Direct monofluoromethylation of O-, S-, N-, and P-nucleophiles with PhSO(NTs)CH2F: the accelerating effect of α-fluorine substitution†
Xiao
Shen
,
Min
Zhou
,
Chuanfa
Ni
,
Wei
Zhang
and
Jinbo
Hu
*
Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Ling-Ling Road, Shanghai, 200032, China. E-mail: jinbohu@sioc.ac.cn; Fax: +86 21-64166128
First published on 10th September 2013
Abstract
An efficient and direct monofluoromethylation of O-, S-, N-, and P-nucleophiles with PhSO(NTs)CH2F 1 has been developed. In contrast to the previously known detrimental effect of α-fluorine substitution on SN2 reactions, the current monofluoromethylation is accelerated by the α-fluorine substitution. Based on a mechanistic study, a new reactivity of sulfoximine (as a radical monofluoromethylation reagent) is disclosed.
Recently, fluorine-containing compounds have attracted increasing attention in pharmaceutical and agrochemical fields, because the incorporation of fluorine atom(s) or fluoroalkyl group(s) (such as CF3, CF2H, and CH2F) into bioactive molecules can often result in profound changes in their chemical and biological properties.1 In this context, monofluoromethyl compounds are particularly valuable, as the CH2F functionality can mimic CH3 and CH2OH groups, which are often encountered in biologically active molecules.2 Nucleophilic monofluoromethylation has been well developed with fluoromethyl phenyl sulfone, fluorobis(phenylsulfonyl)methane, fluoromalonates and other reagents as a powerful strategy for the introduction of the CH2F moiety into organic molecules.3 Direct electrophilic monofluoromethylation has also been reported by using FCH2X (X = I, Br, Cl, OSO2R (R = methyl, tolyl, trifluoromethyl)) and other reagents.3a,4 Our previous study revealed that the reactions of a variety of O-, S-, and N-nucleophiles with FCH2Cl were not sensitive to the presence of a radical scavenger such as nitrobenzene, which supports the operation of an SN2 mechanism rather than a radical mechanism in these reactions.4a Although nucleophilic and electrophilic (via SN2 mechanism) monofluoromethylations have been well established, reports of radical monofluoromethylation are scarce.5,6 In 1971, Raymond and Andrews reported the characterization of the monofluoromethyl radical (CH2F˙) by the matrix reaction of bromofluoromethane with alkali metals, but its synthetic application was not demonstrated.5 Very recently, Baran and co-workers elegantly reported a C–H radical monofluoromethylation using (FCH2SO2)2Zn; however, the method was only applied to N-heteroaromatic compounds.6 Therefore, new radical monofluoromethylation methods are highly desirable.
Given their important physiological and diverse chemical properties, sulfoximines and sulfoximinium salts have been widely used in organic synthesis.7 Recently, the use of fluorinated sulfoximines and sulfoximinium salts as fluoroalkylating agents has attracted much attention.8,9 In our previous work, we found that the carbanion derived from PhSO(NTs)CF2H was highly unstable and readily decomposed to difluorocarbene, which could be captured by a variety of C-, S-, and N-nucleophiles (Scheme 1a).8d In contrast, the carbanion derived from (R)-PhSO(NTs)CH2F [(R)-1] was found to possess good thermal stability and nucleophilicity, and we were therefore able to achieve a highly enantioselective fluorocyclopropanation reaction via a Michael addition–elimination process (Scheme 1b).8b Although both fluorinated and non-fluorinated sulfoximines have been successfully used in nucleophilic or electrophilic (fluoro)alkylation reactions, a reaction involving the production of the (fluoro)alkyl radical via the C–S bond homolysis of a neutral sulfoximine has never been reported.10 Herein, we report a direct monofluoromethylation of O-, S-, N-, and P-nucleophiles using PhSO(NTs)CH2F (1) as a novel monofluoromethylating agent (Scheme 1c). We also disclose a preliminary study into the mechanism which supports the operation of a radical (SRN1) mechanism in this reaction. The accelerating effect of α-fluorine substitution in sulfoximine 1 on the current monofluoromethylation provides additional intriguing insight into the unusual reactivities of fluorinated sulfoximines.8,9
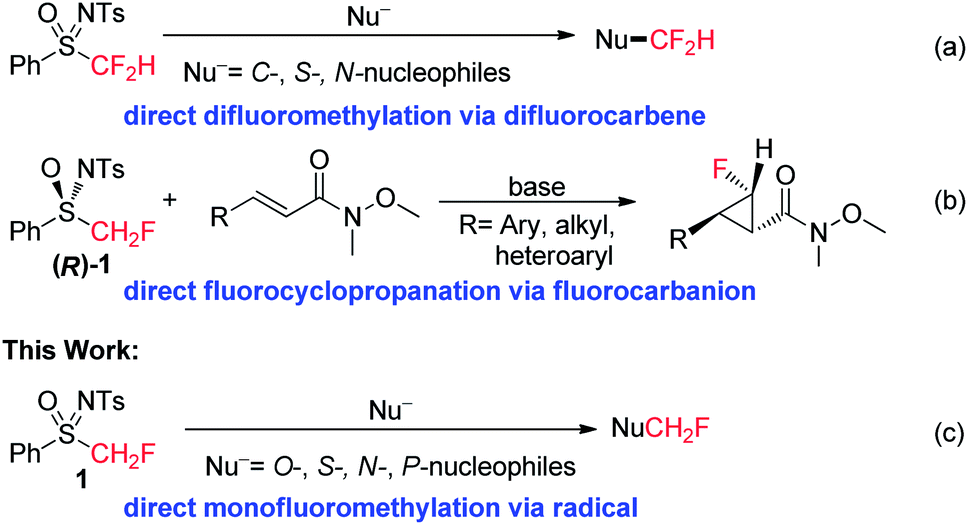 | ||
| Scheme 1 Fluoroalkylations with PhSO(NTs)CF2H and PhSO(NTs)CH2F reagents. | ||
Firstly, we developed a new and efficient synthesis of N-tosyl-S-fluoromethyl-S-phenylsulfoximine (1) on a relatively large scale (Scheme 2). PhSCH2F was prepared according to the reported procedure.11N-Tosyl-S-fluoromethyl-S-phenyl-sulfilimine (2) was readily prepared by imidation of PhSCH2F with chloramine-T·3H2O in 71% yield. Oxidation of 2 (on 300 mmol scale) with H2O2 gave 1 (73.5 g) in 75% yield. It is noteworthy that 1 is a stable white solid, which does not decompose even after being stored under air for a year.
