Rhodium-catalyzed regioselective opening of vinyl epoxides with Et3N·3HF reagent – formation of allylic fluorohydrins†
Qi
Zhang
and
Hien M.
Nguyen
*
University of Iowa, Department of Chemistry, Iowa City, IA 52242, USA. E-mail: hien-nguyen@uiowa.edu
First published on 3rd October 2013
Abstract
A highly regioselective rhodium-catalyzed ring-opening of vinyl epoxides with Et3N·3HF reagent to form branched allylic fluorohydrins is described. The reaction occurs at room temperature under ambient air and relies on RhCOD2BF4 as an effective catalyst, providing the desired 1,2-addition allylic fluorohydrins in moderate to good yields with excellent levels of regioselectivity. Mechanistic studies demonstrate that the regioselective ring-opening of enantiopure vinyl epoxide occurs with inversion of stereochemistry.
Introduction
The many important roles that fluorine-containing compounds play in pharmaceuticals, agrochemicals, medical imaging, and high-performance materials have made the synthesis of this class of molecules a major focus in recent years.1 Consequently, practical and elegant methods have been developed for the construction of aryl–F and alkyl–F bonds.2,3 In contrast, the synthesis of allylic fluorides remains relatively underdeveloped.4 In recent years, a few examples of the transition-metal-catalyzed fluorination of allylic electrophiles with fluoride ions have been reported for the regio- and enantioselective preparation of allylic fluoride products.5 We recently reported that [IrClCOD]2 is an effective catalyst for the regioselective fluorination of allylic trichloroacetimidates with Et3N·3HF, providing secondary and tertiary allylic fluorides with good yields and excellent selectivity (Scheme 1a).6 In an effort to expand the capabilities of our fluorination method, we sought the development of a variant proceeding via the opening of vinyl epoxides, as depicted in Scheme 1b.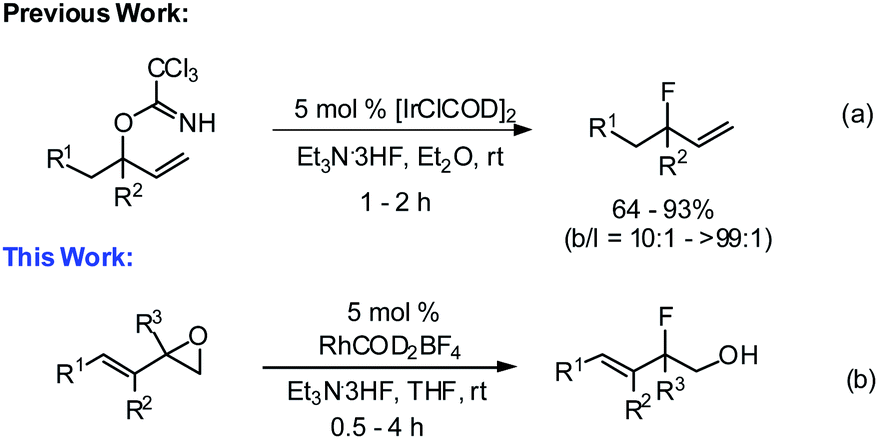 | ||
| Scheme 1 Transition-metal-catalyzed fluorination. | ||
Vinyl epoxides are commonly used building blocks for the synthesis of biologically active targets.7 They are quite reactive in the presence of transition-metal catalysts.8 Palladium is most frequently used to promote the ring-opening of vinyl epoxides with a wide variety of nucleophiles, leading to 1,4-addition products due to the electron-deficient nature of the vinyl epoxide oxygen.9 The 1,2-addition products can be achieved by choice of the ligand and co-catalysts.10 Rhodium has also been reported as an effective catalyst for the ring-opening of vinyl epoxides with both anilines and alcohols to provide trans-1,2-amino alcohols or alkoxy alcohols, respectively, in good yield and with high levels of diastereo- and regio-selectivity.11 Even though the transition-metal-catalyzed asymmetric ring-opening of terminal epoxides1e,12 and oxabicyclic olefins13 using the nucleophilic fluoride ion has been developed, to the best of our knowledge there are no reports on the transition-metal-catalyzed regioselective ring-opening of vinyl epoxides with nucleophilic fluoride reagents.14
