DOI:
10.1039/C4RA11567H
(Paper)
RSC Adv., 2014,
4, 59809-59816
A near-infrared “on–off” fluorescent and colourimetric cyanide chemodosimeter based on phenothiazine with applications in living cell imaging†
Received
30th September 2014
, Accepted 28th October 2014
First published on 29th October 2014
Abstract
A great deal of effort has been devoted to developing easy-to-use near-infrared probes for detecting analytes due to their advantages in the field of biosensing. Herein, a near-infrared “on–off” fluorescent chemodosimeter PTZ based on dicyano-vinyl-functionalised phenothiazine was designed and synthesised. The PTZ compound was shown to efficiently recognise cyanide ions in aqueous media by virtue of the special nucleophilicity of cyanide ions, which affects the intramolecular charge transfer efficiency in the molecule. Furthermore, it was found that it exhibited a rapid colourimetric and quenchable near-infrared (NIR) fluorescent response to cyanide ions with a detection limit as low as 67 nM, and that other anions showed almost no interference even at high concentrations. Optical spectroscopic measurements, 1H NMR and mass spectrometry titrations, and theoretical simulations were carried out to elucidate the sensing mechanism of compound PTZ. Moreover, potential applications of this compound for biosensing have been exemplified by the successful fluorescent microscopic imaging for the detection of cyanide ions in HeLa cells.
Introduction
Cyanide is an important industrial chemical and has widespread uses in various fields such as fibre manufacturing, electroplating, metallurgy, herbicide production, and silver or gold mining.1 Cyanide-containing substances, which have been found in water, polluted air, soil, and vehicle exhaust, are extremely detrimental to mammals even in trace concentrations due to the lethal damage they cause to nervous systems.2 As a result of its high toxicity to human beings, the maximum permissive level of cyanide ions in drinking water for a healthy life is set at as low as 0.07 mg L−1 (1.9 μM) by the World Health Organization (WHO).3 Therefore, the development of convenient and efficient detection techniques for tracing small amounts of cyanide ions has gained considerable attention in recent years; these methods include titrimetric, potentiometric, voltammetric, Raman spectroscopy, chromatography and electrochemical methods.4 Unfortunately, most of these techniques are time-consuming, costly, and involve sophisticated instruments.
Compared to traditional techniques, chemical probes based on absorption or fluorescence changes are more feasible for detecting cyanide ions owing to their many appealing advantages such as high selectivity, hypersensitivity, easy operation, economy and rapid real-time monitoring. Thus, a number of colourimetric or fluorescent probes for cyanide ions have been hitherto reported, and several alternative strategies have been employed to realise recognition. These strategies include utilising hydrogen bonding interactions,5 forming cyanide ion complexes6 or demetalating preassembled complex probes7 by taking advantage of the coordination ability of cyanide ions, and attacking on electrophilic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds,8 activated carbonyl groups,9 polarised CN bonds10 or boron centres11 by virtue of the nucleophilic characteristic of cyanide ions.
C double bonds,8 activated carbonyl groups,9 polarised CN bonds10 or boron centres11 by virtue of the nucleophilic characteristic of cyanide ions.
Among these strategies, chemodosimeters, which take advantage of the unique nucleophilicity of cyanide ions to realise recognition, are much more promising compared to probes based on hydrogen bonds or complexation processes since chemodosimeters can detect analytes through highly selective and irreversible chemical reactions that lead to observable signals with accumulative effects.12 However, despite the considerable progress in the development of chemodosimeters for cyanide ions detection, it remains challenging to design chemodosimeters with simple structures and outstanding properties, for instance, synchronous colourimetric and fluorescent responses in the near-infrared (NIR) region. It is well-known that NIR fluorescence response, ascribed to the emission wavelength of 650–900 nm, is highly desired in bioimaging for deeper penetration into tissues, low damage to living cells, weak scattering, and negligible autofluorescence background signals.13 As notable heterocyclic compounds with electron-rich sulphur and nitrogen moieties, phenothiazine and its derivatives exhibit excellent electro-optical properties and are some of the most promising candidates for applications as multifunctional optoelectronic materials such as dye-sensitised solar cells,14 electrochromic displays,15 photocatalytic materials,16 and organic light-emitting diodes.17 With regard to chemodosimeters, however, reports relating to phenothiazine have been scarce.18
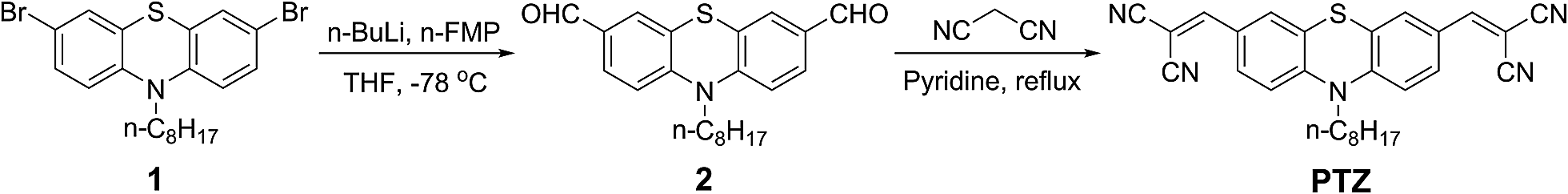
With these considerations, this work aims to design and synthesise a chemodosimeter PTZ (Scheme 1) consisting of an n-octyl-substituted phenothiazine as the signalling moiety and two dicyano-vinyl groups as the reactive subunits to form an intramolecular charge transfer (ICT) system with an acceptor–donor–acceptor (A–D–A) configuration. The chemodosimeter displays high sensitivity and selectivity for cyanide ions over other anions with a rapid response in aqueous media and is successfully applied in the bioimaging of living cells.