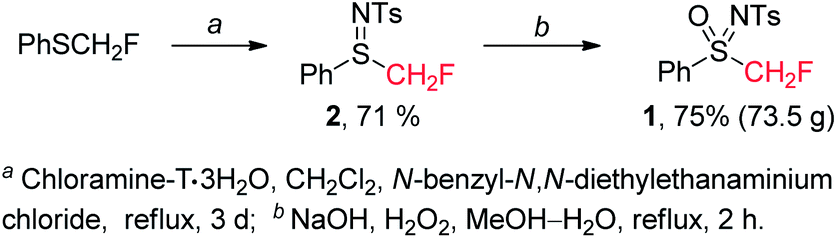 | ||
| Scheme 2 Preparation of N-tosyl-S-fluoromethyl-S-phenylsulfoximine (1). | ||
With compound 1 in hand, we investigated the direct monofluoromethylation of O-, S-, and N-nucleophiles by using (1,1′-biphenyl)-4-ol (3a) as a model substrate, and sodium hydride (NaH) as a base. Typically, 3a was stirred with NaH at room temperature (rt) for 30 min, after which 1 was added and the solution was stirred at a specified temperature for a specified time, as shown in Table 1. It was found that the choice of solvent was important for the reaction (Table 1, entries 1–6). Polar solvents such as DMSO and DMF are beneficial for the reaction. The optimal yield (95%) of 4a was obtained when the reaction was performed in DMSO at 80 °C for 4 h, with the ratio of 3a, 1, and NaH being 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3:1.25 (Table 1, entry 7).
:1.3:1.25 (Table 1, entry 7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Sol |
3a:1:NaH |
T (°C) | t (h) | Yieldb (%) |
| a Under N2, NaH (60% purity) was added to the solution of 3a (51 mg, 0.3 mmol) in solvent (2 mL) at rt; 30 min later, 1 in solvent (1 mL) was added and the solution was stirred at the conditions shown. b Yield determined by 19F NMR spectroscopy. c Isolated yield. | |||||
| 1 | THF | 1:1:1.2 |
60 | 35 | Trace |
| 2 | CH3CN | 1:1:1.2 |
60 | 35 | <10 |
| 3 | NMP | 1:1:1.25 |
60 | 35 | 74 |
| 4 | DMF | 1:1:1.25 |
60 | 35 | 81 |
| 5 | DMSO | 1:1:1.25 |
60 | 35 | 89 |
| 6 | DMSO | 1:1.2:1.3 |
60 | 36 | 94c |
| 7 | DMSO | 1:1.3:1.25 |
80 | 4 | 95c |
| 8 | DMSO | 1:1.2:1.3 |
rt | 36 | 3 |

We chose entry 7 shown in Table 1 as the standard conditions under which to study the scope of the reactions between the O-, S-, and N-nucleophiles 3 and PhSO(NTs)CH2F (1). The results are summarized in Scheme 3. The reaction proved to be general and a variety of structurally diverse phenols were successfully monofluoromethylated by 1 to give the corresponding monofluoromethyl ethers 4 in good to excellent yields (71–95%). The reaction is tolerant of chloro, bromo, and iodo substituents that are useful in transition metal-catalyzed cross-coupling reactions, enabling the subsequent synthesis of more useful CH2F-containing compounds. However, the current reaction conditions were not amenable to the reaction with 2-phenylethanol (3j), and only trace amounts of 4j were formed, with 75% of compound 1 being recovered.12 Moreover, the direct transfer of CH2F to sulfur-nucleophiles under similar reaction conditions (1, NaH, 80 °C, 4 h) was also found to be successful, and a variety of thiophenol and its derivatives