Chemoselective sulfenylation and peptide ligation at tryptophan†
Lara R.
Malins
,
Katie M.
Cergol
and
Richard J.
Payne
*
School of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: richard.payne@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 5877
First published on 10th October 2013
Abstract
Peptide ligation–desulfurization chemistry at 2-thiol tryptophan (Trp) is described for the first time. Installation of a thiol auxiliary was achieved through late-stage chemoselective sulfenylation chemistry at the 2-position of the indole ring of Trp either in solution or on solid support, thus abrogating the need for the preparation of a pre-formed thiolated amino acid. Peptides possessing the 2-thiol Trp functionality on the N-terminus were shown to facilitate high yielding ligation reactions with a variety of C-terminal peptide thiophenyl thioesters. Efficient removal of the 2-thiol Trp auxiliary following the ligation reactions was achieved via reductive desulfurization and provided native peptide products in excellent yields. The utility of the methodology was demonstrated in the synthesis of a glycosylated fragment of the N-terminal extracellular domain of the chemokine receptor CXCR1.
Introduction
Advances in peptide ligation chemistry have expanded the array of synthetic tools available for the construction of complex proteins and glycoproteins.1–3 The most widely used method, native chemical ligation,4 involves the reaction of an N-terminal cysteine (Cys) residue with a C-terminal peptide thioester, in which an initial transthioesterification step is followed by a rapid S to N acyl shift to yield a native peptide bond. The technique is chemoselective in the presence of unprotected amino acid side chains and occurs in aqueous media at neutral pH. The requirement of a Cys residue for the ligation event has led to significant interest in the synthesis of unnatural amino acids bearing reactive thiol or selenol auxiliaries capable of facilitating ligation reactions5via the same mechanism proposed for native chemical ligation. Desulfurization or deselenization following the ligation event,6,7 using either metal8 or free-radical9,10 based procedures, then provides a native amino acid residue at the ligation junction. This strategy, first demonstrated by Dawson and coworkers in the ligation and subsequent conversion of Cys to alanine (Ala),8 has also enabled ligation at phenylalanine (Phe),11–13 valine (Val),14,15 lysine (Lys),16,17 threonine (Thr),18 proline (Pro),19,20 leucine (Leu),21,22 glutamine (Gln),23 arginine (Arg)24 and aspartic acid (Asp).25The development of these ligation approaches has greatly expanded the scope of ligation beyond Cys-containing peptides. However, the main obstacle in utilising ligation methods at amino acids other than Cys is the synthetic challenge of accessing a suitably protected thiol- or selenol-functionalised amino acid building block for direct incorporation into peptides via solid-phase peptide synthesis (SPPS) (Scheme 1A). Indeed, with the exception of Val ligations (achieved using suitably protected, commercially available penicillamine)15 and thiol and selenol Pro-mediated ligations (using commercially available 4-thioproline19 or 4-hydroxyproline precursors,20 respectively) current approaches often require numerous synthetic steps (7–16)5 thus limiting the widespread use of these building blocks in peptide ligation chemistry. We envisioned that an ideal approach to thiol- or selenol-derived amino acids would involve late-stage installation of the ligation auxiliary, following the construction of the peptide (Scheme 1B), thereby eliminating the need for the synthesis of a preformed building block. Given the vast array of functionality represented in the side chains of proteinogenic amino acid residues, chemoselectivity is a major barrier to selective installation of a reactive thiol or selenol auxiliary into a fully synthesised, unprotected peptide.
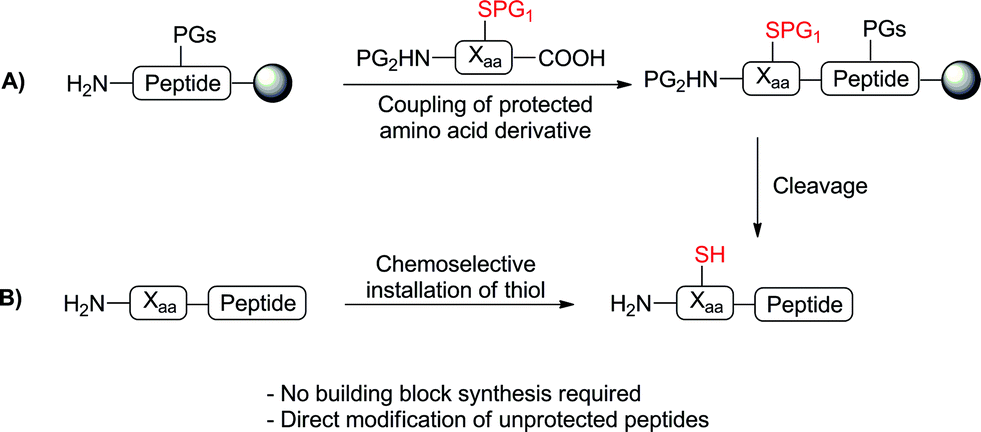 | ||
| Scheme 1 (A) Incorporation of a synthetic thiolated building block via SPPS and (B) direct, chemoselective installation of a thiol onto unprotected peptides; Xaa = any amino acid, PG = protecting group. | ||
We were encouraged by early reports from Scoffone et al. that demonstrated the site-selective modification of the nucleophilic 2-position of the Trp indole ring through electrophilic sulfenylation with various sulfenyl chlorides.26,27 Importantly, these studies showed that in acidic media other nucleophilic amino acid side-chains remained unmodified, as in the case of the ε-amino moiety of Lys and the alcohol functionalities of serine (Ser) and Thr, or reversibly modified in the case of Cys, which forms an asymmetric disulfide that can be easily reduced. Additionally, facile thiolytic cleavage of the resulting 2-Trp thioether peptide derivatives using an external thiol nucleophile can yield the corresponding 2-thiol Trp derivatives.28 To date, this chemistry has been used in a few isolated