Programmed synthesis of arylthiazoles through sequential C–H couplings†
Satoshi
Tani
a,
Takahiro N.
Uehara
a,
Junichiro
Yamaguchi
a and
Kenichiro
Itami
*ab
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Chikusa, Nagoya 464-8602, Japan. E-mail: itami.kenichiro@a.mbox.nagoya-u.ac.jp; Fax: +81 52-788-6098
bInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Chikusa, Nagoya 464-8602, Japan
First published on 17th September 2013
Abstract
A programmed synthesis of privileged arylthiazoles via sequential C–H couplings catalyzed by palladium or nickel catalysts has been accomplished. This versatile protocol can supply all possible arylthiazole substitution patterns (2-aryl, 4-aryl, 5-aryl, 2,4-diaryl, 2,5-diaryl, 4,5-diaryl, and 2,4,5-triaryl) from an unfunctionalized thiazole platform by 11 distinct synthetic routes. We have generated over 150 arylthiazoles by using this methodology. We have applied this method to the rapid synthesis of fatostatin (SREBP inhibitor), and the gram-scale synthesis of triarylthiazoles has been demonstrated.
Introduction
Thiazoles attached with aryl or heteroaryl groups (arylthiazoles) represent privileged structural motifs that are frequently utilized in functional organic materials including organic electroluminescent devices, as well as in bioactive compounds and pharmaceuticals (Fig. 1).1 Thus, an efficient synthetic methodology that allows cost- and step-efficient production of arylthiazoles is in high demand. Moreover, in a situation where the structure–property relationships are not predictable, a uniform and programmable design that allows the synthesis of all possible arylthiazoles (2-aryl, 4-aryl, 5-aryl, 2,4-diaryl, 2,5-diaryl, 4,5-diaryl, and 2,4,5-triaryl) would help accelerate identifying and optimizing the “functional” arylthiazole structures. Therefore, both step economy and programmability need to be implemented in the next-generation synthesis of privileged arylthiazoles.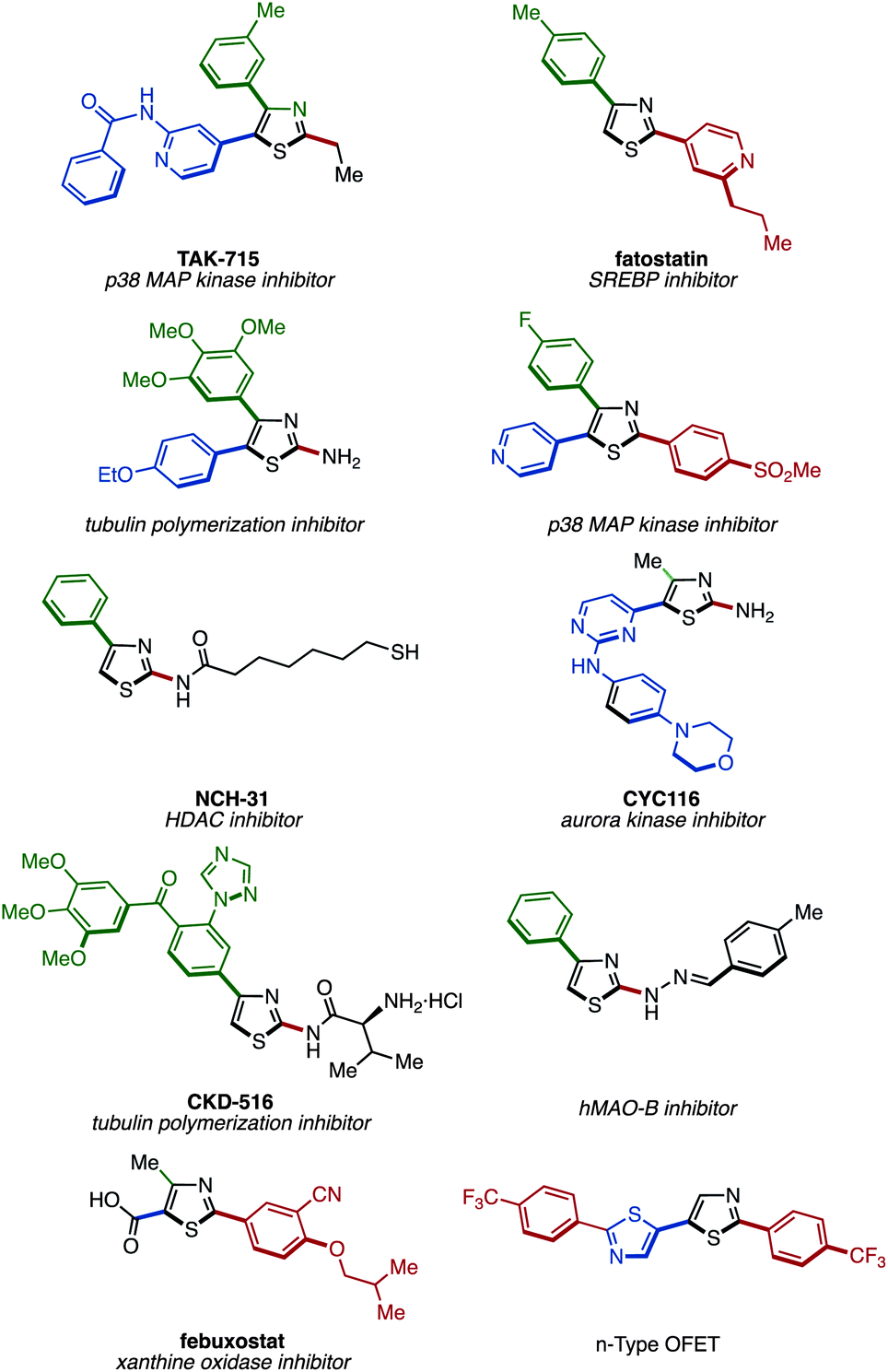 | ||
| Fig. 1 Selected examples of functional arylthiazoles. | ||
To date, the majority of arylthiazoles are synthesized by the classical Hantzsch thiazole synthesis, connecting α-haloketones and thioamides in a convergent manner.2 The advantage of this method is that it is possible to synthesize large amounts of arylthiazoles from inexpensive starting materials. However, the syntheses of both α-haloketones and thioamides typically require several steps from commercially available compounds, rendering the rapid construction of arylthiazole libraries difficult. To address this drawback, various cyclization methods for the synthesis of arylthiazoles have been developed.3 As an alternative and newer method, palladium-catalyzed cross-coupling reactions allow the divergent synthesis of arylthiazoles.4,5 However, superfluous steps are required prior to the cross-coupling; the parent thiazole must be pre-functionalized with a halogen or a metal group at the position where aryl groups are to be installed.
In pursuit of developing a step-economical and programmable arylthiazole synthesis, we have been attracted by the potential of catalytic C–H arylation reactions of thiazoles.6–9 Unlike the cross-coupling method, aryl groups can be installed onto the thiazole core directly in C–H arylation (step economy). Moreover, as the three C–H bonds on thiazole (C2, C4, C5) are chemically nonequivalent, it is in principle possible to acquire bond selectivity (regioselectivity) in aryl-installing reactions and to strategically utilize them in orthogonal functionalization (programmability).
Herein, we report a general and programmable synthetic scheme that can synthesize all possible arylthiazole substitution patterns (2-aryl, 4-aryl, 5-aryl, 2,4-diaryl, 2,5-diaryl, 4,5-diaryl, and 2,4,5-triaryl) from an unfunctionalized thiazole platform (Fig. 2).10 In this article, an application to the rapid synthesis of fatostatin (SREBP inhibitor), a re-optimization of C4-selective arylation of 2-substituted thiazoles, and a gram-scale synthesis of triarylthiazole are also described. Noteworthy features of the present methodology are that (i) all aryl groups assembled stem from readily available aryl halides or arylboronic acids, (ii) the aryl-group-installation at the desired position can be achieved by the choice of catalytic systems, and (iii) due to the regiodivergent nature of the established catalytic conditions, 11 distinct pathways for 7 arylthiazoles have been established.
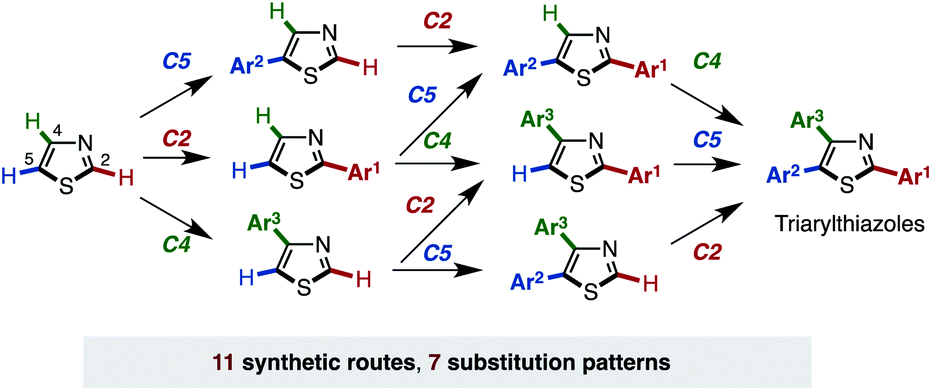 | ||
| Fig. 2 Programmed synthesis of arylthiazoles. | ||
In order to achieve fully programmed synthesis of arylthiazoles as shown in Fig. 2, it is essential to acquire regiodivergency in thiazole C–H arylation by catalyst control.11 This is conceptually different from the previously reported procedures for the selective synthesis of multiply arylated thiazoles, which are mainly based on the inherent reactivity difference of three thiazole C–H bonds (substrate control).12,13 In 2008, Fagnou and coworkers reported a pioneering work on the regioselective synthesis of arylthiazoles via Pd-catalyzed C–H couplings to install three different aryl groups (Fig. 3; i).11a Their method, wherein a thiazole was first oxidized to a thiazole N-oxide, involved C–H arylations of thiazole N-oxides with aryl halides in the order of C2, C5 then C4; these reactions all proceeded regioselectively to give the desired triarylthiazole N-oxides. Finally, reduction of the N-oxides was conducted to afford the target triarylthiazoles. In 2011, Murai, Shibahara, and coworkers reported a sequential C–H arylation of thiazoles using their own Pd catalyst (Fig. 3; ii).11b Thiazoles were coupled with aryl halides in the order of C5, C2, then C4 to afford triarylthiazoles. Although these two protocols successfully provide arylthiazoles in a regioselective manner, only one pathway en route to triarylthiazoles has been developed. Moreover, these protocols allow access to only 3 arylthiazole substitution patterns and the pathways leading to the 4 remaining substitution patterns have not been developed (4-aryl, 5-aryl, 2,4-diaryl, and 4,5-diaryl for the Fagnou method, and 2-aryl, 4-aryl, 2,4-diaryl, and 4,5-diaryl for the Murai–Shibahara method). A similar limitation was also observed in our previous work on the regioselective synthesis of multiply arylated thiophenes.13a
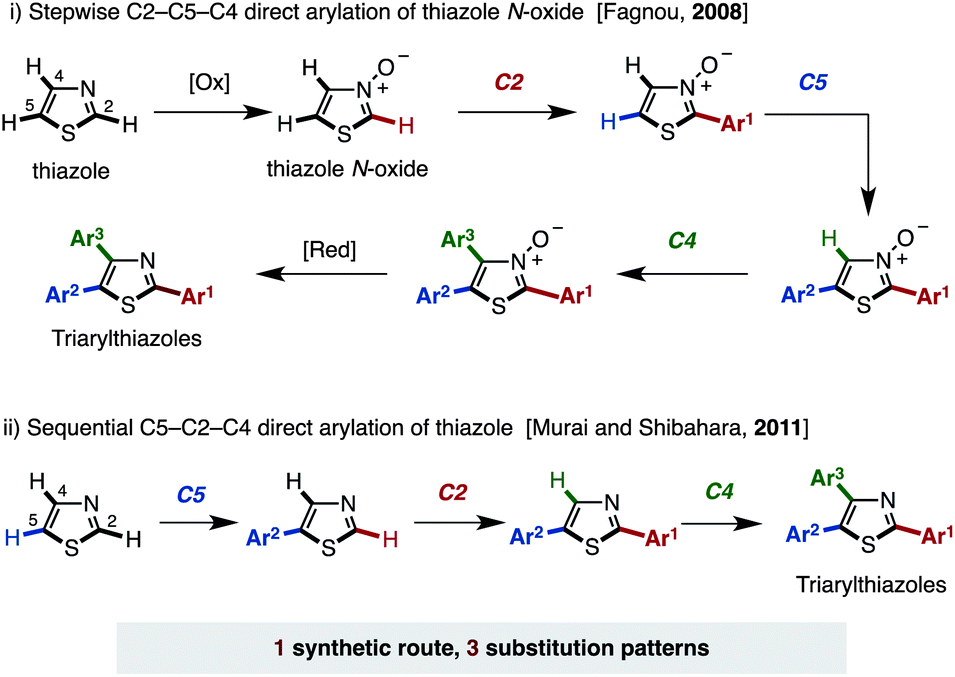 | ||
| Fig. 3 Selective synthesis of multiply arylated thiazoles by Fagnou and Murai–Shibahara. | ||
These seminal works clearly show the limitation of relying solely on substrate control (reactivity difference of C–H bonds of thiazole). Catalyst-controlled regioselectivity (regiodivergency) and selective C–H arylation at the least reactive C4 position have to be established to realize the programmed synthesis of arylthiazoles. The latter issue is evident from the previous reports by Fagnou and Murai–Shibahara as they both conducted the C4 arylation in the last step.