were successfully monofluoromethylated by reagent 1, affording the corresponding monofluoromethyl sulfides in high yields (82–98%). Heteroaryl thiols such as benzo[d]thiazole-2-thiol (3o), 1-(tert-butyl)-1H-tetrazole-5-thiol (3p), and pyridine-2-thiol (3q) were also suitable substrates for the current monofluoromethylation reaction, resulting in the corresponding products 4o (92% yield), 4p (76% yield), and 4q (85% yield), respectively. In contrast to the reaction of 3o with PhSO(NTs)CF2H where both N-difluoromethylation and S-difluoromethylation occurred,8d only S-monofluoromethylation was observed in the current monofluoromethylation reaction, which might indicate the different mechanism of the two reactions. The current reaction conditions were also amenable to the monofluoromethylation of phenylmethanethiol (3r), and the resulting product 4r was obtained in 97% yield. Bicyclic hetereoaryl compounds featuring a 1-(fluoromethyl)-1H-benzo[d]imidazyl group have previously been reported to be effective phosphodiesterase 10 (PDE 10) inhibitors.13 Therefore, we carried out the direct monofluoromethylation of some N-heterocyclic secondary amines. As shown in Scheme 3, 2-phenyl-1H-imidazole (3s), 2-phenyl-1H-benzo[d]imidazole (3t), and 5,6-dimethyl-1H-benzo[d]imida-zole (3u) were successfully monofluoromethylated to give the corresponding products 4s (71% yield), 4t (62% yield), and 4u (86% yield), respectively. When 5-nitro-1H-benzo[d]imidazole (3v) was used as a substrate, a mixture of 4va and 4vb was obtained in 71% yield. It is worth noting that the current method was also applicable to the direct monofluoromethylation of 1H-benzo[d][1,2,3]triazole (3w), affording 4wa (50% yield) and 4wb (24% yield), which could be separated by silica gel column chromatography.
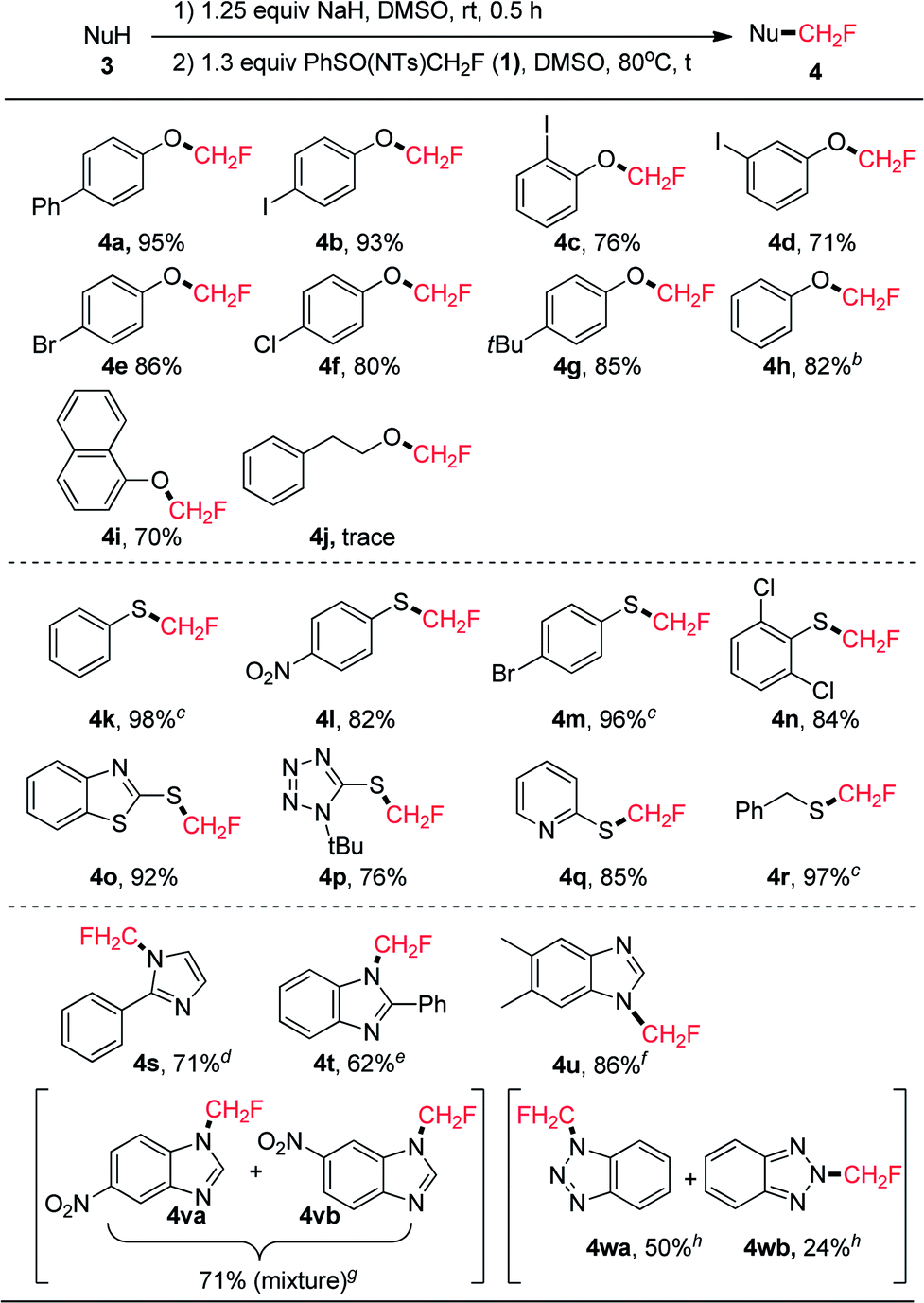 | ||
| Scheme 3 Monofluoromethylation of O-, S-, and N-Nucleophiles with PhSO(NTs)CH2F 1.a All reactions were performed at 80 °C for 4 h, and values indicate isolated yields unless otherwise noted. b Low bp, yield was determined by 19F NMR. c Unstable, yield was determined by 19F NMR. d 8 h. e 9.5 h. f 8.5 h. g 71 h. h 18 h. | ||
To further broaden the scope of this new monofluoromethylation protocol, we applied this method to other nucleophiles (Scheme 4). It was found that diphenylphosphine 5 could also react with sulfoximine 1 under similar conditions to give product 6 in 92% yield, after the addition of hydrogen peroxide to quench the reaction (Scheme 4a). Compound 6 was reported to be an effective reagent for the synthesis of monofluoroalkenes.14 To our delight, even 4-methoxybenzoic acid 7 was also a suitable substrate for the monofluoromethylation reaction, and product 8 was produced in 80% yield when the reaction temperature was raised to 100 °C (Scheme 4b).