examples, specifically to install a 2-sulfhydryl group at the sole Trp residue in the peptide hormones glucagon,29,30 corticotropin,31 and luliberin.32 However, the true scope of this chemistry remains largely unexplored. Given the ease with which a potentially reactive thiol auxiliary can be installed chemoselectively onto Trp residues in fully deprotected peptides and proteins, we were interested in investigating whether 2-thiol Trp containing peptides would be capable of facilitating peptide ligation chemistry with C-terminal peptide thioesters. Current methods for ligation at N-terminal Trp residues include a Pictet–Spengler approach by Tam and co-workers, which results in the formation of an unnatural tetrahydro-β-carboline linkage,33 and the ligation of Trp isopeptide units by N- to N-acyl transfer,34 which is reliant on basic, non-aqueous reaction media and a multi-step synthesis of pre-formed isopeptide ligation precursors which can undergo the requisite rearrangement. Such factors limit the general applicability of these approaches in the synthesis of native peptide and protein targets. We envisioned that ligation chemistry facilitated by 2-thiol Trp-containing peptides would greatly enhance the scope and utility of ligation technologies, especially if peptide precursors could be accessed in a simple and rapid manner. In an approach mechanistically similar to native chemical ligation, we proposed that a reaction with a peptide thioester would proceed via an initial transthioesterification step with the indole 2-thiol functionality followed by an S- to N-acyl shift through a 7-membered ring transition state to generate a native amide bond (Scheme 2). Based on prior reports of auxiliary-mediated ligations,35–39 we envisaged that the S- to N-acyl rearrangement would still occur through the larger ring sized transition state when compared with native chemical ligation (5-membered ring transition state).
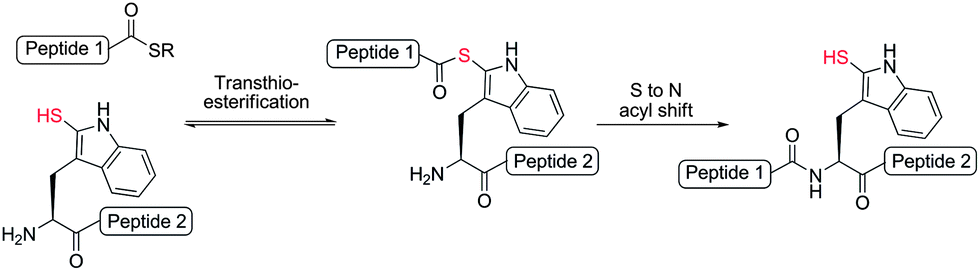 | ||
| Scheme 2 Proposed peptide ligation reaction at 2-thiol Trp. | ||
Results and discussion
Sulfenylation chemistry
Our exploration of 2-thiol Trp for the development of a new peptide ligation reaction began with the synthesis of an appropriate model system bearing the 2-thiol Trp moiety (Scheme 3). To this end, we first constructed the resin-bound peptide 1 through standard Fmoc-SPPS on Rink amide resin (see ESI†). Following cleavage from the resin using an acidic cocktail, the fully deprotected peptide H-WSPGYS-NH22 was obtained in 88% yield. This peptide was then subjected to sulfenylation using commercially available 2,4-dinitrophenylsulfenyl chloride (DNPS-Cl) in AcOH26 for 16 h at ambient temperature to afford the 2-sulfenylated peptide 3 in 56% yield. The chemo- and regioselectivity of the reaction was confirmed via1H NMR spectroscopy (see ESI†), which revealed exclusive reaction at the 2-position of the Trp indole ring.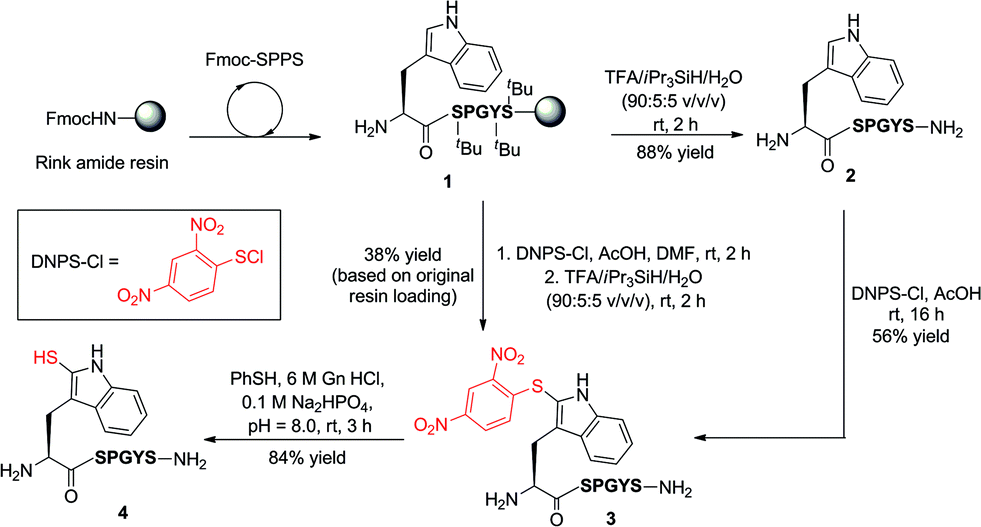 | ||
| Scheme 3 Synthesis of 2-thiol Trp peptide 4via sulfenylation (in solution or on-resin) and subsequent thiolysis. (S = Ser, P = Pro, G = Gly, Y = Tyr.) | ||
To improve the efficiency of the reaction and minimise intermediate purification steps, we also sought to effect sulfenylation of Trp directly on resin. Gratifyingly, we were able to achieve solid-phase sulfenylation through treatment of resin-bound peptide 1 with 20 eq. DNPS-Cl and 20 eq. AcOH in DMF at ambient temperature for 2 h to generate, after acidic cleavage from the resin, peptide 3 in 38% yield based on the original resin loading. Interestingly, the success of the reaction required the presence of an unprotected indole nitrogen. Side-chain Boc-protected Trp residues (often incorporated as standard protection of the side chain indole) were unreactive to the sulfenylation conditions. This suggests that site-selective solid-phase sulfenylation of peptides bearing multiple Trp residues may be possible. Following solution or solid-phase sulfenylation, DNPS removal from peptide 3 was facilitated through thiolysis to unmask the reactive indole thiol functionality. Although previous studies have utilised β-mercaptoethanol to effect thiolysis,28 in our hands, treatment with excess thiophenol in the presence of a buffer comprised of 6 M guanidine hydrochloride (Gn·HCl)/0.1 M Na2HPO4 at pH = 8.0 optimally afforded 2-thiol Trp containing peptide 4 in 84% yield following HPLC purification. It should be noted that this peptide rapidly oxidized to the corresponding disulfide dimer, which was solely isolated following lyophilisation. In addition to NMR spectroscopy and HPLC analysis, our model peptides 2–4 were also characterised by UV absorption spectroscopy to provide a simple and convenient means of following reactions at the 2-position of Trp within these molecules (Fig. S1 and Table S2, see ESI† for details). Importantly, each peptide possessed a characteristic absorption pattern, which may therefore be used to follow transformations in large peptide and protein systems via UV spectroscopic experiments.