Although the catalyst-controlled C4-selective thiazole arylation is critically needed, it has been well documented that C2 and/or C5 positions are preferentially arylated over the C4 position under transition-metal-catalyzed C–H arylation conditions (Fig. 4).7 Under forcing conditions, diarylation occurs at the C2 and C5 positions under the influence of palladium catalysts.7b,8 The reactivity of the three positions of thiazole in C–H arylation (C2, C5 ≫ C4) is in line with the typical reactivity profile of the thiazole ring (Fig. 4). For example, the deprotonation of thiazoles occurs preferentially at the C2 position. Thus, under the C–H arylation conditions using a strong base, C2 arylation tends to take place preferentially. On the other hand, thiazoles react with electrophiles preferentially at the C5 position. Thus, under the C–H arylation conditions where the nucleophilicity of thiazole plays a key role, C5 arylation tends to predominate. Radical reactions of thiazoles are also known, but the C4 position is least reactive. Only one example of reactivity preference for the C4 position of thiazoles is known, but the C2 position has to be substituted and some C5 substitution is also observed.14 Thus, the introduction of an aryl group at the C4 position of a thiazole derivative required a number of synthetic steps.15
 | ||
| Fig. 4 Paucity of C4-selective thiazole C–H arylation and the general reactivity of thiazole. | ||
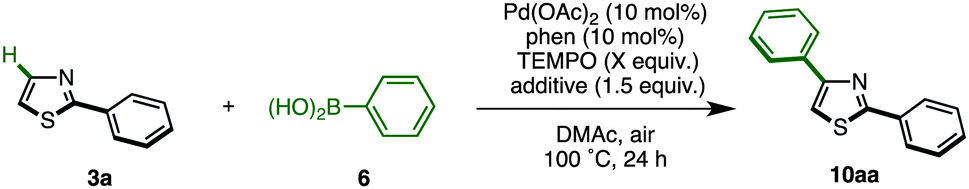
Recently, we discovered the first C4-selective C–H arylation of 2-phenylthiazole with arylboronic acids, catalyzed by a Pd(OAc)2/phen (1,10-phenanthroline)/TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl) system (Scheme 1).9 This previously inaccessible regiochemical outcome and the hitherto unrecognized regiocontrolling elements that have been uncovered in this study should provide a deeper understanding of the reactivity of thiazoles. Although a full understanding of the mechanism is still ongoing, our preliminary experiments and theoretical calculations of related reactions indicated that our C4-selective arylation occurs by a mechanism involving Heck-like concerted arylpalladation across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of thiazoles.16 Although we did not fully explore the scope of this reaction in our previous communication (only three reactions of 2-phenylthiazole were examined), this breakthrough finding finally opened the door for the programmed synthesis of arylthiazoles, providing access to 7 arylthiazole substitution patterns by 11 different synthetic routes.
C bond of thiazoles.16 Although we did not fully explore the scope of this reaction in our previous communication (only three reactions of 2-phenylthiazole were examined), this breakthrough finding finally opened the door for the programmed synthesis of arylthiazoles, providing access to 7 arylthiazole substitution patterns by 11 different synthetic routes.
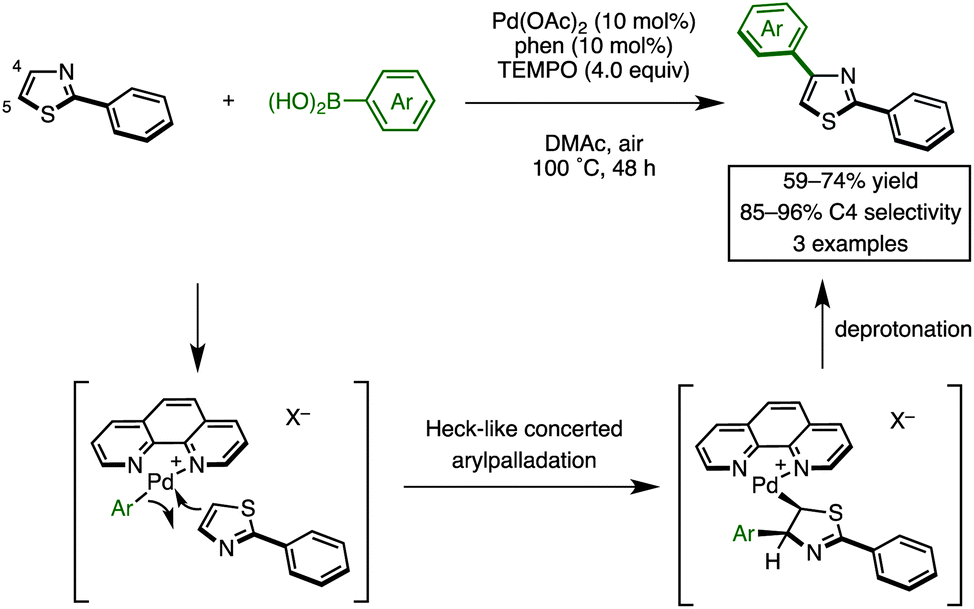 | ||
| Scheme 1 C4-selective C–H arylation of 2-phenylthiazole. | ||
Results and discussion
Transition-metal-catalyzed C–H arylation of thiazoles at the C2 position has already been reported by several groups including our own.7 After extensive screening of various reaction conditions for the preparation of 2-arylthiazoles,17 we came to the conclusion that Rossi's Pd/Cu-based protocol (Method A),7g our Ni-based protocol (Method B),7k,7r and Mori's Pd-based protocol (Method C)7n can effectively convert thiazole (1) to 2-arylthiazoles 3 using aryl halides 2 as an aryl source (Table 1). It should be noted, however, that there is no single, universal catalytic system; all three systems have pros and cons.|
|
|---|
| a Reaction conditions. Method A: 1 (0.8 mmol), 2 (X = I, 0.4 mmol), Pd(OAc)2 (0.02 mmol), CuI (0.8 mmol), DMF (1.0 mL), 140 °C, 40 h. Method B: 1 (0.6 mmol), 2 (X = I, 0.4 mmol), Ni(OAc)2 (0.04 mmol), bipy (0.04 mmol), LiOt-Bu (0.8 mmol), 1,4-dioxane (1.0 mL), 120 °C, 36 h. Method C: 1 (0.6 mmol), 2 (X = Br, 0.4 mmol), Pd[P(t-Bu)3]2 (0.008 mmol), LiOt-Bu (0.6 mmol), 1,4-dioxane (1.2 mL), 100 °C, 12 h. b The reaction was performed for 16 h. |

|
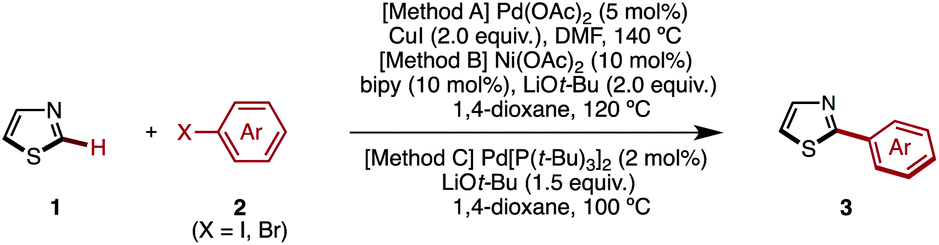
It was found that Rossi's palladium catalyst system [Method A: Pd(OAc)2 and CuI in DMF at 140 °C] was generally preferred for the synthesis of 2-arylthiazole 3 from 1. When a sterically hindered aryl iodide such as 2-methyl-1-iodobenzene (2f) and an electron-deficient heteroaryl iodide such as 3-iodopyridine (2t) were used, our nickel catalytic system [Method B: Ni(OAc)2, bipy (2,2′-bipyridyl) and LiOt-Bu in 1,4-dioxane at 85 °C] was most effective and gave higher yields than Method A (76% for 3f and 49% for 3t). In the synthesis of 2-arylthiazoles 3j, 3p and 3r, Mori's palladium catalyst system [Method C: Pd[P(t-Bu)3]2, LiOt-Bu and 1,4-dioxane at 100 °C] was most effective, affording the corresponding coupling product with complete regioselectivity at the C2 position.
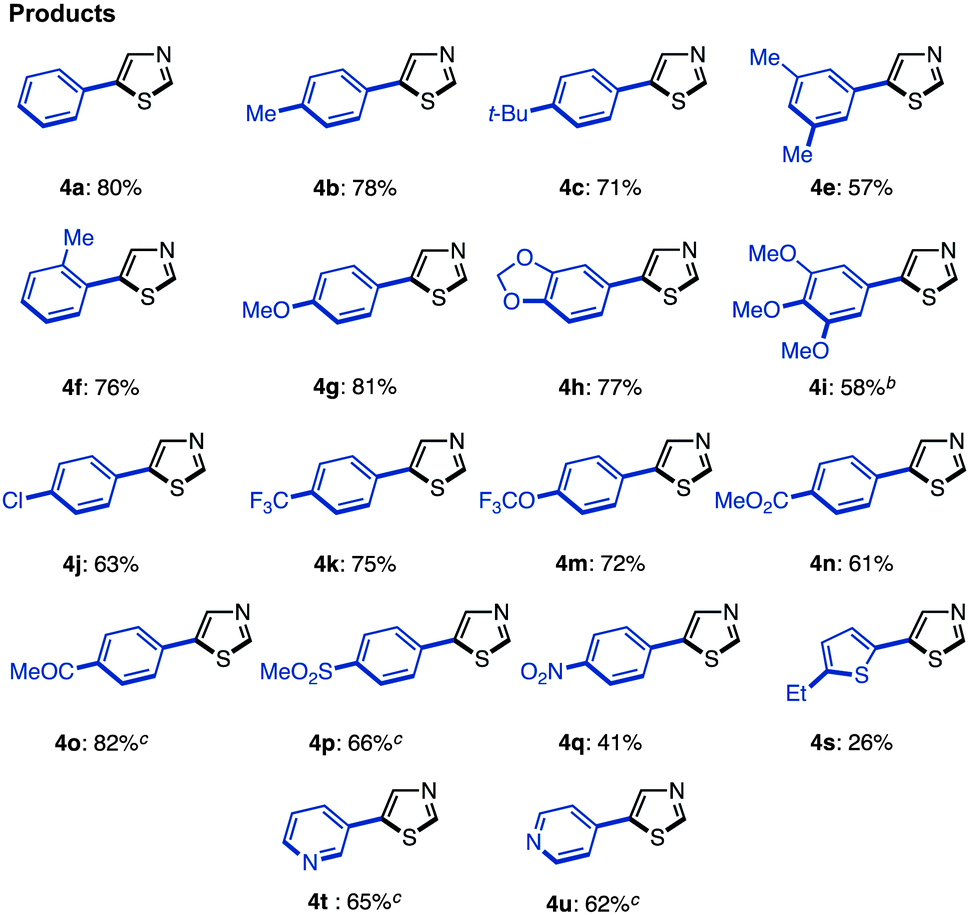
The synthesis of 5-arylthiazoles 4via C–H arylation of thiazoles has already been reported under various reaction conditions.7 However, these conditions had not been optimized for unfunctionalized thiazole (1). Therefore, we began by optimizing the conditions for C5-selective C–H arylation of 1, based on our protocol [PdCl2, bipy in DMF at 120 °C]7q and Fagnou's protocol [Pd(OAc)2, PMe(t-Bu)2·HBF4, Cs2CO3 in t-AmylOH].7m As a result, optimal conditions for the coupling of thiazole (1) and aryl halides 2 have been determined.17 Treatment of iodobenzene (2; X = I) with thiazole (1: 1.5 equiv.) in the presence of Pd(OAc)2 (5 mol%), PMe(t-Bu)2·HBF4 (10 mol%), and Cs2CO3 (1.5 equiv.) in t-AmylOH at 80 °C for 36 h afforded the desired C5-phenylated product 4a in 80% yield with virtually complete regioselectivity (Method D). Under these conditions, we examined the substrate scope of C5-selective C–H arylation of thiazole with aryl halides as shown in Table 2. Aryl iodides bearing an alkyl group at the para, meta and even ortho position underwent C5-selective arylation to give the corresponding products (4b–f) in good yields. Aryl iodides bearing electron-rich substituents (resulting in products 4g–i) and electron-deficient substituents (resulting in products 4j–q) as well as heteroaryl iodides (resulting in products 4s–u) gave the desired products. However, when 2-iodo-5-ethylthiophene (2i) was used as the coupling partner, the yield of product 4i was low (26%) due to the occurrence of homocoupling of 2i as a side reaction. In some cases, aryl bromides were also applicable to give the corresponding coupling products (4i, 4o, 4t and 4u) in good to excellent yields. Since 4′-bromoacetophenone (2o), bromo-4-(methylsulfonyl)benzene (2p), and bromopyridines (2t and 2u) were less reactive under Method D, we changed the reaction conditions to those reported by Doucet and coworkers [Method E: Pd(OAc)2, KOAc in DMAc, 130 °C].7l As a result, these coupling reactions performed well to give the desired products (4o, 4p, 4t, and 4u).
|
|
|---|
| a Reaction conditions. Method D: 1 (0.6 mmol), 2 (X = I, 0.4 mmol), Pd(OAc)2 (0.02 mmol), PMe(t-Bu)2·HBF4 (0.04 mmol), Cs2CO3 (0.6 mmol), t-AmylOH (1.0 mL), 80 °C, 36 h. b Bromobenzene (0.4 mmol) was used instead of iodobenzene, and the reaction was performed at 100 °C for 18 h. c Method E: 1 (0.8 mmol), 2 (X = Br, 0.4 mmol), Pd(OAc)2 (0.004 mmol), KOAc (0.8 mmol), DMAc (1.0 mL), 130 °C, 20 h. |
|
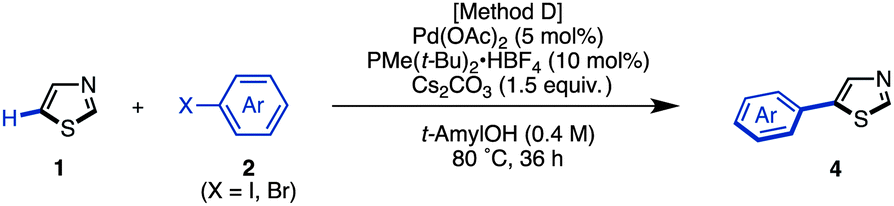
As already described, we have developed a palladium-catalyzed C4-selective C–H arylation of 2-arylthiazoles with arylboronic acids under a Pd(OAc)2/phen/TEMPO system (Scheme 1).9 However, when we applied this first-generation protocol to the C–H arylation of an unfunctionalized thiazole (1) with phenylboronic acid (6a), the yield of the desired coupling product 8a was abysmal (<10% yield). We envisioned that some form of protecting group at the C2 position was necessary. Namely, it was thought that the C4-arylation product 8 could be synthesized by a sequence of (i) protection of 1, (ii) C4-arylation of 2-substituted thiazole 5 with an arylboronic acid 6, and (iii) removal of the C2 substituent from the resulting C4-arylated thiazole 7 (Scheme 2).