 | ||
| Scheme 4 Monofluoromethylation of diphenylphosphine (5) and 4-methoxybenzoic acid (7). | ||
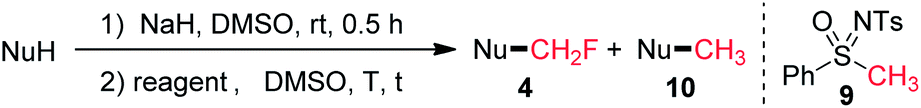
Although Johnson and co-workers reported the nucleophilic methylidene transfer reaction with the anion of N-tosyl-S-methyl-S-phenylsulfoximine (9) in 1970,15 there has been no report on the direct methylation of phenols by using 9 as a methylation reagent. Encouraged by our aforementioned success with the direct monofluoromethylation using sulfoximine 1, we carried out the reaction of 3a with non-fluorinated sulfoximine 9 under similar conditions (80 °C, 4 h). To our surprise, product 10a was only obtained in 3% yield (Table 2, entry 2). When the temperature was raised to 120 °C, 10a was formed in 75% yield (Table 2, entry 3). When 3a was treated with 1.3 equivalents of 1 and 1.3 equivalents of 9 in one pot, the total yield of product 4a and 10a was 89%, with the ratio of 4a/10a being 145/1 (Table 2, entry 4). We also tested the competitive reactions of 1 and 9 with thiol 3o, imidazole 3u, diphenylphosphine 5, and acid 7 as substrates (see ESI†). In all cases, the monofluoromethylation product was obtained as the major product and the methylation product was formed as the minor product. These results suggest that α-fluoro sulfoximine 1 possesses a higher reactivity than the non-fluorinated sulfoximine 9 under the current reaction conditions.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | NuH | Reagent | T (°C) | t (h) | Product | Yield (%) | 4/10 |
| a NaH (1.25 equiv.) and reagent (1.3 equiv.) were used. | |||||||
| 1 |
|
1 | 80 | 4 | 4a | 95 | |
| 2 | 9 | 80 | 4 | 10a | 3 | ||
| 3 | 9 | 120 | 6 | 10a | 75 | ||
| 4 | 1 + 9 | 120 | 6 | 4a + 10a | 89 | 145/1 | |
It is known that α-fluorine substitution can decrease the reactivity of methylene halides in SN2 reactions.16 In 1955, Hine and co-workers reported that the SN2 reactivity of FCH2Br proved to be about 350 times less reactive than CH3Br in its reaction with iodide ions in acetone at 20 °C.16a Very recently, Dolbier and co-workers reported that the substitution of 1-bromononane by azide ions at 50 °C in DMSO was 7.0 times faster than its α-fluorinated analog.16b Therefore, the unusual accelerating effect of α-fluorine substitution in our current monofluoromethylation reaction (Table 2) suggests that the monofluoromethylation reaction proceeds through a different reaction mechanism as opposed to an SN2 pathway.