Results and discussion
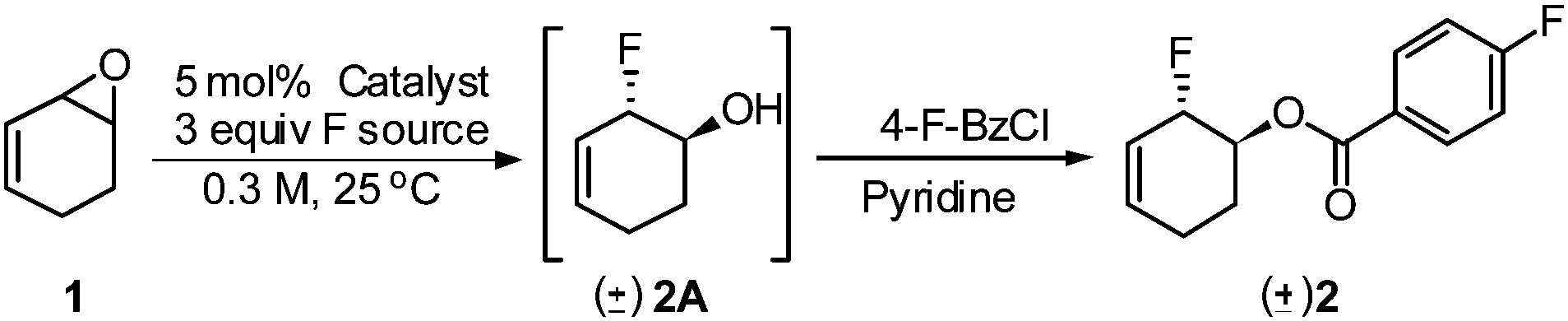
Initial studies focused on establishing the regioselectivity of the opening of vinyl epoxide 1 with Et3N·3HF reagent (Table 1). Using our optimized conditions for the iridium-catalyzed fluorination of allylic trichloroacetimidates,6 we began our investigation with 2.5 mol% [IrCl(COD)]2 (entry 1) in Et2O at room temperature. The 1,2-addition allylic fluorohydrin 2A was obtained in 37% NMR yield. A number of rhodium(I) catalysts were then investigated. RhCOD2BF4 (entry 4) produced the best result to provide an 88% NMR yield of 2A.15 We subsequently screened a number of solvents, and THF (entry 7) provided 2A with the highest NMR yield (92%). In all cases with rhodium catalysts, the ring-opening of 1 proceeded to completion within 30 min, and the undesired 1,4-addition product was not observed. A control experiment (entries 8 and 9) was performed without the catalyst, and a 23–35% NMR yield of allylic fluorohydrin 2A was observed after 18 h.16 Olah's reagent performed poorly under rhodium conditions (entry 10) with only 12% conversion after the reaction had been stirring for 14 h. Other nucleophilic fluoride sources (CsF, TBAT, AgF, and KF) showed no reactivity (entries 11–14). For full analysis and isolated yield determination, crude fluorohydrin 2A was treated with 4-fluorobenzoyl chloride (entry 15), and the corresponding allylic fluoride 2 was isolated in 79% yield over two steps.17 To demonstrate the reproducibility and scalability of this reaction, 10 mmol of epoxide 1 (entry 15) was subjected to similar conditions: fluoride product 2 was obtained in 76% yield, which is comparable to the use of 0.2 mmol of substrate 1.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | F source | Solvent | Time (h) | NMR yield 2Ab (%) | Isolated yield 2c (%) |
| a All reactions were conducted with 0.2 mmol of vinyl epoxide 1. b The NMR yield of allylic fluorohydrin 2A was determined by 19F NMR analysis of the crude reaction mixture using PhCF3 as an internal standard. c For full analyses, crude product 2A was then transformed into the isolable allylic 4-fluorobenzoate 2 and the isolated yield was determined. d The reaction was performed with 10 mmol of vinyl epoxide 1. | |||||||
| 1 | [IrCl(COD)]2 | 2.5 | Et3N·3HF | Et2O | 2 | 37 | |
| 2 | [RhCl(COD)]2 | 2.5 | Et3N·3HF | Et2O | 2 | 55 | |
| 3 | RhCOD2OTf | 5 | Et3N·3HF | Et2O | 0.5 | 70 | |
| 4 | RhCOD2BF4 | 5 | Et3N·3HF | Et2O | 0.5 | 88 | |
| 5 | RhCOD2BF4 | 5 | Et3N·3HF | MTBE | 0.5 | 71 | |