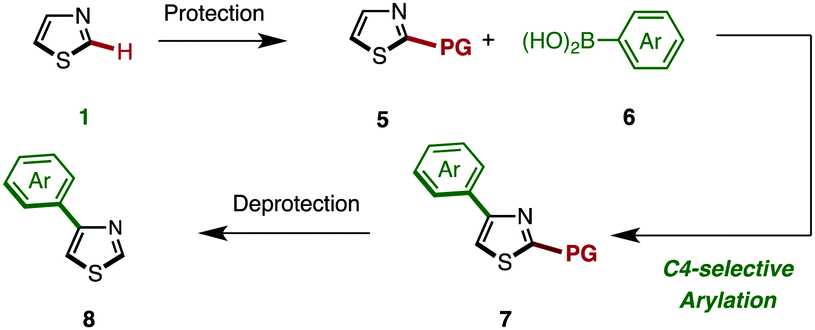 | ||
| Scheme 2 Protection–arylation–deprotection sequence for the synthesis of 4-arylthiazoles 8. | ||
Among many possible protecting groups at the C2-position of thiazole, we decided to apply Mori's procedure using a diphenylmethanol group.18 The protected thiazole 5 was readily prepared by the treatment of 1 with n-BuLi, followed by the reaction with benzophenone in 86% yield (Scheme 3).
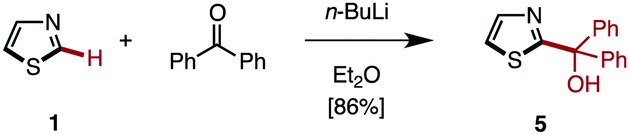 | ||
| Scheme 3 “Protection” of thiazole at the C2-position. | ||
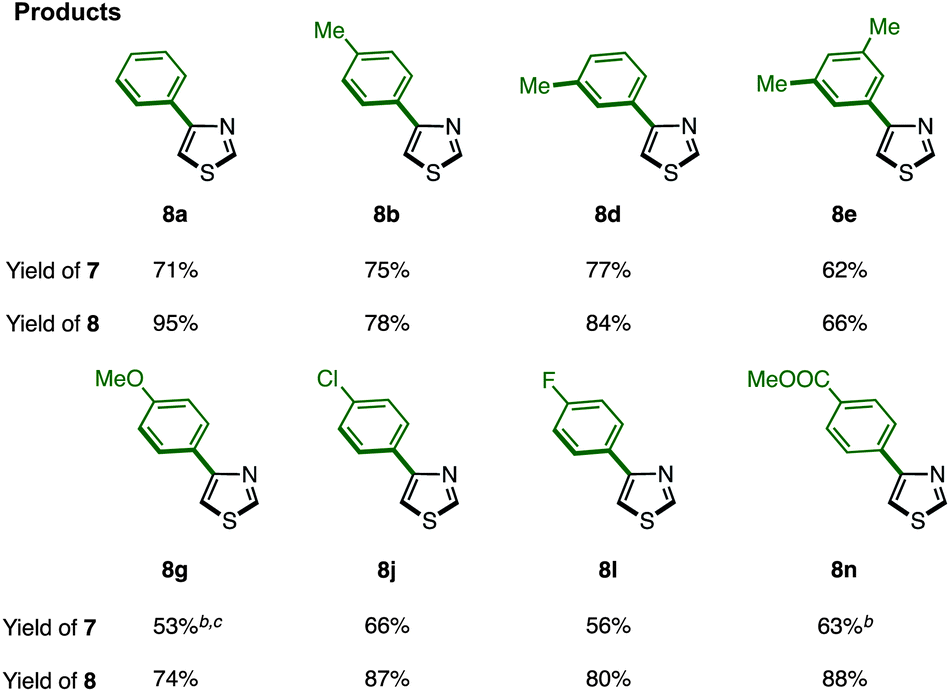
The protected thiazole 5 was then reacted with arylboronic acid under the influence of Pd(OAc)2/phen/TEMPO. During these studies, we re-optimized our C4-selective arylation conditions, employing less amounts of TEMPO but using LiBF4 as an additive (Method F). Details of these conditions are discussed later in the manuscript (Table 7). Thus, the treatment of 5 (1.0 equiv.) and phenylboronic acid (6a: 4.0 equiv.) in the presence of Pd(OAc)2 (10 mol%), phen (10 mol%), TEMPO (50 mol%), and LiBF4 (1.5 equiv.) in DMAc under air at 100 °C afforded the C4-phenylated thiazole 7a in 71% yield with complete regioselectivity (Table 3). Deprotection of the diphenylmethanol group of 7a was accomplished by treatment with Cs2CO3 at 150 °C (ref. 18) to afford 4-phenylthiazole (8a) in 95% yield. The scope of C4-selective C–H arylation of thiazole 5 with respect to various arylboronic acids 6 (followed by diphenylmethanol group removal) is summarized in Table 3. A variety of arylboronic acids reacted smoothly to furnish the coupling products (7b–n), and the desired 4-arylthiazoles (8b–n) were obtained in good yields after deprotection.
|
|
|---|
| a Reaction conditions. Method F: 5 (0.25 mmol), 6 (1.0 mmol), Pd(OAc)2 (0.025 mmol), phen (0.025 mmol), TEMPO (0.125 mmol), LiBF4 (0.38 mmol), DMAc (0.5 mL), air, 100 °C, 48 h; then 7 (0.1–0.2 mmol), Cs2CO3 (1.0 mmol), m-xylene (0.8 mL), 150 °C, 40 h. b TEMPO (0.25 mmol) was used. c The yield was determined by 1H-NMR. |
|
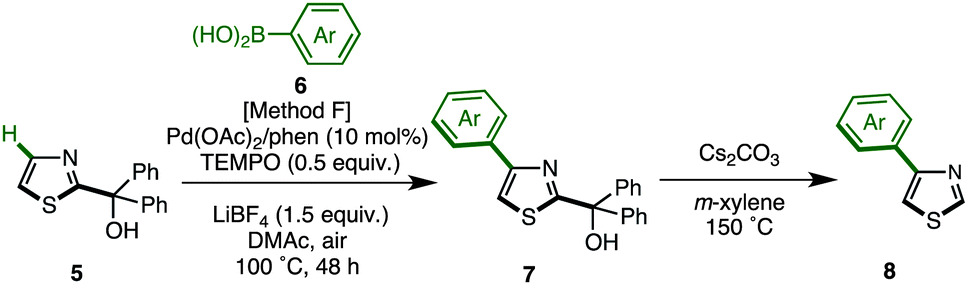
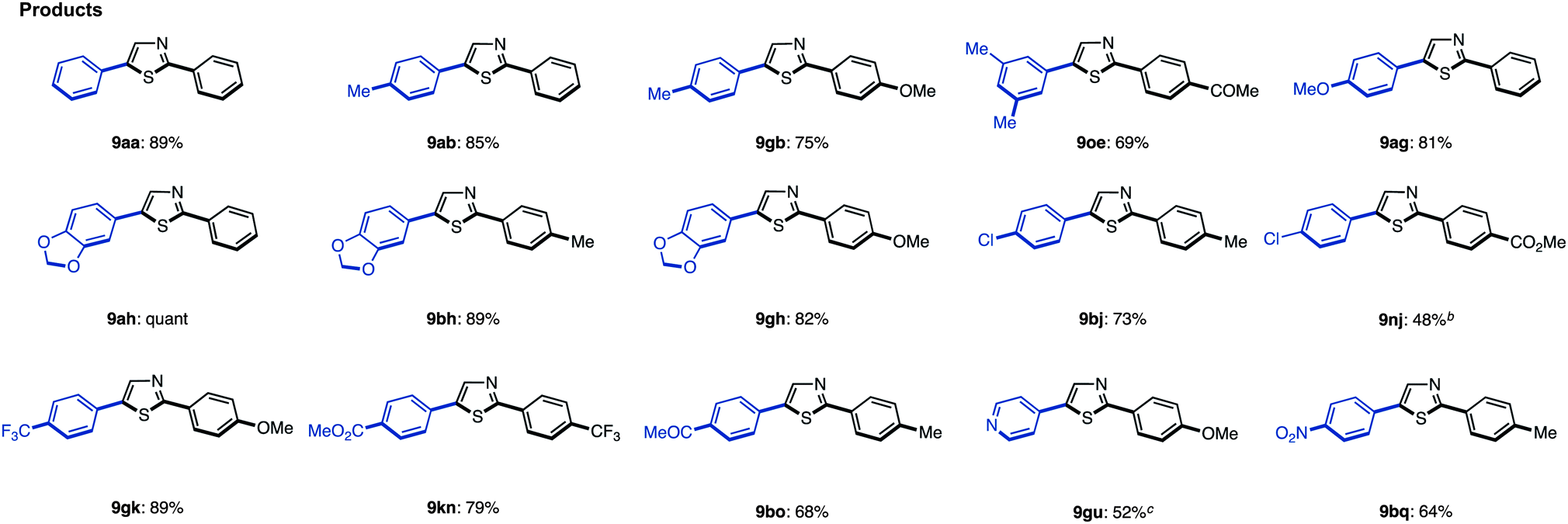
We have established methods for the first arylation of thiazole (1) to obtain mono-arylated thiazoles of three different substitution patterns (3, 4, and 8) with complete regioselectivity. Thus, we then carried out the second arylation, in which the mono-arylated thiazoles were coupled with another aryl coupling partner to obtain diarylthiazoles. We first performed the C5-selective arylation of 2-arylthiazoles 3 with aryl iodides 2 under our developed conditions [PdCl2(bipy), Ag2CO3 in 1,4-dioxane at 120 °C].7q After a slight modification of reaction conditions (Method G), we turned our attention to defining the scope and functional group tolerance of the aryl iodide coupling partner in the synthesis of 2,5-diarylthiazoles 9 from 2-arylthiazoles 3 (Table 4). Numerous aryl iodides and 2-arylthiazoles were coupled with each other to give 2,5-diarylthiazole products 9 in excellent yields. When coupling reactions between 4-iodopyridine (2u) and 2-(4-methoxyphenyl)thiazole (3g) as well as chloropyridine (2j) and methyl 4-(2-thiazolyl)benzoate (3n) were conducted, the yields of the resulting diarylthiazoles (9gu and 9nj) were low under our standard conditions (Method G). After increasing the reaction temperature and time, these yields were rendered moderate (48% and 52% yield, respectively).