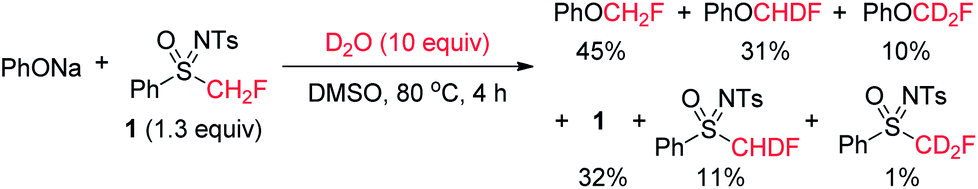 | ||
| Scheme 5 Monofluoromethylation of PhONa in the presence of D2O. | ||
In our previous work, a difluorocarbene mechanism was proposed for the difluoromethylation of PhSNa with PhSO(NTs)CF2H, based on deuterium-labeling experiments.8d It was found that PhSCF2D was obtained as the major product (PhSCF2D/PhSCF2H = 6/1) in the presence of 10 equivalents of D2O.8d We subsequently investigated the reaction of PhONa with sulfoximine 1 in the presence of 10 equivalents of D2O (Scheme 5). Since an excess amount of D2O was present in the reaction mixture, the deuterated monofluoromethylation product should be the major product, if the monofluorocarbene pathway was dominant in the monofluoromethylation reaction. However, the monofluoromethylation yielded non-deuterated PhOCH2F as the major product (PhOCH2F, 45% yield; PhOCHDF, 31% yield; PhOCD2F, 10% yield). Note that the deuterated products might result from the reactions of the deuterated sulfoximines, because substantial amounts of PhSO(NTs)CHDF (11% yield) and PhSO(NTs)CD2F (1% yield) were detected in the reaction mixture. Furthermore, we also attempted to trap monofluorocarbene with 2,3-dimethylbut-2-ene in the presence of a phenolate. It was reported that monofluorocarbene could readily react with 2,3-dimethylbut-2-ene to form 3-fluoro-1,1,2,2-tetramethylcyclopropane.17 However, no 3-fluoro-1,1,2,2-tetramethylcyclopropane was obtained when 2,3-dimethylbut-2-ene was added into the monfluoromethylation reaction of 3a, and product 4a was obtained in 96% yield (Scheme 6). These results indicate that the monofluorocarbene mechanism is not likely to be the major pathway for the current monofluoromethylation reaction.
 | ||
| Scheme 6 Monofluoromethylation of 3a in the presence of 2,3-dimethylbut-2-ene. | ||
In 1969, Sangster and Thynne reported that CH2F˙ is six times more reactive towards ethylene than CH3˙,18 which is somewhat consistent with the accelerating effect of α-fluorine substitution in our monofluoromethylation reaction. In order to probe the possibility of a radical mechanism in our monofluoromethylation reaction, we added radical scavengers into the reaction mixtures.19 The results are shown in Table 3. When nitrobenzene was added as an additive, the yield of 4a was decreased to 72%, and compound 1 was recovered in 47% yield (Table 3, entry 1). When the better electron acceptor, 1,4-dinitrobenzene, was employed, the monofluoromethylation was totally inhibited and 4-(4-nitrophenoxy)-1,1′-biphenyl (probably resulting from the reaction of ArO˙ with 1,4-dinitrobenzene) was obtained in 78% yield, while compound 1 was recovered in 120% yield (Table 3, entry 2).20 When benzoquinone was added, 4a was obtained in only 5% yield, and 3a was recovered in 87% yield (Table 3, entry 3). It is worth noting that a similar inhibitory effect was observed when PhONa was directly used as the substrate instead of using the 3a/NaH system (for details, see ESI†). Moreover, the yield of 4a was increased from 5% to 35% when the reaction time was extended from 4 to 8 hours (Table 3, entries 3 and 4), which is in accordance with the inhibiting effect of benzoquinone in SRN1 reactions. Further study showed that the yields of the reactions of 3o, 3u, 5, and 7 with 1 were also largely decreased in the presence of benzoquinone (Table 3, entries 5–8). It is noteworthy that the reaction of 3a with 9 could also be suppressed by benzoquinone, as the yield of product 10 decreased to 25% (Table 3, entry 9).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | NuH | Additive | T (°C) | t (h) | Yield (%) | Unreactede1 (%) |
| a Yield was determined by 19F NMR. b 4-(4-Nitrophenoxy)-1,1′-biphenyl was isolated in 78% yield. c 3a was recovered in 87% yield. d 9 was used instead of 1, the yield of methylation product refers to the isolated yield, and the yield of residual 9 was not determined (ND). e 130% of 1 (based on the amount of NuH) was added as starting material. | ||||||
| 1 | 3a | Nitrobenzene | 80 | 4 | 72 | 47 |
| 2b | 3a | 1,4-Dinitrobenzene | 80 | 4 | 0 | 120 |
| 3c | 3a | Benzoquinone | 80 | 4 | 5 | 110 |
| 4 | 3a | Benzoquinone | 80 | 8 | 35 | 94 |
| 5 | 3o | Benzoquinone | 80 | 4 | 7 | 120 |
| 6 | 3u | Benzoquinone | 80 | 8.5 | 0 | 71 |
| 7 | 5 | Benzoquinone | 80 | 6 | 20 | 60 |
| 8 | 7 | Benzoquinone | 100 | 12 | 52 | 54 |
| 9d | 3a | Benzoquinone | 120 | 6 | 25 | ND |
Based on the aforementioned experimental results, an SRN1 mechanism19 was proposed as shown in Scheme 7, though further mechanistic investigation is necessary to gain more details. An SET (single electron transfer) from the nucleophile to 1 afforded a radical anion A,21 followed by the elimination of B8d,15 (concerted or stepwise) to afford the monofluoromethyl radical, which combined with another nucleophile to form radical anion intermediate C. The product NuCH2F was formed after the SET from C to 1 with the formation of intermediate A.