Ligation at 2-thiol Trp
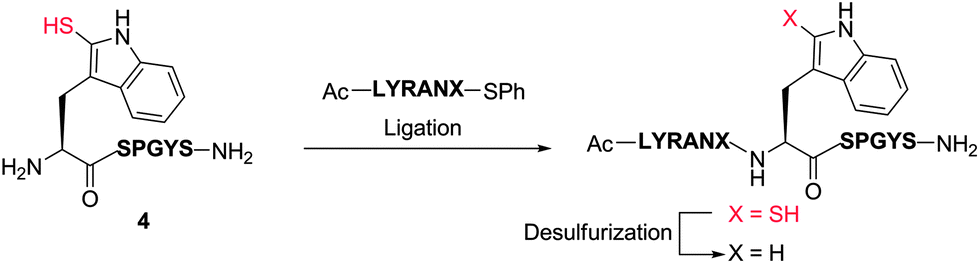
With model peptide 4 bearing an N-terminal 2-thiol Trp residue in hand, we were next interested in exploring the scope of ligation chemistry at the thiol-derived Trp residue. To this end, a variety of C-terminal peptide alkyl thioesters [Ac-LYRANX-S(CH2)2CO2Et, where X = glycine (Gly), Ala, methionine (Met), Phe, Pro] were synthesised according to literature methods.40 A model Gly thioester (Ac-LYRANG-S(CH2)2CO2Et) was chosen for the initial screening and optimisation studies. Under standard native chemical ligation conditions (6 M Gn·HCl/0.1 M Na2HPO4/100 mM tris(2-carboxyethyl)phosphine (TCEP), 5 mM concentration with respect to 4, pH = 7.3–7.5, 2 vol% PhSH, 37 °C)4,41 we were disappointed to observe only a small amount of the desired ligation product after 24 h, with the major by-product corresponding to the starting peptide bearing a phenyl thioether at the indole 2-position (see Fig. S6, ESI†). We hypothesised that this by-product arose following the initial transthioesterification step, in which the newly formed electrophilic thioester intermediate was intercepted at the C-2 position of the indole with excess exogenous aryl thiol before the S- to N-acyl shift event could occur. The resulting thiophenyl-derived indole was then incapable of facilitating ligation. To overcome this issue, we decided to synthesise a pre-formed activated thiophenyl thioester (Ac-LYRANG-SPh, 5) for use in the ligation reaction (see ESI† for synthetic details). This aryl thioester was chosen based on the previous success of this functionality in kinetically controlled native chemical ligation.42 In addition, we chose to perform the ligation reaction at a lower concentration (1.6 mM with respect to 4) to minimise attack of the indole C-2 position by the one molar equivalent of thiophenol displaced in the initial transthioesterification step. We were pleased to find that under these modified conditions, reaction of 4 with Gly thiophenyl thioester 5 resulted in conversion to the desired ligation product (along with substantial thioester hydrolysis) via HPLC-MS without any observed formation of the 2-thiophenyl Trp by-product.Having demonstrated that ligation at Trp could be effected at a C-terminal Gly thioester 5, we next conducted a reaction with a model Ala thiophenyl thioester (Ac-LYRANA-SPh 6) to assess the effects of increased steric demand at the ligation junction. Gratifyingly, this reaction also proceeded without formation of the thiophenyl ether by-product under the same conditions used for the ligation at the Gly thioester. However, in this case we noted the appearance of a small amount (ca. 10%) of epimerized thiophenyl thioester during the course of the reaction. Interestingly, despite the observed epimerization of the starting material, the stereochemical integrity of the ligation product was retained (as judged by HPLC-MS analysis). Given this surprising result, we decided to probe the potential role of the 2-thiol Trp moiety within peptide 4 on the observed epimerization. To this end, we first incubated the Ala thiophenyl thioester 6 under ligation conditions in the absence of 4 (see ESI†, Fig. S40). To our surprise, at pH 7, the model Ala thiophenyl thioester underwent significant epimerization (ca. 15% at t = 1 h) along with substantial hydrolysis over time. Epimerization was also observed upon incubation of 6 with ligation buffer in the absence of TCEP (Fig. S42†). In order to avoid epimerization of the acyl donor peptide we subsequently screened a variety of pre-formed activated thioesters with varying reactivities (as estimated by the pKa value and corresponding leaving group propensity of the thiol component).43 These included peptide thioesters bearing C-terminal 4-mercaptophenylacetic acid (MPAA), 4-(amino)thiophenol, 2-(hydroxy)thiophenol and 2-mercaptoethanesulfonate sodium salt (MESNa) moieties. The more reactive aryl thioesters (lower pKa values) all led to enhanced hydrolysis and/or epimerization upon incubation, while the MESNa-derived alkyl thioester did not epimerize but was highly unreactive in 2-thiol Trp-mediated peptide ligations. To the best of our knowledge, epimerization of pre-formed aryl thioesters in ligation reactions at N-terminal Cys or thiol-derived amino acids has not been previously observed. It is highly probable that ligations at Cys residues (and most thiolated amino acids) with the majority of activated C-terminal peptide thioesters proceed extremely rapidly and without thioester epimerization. However, slower ligation systems, particularly those involving more sterically demanding or weakly nucleophilic N-terminal Cys surrogates and/or bulky C-terminal thioesters requiring lengthy reaction times41 may be prone to epimerization by-products. It is therefore recommended that such tendencies are carefully considered in the design of new ligation methodologies and in experiments employing pre-formed activated thioesters.