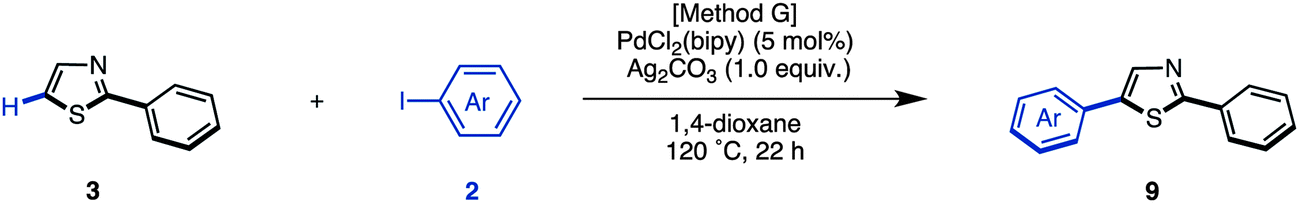
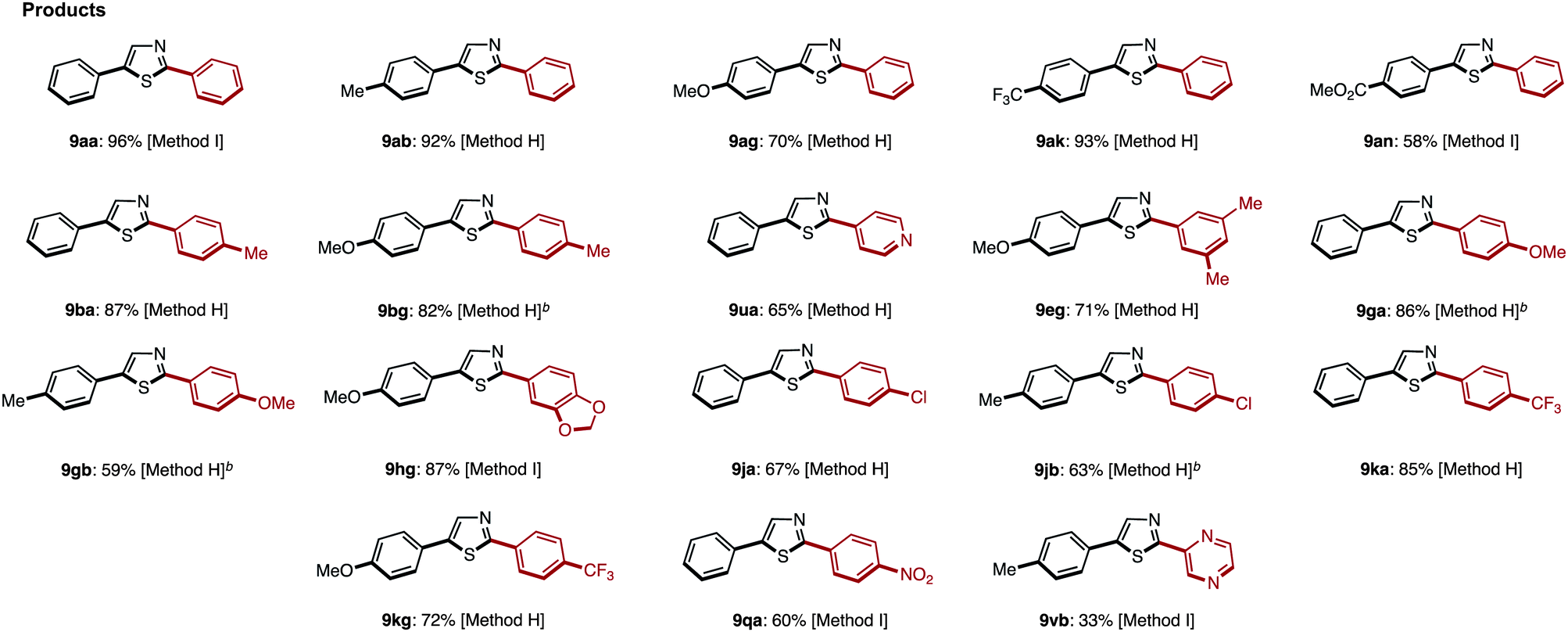
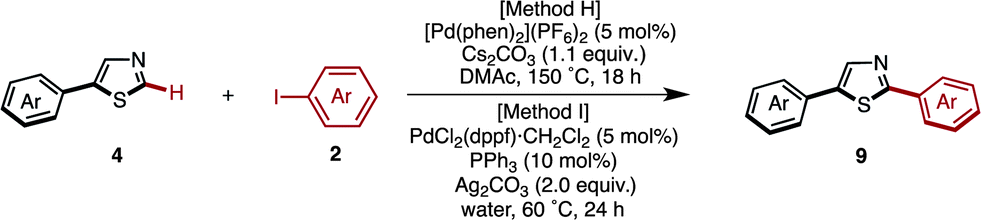
Starting from 5-arylthiazoles 4, we found that C2-selective arylation was possible using two different methods, namely the protocols originally developed by Murai–Shibahara8d and Greaney.7h For example, when Murai's protocol [Method H: Pd(phen)2(PF6)2, Cs2CO3, DMAc, 150 °C] was used for the coupling of 2-phenylthiazole (3a) with iodobenzene, 2,5-diphenylthiazole (9aa) was obtained in 83% yield, whereas Greaney's protocol [Method I: PdCl2(dppf)·CH2Cl2, Ph3P, Ag2CO3, water, 60 °C] gave a 96% yield. Employing these conditions, the C2-selective arylation of 4 was conducted with various aryl iodides (Table 5). Although most of the coupling reactions proceeded by Method H to give the corresponding products in good yields, the yields of 9an, 9hg, 9qa, and 9vb were low due to competing hydrolysis of the substituents or functional group intolerance. After changing the reaction conditions to Method I, these products were generated in moderate yields. In the case of 9bg, 9ga, 9gb and 9jb, the yields were better at lower temperatures (130 °C) to prevent decomposition of the product.
|
|
|---|
| a Reaction conditions. Method H: 4 (0.2 mmol), 2 (0.22 mmol), Pd(phen)2(PF6)2 (0.01 mmol), Cs2CO3 (0.22 mmol), DMAc (0.8 mL), 150 °C, 18 h. Method I: 4 (0.2 mmol), 2 (0.24 mmol), PdCl2(dppf)·CH2Cl2 (0.01 mmol), Ph3P (0.02 mmol), Ag2CO3 (0.4 mmol), water (1.0 mL), 60 °C, 24 h. b The reaction was performed at 130 °C. |
|
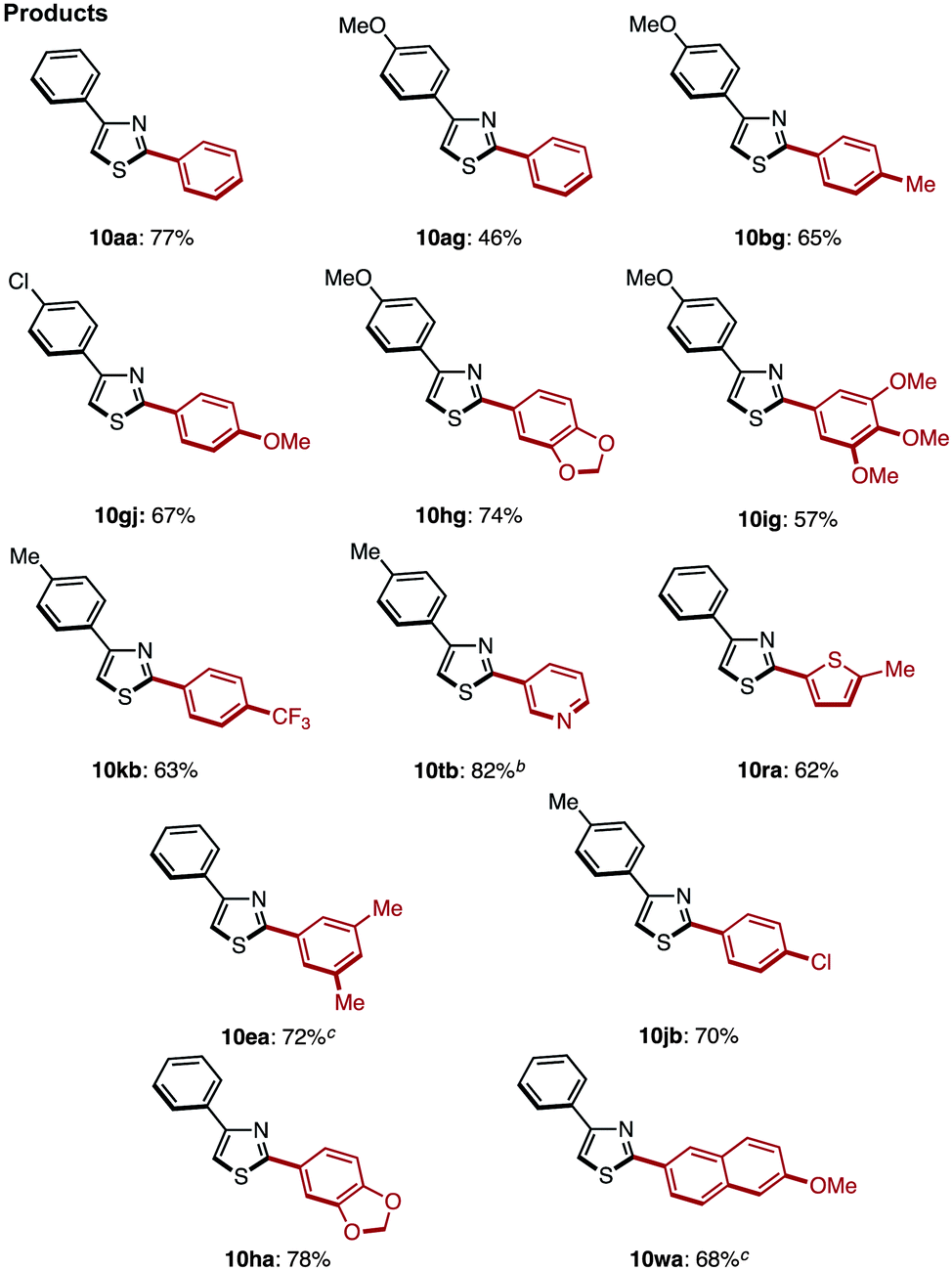
The C2-selective C–H arylation of 4-arylthiazoles 8 has not been reported thus far. Thus, without literature precedent, we attempted the coupling under several reaction conditions.17 When the coupling reaction between 4-phenylthiazole (8a) and iodobenzene was conducted with our Ni catalyst7k,7r or under Pd catalysis reported by Rossi7g and Hoarau,7j the coupling product 10aa was obtained in low yield (22% and 6% yield, respectively). We then found that Mori's protocol [Method C: Pd[P(t-Bu)3]2 and LiOt-Bu in 1,4-dioxane]7n was superior to others for the C–H arylation; 2,4-diphenylthiazole (10aa) was obtained in 55% yield from 8a and bromobenzene, and the yield was increased to 77% after the slight modification of conditions (Method C′: reaction temperature was lowered to 80 °C).17 Using these optimal conditions, we further investigated the coupling of various 4-arylthiazoles and bromoarenes as shown in Table 6. Gratifyingly, we found that a relatively broad range of 4-arylthiazoles and bromoarenes can couple with complete C2 selectivity and in good yields.
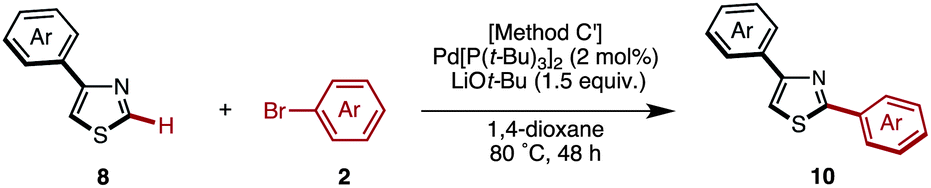
We subsequently applied this protocol to the synthesis of fatostatin, which was reported as a sterol regulatory element-binding protein (SREBP) by Uesugi and coworkers.19 4-(p-Tolyl)thiazole (8b) was reacted with 4-bromo-2-propylpyridine (11)20 under the influence of Pd[P(t-Bu)3]2/LiOt-Bu (Method C) to give fatostatin in 53% yield (Scheme 4). Although this synthesis is not shorter than that of the original report,19 it could easily bring structural diversity to the aryl substituent at the C2 position of the thiazole core.
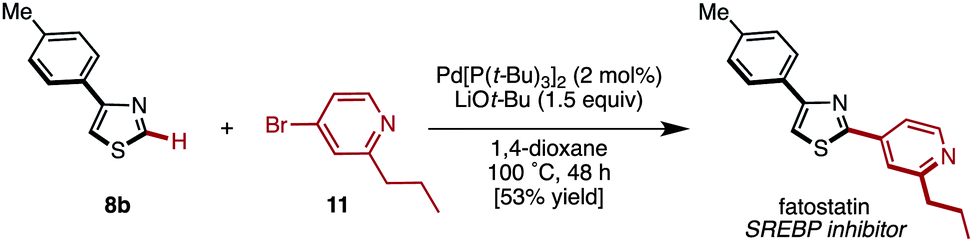 | ||
| Scheme 4 Synthesis of fatostatin by C–H arylation. | ||
Next, a C4-selective C–H arylation of 2-arylthiazoles 3 was investigated. Following our previously reported conditions for the C4-selective arylation of 2-arylthiazoles,9 2-phenylthiazole (3a) was treated with 4.0 equiv. of phenylboronic acid (2a) in the presence of Pd(OAc)2/phen catalyst and TEMPO (4.0 equiv.) to give diphenylthiazole in 85% combined yield (10aa and its regioisomer) with 86% regioselectivity at the C4 position of 3a (74% isolated yield; Table 7, entry 1). Although 10aa could be synthesized, the reaction requires an excess amount of TEMPO (4.0 equiv. to thiazole substrate), and the regioselectivity was not perfect. Therefore, a re-optimization of the coupling reaction was conducted (Table 7). The amount of TEMPO was eventually reduced to 0.5 equiv. (entry 2) as well as to 0.2 equiv. (entry 3) under an atmosphere of air. Surprisingly, the coupling reaction proceeded well without TEMPO to give the corresponding coupling product 10aa in 71% isolated yield (entry 4). We hypothesized that the oxidation of Pd(0) to Pd(II) might not always require any co-oxidants, and also worked under air (molecular oxygen) as a terminal oxidant in the case of 2-phenylthiazole (3a). However, we continued to use 0.5 equiv. of TEMPO, since it was often effective for the C–H arylation of 2-arylthiazoles 3 (see Table 8). It should be noted that C4-selective C–H arylation of thiophenes necessitates the use of TEMPO as a co-oxidant under air; otherwise the products were produced in low yield.9 Next, after screening additives, LiBF4 (1.5 equiv.) was identified as the optimal additive; both the reaction yield (77%) and regioselectivity (90%) were slightly improved (entry 5). Although the role of LiBF4 remains unclear, we assume that the lithium cation coordinates with the nitrogen atom of 2-arylthiazole to prevent over-arylation as well as to increase regioselectivity. In contrast, the use of lithium base additives such as LiOAc (entry 6) and LiOH (entry 7) decreased the yield and regioselectivity.