 |
| | Scheme 1 Synthetic routine of compound PTZ. | |
Experimental
Materials and instrumentation
3,7-Dibromo-10-octyl-10H-phenothiazine (compound 1) was prepared and purified according to procedures in the literature.19 n-Butyl lithium (2.5 M solution in hexane), N-formylmorpholine (N-FMP) and malononitrile were purchased from Sigma-Aldrich Co. and used as received. Tetrahydrofuran (THF) was purchased from Aladdin Industrial Inc. (Shanghai, China) and used after distillation from a boiling solution containing sodium metal. All other reagents were of analytical grade, purchased from J&K Scientific Ltd. and used without further treatment. Thin-layer chromatography (TLC) analyses were performed on silica-gel plates, and flash chromatography was conducted using silica-gel column packages purchased from Qing-dao Haiyang Chemical Company, China.
1H NMR and 13C NMR spectra in CDCl3 or DMSO-d6 were recorded on Bruker AM-400 spectrometers with tetramethylsilane (TMS) as the internal standard. Mass spectra (MS) were recorded on a Waters LCT Premier XE spectrometer with acetonitrile as solvent. UV-vis absorption spectra were performed on a Varian Cray 500 spectrophotometer, and fluorescence spectra were recorded on a Horiba Fluoromax-4 fluorescence spectrometer. The pH values of the test solutions were measured by a pH-meter (Mettler Toledo FE20). All the measurements were performed at room temperature.
1H NMR titration
1H NMR titration spectra were carried out by the addition of 0–2.2 equiv. of tetrabutylammonium (TBA) cyanide (1.2 M in DMSO-d6) to chemodosimeter PTZ (25 mM) in DMSO-d6 at room temperature.
Procedure for anion sensing
Doubly distilled water and a spectroscopic grade of DMSO were used as the solvents throughout all experiments. Compound PTZ was dissolved in DMSO to afford a stock solution with a concentration of 10 mM, which was diluted with DMSO–H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution to 10 μM. A stock solution of CN− with a concentration of 5 mM was prepared by adding NaCN into doubly distilled water in a 5 mL volumetric flask. Salts including KF, NaCl, NaBr, KI, NaNO2, KNO3, NaOAc·3H2O, Na3PO4·12H2O, Na2HPO4·12H2O, NaH2PO4, NaHCO3, K2CO3, KSCN, Na2S and Na2SO4 were used to prepare 50 mM stock solutions of other competitive anions in 5 mL volumetric flasks with doubly distilled water. The changes in the absorption and fluorescence spectra caused by CN− (22 μM) and miscellaneous interfering anions (200 μM) with their K+ or Na+ salts in water solutions were recorded. Each spectrum was acquired 2 min after anion addition.
:1) solution to 10 μM. A stock solution of CN− with a concentration of 5 mM was prepared by adding NaCN into doubly distilled water in a 5 mL volumetric flask. Salts including KF, NaCl, NaBr, KI, NaNO2, KNO3, NaOAc·3H2O, Na3PO4·12H2O, Na2HPO4·12H2O, NaH2PO4, NaHCO3, K2CO3, KSCN, Na2S and Na2SO4 were used to prepare 50 mM stock solutions of other competitive anions in 5 mL volumetric flasks with doubly distilled water. The changes in the absorption and fluorescence spectra caused by CN− (22 μM) and miscellaneous interfering anions (200 μM) with their K+ or Na+ salts in water solutions were recorded. Each spectrum was acquired 2 min after anion addition.
Computational details
Density functional theory (DFT) calculations using the hybrid PBE0 functional20 and the double-zeta basis set 6-31G(d,p)21 were employed to optimise the geometries of compound PTZ and its adduct with cyanide ions. The long alkyl chain of compound PTZ was replaced by a methyl group to save computational time without diminishing the reliability of the results. Frequency analyses were performed on the optimised geometries to confirm that the geometries are true minima on the potential energy surface. Time-dependent (TD) DFT calculations using the PBE0 functional and the triple-zeta 6-311+G(2d,p) basis set22 that includes diffuse basis functions were then carried out to provide insight into the molecular orbital compositions of the excited singlet states. For compound PTZ, its geometry in the first excited state was also optimised using TD-DFT calculations at the TD-PBE0/6-31G(d,p) level of theory followed by a single point calculation at the TD-PBE0/6-311+G(2d,p) level of theory. In both DFT and TD-DFT calculations, the solvent effect of DMSO was taken into account by the polarisable continuum model (PCM).23 All calculations were carried out using the Gaussian 09 program package.24
Synthesis
The synthetic route of compound PTZ is shown in Scheme 1.