In light of these considerations and having demonstrated previously that ligation chemistry at 2-thiol Trp was possible using pre-formed Gly and Ala thiophenyl thioesters 5 and 6, respectively, we proceeded to further optimise this system to ensure the stereochemical integrity of the products and to improve the yield of the reaction. Specifically, to reduce the likelihood of epimerization in the ligated product, we first performed ligation reactions at reduced pH (6.5–6.7). In addition, an excess of peptide thioester (1.5–2.0 eq. total) was added over two separate portions (see Table 1) to reduce thioester hydrolysis and to simultaneously minimise the amount of time the thiophenyl thioester was incubated with the ligation buffer. The ligation reactions were also performed at a 4 mM concentration with respect to peptide 4, which further increased the rate of ligation whilst suppressing the thiophenol ether by-product previously observed. Under our optimised conditions, we were able to successfully ligate 4 with model Gly and Ala thiophenyl thioesters 5 and 6, with the desired ligation products isolated in excellent yields by reverse-phase HPLC purification following a 24 h reaction time (71% and 81%, respectively, see Table 1, entries 1 and 2). Importantly, the reaction with C-terminal Ala thioester 6 did not lead to any detectable epimerization in the ligation product (see Fig. 1A). Interestingly, in both cases, the 2-thiol Trp ligation products quickly oxidized in the presence of air to generate the corresponding disulfides. The high oxidation potential is evident in Fig. 1A, where peak (a) represents the free thiol ligation product and peak (b) the formation of the corresponding disulfide, despite the presence of an excess of the phosphine reducing agent (TCEP) in the ligation buffer.
|
|
|||
|---|---|---|---|
| Entry | Thioester (X =) | Ligationa | Desulfurizationa |
| a Isolated yields. b Additional thioester added at t = 8 h. c Additional thioester added at t = 15 h. d t = 30 h reaction time; ligation conditions: buffer (6 M Gn·HCl/0.1 M Na2HPO4/100 mM TCEP, 4 mM concentration with respect to 4), final pH = 6.5–6.7, 37 °C, 24 h; desulfurization conditions: Pd/Al2O3, buffer (6 M Gn·HCl/0.1 M Na2HPO4, adjusted to pH 5.8), H2 gas, 0 °C, 4 h. (L = Leu, R = Arg, N = Asn.) | |||
| 1 | 5: Glyb | 71% | 71% |
| 2 | 6: Alac | 81% | 89% |
| 3 | 7: Met | 80% | 61% |
| 4 | 8: Phe | 65% | 72% |
| 5 | 9: Prod | 58% | 82% |
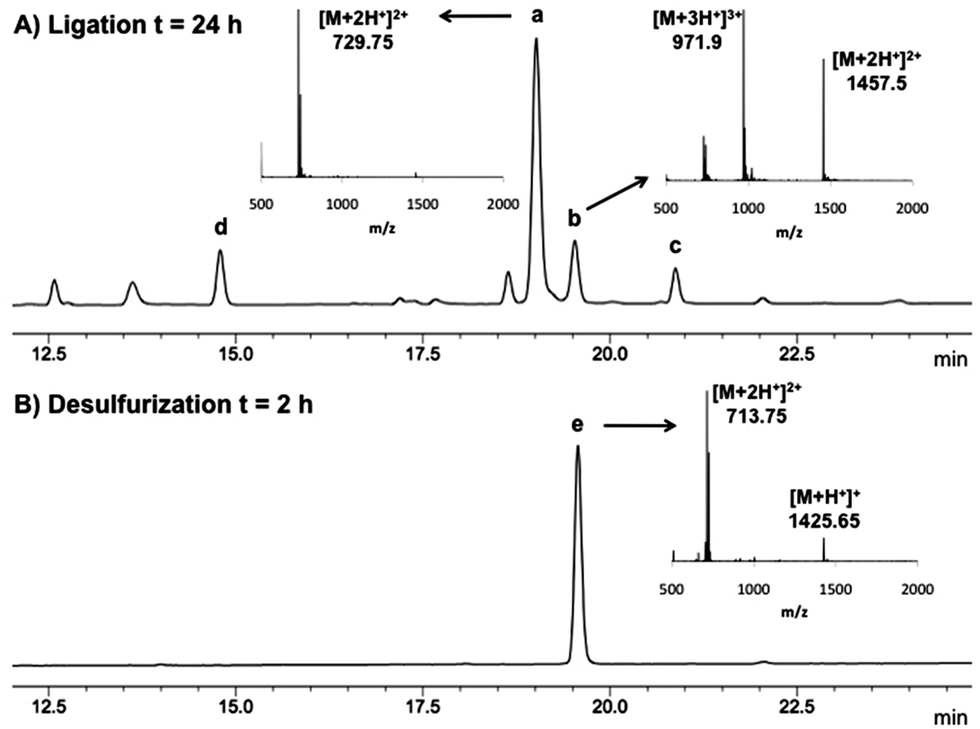 | ||
| Fig. 1 (A) Analytical HPLC trace (λ = 280 nm) of a representative crude ligation reaction between 2-thiol Trp peptide 4 and Ac-LYRANA-SPh 6; peak a = ligation product (free thiol) [M + 2H+]2+ = 729.75; b = ligation product (disulfide) [M + 2H+]2+ = 1457.5; c = Ac-LYRANA-SPh 6 [M + H+]+ = 841.4; d = hydrolysed thioester [M + H+]+ = 749.5. (B) Analytical HPLC trace (λ = 280 nm) of a crude desulfurization reaction of the ligation product shown in (A); e = desulfurization product [M + H+]+ = 1425.65. | ||
We were next interested in gauging the scope of the reaction at peptide thioesters bearing a range of C-terminal residues with varying degrees of steric bulk. For this purpose, thiophenyl thioesters (Ac-LYRANX-SPh) bearing C-terminal Met 7, Phe 8 and Pro 9 residues (in addition to thioesters 5 and 6 described above) were synthesised (see ESI†). Under the optimised conditions developed previously (6 M Gn·HCl/0.1 M Na2HPO4/100 mM TCEP, 4 mM concentration with respect to 4, pH = 6.5–6.7, 37 °C), we were able to successfully ligate peptide 4 to the model Met and Phe thiophenyl thioesters in good yields (80% and 65%, respectively) following HPLC purification and without detectable epimerization of the ligation products. Reactions were allowed to proceed for 24 h although the ligations had progressed considerably within the first 4 h of reaction time (see Fig. S16 and S18, ESI†). Notably, the ligation reaction with model C-terminal Pro thioester 9 was run for 30 h to provide the desired ligation product in 58% yield after HPLC purification (Table 1, entry 5). The notoriously sluggish reactivity of Pro thioesters has been attributed to a decrease in electrophilicity of the thioester carbonyl carbon caused by an orbital interaction with the Pro amide oxygen.44 By pre-forming a highly activated thiophenyl thioester, we were able to increase the reactivity of the thioester carbonyl carbon to effectively facilitate the ligation reaction.