|
|
||||
|---|---|---|---|---|
| Entry | TEMPO/equiv. | Additive | C4/C5 | Yieldb (%) |
| a Reaction conditions: 3a (0.25 mmol), 6 (1.0 mmol), Pd(OAc)2 (0.025 mmol), phen (0.025 mmol), TEMPO (none to 1.0 mmol), additive (0.38 mmol), DMAc (0.5 mL), air, 100 °C, 24 h. b The combined yield of regioisomers is given. Isolated yield of 10aa is given in parenthesis. | ||||
| 1 | 4.0 | — | 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14 :14 |
85 (74) |
| 2 | 0.5 | — | 86:14 |
85 (74) |
| 3 | 0.2 | — | 88:12 |
77 |
| 4 | — | — | 88:12 |
83 (71) |
| 5 | 0.5 | LiBF4 | 90:10 |
93 (83) |
| 6 | 0.5 | LiOAc | 58:42 |
40 |
| 7 | 0.5 | LiOH | 9:91 |
68 |
|
|
|---|
| a Reaction conditions. Method F: 3 (0.25 mmol), 6 (1.0 mmol), Pd(OAc)2 (0.025 mmol), phen (0.025 mmol), TEMPO (0.125 mmol), LiBF4 (0.38 mmol), DMAc (0.5 mL), air, 100 °C, 24 h. Yield of the isolated product and regioselectivity (in parenthesis) are given. b The reaction was performed for 48 h. c 4-MeO-TEMPO (0.125 mmol) was used instead of TEMPO. d TEMPO (0.25 mmol) was used. |
|
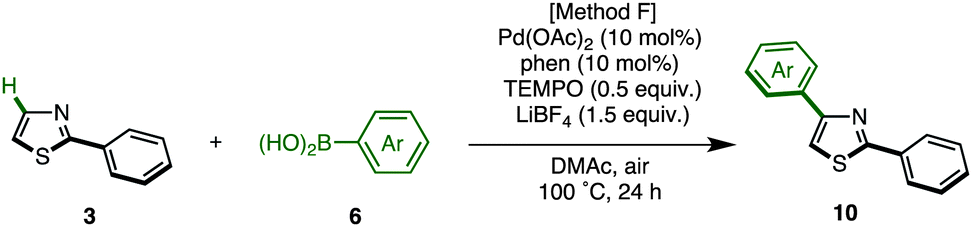
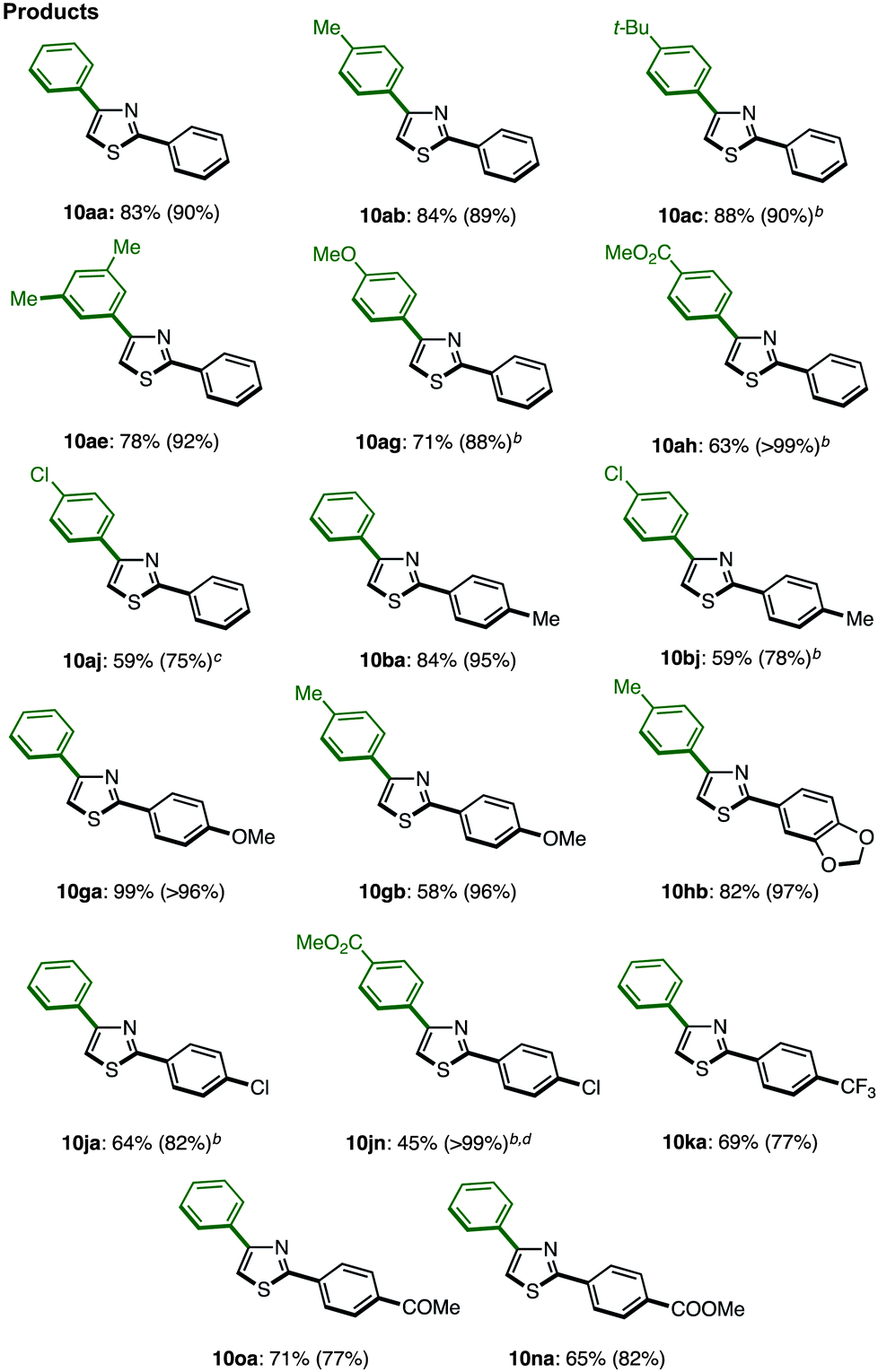
With these optimal conditions in hand (Method F), a range of 2,4-diarylthiazoles 10 were synthesized from 2-arylthiazoles 3 (Table 8). 2-Arylthiazoles bearing electron-donating groups (such as 4-methyl, 4-tert-butyl, 3,5-dimethyl, and 4-methoxy groups) on the aryl substituent were coupled with various arylboronic acids 6 to give the corresponding products 10aa–ag in good yields. 2-Arylthiazoles with electron-deficient groups (such as 4-methylester and 4-chloro groups) on the aryl substituent also gave products 10ah–aj in moderate to good yields after slight modification of our standard conditions. Additionally, a broad range of arylboronic acids 6 was also successfully employed in the C4-selective arylation. Although some reactions displayed incomplete regioselectivity, it is noteworthy that 2,4-diarylthiazoles 10 can be readily separated from the minor regioisomer (2,5-diarylthiazoles) by flash column chromatography.
The synthesis of 4,5-diarylthiazoles 12 by direct arylation has also not been reported thus far. For the synthesis of this motif using C–H coupling methodology, it is possible to select two synthetic routes: (i) C5-selective C–H arylation of 4-arylthiazoles 8 (route A, Scheme 5) and (ii) C4-selective C–H arylation of 5-arylthiazoles 4 (route B; Scheme 5). However, we only attempted route A since route B has synthetic problems in that 5-substituted thiazoles show a low reactivity at the C4 position due to steric reasons. Since the C2-arylation of 5-arylthiazoles is preferred, the substrate might need to be protected at C2, reacted at C4, and subsequently deprotected (see Table 3).
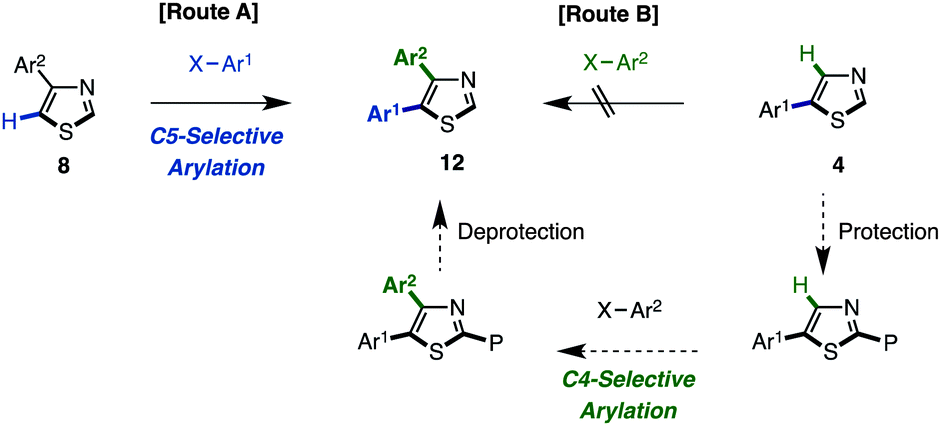 | ||
| Scheme 5 Two possible routes for the synthesis of 2,5-diarylthiazoles. | ||
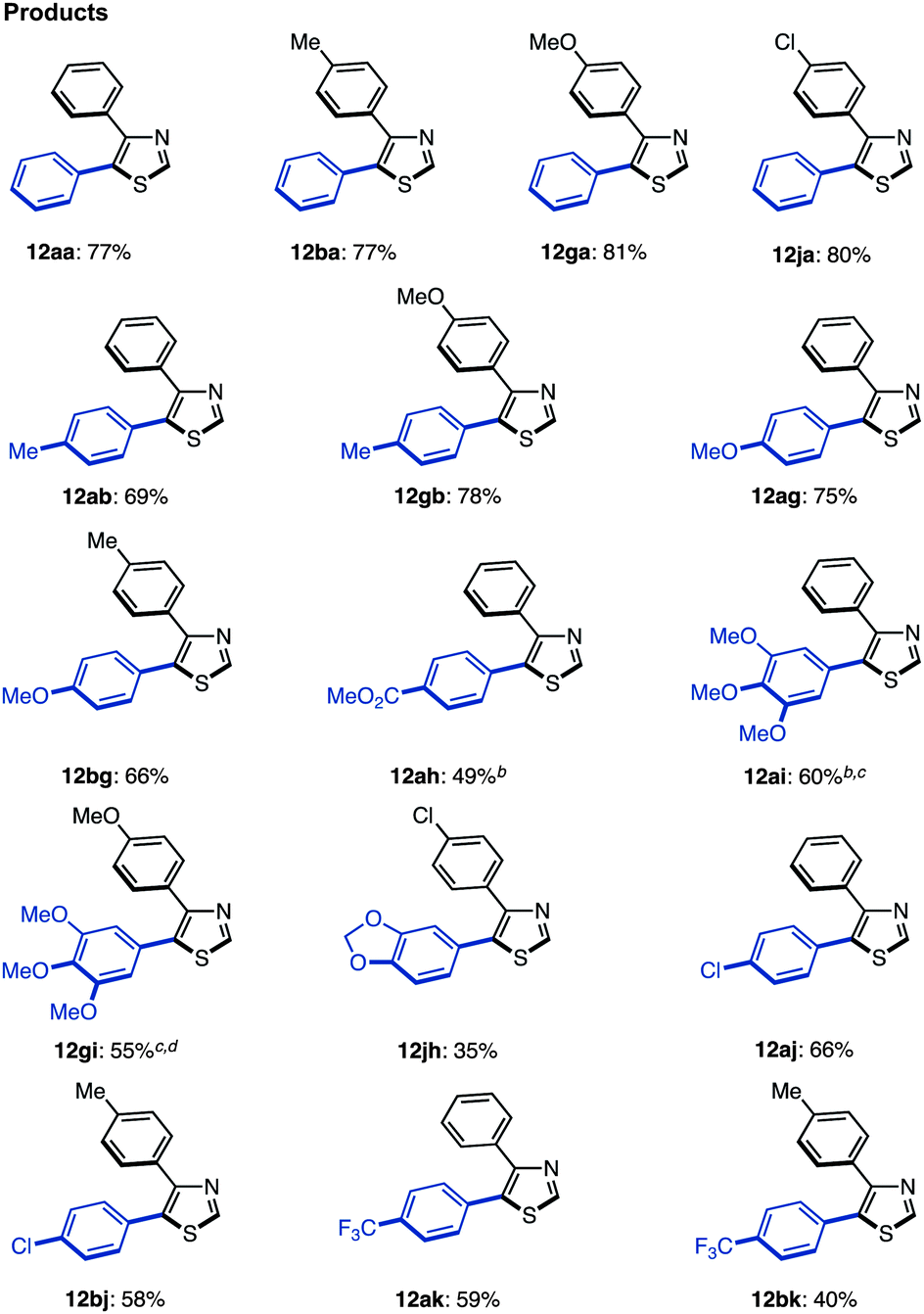
Thus, we investigated coupling reaction conditions for the synthesis of 2,5-diarylthiazoles via route A. As described in Table 2, we had already optimized the reaction conditions (Method D) for the C5-selective C–H arylation of thiazole (1). Using those conditions with slight modifications,17 the coupling reaction of 4-phenylthiazole (8a) with iodobenzene (2) in the presence of Pd(OAc)2 (10 mol%), PMe(t-Bu)2·HBF4 (10 mol%) and Cs2CO3 (1.5 equiv.) in t-AmylOH at 80 °C for 36 h afforded 4,5-diphenylthiazole (12aa) in 77% yield. Under these conditions, an over-arylation product (triphenylthiazole) was obtained in 14% yield. The substrate scope for the C5-selective C–H arylation of 4-arylthiazoles was further investigated as shown in Table 9. Although each reaction produced triarylthiazoles in less than 10% yield, the desired 2,5-diarylthiazoles 12 were only obtained in moderate yields, since the reactivity was similar to the C5-selective C–H arylation of unfunctionalized thiazole (see Table 2). To obtain 12ah, 12ai, and 12gi in better yields, higher temperatures were required, and in the case of an electron-rich aryl coupling partner such as 3,4,5-trimethoxyphenyl halide, the use of the aryl bromide instead of the aryl iodide provided a better result to give 12gi in 55% yield.