Synthesis of 10-octyl-10H-phenothiazine-3,7-dicarbaldehyde 2. 3,7-Dibromo-10-octyl-10H-phenothiazine (0.469 g, 1.0 mmol) was dissolved in anhydrous THF (20.0 mL), and n-butyl lithium (0.960 mL of 2.5 M solution in hexane, 2.40 mmol) was added dropwise under an argon atmosphere at −78 °C using a syringe. The mixture was stirred for 1 h at −78 °C, and N-FMP (0.172 g, 1.50 mmol) was then added dropwise using a syringe. The mixture was stirred for an additional hour at −78 °C. After warming to room temperature, the mixture was stirred overnight and then poured into hydrochloric acid (1.0 M, 10.0 mL). The mixture was extracted with dichloromethane (20.0 mL × 2). The organic layer was washed with brine (20.0 mL), dried over MgSO4, filtrated, and concentrated. The residue was purified by column chromatography (silica gel, petroleum ether–dichloromethane 1:1) to give compound 2 (0.213 g, 58%) as a yellow powder. 1H NMR (400 MHz, CDCl3): δ 0.86 (t, J = 6.6 Hz, 3H, –CH3), 1.25–1.34 (m, 8H, –CH2–), 1.41–1.46 (m, 2H, –CH2–), 1.78–1.85 (m, 2H, –CH2–), 3.92 (t, J = 7.2 Hz, 2H, N–CH2–), 6.94 (d, J = 8.4 Hz, 2H, Ar-H), 7.55 (s, 2H, Ar-H), 7.65 (d, J = 8.4 Hz, 2H, Ar-H), 9.81 (s, 2H, –CHO). 13C NMR (100 MHz, CDCl3): δ 14.0, 22.6, 26.6, 26.7, 29.0, 29.1, 31.7, 48.5, 115.6, 124.5, 128.4, 130.1, 132.0, 148.9, 189.8.
Synthesis of 2,2′-((10-octyl-10H-phenothiazine-3,7-diyl)bis(methanylydiene))dimalononitrile PTZ. A mixture of compound 2 (0.10 g, 0.272 mmol), pyridine (15.0 mL), and malononitrile (0.066 g, 1.0 mmol) was refluxed under an argon atmosphere for 2 h. After cooling to room temperature, the mixture was poured into water (30.0 mL). The mixture was extracted with dichloromethane (20.0 mL × 2). The organic layer was washed with brine (20.0 mL × 2), dried over MgSO4, filtrated, and concentrated. The residue was purified by column chromatography (silica gel, petroleum ether–dichloromethane 1:1) to give the compound PTZ (0.099 g, 79%) as a purple-red solid. 1H NMR (400 MHz, DMSO-d6): δ 0.76 (t, J = 6.8 Hz, 3H, –CH3), 1.14–1.17 (m, 8H, –CH2–), 1.27–1.32 (m, 2H, –CH2–), 1.58–1.65 (m, 2H, –CH2–), 3.91 (t, J = 6.8 Hz, 2H, N–CH2–), 7.21 (d, J = 8.8 Hz, 2H, Ar-H), 7.58 (s, 2H, Ar-H), 7.78 (dd, J = 8.8 1.6 Hz, 2H, Ar-H), 8.21 (s, 2H, = CH–). 13C NMR (100 MHz, CDCl3): δ 13.1, 21.6, 25.5, 25.7, 28.0, 28.1, 30.6, 47.7, 78.8, 112.0, 113.1, 114.9, 123.0, 125.6, 128.6, 130.2, 147.0, 156.0. HRMS (EI): m/z, [M]+ calcd. for C28H25N5S, 463.1831; found, 463.1832.
Cell culture
HeLa cells (human neuroblastoma cells) were grown in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) at 37 °C in a humidified 5% CO2 atmosphere. Cells (1 × 105 cells per mL) were plated on 14 mm glass cover slips and allowed to adhere for 12 h.
Living cell imaging
Experiments to assess CN− uptake were performed over 20 min in RPMI-1640/DMSO (99:1, v/v) solution supplemented with 20 μM TBACN (tetrabutylammonium cyanide). Before the experiments, HeLa cells (10 μM) were washed with phosphate buffered saline (PBS, pH = 7.4), and then incubated with compound PTZ (10 μM) in RPMI-1640/DMSO (99:1, v/v) solution for 30 min at 37 °C. Cell imaging was then carried out after washing cells with PBS. Confocal fluorescence imaging was performed with an OLYMPUS IX81 laser scanning microscope with a 60P oil-immersion objective lens. Fluorescence images of compound PTZ-loaded cells were monitored at 650–750 nm with an excitation wavelength of 488 nm using a HeNe laser.
Results and discussion
Synthesis and characterisation
The target compound PTZ was prepared simply by the general Knoevenagel condensation reaction between compound 2 and excess malononitrile, as illustrated in Scheme 1. Compound PTZ exhibited good solubility in common organic solvents such as DMSO, DMF, THF, and CH3CN. The chemical structure of compound PTZ was well confirmed by 1H NMR, 13C NMR, and HRMS (see the Experimental section and ESI† for details).
Absorption spectral response to cyanide ions
The changes in the absorption spectrum of compound PTZ with the gradual addition of aqueous CN− were first explored. Considering the biological and environmental applications, the solution of compound PTZ in mixed DMSO–H2O (9:1, v/v) solvent was selected as the testing system to investigate the optical responses at room temperature. As depicted in Fig. 1, compound PTZ displayed two main absorption bands at 321 nm and 488 nm. The absorption band centred at 488 nm, which was attributed to the intramolecular charge transfer (ICT) transition in the molecule, decreased evidently upon the gradual addition of CN− accompanied by the emergence of a new absorption band at 260 nm with an isosbestic point at 274 nm during the titration. When the amount of CN− was 2.2 equiv., the absorption intensity at 488 nm was the weakest. This intense reduction in the absorption resulted in the colour change of the solution from pink to colourless (inset of Fig. 1), indicating that compound PTZ could serve as a colourimetric probe to detect CN− by the naked eye.