Desulfurization
Having successfully demonstrated that peptide ligation reactions could be conducted at an N-terminal 2-thiol Trp-containing peptide, we next sought to remove the thiol auxiliary to generate native peptide products. Commonly employed metal-free radical desulfurization of the ligation products using VA-044 as an initiator9 was first conducted. Unfortunately, these conditions did not facilitate desulfurization, likely due to the relative strength of the carbon–sulfur bond in the 2-thiol indole moiety and significant contributions of the indoline-2-thione tautomeric form.45 On the basis of literature precedent for the reductive desulfurization of indoline-2-thiones,46 we reasoned that standard metal-based reductive desulfurization protocols could be used to facilitate removal of the thiol auxiliary. Indeed, we were pleased to find that treatment with Pd on Al2O3 in buffer (6 M Gn·HCl/0.1 M Na2HPO4, adjusted to pH 5.8) in the presence of H2 gas at 0 °C8 yielded cleanly desulfurized peptides (61–89% isolated yield) in under 4 h without any observable over-reduction of the Met, Trp, Tyr, or Phe residues (see Table 1 and Fig. 1B). Importantly, as further confirmation of the stereochemical integrity of our 2-thiol Trp ligation–desulfurization protocol, desulfurized products derived from potentially epimerizable thioesters (X = Ala, Met, Phe) were compared to analytical standards synthesised through standard Fmoc-SPPS containing both L- and D-amino acids at the C-terminal thioester ligation junction. 1H NMR spectroscopy and analytical HPLC co-elution studies (see ESI†) confirmed the successful preparation of the desired ligation products with complete retention of stereochemistry of the residue corresponding to the C-terminal amino acid of the peptide thioester component. This confirmation of stereochemical integrity suggests that under the optimised ligation conditions, the rate of ligation is substantially faster than the rate of epimerization of the activated thiophenyl thioester.Following the success of the ligation–desulfurization protocol employing model 2-thiol Trp peptide 4, we were next interested in further gauging the compatibility of the methodology in the presence of additional Met and Cys residues within a given peptide sequence. To this end, we synthesised peptide 10, bearing a C-terminal Met residue and an N-terminal 2-thiol Trp residue. Assembly of the peptide by standard Fmoc-SPPS, followed by solid-phase sulfenylation and cleavage from the resin provided peptide 11 in 34% yield based on the original resin loading (Scheme 4). Thiolysis of peptide 11 subsequently afforded peptide 10 bearing a 2-thiol Trp moiety in 71% yield. This model peptide was next ligated with thiophenyl thioester 8, containing a C-terminal Phe residue, under the optimised ligation conditions to provide 12 in 78% yield following HPLC purification. Removal of the thiol ligation auxiliary via reductive desulfurization then afforded the native peptide product 13 in 99% isolated yield with no evidence of Met desulfurization. Peptide 10 was similarly ligated with an additional model peptide thioester, Ac-LYRC(Acm)NG-SPh 14, containing an internal Cys(Acm)47 residue, providing 15 in 61% yield. Protection of the Cys residue was necessary to prevent the conversion of Cys to Ala upon reductive desulfurization, as has been reported for native chemical ligation–desulfurization.8 Treatment of 15 with H2 and Pd on Al2O3 successfully effected removal of the 2-thiol Trp auxiliary, without reduction of the Cys or Met residue, to afford 16 in 66% yield following isolation by reverse-phase HPLC.