|
|
|---|
| a Reaction conditions. Method D: 8 (0.2 mmol), 2 (X = I, 0.2 mmol), Pd(OAc)2 (0.01 mmol), PMe(t-Bu)2·HBF4 (0.02 mmol), Cs2CO3 (0.3 mmol), t-AmylOH (0.5 mL), 80 °C, 36 h. b The reaction was performed at 100 °C for 18 h. c 2 (X = Br, 0.2 mmol) was used. d The reaction was performed at 100 °C for 36 h. |
|
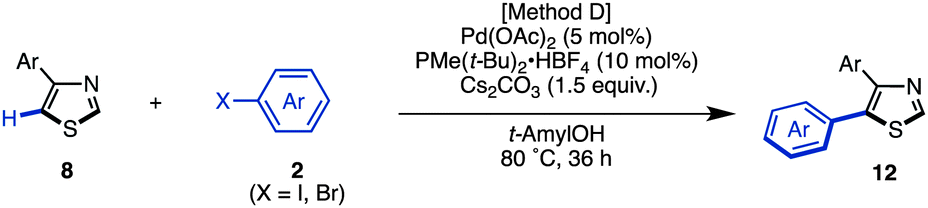
C–H arylation of 2,5-diarylthiazoles at the C4 position has only been reported by Shibahara, Murai and co-workers, which uses Pd(phen)2(PF6)2 as a catalyst, and Cs2CO3 as a base in DMAc.8d These reaction conditions were modified with respect to temperature, time, and equivalents of aryl iodide, and the best results were obtained at 130 °C for 40 h using 1.5 equiv. of aryl iodide (68% isolated yield of 13aaa). Under the optimized conditions (Method H′), we carried out a direct C4-arylation using various 2,5-diarylthiazoles 9 and aryl iodides 2 (Table 10). Not only were triarylthiazoles generated using iodobenzene (2; to give 13aaa and 13gak), but also substituted iodobenzenes (methyl, dimethyl, methoxy, trifluoromethyl substituents) and even heteroaromatics led to the corresponding triarylthiazoles 13aba, 13gek, 13aga, 13aka, and 13bea.
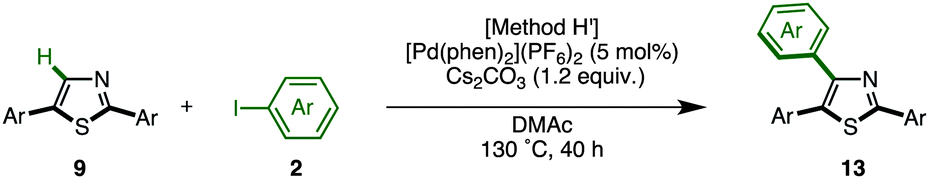
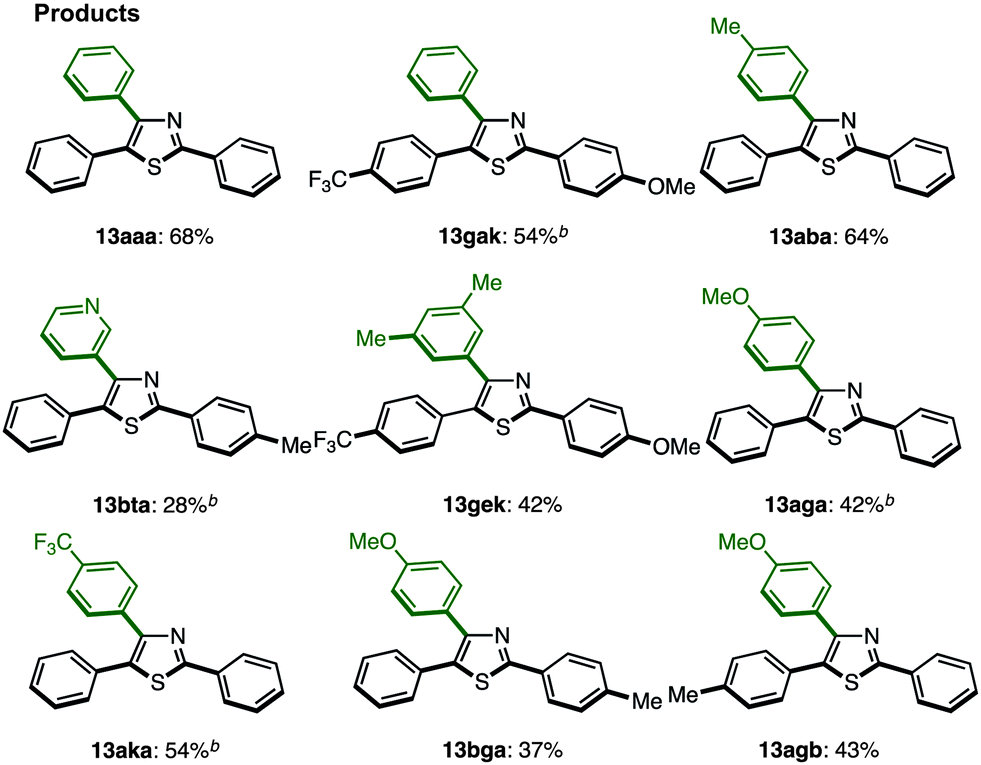
The somewhat lower yields of the coupling reaction were analyzed by GC analysis, and it was found that the arylation at the aryl groups on the thiazole cores took place in these particular reactions (Scheme 6). Thus, the possible side reactions are (i) overarylation of triarylthiazole 13 at the ortho position of the C2- or C4-aryl substituents after/before C4-arylation of 9 and (ii) further C–H arylation from 2,5-diarylthiazoles at the C4 or C2 position to give multi-arylated products.
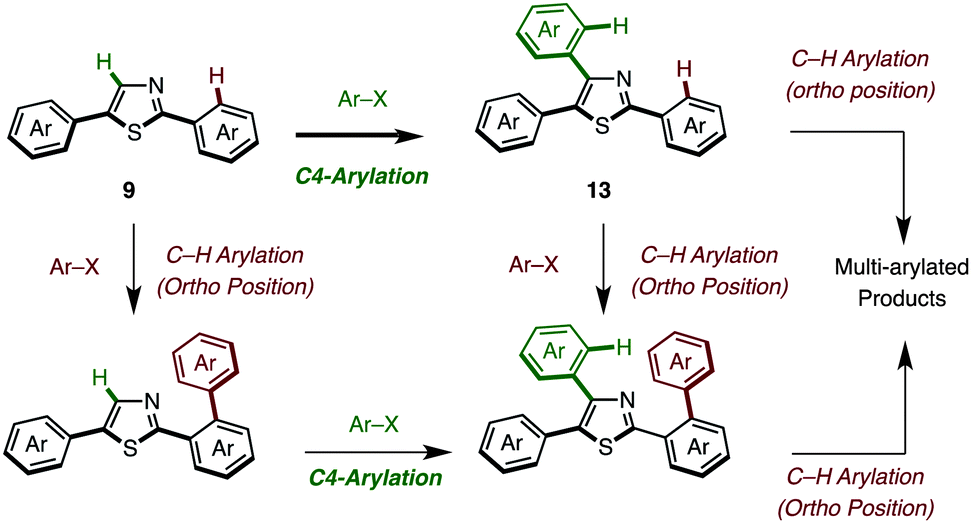 | ||
| Scheme 6 Potential side reactions in the C4-arylation of 2,5-diarylthiazoles. | ||
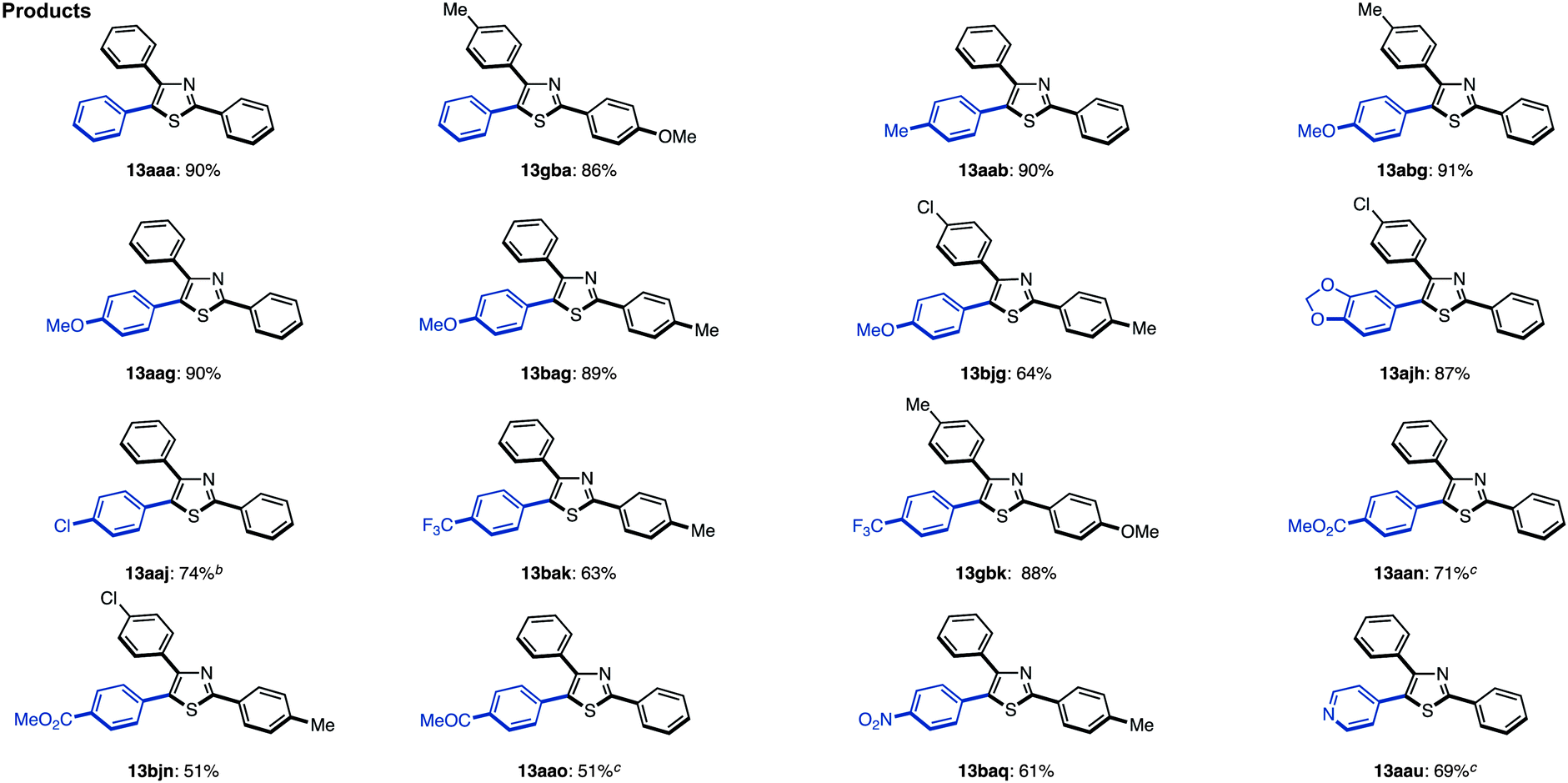
When the reaction conditions of C5-selective C–H arylation of 2-arylthiazoles were optimized (see Table 4), the best results were obtained using our previous conditions for palladium catalysis, PdCl2(bipy)/Ag2CO3 in 1,4-dioxane (Method G).7q We were pleased to find that a wide range of 2,4-diarylthiazoles 10 and aryl iodides 2 can be coupled to give triarylthiazoles 13 under those conditions (Table 11). Both electron-poor and electron-rich aryl iodides can be used in the C–H coupling reaction, but the reactions using more electron-poor aryl iodides (those containing chloro, ester and acetyl groups) as well as iodopyridine required longer reaction times and higher temperatures. When electron-deficient 2,4-diarylthiazoles were used, the corresponding triarylthiazoles were formed in relatively low yields (13bjg and 13njn). To improve the yields of 13aaj, 13aan, 13aao and 13aau, the reactions were conducted over longer time periods (12 h to 22 h) and/or at elevated temperatures (120 °C to 140 °C).