 |
| | Fig. 1 UV-vis absorption spectral changes of compound PTZ (10 μM) in the presence of increasing concentrations of CN− (0–22 μM) in DMSO–H2O (9:1, v/v) solution at 25 °C. Each spectrum was acquired 2 min after CN− addition. Inset: colour change of compound PTZ (10 μM) in the absence and presence of CN− (22 μM) under a daylight lamp. | |
Fluorescence spectral response to cyanide ions
Probes are usually disturbed by the protons in solution; thus, their low sensitivity to the operational pH is extremely important. This sensitivity was first investigated by determining the effects of pH on the fluorescence intensity of compound PTZ in the absence and presence of CN−. Fig. 2 shows that compound PTZ can detect CN− within a wide range of pH values (6.0–10.0); in this region, compound PTZ with CN− exhibited dramatic decreases in the fluorescence intensity, whereas compound PTZ without CN− showed no apparent changes in the fluorescence intensity at 702 nm. This result suggested that no buffer solutions are required for the detection of CN−, which is convenient for the practical detection of CN−. In consideration of the environmental and biological applications, the pH value of 7.0 was chosen as the testing condition.
 |
| | Fig. 2 Fluorescence intensity at 702 nm of free PTZ (10 μM) and PTZ + 2.2 equiv. of CN− in DMSO–H2O (9:1, v/v) solution under different pH conditions at 25 °C. λex = 488 nm. Slits: 5 nm/5 nm. Each spectrum was acquired 2 min after CN− addition. | |
The concentration-dependent change of the fluorescence spectrum was explored by adding different amounts of CN− to the solutions of compound PTZ. As shown in Fig. 3, compound PTZ exhibits a strong NIR fluorescence band centred at 702 nm, which arises from the ICT-involved electron-donor group (phenothiazine core) and electron-withdrawing groups (dicyano-vinyl).25 The fluorescence at 702 nm decreased sharply with the gradual addition of CN−, and the fluorescence was completely quenched when 2.2 equiv. of CN− was added to the solution of compound PTZ, indicating that compound PTZ worked as a fluorescent probe in “turn-off” mode. The inset in Fig. 3 demonstrates that the change in the fluorescence colour from red to dark could be easily distinguished by the naked eye using a normal UV lamp. The disappearance of the NIR emission peak should be attributed to the nucleophilic attack by equivalent CN− toward the electrophilic C of the CC double bond to inhibit the ICT effect,8a–d,25 as illustrated in Scheme 2. During the addition process, two electron-withdrawing dicyano-vinyl groups are expected to transform into anionic electron-rich groups along with the destruction of the CC double bonds and the simultaneous emergence of the negative charges.
 |
| | Fig. 3 Fluorescence spectral changes of compound PTZ (10 μM) in the presence of increasing concentrations of CN− (0–22 μM) in DMSO–H2O (9:1, v/v) solution at 25 °C. λex = 488 nm. Slits: 5 nm/5 nm. Each spectrum was acquired 2 min after CN− addition. Inset: fluorescence change of compound PTZ (10 μM) in the absence and presence of CN− (22 μM) under a UV lamp at 365 nm. | |
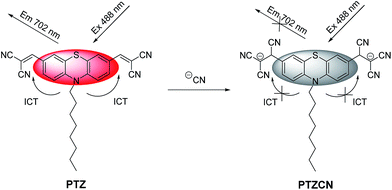 |
| | Scheme 2 Proposed mechanism for the nucleophilic addition of CN− to compound PTZ. | |
To better visualise the sensing process, we plotted the fluorescence intensity at 702 nm with respect to the concentration of CN− (Fig. 4). The fluorescence intensity decreased with CN− concentration and reached saturation at a relatively low CN− concentration of 2.2 equiv. The inset of Fig. 4 shows that the fluorescence intensity at 702 nm varied almost linearly with the concentration of CN− in the range of 0–14 μM, with a coefficient R2 = 0.994. The detection limit for CN− was calculated by the formula (3σ/k),26 where σ is the standard deviation of the blank solution, and k is the slope of the calibration curve obtained from the inset of Fig. 4. The detection limit was determined to be 67 nM, which amounts to one twenty-eighth of the maximum contaminant level (MCL) for CN− in drinking water (1.9 μM) set by the WHO.3 As a NIR fluorescence chemodosimeter, compound PTZ provides a new opportunity to achieve the practical calibration and determination of CN− concentrations in aqueous solutions.
 |
| | Fig. 4 Plot of fluorescence intensity at 702 nm of compound PTZ (10 μM) with the addition of CN− (0–30 μM) in DMSO–H2O (9:1, v/v) solution at 25 °C. λex = 488 nm. Slits: 5 nm/5 nm. Each spectrum was acquired 2 min after CN− addition. Inset: fluorescence intensity at 702 nm as a linear function of CN− concentration from 0 to 14 μM. | |
1H NMR titrations and MS experiments
To elucidate the sensing mechanism of compound PTZ to CN−, 1H NMR titration experiments were carried out in DMSO-d6 (Fig. 5). With the addition of CN−, the peaks of the resonance α-H corresponding to the vinylic protons (Ha and Hb) at 8.21 ppm diminished gradually and finally disappeared at the CN− concentration of 2.2 equiv. Meanwhile, a new peak at 4.04 ppm corresponding to the reaction product tricyano-ethyl protons (H′′a and H′′b) emerged and grew. Moreover, the signals for the aromatic protons (H′′d, H′′f, and H′′h) of the phenothiazine core shifted upfield as a result of the sharp decrease in the electron-withdrawing effect of the dicyano-vinyl groups. These results indicated that the transformation from PTZ to PTZCN occurred (Fig. 5), which was further confirmed by MS (ESI-) experiments. When 2.2 equiv. of CN− was introduced to the PTZ solution, a new peak appeared at m/z 757.47 (Fig. S1 in the ESI†) that was assigned to the [PTZCN + TBA]− adduct (the calculated [C46H61N8S]+ value was 757.47). To determine the stoichiometry of compound PTZ and CN−, the method of continuous variations (Job's plot) was used. When the molar fraction of CN− was around 0.66, the absorbance value approached a maximum, indicating a definite 1:2 reaction mode between compound PTZ and CN− (Fig. 6).