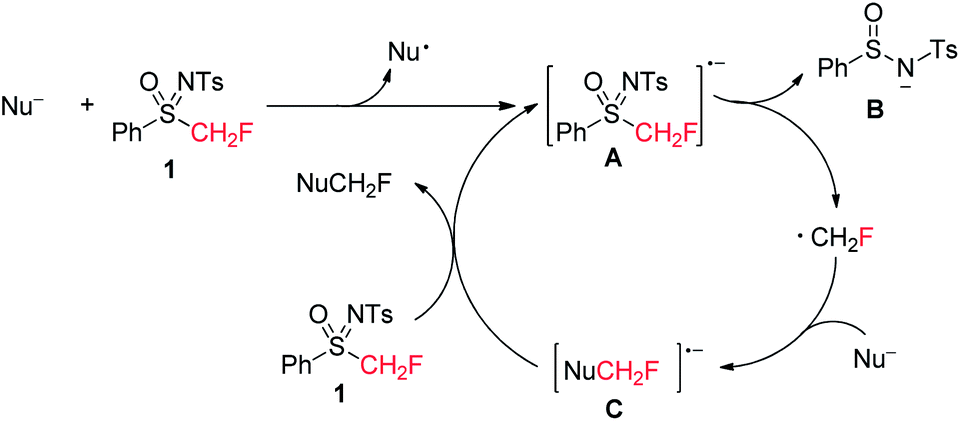 | ||
| Scheme 7 Proposed reaction mechanism. | ||
Subsequently, we tested the possibility of direct trifluoromethylation of 3a with PhSO(NTs)CF311 (Scheme 8a). It is interesting that the expected product, 4-(trifluoromethoxy)-1,1′-biphenyl (12), was not formed, while a large amount of CF3H was afforded, and the starting material 3a was isolated in 88% yield. It was found that when 11 was treated with PhONa in the presence of 4-bromobenzaldehyde (13), the nucleophilic trifluoromethylation product 14 was obtained in 50% yield, and CF3H was afforded in no less than 19% yield accompanied by the recovery of sulfoximine 11 in 65% yield (Scheme 8b).22 To the best of our knowledge, [CF3˙] is a typical electrophilic radical and there has been no report on the free radical trifluoromethylation of electrophilic aldehydes.23 Therefore, a trifluoromethyl anion (“CF3−”) could possibly be involved in the reaction. The process of [CF3−] production from PhSO(NTs)CF3 is somewhat similar to the reaction of PhSO2CF3 with alkoxides.24 Therefore, it can be concluded that the number of fluorine substituents significantly affects the reactivity of mono-, di-,8d and trifluoromethyl sulfoximines. In contrast to direct electrophilic monofluoromethylation with PhSO(NTs)CH2F via [CH2F˙] and direct electrophilic difluoromethylation with PhSO(NTs)CF2H via [:CF2],8d PhSO(NTs)CF3 was found to be a potential direct nucleophilic trifluoromethylation reagent via a [CF3−] intermediate (Scheme 9).
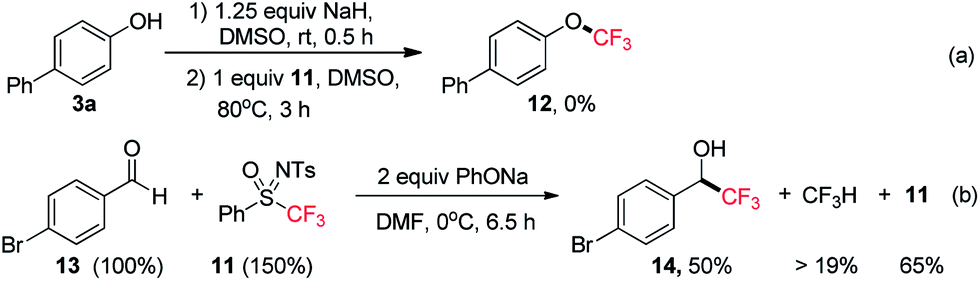 | ||
| Scheme 8 Reactions with reagent 11. | ||
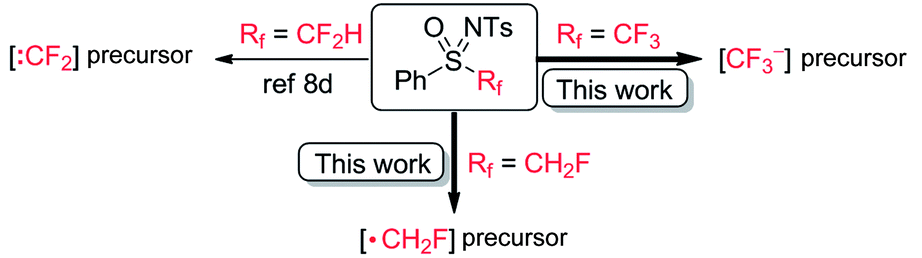 | ||
| Scheme 9 Diversified reactivities of fluoroalkyl sulfoximines. | ||
In conclusion, N-tosyl-S-fluoromethyl-S-phenylsulfoximine (1) was conveniently prepared and used as a new efficient monofluoromethylating agent for O-, S-, N-, and P-nucleophiles. In contrast to the previously known detrimental effect of α-fluorine substitution on SN2 reactions, the current monofluoromethylation with 1 was accelerated by the α-fluorine substitution. The preliminary mechanistic study suggests a radical mechanism involving an SET process. To the best of our knowledge, this is the first example of a fluoroalkylation reaction using a sulfoximine as a fluoroalkyl radical precursor. Moreover, PhSO(NTs)CF3 was found to be a nucleophilic trifluoromethylating agent via [CF3−] which further highlights the diverse reactivities of fluoroalkyl sulfoximines.