| 6 | RhCOD2BF4 | 5 | Et3N·3HF | CH2Cl2 | 0.5 | 79 | |
| 7 | RhCOD2BF4 | 5 | Et3N·3HF | THF | 0.5 | 92 | |
| 8 | None | 0 | Et3N·3HF | Et2O | 18 | 23 | |
| 9 | None | 0 | Et3N·3HF | THF | 18 | 35 | |
| 10 | RhCOD2BF4 | 5 | Pyridine·HF | THF | 14 | 12 | |
| 11 | RhCOD2BF4 | 5 | CsF | THF | 18 | 0 | |
| 12 | RhCOD2BF4 | 5 | TBAT | THF | 18 | 0 | |
| 13 | RhCOD2BF4 | 5 | AgF | THF | 18 | 0 | |
| 14 | RhCOD2BF4 | 5 | KF | THF | 18 | 0 | |
| 15 | RhCOD2BF4 | 5 | Et3N·3HF | Et2O | 0.5 | 89 (88)d | 79 (76)d |
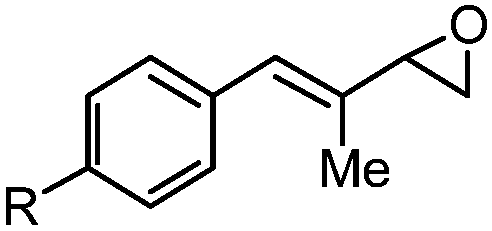
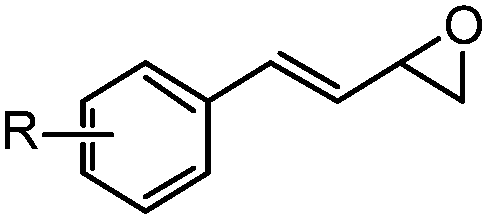
With the optimized conditions in hand, we proceeded to explore the scope and limitation of the rhodium-catalyzed regioselective ring-opening of vinyl epoxides (Table 2). We discovered that five- and seven-membered ring vinyl epoxides performed poorly under the same reaction protocol. We reasoned that acyclic vinyl epoxides possessing greater conformational flexibility may undergo ring-opening in the presence of RhCOD2BF4 and Et3N·3HF reagent. To our excitement, various acylic vinyl epoxide substrates 3–13 (Table 2) proceeded smoothly under rhodium conditions. Because most epoxide substrates are too volatile for isolation, all allylic fluorohydrins 14A–24A were converted into the 4-fluoro-benzoate derivatives 14–24 for full analyses and isolated yield calculation. Overall, the reactivity of the vinyl epoxides is dependent on both the steric and electronic nature of the substituents at the C(1)- and C(2)-position of the allyl moiety. For instance, the presence of the methyl group at the C(2)-position of vinyl epoxides 3 and 4 (Table 2, entries 1 and 2) provided allylic fluorides 14 and 15 in 60% and 62% yield, respectively, compared to their epoxide counterparts 5–8 (43–58%, entries 3–6). We discovered that vinyl epoxides containing the C(1)-electron-donating groups (e.g. 4-methoxy-phenyl) only resulted in decomposition. In contrast, electron-withdrawing substituents at the C(1)-position of vinyl epoxides 4–7 (entries 2–5) provided allylic fluorohydrins 15A–18A in 50–76% NMR yield (43–62% of their isolable fluoride derivatives). On the other hand, increasing the steric hindrance at the C(2)-position (entry 7) requires a significantly longer reaction time (10 h vs. 1 h) and allylic fluoride 20 was isolated in 59% yield. Vinyl epoxide 10 (entry 8) bearing the primary alkyl functionality also performed adequately to provide the allylic fluorohydrin 21A in 61% NMR yield.
|
|
||||
|---|---|---|---|---|
| Entry | Vinyl epoxides | Time (h) | Allylic fluorohydrins NMR yielda | Allylic fluorides isolated yield |
a NMR yields of allylic fluorohydrins 14A–24A were determined by 19F NMR analysis of the reaction mixture using PhCF3 as an internal standard.
b Reactions were performed using 0.2 mmol of vinyl epoxides.