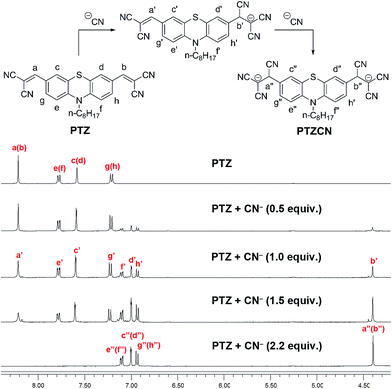 |
| | Fig. 5 Partial 1H NMR (400 MHz) titrations of the chemodosimeter PTZ in DMSO-d6 (25 mM) with the addition of CN− (as TBA salts in DMSO-d6) at 25 °C. Each spectrum was acquired 2 min after CN− addition. | |
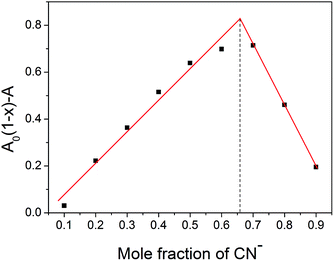 |
| | Fig. 6 Job's plot of compound PTZ and CN−. A and A0 are the absorbance values at 488 nm of compound PTZ in the presence and absence of CN−, respectively. The total concentration of compound PTZ and CN− is 0.10 mM in DMSO–H2O (9:1, v/v) solution at 25 °C. | |
Theoretical calculations
To gain insight into the sensing mechanism, we carried out DFT and TD-DFT calculations on compound PTZ and its adduct with CN−. As shown in Fig. S2 in the ESI,† upon reaction with CN−, the energy levels of both the HOMO and LUMO are lifted; this effect on the LUMO is very significant and leads to an enlarged HOMO–LUMO gap of 4.58 eV for the compound PTZCN. This is expected since the attack of CN− breaks the π-conjugation between the central phenothiazine unit and the peripheral dicyano-vinyl groups. As a consequence, the absorption spectrum of compound PTZ is notably narrowed during the reaction with CN−, and the absorption in the visible region almost vanishes. TD-DFT calculations provide the molecular orbital contributions to the absorption spectra, as shown in Table S1 in the ESI.† The HOMO → LUMO transition of compound PTZ gives rise to the absorption band at the longest wavelength, which corresponds to the charge transfer from the central phenothiazine unit to the peripheral dicyano-vinyl groups. Population analysis suggests that this excitation process involves a charge transfer of 0.217e. The relatively weak absorption band next to the absorption band at the maximum wavelength arises from the HOMO → LUMO+1 transition, which also exhibits a charge transfer character. The two stronger absorption bands at shorter wavelengths correspond to the charge transfer HOMO−1 → LUMO transition and the local HOMO → LUMO+3 transition. To verify the reliability of these results, we have also carried out TD-DFT calculations using the range-separated CAM-B3LYP functional;27 these calculations resulted in very similar molecular orbital contributions and charge transfer characters (Table S2 in the ESI†). Notably, the PBE0 functional underestimates the S0 → S1 excitation energy by ∼0.2 eV, while the CAM-B3LYP functional overestimates the excitation energy by ∼0.2 eV. Since the PBE0 functional has been demonstrated to adequately describe the charge transfer character of a similar cyanide probe based on dihydrophenazine,25 we base our discussion of the results on the TD-DFT calculations using the PBE0 functional.
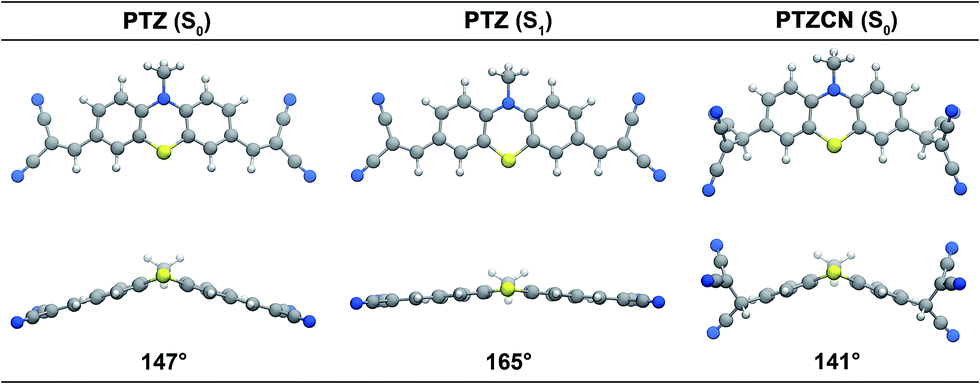
The ground state of compound PTZ has a butterfly-shaped geometry (Fig. 7) that becomes much more flat in the optimised geometry of the first excited state (S1), as reflected by the increase in the bent angle from 147° to 165°. The vertical excitation energy at the optimised S1 is computed as 1.78 eV (Table S1 in the ESI†), in good agreement with experimentally observed fluorescence. This emission arises from the electronic transition between the HOMO and LUMO of compound PTZ, which corresponds to the charge transfer between the central phenothiazine unit and the peripheral dicyano-vinyl groups. Similar to the previously reported dihydrophenazine-based cyanide probe,25 the fluorescence of compound PTZ is of ICT character.