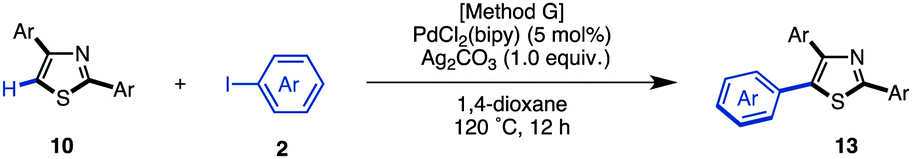
To demonstrate the utility of the C5-arylation of 2,4-diarylthiazoles, a gram-scale synthesis of triarylthiazole 13abh was undertaken (Scheme 7). Thiazole (1) was coupled with iodobenzene under Pd(OAc)2/CuI catalysis (Method A) to give 2-phenylthiazole (3a) in 72% yield (1.38 g). Afterwards, palladium-catalyzed C4-selective C–H arylation of 3a with p-tolylboronic acid was conducted under the influence of Pd(OAc)2/phen/TEMPO/LiBF4 (Method F) to furnish the corresponding 2,4-diarylthiazole 10ab in 75% yield with >95% regioselectivity (1.51 g). Finally, the C–H arylation of 10ab at the C5-position with aryl iodide in the presence of PdCl2(bipy)/Ag2CO3 catalyst (Method G) gave triarylthiazole 13abh in 92% yield (1.50 g).
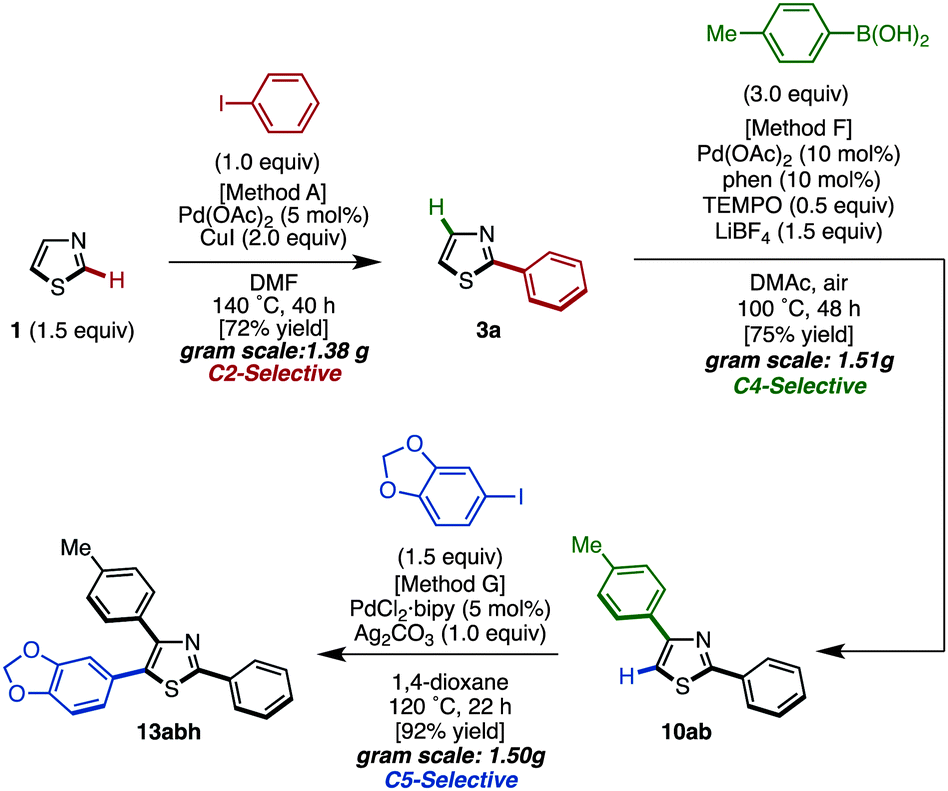 | ||
| Scheme 7 Gram-scale synthesis of triarylthiazole by sequential C–H arylations. | ||
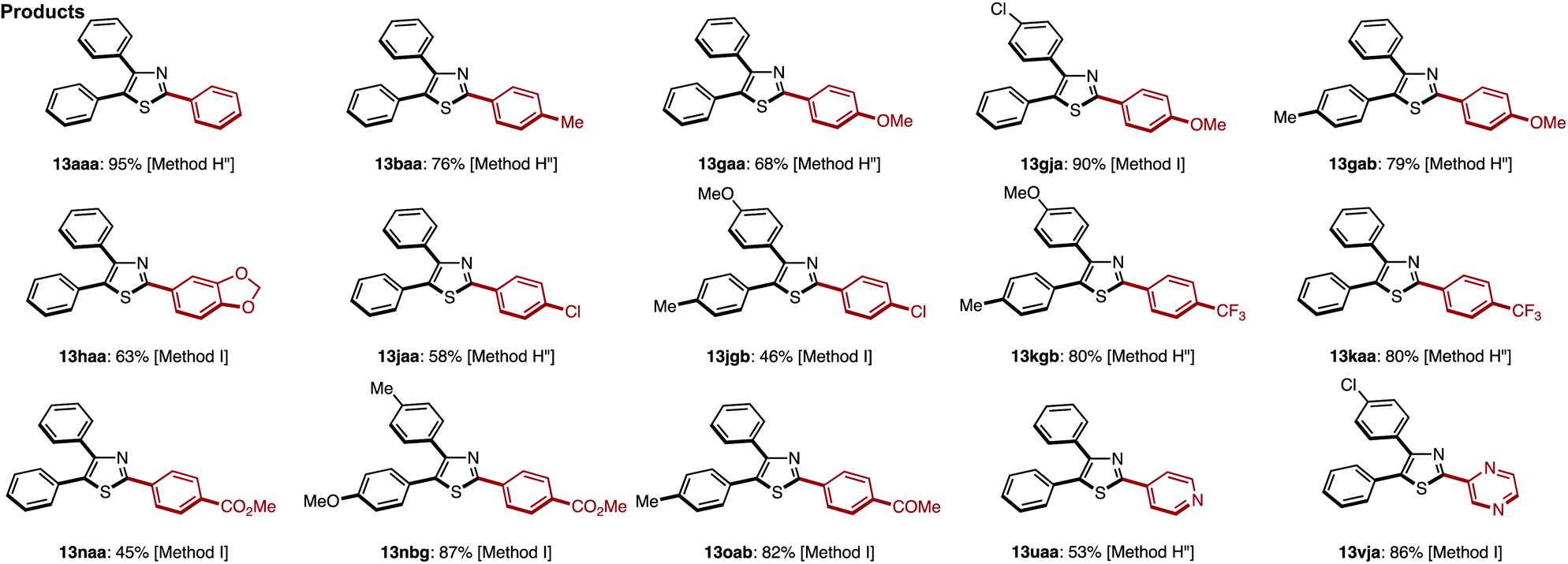
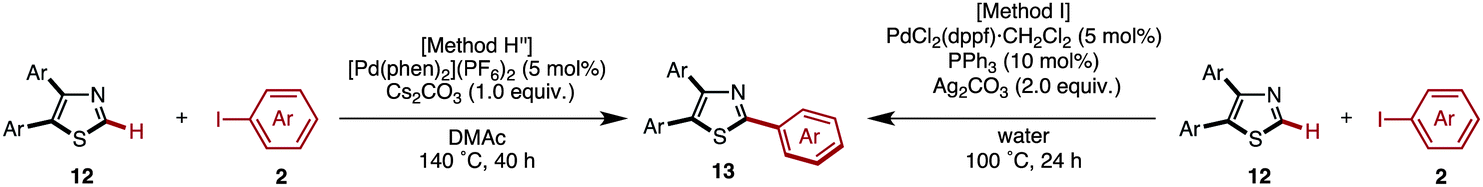
We first selected two representative catalyst systems [our own conditions (Method G)7q and Murai's conditions (Method H)8d] for the C–H arylation of 4,5-diarylthiazoles to provide triarylthiazoles 13 (Table 12). The coupling of 4,5-diphenylthiazole (12aa) with iodobenzene (2) under these two conditions gave an identical yield (66% yield) of the desired triphenylthiazole (13aaa). However, the starting material 12aa still remained under Murai's conditions, whereas 12aa was consumed using our protocol. After some modification of Murai's conditions with respect to the equivalents of base and iodobenzene (Method H′′), the yield of 13aaa was increased to 85% (isolated yield). When the same reaction was performed under Greaney's conditions (Method I),7h13aaa was obtained in 95% yield. These two different conditions for the C–H arylation of 12aa led to an extension of the reaction scope with respect to the 4,5-diarylthiazoles and aryl halide substrates (Table 12).
|
|
|---|
| a Reaction conditions. Method H′′: 12 (0.2 mmol), 2 (0.3 mmol), Pd(phen)2(PF6)2 (0.01 mmol), Cs2CO3 (0.20 mmol), DMAc (0.8 mL), 140 °C, 40 h. Method I: 12 (0.125–0.15 mmol), 2 (0.15–0.18 mmol), PdCl2(dppf)·CH2Cl2 (5 mol%), Ph3P (10 mol%), Ag2CO3 (0.25–0.3 mmol), water (1.0 mL), 100 °C, 24 h. |
|
Conclusions
In summary, we have developed a programmed synthesis of all substitution patterns of arylthiazoles (2-aryl, 4-aryl, 5-aryl, 2,4-diaryl, 2,5-diaryl, 4,5-diaryl, and 2,4,5-triaryl) via sequential C–H coupling. Representative reaction conditions are summarized in Scheme 8. Noteworthy features of our method are: (i) the synthetic expediency in introducing aryl groups by step-economical, direct C–H arylation, (ii) all aryl groups assembled stem from readily available aryl halides or arylboronic acids, (iii) the aryl-group-installation at the desired position can be achieved by the choice of catalytic systems, and (iv) the possibility of synthesizing all substitution patterns of arylthiazoles by 11 distinct pathways. Using this protocol, we have accomplished the synthesis of over 150 different arylthiazoles, among which exists fatostatin, known as a SREBP inhibitor. This method can be applied to a gram-scale synthesis of triarylthiazoles. Testing the biological activity of the generated library of arylthiazoles for agrochemical and pharmaceutical research is now the focus of our ongoing efforts.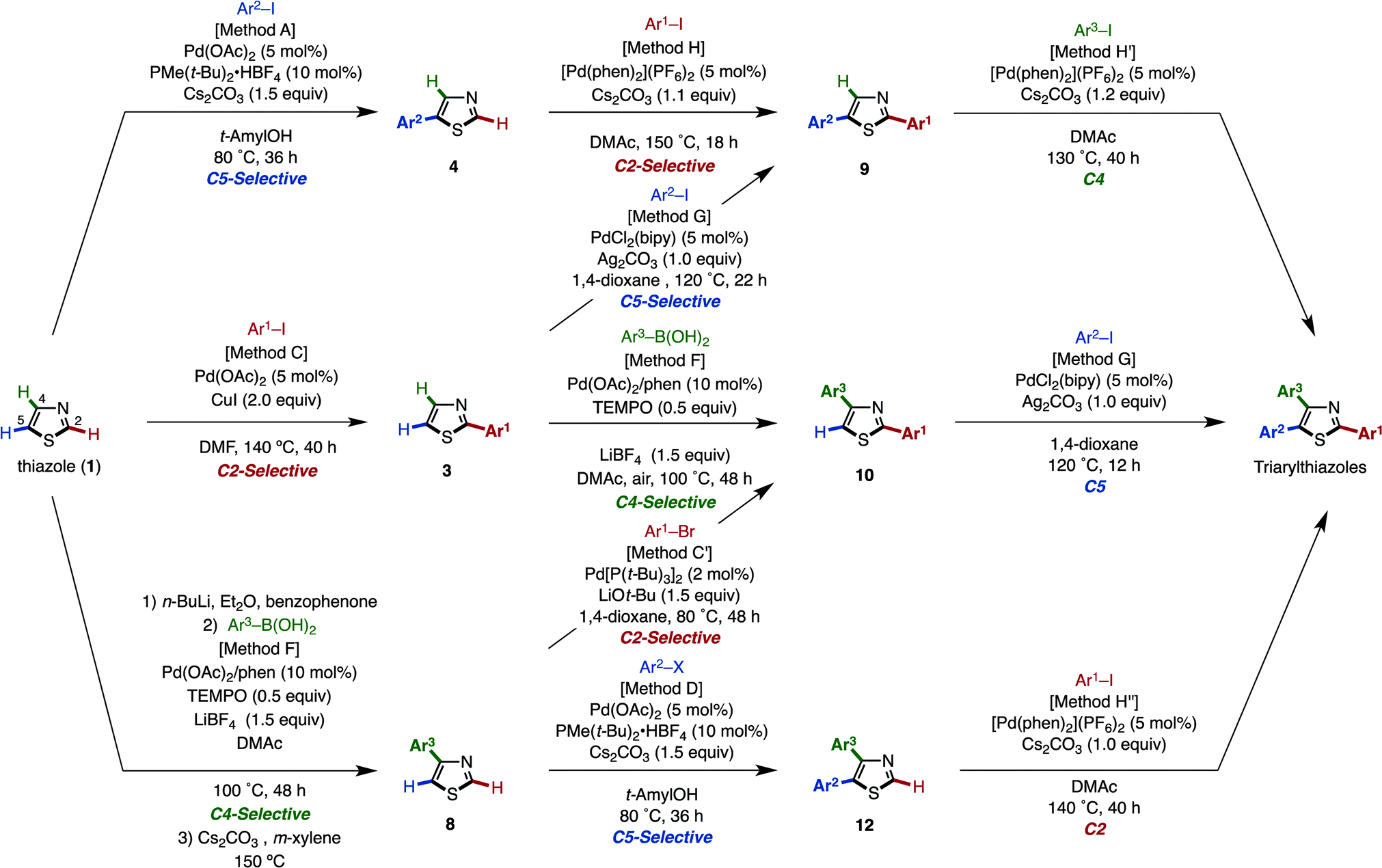 | ||
| Scheme 8 Programmed synthesis of all substitution patterns of thiazoles. | ||
Acknowledgements
This work was supported by the Funding Program for Next Generation World-Leading Researchers from JSPS (220GR049 to K.I.), and Grants-in-Aid for Scientific Research on Innovative Areas “Molecular Activation Directed toward Straightforward Synthesis” (25105720 to J.Y.) and KAKENHI (25708005 to J.Y.) from MEXT. ITbM is supported by the World Premier International Research Center (WPI) Initiative, Japan.Notes and references
- (a) S. Miwatashi, Y. Arikawa, E. Kotani, M. Miyamoto, K. Naruo, H. Kimura, T. Tanaka, S. Asahi and S. Ohkawa, J. Med. Chem., 2005, 48, 5966 CrossRef CAS PubMed; (b) Y. Choi, Y. Kawazoe, K. Murakami, H. Misawa and M. Uesugi, J. Biol. Chem., 2003, 278, 7320 CrossRef CAS PubMed; (c) J. Lee, S. J. Kim, H. Choi, Y. H. Kim, I. T. Lim, H. Yang, C. S. Lee, H. R. Kang, S. K. Ahn, S. K. Moon, D.-H. Kim, S. Lee, N. S. Choi and K. J. Lee, J. Med. Chem., 2010, 53, 6337 CrossRef CAS PubMed; (d) T. Suzuki, S. Hisakawa, Y. Itoh, N. Suzuki, K. Takahashi, M. Kawahata, K. Yamaguchi, H. Nakagawa and N. Miyata, Bioorg. Med. Chem. Lett., 2007, 17, 4208 CrossRef CAS PubMed; (e) M. S. Alam, L. Liu, Y.-E. Lee and D.-U. Lee, Chem. Pharm. Bull., 2011, 59, 568 CrossRef CAS; (f) D. Zwilling, S.-Y. Huang, K. V. Sathyasaikumar, F. M. Notarangelo, P. Guidetti, H.-Q. Wu, J. Lee, J. Truong, Y. Andrews-Zwilling, E. W. Hsieh, J. Y. Louie, T. Wu, K. Scearce-Levie, C. Patrick, A. Adame, F. Giorgini, S. Moussaoui, G. Laue, A. Rassoulpour, G. Flik, Y. Huang, J. M. Muchowski, E. Masliah, R. Schwarcz and P. J. Muchowski, Cell, 2011, 145, 863 CrossRef CAS PubMed.