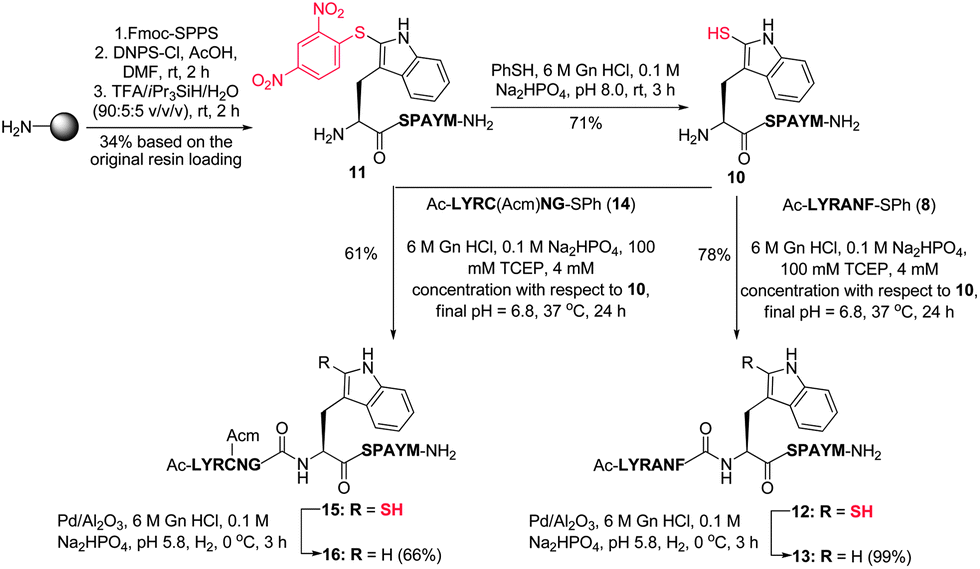 | ||
| Scheme 4 Synthesis of 2-thiol Trp peptide 10via solid-phase sulfenylation and subsequent ligation with thioesters 8 and 14. (M = Met, C = Cys.) | ||
Having demonstrated the feasibility of the ligation–desulfurization protocol on a variety of model peptides, we were next interested in applying the methodology to the synthesis of a more complex target. Specifically, we chose to assemble a glycopeptide fragment of the N-terminal extracellular domain of the chemokine receptor CXCR1, which bears a single Trp residue. We envisaged that the target peptide, CXCR1 (1–28) 17, containing an N-linked GlcNAc moiety β-linked to an Asn residue and three internal Met residues, could be accessed via ligation–desulfurization chemistry between glycopeptide 18 bearing an N-terminal 2-thiol Trp moiety, corresponding to CXCR1 (10–28), and peptide thioester 19 corresponding to residues 1–9 of CXCR1 (Scheme 5). CXCR1 (10–28) 20 was first assembled on resin via standard Fmoc-SPPS, which included coupling of glycosylamino acid cassette 21. This was followed by on-resin deacetylation of the acetate protecting groups on the carbohydrate and solid-phase sulfenylation to afford 20 in 9% yield after cleavage and purification (see ESI† for details). Thiolysis of peptide 20 then afforded the 2-thiol Trp glycopeptide 18 in 60% yield. Subsequent ligation with the CXCR1 (1–9) thiophenyl thioester 19 proceeded to completion within 5 h under the ligation conditions described earlier (see ESI†) and following HPLC purification, the desired product 22 was isolated in excellent yield (91%). Initial desulfurization of 22 under standard reductive desulfurization conditions (0 °C, 3 h) resulted in the partial desulfurization (∼15–20%) of a single, highly labile Met residue, a by-product that was not previously observed in the model peptide examples. The reaction was subsequently performed for a shorter period of time (5 min), which effected quantitative conversion to CXCR1 (1–28) 17 as judged by HPLC-MS analysis with no evidence of the concomitant reduction of Met. Unfortunately, isolation of 17via preparative HPLC following reductive desulfurization was complicated by adsorption of the peptide to the Pd catalyst. However, treatment with excess thiourea prior to purification facilitated isolation of the target glycopeptide 17 in excellent yield (79%). Importantly, the application of the 2-thiol Trp ligation–desulfurization protocol in the high-yielding synthesis of CXCR1 (1–28) highlights the utility of this methodology for the construction of complex peptide targets.