 |
| | Fig. 7 Optimised geometries of compounds PTZ (ground state and excited state) and PTZCN (ground state). | |
The TD-DFT calculations also suggest that the maximum absorption wavelength of compound PTZCN in DMSO appears at around 336 nm (Table S3 in the ESI†), in agreement with experimental observations. Inspection into the frontier molecular orbitals of compound PTZCN reveals that HOMO−1 and HOMO are largely located on the reaction product of CN− and the dicyano-vinyl group, while HOMO−2 and the virtual orbitals are localised on the central phenothiazine unit. As a result, the electronic transitions from HOMO−1 and/or HOMO to the virtual orbitals exhibit rather low oscillator strengths (Table S3 in the ESI†), and only excitations involving the HOMO−2 → LUMO+2 transition show rather strong oscillator strengths. Therefore, due to the breaking of the π-conjugation and the localisation of HOMO−1 and HOMO on the peripheral groups, the absorption spectrum of compound PTZCN is significantly narrowed compared to that of compound PTZ. Upon the addition of CN−, the reaction product PTZCN is almost transparent at 488 nm, and the original fluorescence of compound PTZ disappears in turn.
Selective and competitive experiments
To obtain a promising probe, high selectivity and anti-interference ability toward the analyte over other competitive species are necessary. The fluorescence behaviours of chemodosimeter PTZ to anions (including CN−, F−, Cl−, Br−, I−, NO2−, NO3−, OAc−, PO43−, HPO42−, H2PO4−, HCO3−, CO32−, SCN−, S2−, and SO42−) were tested in DMSO–H2O (9:1, v/v) solution. The fluorescence quenching efficiencies (I0 − I)/I0, where I0 and I respectively represent the fluorescence intensity of compound PTZ in the absence and presence of anions at 702 nm, are displayed in Fig. 8. These competitive anions, some of which often show strong interference in CN− detection, did not bring about any distinct changes in the fluorescence quenching efficiency, even in high equivalents. The competitive experiments also showed that the fluorescence of compound PTZ with CN− was not disturbed by the addition of excess competitive anions. Meanwhile, no significant changes in visual and emission colours were observed in parallel experiments with other anions (Fig. 9). These results further confirmed that only CN− could lead to the apparent colour and fluorescence changes of compound PTZ. In general, compound PTZ exhibits extraordinary selectivity and anti-interference ability for CN− over high concentrations of other investigated anions and can therefore be employed as an excellent optical probe for CN− detection.
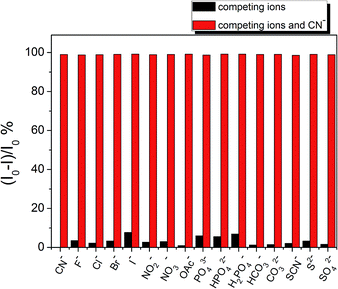 |
| | Fig. 8 The fluorescence quenching efficiency (I0 − I)/I0 of compound PTZ (10 μM) at 702 nm with the addition of various anions in DMSO–H2O (9:1, v/v) solution at 25 °C. λex = 488 nm. Slits: 5 nm/5 nm. The miscellaneous interfering anions are at concentrations of 200 μM, while the concentration of CN− is 22 μM. | |
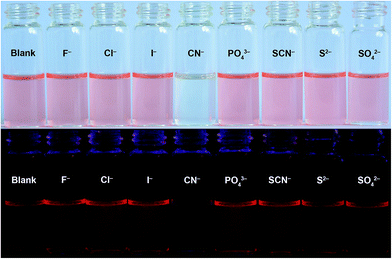 |
| | Fig. 9 Naked-eye visible (above) and fluorescence (below, λex = 365 nm) images of compound PTZ (10 μM) upon the addition of 2.2 equiv. of CN− and 20 equiv. of various interfering anions in DMSO–H2O (9:1, v/v) solution at 25 °C. | |
Fluorescence bioimaging of intracellular cyanide ions
To assess the feasibility of compound PTZ as a NIR probe for biological applications, confocal laser scanning experiments were carried out using HeLa cells to imaging intracellular CN−. Fig. 10 shows the fluorescence and bright field images of HeLa cells. Fig. 10(a)–(c) show that HeLa cells incubated with compound PTZ (10 μM) in RPMI-1640/DMSO (99:1, v/v) solution for 30 min at 37 °C exhibited strong intracellular fluorescence. When cells were supplemented with 20 μM CN− in the growth medium for 20 min at 37 °C followed by subsequent staining with compound PTZ for 30 min at 37 °C, an arresting switch-off fluorescence was observed in the intracellular region (Fig. 10(d)–(f)). These results agreed with the results of the titration experiments. Notably, the bright field images of cells with or without the CN− treatment did not show any gross morphological differences, suggesting that cells were viable throughout the imaging experiments. The overlay of fluorescence and bright field images indicated that fluorescence signals were localised in the cytosol region, suggesting that the probe could efficiently permeate into the living cells from the growth medium. The distribution of PTZ dyes in the cells was further confirmed by Z-scan luminescence imaging of living HeLa cells treated with compound PTZ (Fig. S5 in the ESI†). Moreover, living cell imaging experiments suggested that the intrinsic substances in the cell did not quench the fluorescence of compound PTZ, while a remarkable “on–off” fluorescence was observed in the intracellular region after CN− was introduced to the system. Thus, we conclude that the chemodosimeter PTZ can be utilised to readily monitor CN− within living cells.
