eversole, R. M. Milton, T. B. Reed and T. L. Thomas, J. Am. Chem. Soc., 1956, 78, 5963–5972 CrossRef CAS; (b) T. B. Reed and D. W. Breck, J. Am. Chem. Soc., 1956, 78, 5972–5977 CrossRef CAS.
- (a) S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC; (b) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed; (c) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Cote, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- R. W. Tilford, W. R. Gemmill, H.-C. zur Loye and J. J. Lavigne, Chem. Mater., 2006, 18, 5296–5301 CrossRef CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981–2988 CrossRef CAS.
- S. Fischer, J. Schmidt, P. Strauch and A. Thomas, Angew. Chem., Int. Ed., 2013, 52, 12174–12178 CrossRef CAS PubMed.
- (a) A. Trewin and A. I. Cooper, Angew. Chem., Int. Ed., 2010, 49, 1533–1535 CrossRef CAS PubMed; (b) R. Dawson, E. Stöckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239 RSC; (c) N. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372–375 CrossRef CAS PubMed.
- (a) T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed; (b) W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS.
- (a) J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed; (b) M. Nanasawa, L. Hu and O. Vogl, Polym. J., 1986, 18, 681–687 CrossRef CAS.
- C. L. Brown, D. Philp and J. F. Stoddart, Synlett, 1991, 462–464 CrossRef CAS PubMed.
- J. C. Barnes, M. Juricek, N. L. Strutt, M. Frasconi, S. Sampath, M. A. Giesener, P. L. McGrier, C. J. Bruns, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 183–192 CrossRef CAS PubMed.
- S. Zulfiqar, M. I. Sarwar and C. T. Yavuz, RSC Adv., 2014, 94, 52263–52269 RSC.
- T. Akhter, O. O. Park, H. M. Siddiqi, S. Saeed and K. M. Saoud, RSC Adv., 2014, 87, 46587–46594 RSC.
- K. T. Jackson, T. E. Reich and H. M. El-Kaderi, Chem. Commun., 2012, 48, 8823–8825 RSC.
- (a) J. F. Brennecke, Nature, 1997, 389, 333–334 CrossRef CAS PubMed; (b) J. Sherman, B. Chin, P. D. T. Huibers, R. Garcia-Valls and T. A. Hatton, Environ. Health Perspect., 1998, 106, 253–271 CrossRef CAS; (c) P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- G. R. Desiraju, Solid State Ionics, 1997, 101, 839–842 CrossRef.
- J. S. Park, A. Reina, R. Saito, J. Kong, G. Dresselhaus and M. S. Dresselhaus, Carbon, 2009, 47, 1303–1310 CrossRef CAS PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS PubMed.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS PubMed.
- K. Sakaushi, E. Hosono, G. Nickerl, T. Gemming, H. Zhou, S. Kaskel and J. Eckert, Nat. Commun., 2013, 4, 1485 CrossRef PubMed.
- C. N. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed.
- (a) D. Yuan, W. Lu, D. Zhao and H.-C. Zhou, Adv. Mater., 2011, 23, 3723–3725 CrossRef CAS PubMed; (b) H. A. Patel, F. Karadas, J. Byun, J. Park, E. Deniz, A. Canlier, Y. Jung, M. Atilhan and C. T. Yavuz, Adv. Funct. Mater., 2013, 23, 2270–2276 CrossRef CAS; (c) H. A. Patel, F. Karadas, A. Canlier, J. Park, E. Deniz, Y. Jung, M. Atilhan and C. T. Yavuz, J. Mater. Chem., 2012, 22, 8431–8437 RSC; (d) H. A. Patel, S. Hyun Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed; (e) H. A. Patel, S. H. Je, J. Park, Y. Jung, A. Coskun and C. T. Yavuz, Chem.–Eur. J., 2014, 20, 772–780 CrossRef CAS PubMed.
- (a) N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804–1807 CrossRef CAS PubMed; (b) H. A. Patel and C. T. Yavuz, Chem. Commun., 2012, 48, 9989–9991 RSC.
- (a) P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC; (b) P. M. Budd, N. B. McKeown and D. Fritsch, J. Mater. Chem., 2005, 15, 1977 RSC; (c) N. B. McKeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem.–Eur. J., 2005, 11, 2610–2620 CrossRef CAS PubMed.
- J. Weber, Q. Su, M. Antonietti and A. Thomas, Macromol. Rapid Commun., 2007, 28, 1871–1876 CrossRef CAS.
- M. Rose, ChemCatChem, 2014, 6, 1166–1182 CAS.
- A. Corma and H. Garcia, Eur. J. Inorg. Chem., 2004, 1143–1164 CrossRef CAS.
- S. Srinivasan, W. H. Shin, J. W. Choi and A. Coskun, J. Mater. Chem. A, 2013, 1, 43 CAS.
- (a) H. R. Moon and M. P. Suh, Eur. J. Inorg. Chem., 2010, 3795–3803 CrossRef CAS; (b) K. Kishida, S. Horike, K. Kongpatpanich and S. Kitagawa, Aust. J. Chem., 2013, 66, 464 CrossRef CAS; (c) R. Ohtani, M. Arai, A. Hori, M. Takata, S. Kitao, M. Seto, S. Kitagawa and M. Ohba, J. Inorg. Organomet. Polym. Mater., 2012, 23, 104–110 CrossRef.
- D. H. Hong and M. P. Suh, Chem.–Eur. J., 2014, 20, 426–434 CrossRef CAS PubMed.
- H. S. Choi and M. P. Suh, Angew. Chem., Int. Ed., 2009, 48, 6865–6869 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For detailed synthesis for all polymers, their characterization, solid-state synthesis, screening for crystallinity, ion exchange. See DOI: 10.1039/c4ra10594j |
| This journal is © The Royal Society of Chemistry 2014 |