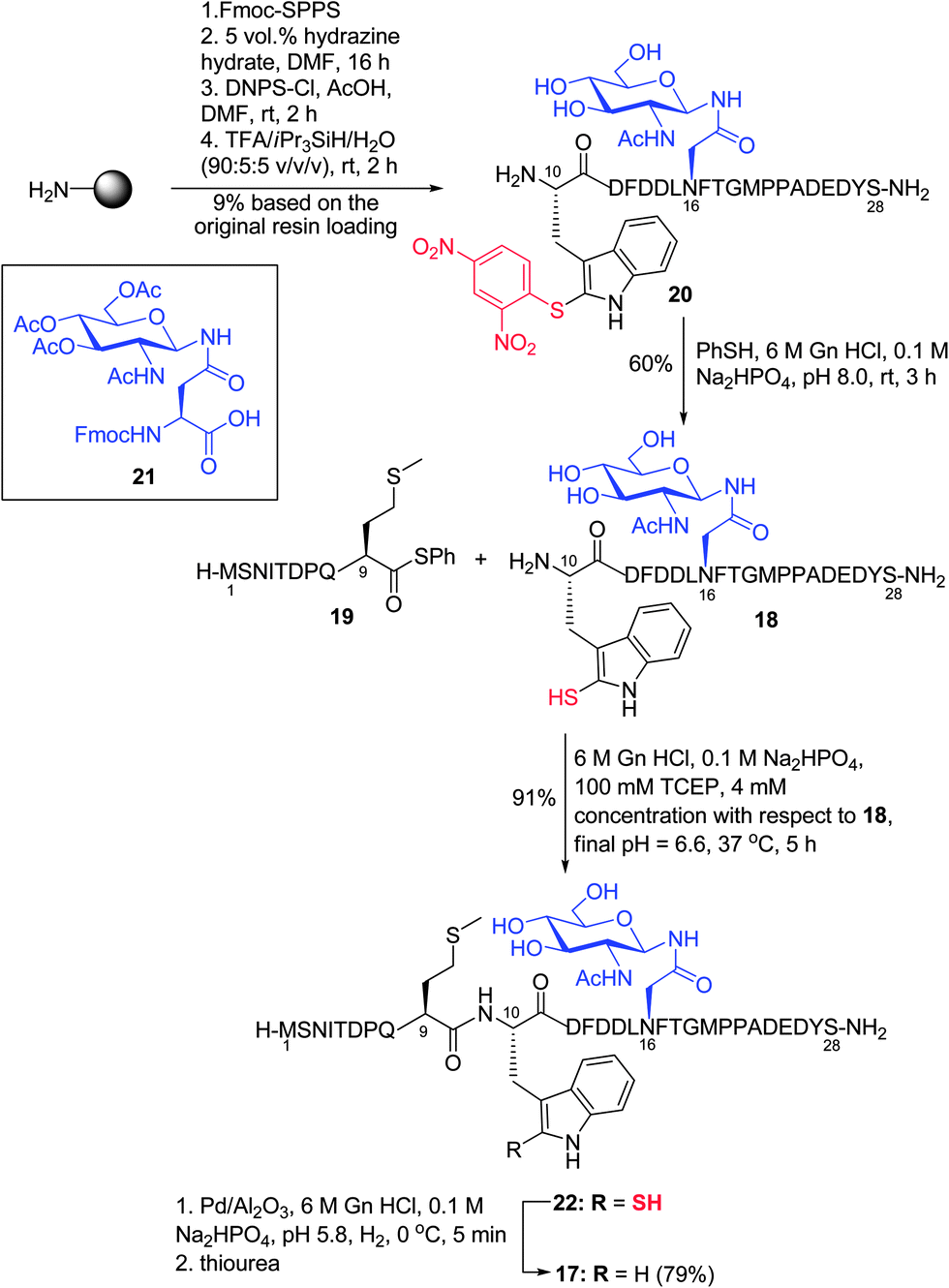 | ||
| Scheme 5 Synthesis of CXCR1 (1–28) via ligation at 2-thiol Trp. (D = Asp, T = Thr, E = Glu, I = Ile, Q = Gln.) | ||
Conclusions
In summary, we have successfully developed the first example of ligation–desulfurization chemistry at Trp. In contrast to the majority of ligation–desulfurization approaches developed at thiol-derived proteinogenic amino acids that require the multi-step synthesis of protected amino acid derivatives, our approach involves the convergent and chemoselective solution or solid-phase sulfenylation of Trp-containing peptides and subsequent conversion to the 2-thiol Trp derivative in completely unprotected peptide fragments. The efficient ligation of model peptides bearing a 2-thiol Trp residue with a variety of pre-formed thiophenyl thioesters, including Pro, has been demonstrated, along with the clean desulfurization of the resulting ligation products to afford native peptides. Importantly, the desulfurization method was shown to be generally tolerant of aromatic side-chains, Met residues and Cys(Acm) groups. Further application of the ligation methodology to the synthesis of the glycopeptide CXCR1 (1–28) demonstrated the utility of the approach in larger peptide systems. Given the ease of installation of the reactive thiol auxiliary as well as the characteristic UV spectral properties of 2-thiol Trp peptides, we envisage that this methodology will find widespread use, particularly in the site-specific modification of expressed protein fragments for ligation and bioconjugation. Studies of this nature are currently underway in our laboratory and will be reported in due course.Acknowledgements
We would like to acknowledge the Australian Research Council for funding (DP130101984), the John Lamberton Research Scholarship and an International Postgraduate Research Scholarship (IPRS) for PhD funding (LRM). We would also like to thank Dr Nick Proschogo for technical support.Notes and references
- S. B. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef CAS PubMed.
- R. J. Payne and C. H. Wong, Chem. Commun., 2010, 46, 21–43 RSC.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CAS.
- C. T. T. Wong, C. L. Tung and X. Li, Mol. BioSyst., 2013, 9, 826–833 RSC.
- P. E. Dawson, Isr. J. Chem., 2011, 51, 862–867 CrossRef CAS.
- H. Rohde and O. Seitz, Biopolymers, 2010, 94, 551–559 CrossRef CAS PubMed.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- N. Metanis, E. Keinan and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 7049–7053 CrossRef CAS PubMed.