 |
| | Fig. 10 Confocal fluorescence and bright field images of HeLa cells. (a) Fluorescence image, (b) bright field image, and (c) overlay image of HeLa cells incubated with compound PTZ (10 μM) in the growth media for 30 min at 37 °C. (d) Fluorescence image, (e) bright field image, and (f) overlay image of HeLa cells supplemented with 20 μM CN− in the growth media for 20 min at 37 °C and then incubated with compound PTZ (10 μM) for 30 min at 37 °C. λex = 488 nm. Collecting region: 650–750 nm. | |
Conclusions
A structurally simple yet efficient chemodosimeter PTZ based on 10-octyl-10H-phenothiazine has been developed for colourimetric and fluorescent detection of cyanide ions in aqueous solution. Benefitting from the nucleophilic addition of the cyanide ions to the α-position of two dicyano-vinyl groups, which blocks the internal charge transfer process, the chemodosimeter PTZ was found to serve as a colourimetric and near-infrared fluorescent probe for the detection of cyanide ions. This probe featured several inherent merits such as high sensitivity and selectivity for cyanide ions, good solubility in aqueous media, and a rapid “turn-off” fluorescence response to cyanide ions. It has also been successfully applied in bioimaging. Moreover, the mechanism of the sensing process was interpreted by 1H NMR titrations, mass spectrometry experiments, and theoretical simulations. Compound PTZ indeed serves as a promising platform for the further development of novel probes with advanced performances. We envisage that this work lays a solid foundation for developing dual-mode anion probes with outstanding optical characteristics.
Acknowledgements
This work was supported by NSFC/China (21406137, 21404068), the Science and Technology Commission of Shanghai Municipality (14YF1410900), the Innovation Program of Shanghai Municipal Education Commission (14YZ128), the Startup Fund of Shanghai University of Electric Power, and the Open Project Fund of Key Labs for Advanced Materials, East China Univ. Science & Technology.
Notes and references
-
(a) Z. C. Xu, X. Q. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127 RSC;
(b) D. W. Boening and C. M. Chew, Water, Air, Soil Pollut., 1999, 109, 67 CrossRef CAS.
- J. L. Gerberding, Toxicological profile for cyanide, U.S. Department of Health and Human Services, Atlanta, 2006 Search PubMed.
- World Health Organization, in Guidelines for drinking-water quality, World Health Organization, Geneva, Switzerland, 2008, vol. 1 Search PubMed.
-
(a) T. Suzuki, A. Hioki and M. Kurahashi, Anal. Chim. Acta, 2003, 476, 159 CrossRef CAS;
(b) B. Vallejo-Pecharromána and M. D. Luque de Castro, Analyst, 2002, 127, 267 RSC;
(c) A. J. Curtis, C. C. Grayless and R. Fall, Analyst, 2002, 127, 1446 RSC;
(d) K. H. Yea, S. Lee, J. B. Kyong, J. Choo, E. K. Lee, S. W. Joo and S. Lee, Analyst, 2005, 130, 1009 RSC;
(e) G. Q. Ding, H. Zhou, J. W. Xu and X. H. Lu, Chem. Commun., 2014, 50, 655 RSC.
- H. Miyaji and J. L. Sessler, Angew. Chem., Int. Ed., 2001, 40, 154 CrossRef CAS.
-
(a) P. Anzenbacher, D. S. Tyson, K. Jursíková and F. N. Castellano, J. Am. Chem. Soc., 2002, 124, 6232 CrossRef CAS PubMed;
(b) Y. H. Kim and J. I. Hong, Chem. Commun., 2002, 512 RSC;
(c) C. Männel-Croisé and F. Zelder, Inorg. Chem., 2009, 48, 1272 CrossRef PubMed;
(d) J. Wang and C. S. Ha, Analyst, 2011, 136, 1627 RSC.
-
(a) S. Das, S. Biswas, S. Mukherjee, J. Bandyopadhyay, S. Samanta, I. Bhowmick, D. K. Hazra, A. Ray and P. P. Parui, RSC Adv., 2014, 4, 9656 RSC;
(b) C. F. Chow, M. H. Lam and W. Y. Wong, Inorg. Chem., 2004, 43, 8387 CrossRef CAS PubMed;
(c) X. D. Lou, L. Y. Zhang, J. Q. Qin and Z. Li, Chem. Commun., 2008, 5848 RSC;
(d) H. S. Jung, J. H. Han, Z. H. Kim, C. Kang and J. S. Kim, Org. Lett., 2011, 13, 5056 CrossRef CAS PubMed.
-
(a) P. B. Pati and S. S. Zade, RSC Adv., 2013, 3, 13457 RSC;
(b) X. S. Zhou, X. Lv, J. S. Hao, D. S. Liu and W. Guo, Dyes Pigm., 2012, 95, 168 CrossRef CAS PubMed;
(c) C. H. Lee, H. J. Yoon, J. S. Shim and W. D. Jang, Chem.–Eur. J., 2012, 18, 4513 CrossRef CAS PubMed;
(d) W. C. Lin, S. K. Fang, J. W. Hu, H. Y. Tsai and K. Y. Chen, Anal. Chem., 2014, 86, 4648 CrossRef CAS PubMed;
(e) X. H. Cheng, R. L. Tang, H. Z. Jia, J. Feng, J. G. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2012, 4, 4387 CrossRef CAS PubMed;
(f) H. D. Li, Z. Wen, L. Y. Jin, Y. H. Kan and B. Z. Yin, Chem. Commun., 2012, 48, 11659 RSC;
(g) B. Chen, Y. B. Ding, X. Li, W. H. Zhu, J. P. Hill, K. Ariga and Y. S. Xie, Chem. Commun., 2013, 49, 10136 RSC.