- (a) A. Hantzsch, Justus Liebigs Ann. Chem., 1889, 250, 257 CrossRef; (b) R. Marbet, H. Scheukel and H. Erlenmeyer, Helv. Chim. Acta, 1945, 28, 924 CrossRef.
- For selected recent examples, see: (a) P. W. Sheldrake, M. Matteucci and E. McDonald, Synlett, 2006, 460 CrossRef CAS PubMed; (b) Y. Ishiwata and H. Togo, Synlett, 2008, 2637 CAS; (c) J. F. Sanz-Cervera, R. Blasco, J. Piera, M. Cynamon, I. Ibáñez, M. Murguía and S. Fustero, J. Org. Chem., 2009, 74, 8988 CrossRef CAS PubMed; (d) G. S. Lingaraju, T. R. Swaroop, A. C. Vinayaka, K. S. S. Kumar, M. P. Sadashiva and K. S. Rangappa, Synthesis, 2012, 1373 CAS.
- (a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed; (b) Topics in Current Chemistry, Vol. 219, Cross-Coupling Reactions: A Practical Guide, ed. N. Miyaura, Springer, Berlin, 2002 Search PubMed; (c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed.
- For selected examples, see: (a) D. Guianvarc'h, J.-L. Fourrey, R. Maurisse, J.-S. Sun and R. Benhida, Org. Lett., 2002, 4, 4209 CrossRef CAS PubMed; (b) G. A. Molander and B. Biolatto, J. Org. Chem., 2003, 68, 4302 CrossRef CAS PubMed; (c) E. L. Stangeland and T. Sammakia, J. Org. Chem., 2004, 69, 2381 CrossRef CAS PubMed; (d) B. H. Lipshutz, B. Frieman and H. Birkedal, Org. Lett., 2004, 6, 2305 CrossRef CAS PubMed; (e) P. Vachal and L. M. Toth, Tetrahedron Lett., 2004, 45, 7157 CrossRef CAS PubMed; (f) P. Stanetty, M. Schnürch and M. D. Mihovilovic, J. Org. Chem., 2006, 71, 3754 CrossRef CAS PubMed.
- Recent reviews on catalytic C–H arylation, see: (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (b) F. Kakiuchi and T. Kochi, Synthesis, 2008, 3013 CrossRef CAS PubMed; (c) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed; (d) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (e) T. Satoh and M. Miura, Synthesis, 2010, 3395 CrossRef CAS PubMed; (f) J. Bouffard and K. Itami, Top. Curr. Chem., 2010, 292, 231 CAS; (g) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (h) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- For selected examples of C2 or C5 selective arylation of thiazoles, see: (a) Y. Aoyagi, A. Inoue, I. Koizumi, R. Hashimoto, K. Tokunaga, K. Gohma, J. Komatsu, K. Sekine, A. Miyafuji, J. Kunoh, R. Honma, Y. Akita and A. Ohta, Heterocycles, 1992, 33, 257 CrossRef CAS; (b) S. Pivsa-Art, T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1998, 71, 467 CrossRef CAS; (c) Y. Kondo, T. Komine and T. Sakamoto, Org. Lett., 2000, 2, 3111 CrossRef CAS PubMed; (d) A. Mori, A. Sekiguchi, K. Masui, T. Shimada, M. Horie, K. Osakada, M. Kawamoto and T. Ikeda, J. Am. Chem. Soc., 2003, 125, 1700 CrossRef CAS PubMed; (e) K. Masui, A. Mori, K. Okano, K. Takamura, M. Kinoshita and T. Ikeda, Org. Lett., 2004, 6, 2011 CrossRef CAS PubMed; (f) M. Parisien, D. Valette and K. Fagnou, J. Org. Chem., 2005, 70, 7578 CrossRef CAS PubMed; (g) F. Bellina, S. Cauteruccio and R. Rossi, Eur. J. Org. Chem., 2006, 1379 CrossRef CAS; (h) G. L. Turner, J. A. Morris and M. F. Greaney, Angew. Chem., Int. Ed., 2007, 46, 7996 CrossRef CAS PubMed; (i) H. A. Chiong and O. Daugulis, Org. Lett., 2007, 9, 1449 CrossRef CAS PubMed; (j) T. Martin, C. Verrier, C. Hoarau and F. Marsais, Org. Lett., 2008, 10, 2909 CrossRef CAS PubMed; (k) J. Canivet, J. Yamaguchi, I. Ban and K. Itami, Org. Lett., 2009, 11, 1733 CrossRef CAS PubMed; (l) J. Roger, F. Požgan and H. Doucet, J. Org. Chem., 2009, 74, 1179 CrossRef CAS PubMed; (m) B. Liégault, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, J. Org. Chem., 2009, 74, 1826 CrossRef PubMed; (n) S. Tamba, Y. Okubo, S. Tanaka, D. Monguchi and A. Mori, J. Org. Chem., 2010, 75, 6998 CrossRef CAS PubMed; (o) H. Hachiya, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2010, 49, 2202 CAS; (p) D. Lapointe, T. Markiewicz, C. J. Whipp, A. Toderian and K. Fagnou, J. Org. Chem., 2011, 76, 749 CrossRef CAS PubMed; (q) S. Yanagisawa and K. Itami, Tetrahedron, 2011, 67, 4425 CrossRef CAS PubMed; (r) T. Yamamoto, K. Muto, M. Komiyama, J. Canivet, J. Yamaguchi and K. Itami, Chem.–Eur. J., 2011, 17, 10113 CrossRef CAS PubMed; (s) K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 169 CrossRef CAS PubMed; (t) K. Amaike, K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 13573 CrossRef CAS PubMed.
- C–H diarylation of thiazole on C2 and C5 positions: (a) Ref. 7b ; (b) A. Yokooji, T. Okazawa, T. Satoh, M. Miura and M. Nomura, Tetrahedron, 2003, 59, 5685 CrossRef CAS; (c) H.-Q. Do, R. M. K. Khan and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 15185 CrossRef CAS PubMed; (d) F. Shibahara, E. Yamaguchi and T. Murai, J. Org. Chem., 2011, 76, 2680 CrossRef CAS PubMed; (e) F. Shibahara, T. Yamauchi, E. Yamaguchi and T. Murai, J. Org. Chem., 2012, 77, 8815 CrossRef CAS PubMed.
- S. Kirchberg, S. Tani, K. Ueda, J. Yamaguchi, A. Studer and K. Itami, Angew. Chem., Int. Ed., 2011, 50, 2387 CrossRef CAS PubMed.
- For a review on platform synthesis, see: (a) K. Itami and J. Yoshida, Chem.–Eur. J., 2006, 12, 3966 CrossRef CAS PubMed ; Multisubstituted olefins; (b) K. Itami, T. Nokami, Y. Ishimura, K. Mitsudo, T. Kamei and J. Yoshida, J. Am. Chem. Soc., 2001, 123, 11577 CrossRef CAS PubMed; (c) K. Itami, T. Kamei and J. Yoshida, J. Am. Chem. Soc., 2003, 125, 14670 CrossRef CAS PubMed; (d) K. Itami, M. Mineno, N. Muraoka and J. Yoshida, J. Am. Chem. Soc., 2004, 126, 11778 CrossRef CAS PubMed; (e) K. Itami, Y. Ohashi and J. Yoshida, J. Org. Chem., 2005, 70, 2778 CrossRef CAS PubMed; (f) K. Tonogaki, K. Itami and J. Yoshida, J. Am. Chem. Soc., 2006, 128, 1464 CrossRef CAS PubMed ; Multisubstituted heteroarenes; (g) K. Itami, D. Yamazaki and J. Yoshida, J. Am. Chem. Soc., 2004, 126, 15396 CrossRef CAS PubMed; (h) S. Yanagisawa, K. Ueda, H. Sekizawa and K. Itami, J. Am. Chem. Soc., 2009, 131, 14622 CrossRef CAS PubMed.
- (a) L.-C. Campeau, M. Bertrand-Laperle, J.-P. Leclerc, E. Villemure, S. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3276 CrossRef CAS PubMed; (b) See ref. 8d.
- T. Okazawa, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2002, 124, 5286 CrossRef CAS PubMed.
- Multiple C–H arylation of heteroarenes to install all different aryl groups, see: (a) Ref. 10h ; (b) L.-C. Campeau, D. R. Stuart, J.-P. Leclerc, M. Bertrand-Laperle, E. Villemure, H.-Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291 CrossRef CAS PubMed; (c) R. Goikhman, T. L. Jacques and D. Sames, J. Am. Chem. Soc., 2009, 131, 3042 CrossRef CAS PubMed.
- N. Chatani, T. Fukuyama, H. Tatamidani, F. Kakiuchi and S. Murai, J. Org. Chem., 2000, 65, 4039 CrossRef CAS PubMed.
- (a) J. Hämmerle, M. Spina, M. Schnürch, M. D. Mihovilovic and P. Stanetty, Synthesis, 2008, 3099 Search PubMed; (b) B. Liégault, I. Petrov, S. I. Gorelsky and K. Fagnou, J. Org. Chem., 2010, 75, 1047 CrossRef PubMed.
- M. Steinmetz, K. Ueda, S. Grimme, J. Yamaguchi, S. Kirchberg, K. Itami and A. Studer, Chem.–Asian J., 2012, 7, 1256 CrossRef CAS PubMed.
- See the ESI† for the details and further screening of conditions.
- H. Furukawa, S. Matsumura, A. Sugie, D. Monguchi and A. Mori, Heterocycles, 2009, 79, 303 CrossRef CAS PubMed.
- S. Kamisuki, Q. Mao, L. Abu-Elheiga, Z. Gu, A. Kugimiya, Y. Kwon, T. Shinohara, Y. Kawazoe, S. Sato, K. Asakura, H.-Y. P. Choo, J. Sakai, S. J. Wakil and M. Uesugi, Chem. Biol., 2009, 16, 882 CrossRef CAS PubMed.
- D. L. Comins and N. B. Mantlo, J. Org. Chem., 1985, 50, 4410 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectral data for all compounds, including scanned images of 1H and 13C NMR spectra. See DOI: 10.1039/c3sc52199k |
| This journal is © The Royal Society of Chemistry 2014 |