c Reactions were performed using 0.5 mmol of vinyl epoxides.
d An 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1,2-addition products 14A and 15A and their undesired 1,4-addition products was observed.
e A 6:1 mixture of 1,2-addition product 20A and its undesired 1,4-addition product was observed.
f A 1.7:1 mixture of 1,2-addition product 21A and undesired 1,4-addition product was observed. :1 mixture of 1,2-addition products 14A and 15A and their undesired 1,4-addition products was observed.
e A 6:1 mixture of 1,2-addition product 20A and its undesired 1,4-addition product was observed.
f A 1.7:1 mixture of 1,2-addition product 21A and undesired 1,4-addition product was observed.
|
||||
|
||||
| 1 | 3: R = H | 1 | 14A (82%)d | 14 (60%)b |
| 2 | 4: R = Cl | 1 | 15A (76%)d | 15 (62%)c |
|
||||
| 3 | 5: R = 4-Cl | 1 | 16A (64%) | 16 (46%)b |
| 4 | 6: R = 3-Br | 1 | 17A (63%) | 17 (43%)c |
| 5 | 7: R = 4-CF3 | 4 | 18A (50%) | 18 (46%)b |
| 6 | 8: R = H | 1 | 19A (69%) | 19 (58%)b |
| 7 |
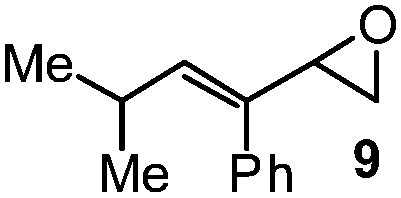
|
10 | 20A (73%)e | 20 (59%)b |
| 8 |
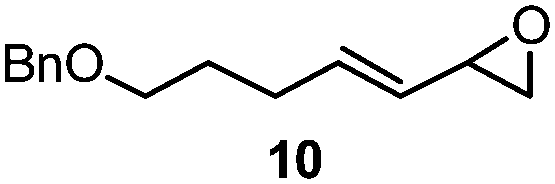
|
3 | 21A (61%)f | 21 (48%)b |
| 9 |
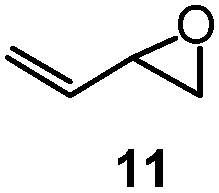
|
1 | 22A (50%) | 22 (49%)b |
|
22A (50%) | 22 (44%)c | ||
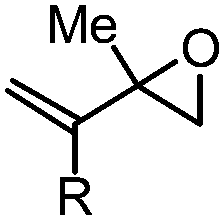
| 10 | 12: R = H | 0.5 | 23A (80%) | 23 (70%)b |
| 11 | 13: R = Me | 0.5 | 24A (65%) | 24 (56%)b |
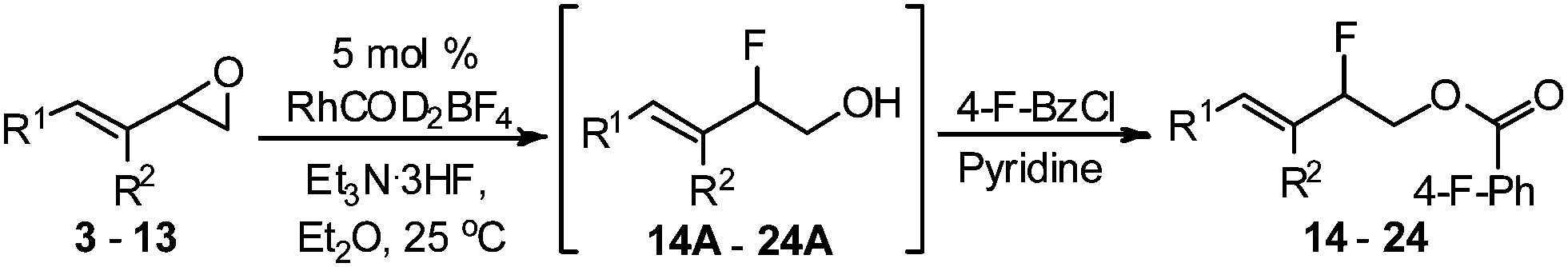
The success of butadiene monoepoxide 11 (Table 2, entry 9) to form fluorohydrin 22A (50% NMR yield) and its isolable fluoride 22 (44–49%) led to our studies of isoprene monoepoxide 12 (entry 10) and C(2)-substituted methyl substrate 13 (entry 11). The reactions were fast (0.5 h) and proceeded with complete regioselectivity, providing tertiary allylic fluorohydrins 23A and 24A, respectively, in good NMR yields (65–80%). As expected, the more hindered vinyl epoxide 13 provided allylic fluorohydrin 24A in lower yield than that of substrate 12. These results are consistent with what has been observed when comparing the ring-opening of vinyl epoxide 3 (entry 1) to that of the more hindered 9 (entry 7), where the NMR yield is 82% vs. 73%.