Acknowledgements
Support of our work by the National Basic Research Program of China (2012CB215500 and 2012CB821600), the National Natural Science Foundation of China (20825209 and 21202189), the Chinese Academy of Sciences, and the Syngenta PhD Studentship (to X.S.) is gratefully acknowledged.Notes and references
- (a) H. J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637 CrossRef PubMed; (b) M. Sani, A. Volonterio and M. Zanda, ChemMedChem, 2007, 2, 1693 CrossRef CAS PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (d) C. Fäh, R. Mathys, L. A. Hardegger, S. Meyer, D. Bur and F. Diederich, Eur. J. Org. Chem., 2010, 4617 CrossRef.
- For selected examples, see (a) J. Kollonitsch, A. A. Patchett, S. Marburg, A. L. Maycock, L. M. Perkins, G. A. Doldouras, D. E. Duggan and S. D. Aster, Nature, 1978, 274, 906 CrossRef CAS PubMed; (b) K. Mckeage and S. J. Keam, Drugs, 2009, 69, 1799 CrossRef CAS PubMed; (c) R. A. Rojas, I. Paluga, C. H. Goldfrad, M. T. Duggan and N. Barnes, J. Asthma, 2007, 44, 437 CrossRef CAS PubMed.
- For a feature article, see: (a) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC; for selected examples, see (b) Y. Li, C. Ni, J. Liu, L. Zhang, J. Zheng, L. Zhu and J. Hu, Org. Lett., 2006, 8, 1693 CrossRef CAS PubMed; (c) C. Ni, Y. Li and J. Hu, J. Org. Chem., 2006, 71, 6829 CrossRef CAS PubMed; (d) T. Fukuzumi, N. Shibata, M. Sugiura, H. Yasui, S. Nakamura and T. Toru, Angew. Chem., Int. Ed., 2006, 45, 4973 CrossRef CAS PubMed; (e) J. T. Palmer, Eur. Patent Appl. 0442754A2, 1991; (f) T. Koizumi, T. Hagi, Y. Horie and Y. Takeuchi, Chem. Pharm. Bull., 1987, 35, 3959 CrossRef CAS.
- (a) W. Zhang, L. Zhu and J. Hu, Tetrahedron, 2007, 63, 10569 CrossRef CAS ; and the references therein; (b) G. K. S. Prakash, S. C. Istvan and G. A. Olah, Org. Lett., 2008, 10, 557 CrossRef CAS PubMed; (c) G. A. Olah and A. Pavlath, Acta Chim. Acad. Sci. Hung., 1953, 3, 425 CAS; (d) Y. Nomura, E. Tokunaga and N. Shibata, Angew. Chem., Int. Ed., 2011, 50, 1885 CrossRef CAS PubMed.
- J. I. Raymond and L. Andews, J. Phys. Chem., 1971, 75, 3235 CrossRef CAS.
- Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herle, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95 CrossRef CAS PubMed.
- For reviews, see: (a) M. Reggelin and C. Zur, Synthesis, 2000, 1 CrossRef CAS; (b) H. Okamura and C. Bolm, Chem. Lett., 2004, 33, 482 CrossRef CAS; (c) M. Harmata, Chemtracts: Org. Chem., 2003, 16, 660 CAS; (d) C. R. Johnson, Acc. Chem. Res., 1973, 6, 341 CrossRef CAS.
- (a) X. Shen, W. Zhang, C. Ni, Y. Gu and J. Hu, J. Am. Chem. Soc., 2012, 134, 16999 CrossRef CAS PubMed; (b) X. Shen, W. Zhang, L. Zhang, T. Luo, X. Wan, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 6966 CrossRef CAS PubMed; (c) W. Zhang, W. Huang and J. Hu, Angew. Chem., Int. Ed., 2009, 48, 9858 CrossRef CAS PubMed; (d) W. Zhang, F. Wang and J. Hu, Org. Lett., 2009, 11, 2109 CrossRef CAS PubMed; (e) W. Zhang and J. Hu, Adv. Synth. Catal., 2010, 352, 2799 CrossRef CAS.