- D. Crich and A. Banerjee, J. Am. Chem. Soc., 2007, 129, 10064–10065 CrossRef CAS PubMed.
- L. R. Malins and R. J. Payne, Org. Lett., 2012, 14, 3142–3145 CrossRef CAS PubMed.
- P. Botti and S. Tchertchian, WO/2006/133962, 2006.
- J. Chen, Q. Wan, Y. Yuan, J. Zhu and S. J. Danishefsky, Angew. Chem., Int. Ed., 2008, 47, 8521–8524 CrossRef CAS PubMed.
- C. Haase, H. Rohde and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef CAS PubMed.
- R. Yang, K. K. Pasunooti, F. Li, X. W. Liu and C. F. Liu, J. Am. Chem. Soc., 2009, 131, 13592–13593 CrossRef CAS PubMed.
- K. S. Ajish Kumar, M. Haj-Yahya, D. Olschewski, H. A. Lashuel and A. Brik, Angew. Chem., Int. Ed., 2009, 48, 8090–8094 CrossRef CAS PubMed.
- J. Chen, P. Wang, J. Zhu, Q. Wan and S. J. Danishefsky, Tetrahedron, 2010, 66, 2277–2283 CrossRef CAS PubMed.
- S. Shang, Z. Tan, S. Dong and S. J. Danishefsky, J. Am. Chem. Soc., 2011, 133, 10784–10786 CrossRef CAS PubMed.
- S. D. Townsend, Z. Tan, S. Dong, S. Shang, J. A. Brailsford and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 3912–3916 CrossRef CAS PubMed.
- Z. Harpaz, P. Siman, K. S. Kumar and A. Brik, ChemBioChem, 2010, 11, 1232–1235 CrossRef CAS PubMed.
- Z. Tan, S. Shang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2010, 49, 9500–9503 CrossRef CAS PubMed.
- P. Siman, S. V. Karthikeyan and A. Brik, Org. Lett., 2012, 14, 1520–1523 CrossRef CAS PubMed.
- L. R. Malins, K. M. Cergol and R. J. Payne, ChemBioChem, 2013, 14, 559–563 CrossRef CAS PubMed.
- R. E. Thompson, B. Chan, L. Radom, K. A. Jolliffe and R. J. Payne, Angew. Chem., Int. Ed., 2013, 52, 9723–9727 CrossRef CAS PubMed.
- E. Scoffone, A. Fontana and R. Rocchi, Biochemistry, 1968, 7, 971–979 CrossRef CAS.
- E. Scoffone, A. Fontana and R. Rocchi, Biochem. Biophys. Res. Commun., 1966, 25, 170–174 CrossRef CAS.
- M. Wilchek and T. Miron, Biochem. Biophys. Res. Commun., 1972, 47, 1015–1020 CrossRef CAS.
- D. E. Wright and M. Rodbell, J. Biol. Chem., 1980, 255, 10884–10887 CAS.
- H. Heithier, L. D. Ward, R. C. Cantrill, H. W. Klein, M. J. Im, G. Pollak, B. Freeman, E. Schiltz, R. Peters and E. J. Helmreich, Biochim. Biophys. Acta, 1988, 971, 298–306 CAS.
- E. Canova-Davis and J. Ramachandran, Biochemistry, 1980, 19, 3275–3280 CrossRef CAS.
- E. Hazum, M. Fridkin, R. Meidan and Y. Koch, Eur. J. Biochem., 1977, 79, 269–273 CrossRef CAS.
- X. F. Li, L. S. Zhang, S. E. Hall and J. P. Tam, Tetrahedron Lett., 2000, 41, 4069–4073 CrossRef CAS.
- V. Popov, S. S. Panda and A. R. Katritzky, Org. Biomol. Chem., 2013, 11, 1594–1597 CAS.
- M.-Y. Lutsky, N. Nepomniaschiy and A. Brik, Chem. Commun., 2008, 1229–1231 RSC.
- A. Brik, Y. Y. Yang, S. Ficht and C. H. Wong, J. Am. Chem. Soc., 2006, 128, 5626–5627 CrossRef CAS PubMed.
- S. Ficht, R. J. Payne, A. Brik and C. H. Wong, Angew. Chem., Int. Ed., 2007, 46, 5975–5979 CrossRef CAS PubMed.
- R. J. Payne, S. Ficht, S. Tang, A. Brik, Y.-Y. Yang, D. A. Case and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 13527–13536 CrossRef CAS PubMed.
- C. S. Bennett, S. M. Dean, R. J. Payne, S. Ficht, A. Brik and C.-H. Wong, J. Am. Chem. Soc., 2008, 130, 11945–11952 CrossRef CAS PubMed.
- Y. Kajihara, A. Yoshihara, K. Hirano and N. Yamamoto, Carbohydr. Res., 2006, 341, 1333–1340 CrossRef CAS PubMed.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS.
- D. Bang, B. L. Pentelute and S. B. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985–3988 CrossRef CAS PubMed.
- E. C. Johnson and S. B. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed.
- S. B. Pollock and S. B. Kent, Chem. Commun., 2011, 47, 2342–2344 RSC.
- T. Hino, K. Tsuneoka, M. Nakagawa and S. Akaboshi, Chem. Pharm. Bull., 1969, 17, 550–558 CrossRef CAS.
- T. Nishio, N. Okuda and C. Kashima, Helv. Chim. Acta, 1990, 73, 1719–1723 CrossRef CAS.
- B. L. Pentelute and S. B. H. Kent, Org. Lett., 2007, 9, 687–690 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of synthetic experiments and characterisation of peptides and ligation products. See DOI: 10.1039/c3sc51497h |
| This journal is © The Royal Society of Chemistry 2014 |