With the ability to access a number of allylic fluorohydrins in moderate to good yields and with excellent regioselectivity, we next sought to establish the utility of this method by transforming tertiary allylic fluorohydrins into the tertiary alkyl fluorides 25 and 26 (Scheme 2), which could be potentially used as structural motifs of anticancer ether phospholipid compounds.18 One-pot regioselective ring-opening of vinyl epoxides 12 and 13 followed by hydrogenation and benzoylation furnished tertiary fluorides 25 and 26 in 52% and 50% yield, respectively.
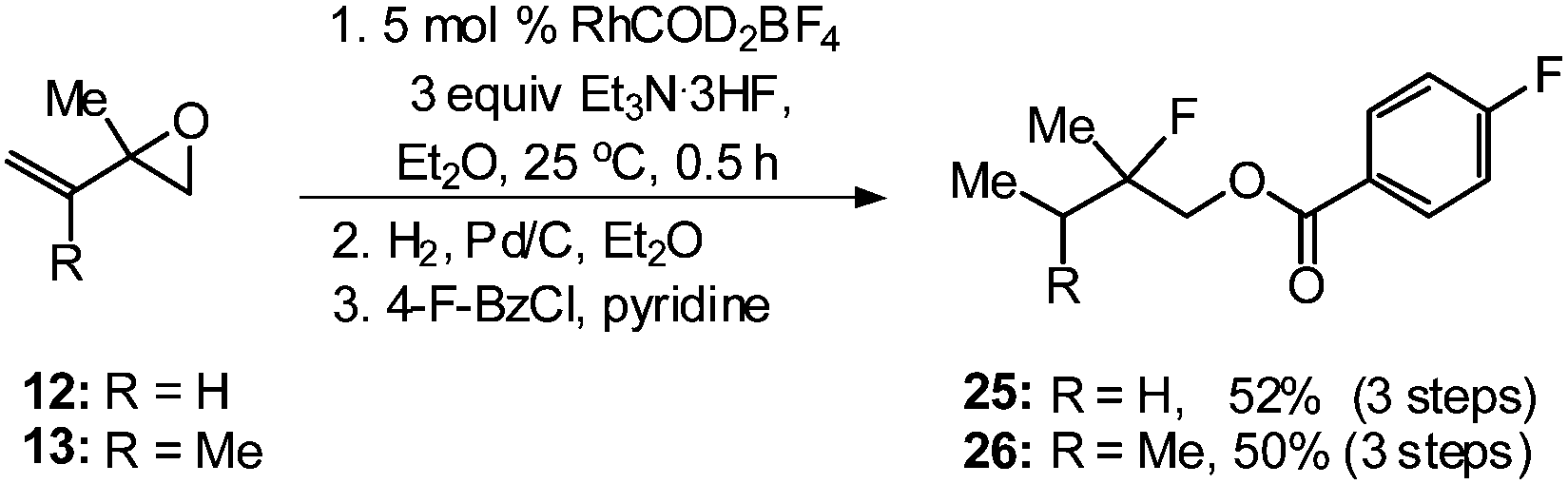 | ||
| Scheme 2 Conversion of allylic fluorohydrins into tertiary alkyl fluorides. | ||
To determine if Lewis acid behavior alone was responsible for reactivity with RhCOD2BF4, we conducted control experiments with cyclohexyl epoxide 27 and styrene oxide 28 (Scheme 3) under the same rhodium conditions. No products were observed after 18 