- (a) Y. Mace and E. Magnier, Eur. J. Org. Chem., 2012, 2479 CrossRef CAS; (b) R. Kowalczyk, A. J. F. Edmunds, R. G. Hall and C. Bolm, Org. Lett., 2011, 13, 768 CrossRef CAS PubMed; (c) G. K. S. Prakash, Z. Zhang, F. Wang, C. Ni and G. A. Olah, J. Fluorine Chem., 2011, 132, 792 CrossRef CAS; (d) C. Urban, F. Cadoret, J.-C. Blazejewski and E. Magnier, Eur. J. Org. Chem., 2011, 4862 CAS; (e) S. Noritake, N. Shibata, S. Nakamura, T. Toru and M. Shiro, Eur. J. Org. Chem., 2008, 3465 CrossRef CAS.
- A computational study on the reaction of β-ketoesters with a sulfoximinium salt without experimental evidence suggests a radical-like species, see Y.-D. Yang, X. Lu, G. Liu, E. Tokunaga, S. Tsuzuki and N. Shibata, ChemistryOpen, 2012, 1, 221 CrossRef CAS PubMed.
- (a) P. G. Theobald and W. H. Okamura, J. Org. Chem., 1990, 55, 741 CrossRef CAS; (b) K. M. More and J. Wemple, Synthesis, 1977, 791 CrossRef CAS.
- The failure of the reaction between 2-phenylethanol and 1 might be explained by the following two points: (1) the relatively poorer electron donating ability of 2-phenylethanolate than phenolate; (2) the deprotonation and subsequent decomposition of 1 resulting from the strong basicity of 2-phenylethanolate.
- P. R. Verhoest and C. J. Helal, US Pat., 0155779 A1, 2007.
- J. H. van Steenis and A. van der Gen, Eur. J. Org. Chem., 2001, 897 CrossRef CAS.
- C. R. Johnson and G. F. Katekar, J. Am. Chem. Soc., 1970, 92, 5753 CrossRef CAS.
- (a) J. Hine, C. H. Thomas and S. J. Ehrenson, J. Am. Chem. Soc., 1955, 77, 3886 CrossRef CAS; (b) H. Martinez, A. Rebeyrol, T. B. Nelms and W. R. , Dolbier, Jr, J. Fluorine Chem., 2012, 135, 167 CrossRef CAS.
- (a) J. L. Hahnfeld and D. J. Burton, Tetrahedron Lett., 1975, 1819 CrossRef CAS; (b) D. L. S. Brahms and W. P. Dailey, Chem. Rev., 1996, 96, 1585 CrossRef CAS PubMed.
- We thank one of the referees for providing the report that we did not notice when we were preparing the paper, see J. M. Sangster and J. C. J. Thynne, Trans. Faraday Soc., 1969, 65, 2110 RSC.
- R. A. Rossi, A. B. Pierini and A. B. Penenory, Chem. Rev., 2003, 103, 71 CrossRef CAS PubMed.
- Previously, the reaction of PhSNa with 1,4-dinitrobenzene in DMSO to form (4-nitrophenyl)(phenyl)sulfane was proposed to proceed via a SRN1 mechanism; see: (a) Y. Liu and W. Zhao, Acta Chim. Sin., 1991, 49, 615 CAS; (b) I. H. Leaver and G. C. Ramsay, Tetrahedron, 1969, 25, 5669 CrossRef CAS.
- We tested the possibility of the homolytic thermal decomposition of 1 generating the CH2F radical. After the heating of PhSO(NTs)CH2F in DMSO at 80 °C for 4 hours, 99% of 1 was recovered. After heating sulfoximine 1 and benzoquinone in DMSO at 80 °C for 4 hours, 96% of 1 and 50% of benzoquinone were recovered. The above results indicate that the homolytic thermal decomposition of 1 generating the CH2F radical is less likely, though it can not be completely ruled out. We thank one of the referees for suggesting this possible mechanistic pathway.
- We performed the similar reactions (as shown in Scheme 8a) with PhSO(NTs)CH2F and PhSO(NTs)CF2H reagents, and no 1-(4-bromophenyl)-2-fluoroethanol or 1-(4-bromophenyl)-2,2-difluoroethanol was obtained.
- (a) W. R. Dolbier Jr, Chem. Rev., 1996, 96, 1557 CrossRef; (b) W. R. Dolbier Jr, in Topics in Current Chemistry, ed. R. D. Chambers, Springer, Heidelberg, 1997, vol. 192, pp. 97–163 Search PubMed; (c) S. Barata-Vallejo and A. Postigo, Eur. J. Org. Chem., 2012, 1889 CrossRef CAS; (d) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed.
- G. K. S. Prakash, J. Hu and G. A. Olah, Org. Lett., 2003, 5, 3253 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sc51831k |
| This journal is © The Royal Society of Chemistry 2014 |