-
(a) S. Madhu, S. K. Basu, S. Jadhav and M. Ravikanth, Analyst, 2013, 138, 299 RSC;
(b) Y. L. Duan and Y. S. Zheng, Talanta, 2013, 107, 332 CrossRef CAS PubMed;
(c) S. J. Hong, J. Yoo, S. H. Kim, J. S. Kim, J. Yoon and C. H. Lee, Chem. Commun., 2009, 189 RSC;
(d) D. G. Cho, J. H. Kim and J. L. Sessler, J. Am. Chem. Soc., 2008, 130, 12163 CrossRef CAS PubMed;
(e) Z. Ekmekci, M. D. Yilmaz and E. U. Akkaya, Org. Lett., 2008, 10, 461 CrossRef CAS PubMed;
(f) M. K. Bera, C. Chakraborty, P. K. Singh, C. Sahu, K. Sen, S. Maji, A. K. Das and S. Malik, J. Mater. Chem. B, 2014, 2, 4733 RSC.
-
(a) J. T. Zhang, S. L. Zhu, L. Valenzano, F. T. Luo and H. Y. Liu, RSC Adv., 2013, 3, 68 RSC;
(b) Y. Zhang, D. H. Yu and G. Q. Feng, RSC Adv., 2014, 4, 14752 RSC;
(c) M. J. Peng, Y. Guo, X. F. Yang, F. Suzenet, J. Li, C. W. Li and Y. W. Duan, RSC Adv., 2014, 4, 19077 RSC;
(d) S. Kumar, P. Singh, G. Hundal, M. S. Hundal and S. Kumar, Chem. Commun., 2013, 49, 2667 RSC;
(e) Y. T. Yang, C. X. Yin, F. J. Huo, J. B. Chao, Y. B. Zhang and F. Q. Cheng, Sens. Actuators, B, 2014, 193, 220 CrossRef CAS PubMed.
-
(a) M. Jamkratoke, V. Ruangpornvisuti, G. Tumcharern, T. Tuntulani and B. Tomapatanaget, J. Org. Chem., 2009, 74, 3919 CrossRef CAS PubMed;
(b) R. Badugu, J. R. Lakowicz and C. D. Geddes, J. Am. Chem. Soc., 2005, 127, 3635 CrossRef CAS PubMed;
(c) T. W. Huanall and F. P. Gabbaï, J. Am. Chem. Soc., 2007, 129, 11978 CrossRef PubMed.
-
(a) Q. Zou, L. Zou and H. Tian, J. Mater. Chem., 2011, 21, 14441 RSC;
(b) Q. Zou and H. Tian, Sens. Actuators, B, 2010, 149, 20 CrossRef CAS PubMed;
(c) J. Q. Ren, W. H. Zhu and H. Tian, Talanta, 2008, 75, 760 CrossRef CAS PubMed.
-
(a) C. Y. Li, M. X. Yu, Y. Sun, Y. Q. Wu, C. H. Huang and F. Y. Li, J. Am. Chem. Soc., 2011, 133, 11231 CrossRef CAS PubMed;
(b) W. H. Zhu, X. M. Huang, Z. Q. Guo, X. M. Wu, H. H. Yu and H. Tian, Chem. Commun., 2012, 48, 1784 RSC.
-
(a) W. J. Wu, J. B. Yang, J. L. Hua, J. Tang, L. Zhang, Y. T. Long and H. Tian, J. Mater. Chem., 2010, 20, 1772 RSC;
(b) S. H. Bae, K. D. Seo, W. S. Choi, J. Y. Hong and H. K. Kim, Dyes Pigm., 2015, 113, 18 CrossRef CAS PubMed.
- D. Cummins, G. Boschloo, M. Ryan, D. Corr, S. N. Rao and D. Fitzmaurice, J. Phys. Chem. B, 2000, 104, 11449 CrossRef CAS.
- J. Lee, J. Kwak, K. C. Ko, J. H. Park, J. H. Ko, N. Park, E. Kim, D. H. Ryu, T. K. Ahn, J. Y. Lee and S. U. Son, Chem. Commun., 2012, 48, 11431 RSC.
- Y. L. Liu, H. Y. Cao, J. H. Li, Z. J. Chen, S. K. Cao, L. X. Xiao, S. G. Xu and Q. H. Gong, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4867 CrossRef CAS.
- M. D. Sun, J. Guo, Q. B. Yang, N. Xiao and Y. X. Li, J. Mater. Chem. B, 2014, 2, 1846 RSC.
- G. Y. Sang, Y. P. Zou and Y. F. Li, J. Phys. Chem. C, 2008, 112, 12058 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 77, 3865 CrossRef ; Errata, Phys. Rev. Lett., 1996, 78, 1396.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani and V. Barone, et al., Gaussian 09, Revision A.2, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- L. Yang, X. Li, J. B. Yang, Y. Qu and J. L. Hua, ACS Appl. Mater. Interfaces, 2013, 5, 1317 CAS.
- Q. Zou, X. Li, J. J. Zhang, J. Zhou, B. B. Sun and H. Tian, Chem. Commun., 2012, 48, 2095 RSC.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of supplemental spectra, DFT/TDDFT calculations. See DOI: 10.1039/c4ra11567h |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.