h, suggesting that RhCOD2BF4 is likely not acting as a Lewis acid and the presence of an alkene unit is required for the reaction to occur. These results are consistent with previously reported results for the rhodium-catalyzed regioselective ring-opening of vinyl epoxides with alcohols and anilines.11
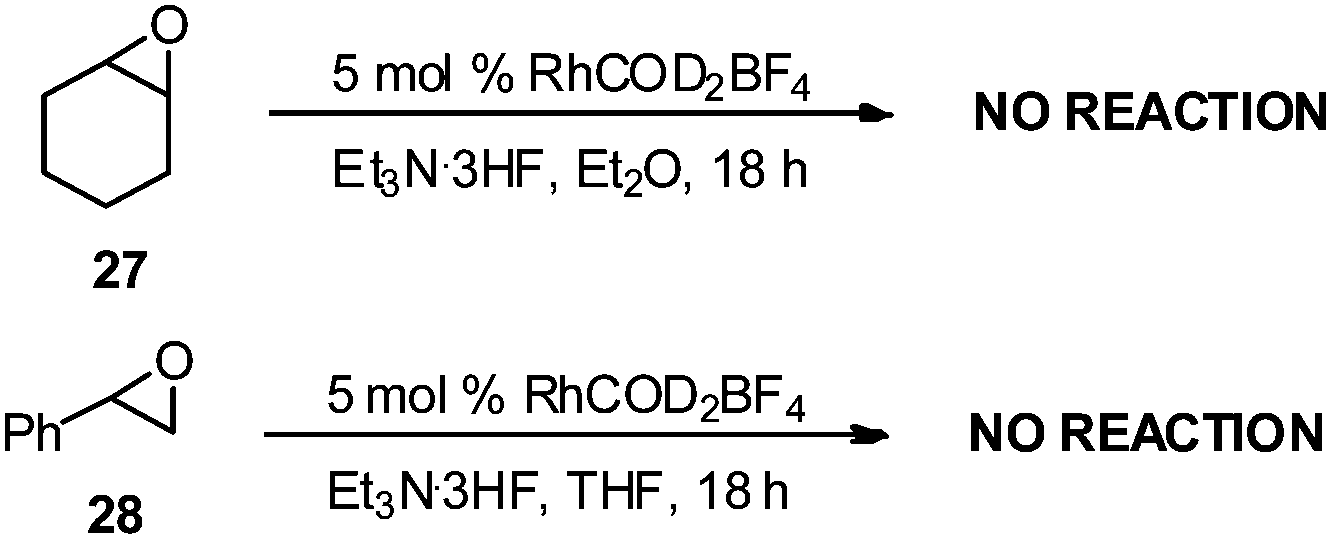 | ||
| Scheme 3 Control experiments. | ||
To gain some mechanistic insight, enantiopure epoxide (S)-11 was subjected to standard rhodium conditions (Scheme 4a). Allylic fluoride 22 was isolated in 43% yield with 69% ee. When the ring opening of (S)-11 was conducted in the absence of the catalyst, 22 was isolated in only 14% yield, albeit with a higher enantioselectivity (80% ee).14b,19,20 On the other hand, opening of enantiopure epoxide (R)-8 containing the C(1)-phenyl group, with and without the rhodium catalyst (Scheme 4b), resulted in significant racemization of allylic fluoride 19 (10–13% ee).
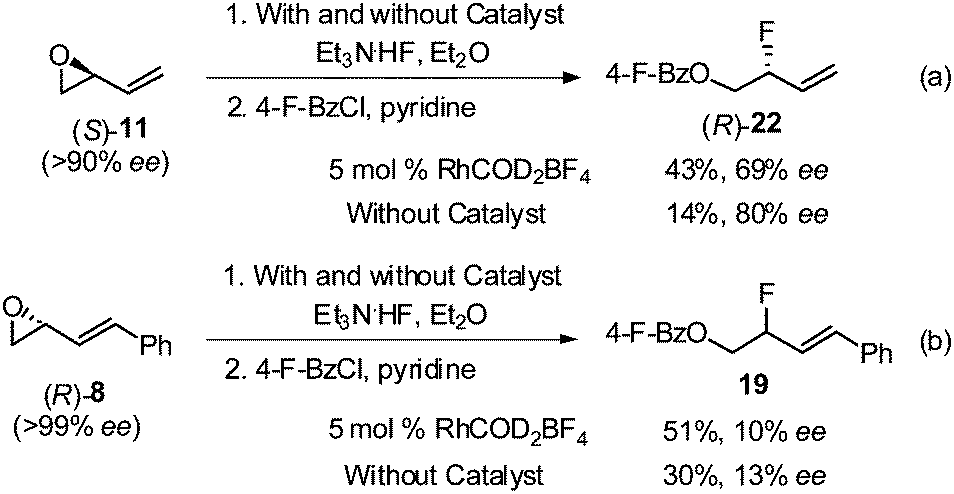 | ||
| Scheme 4 Stereospecific ring-opening of vinyl epoxides. | ||
To establish the absolute stereochemistry of the allylic fluoride product, 22 was subjected to cross-metathesis with 4-bromo-styrene 29 (Scheme 5). The major enantiomer of diene product 30 (67% ee) was shown by X-ray crystallographic analysis to be R-configured.20 Collectively, the data illustrates that the rhodium-catalyzed regioselective opening of vinyl epoxide with Et3N·3HF proceeds with inversion of stereochemical configuration.21 This is opposite to what has been reported for palladium-catalyzed allylic fluorination.5a,f
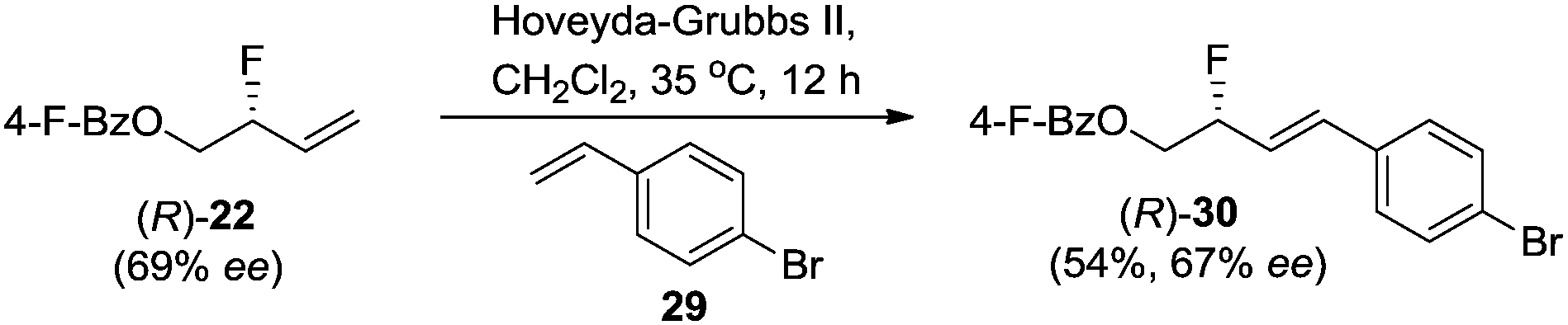 | ||
| Scheme 5 Derivatization of fluoride 22 for X-ray analysis. | ||
Based on the results obtained in Schemes 4 and 5, we propose the following mechanistic rationale for the regioselective ring-opening of vinyl epoxides lacking the C(1)-functional group.22 The rhodium catalyst first coordinates to the olefin of (S)-11 to form rhodium–alkene complex 31 (Scheme 6). Subsequent ionization of 31 provides the π-allylrhodium intermediate 32.23 We hypothesize that hydrogen fluoride may first attack the rhodium center of 32 to form the corresponding fluoride–rhodium complex 33. Fluoride is widely regarded as a hard nucleophile,24 which has been known to bind to the metal center of the allyl–metal complex in allylic substitution.25 Protonation of the alkoxide followed by reductive elimination and benzoylation affords the corresponding (R)-allylic fluoride 22 with retention of stereochemistry, which is confirmed by X-ray crystallographic analysis.20 To rationalize the excellent levels of regioselectivity for the 1,2-addition products, we hypothesize that the fluoride ion selectively attacks the more substituted allyl position of 34. Because (R)-22 was generated in 69% ee (Scheme 4a), we propose that nucleophilic attack by fluoride onto the π-allylrhodium complex 32 (Scheme 6) must occur faster than isomerization of complex 32 to its corresponding ent-32. Alternatively, the erosion of enantioselectivity observed in product 22 could arise if the allylic fluorohydrin intermediate is undergoing a degenerate substitution reaction with the Et3N·3HF reagent.26
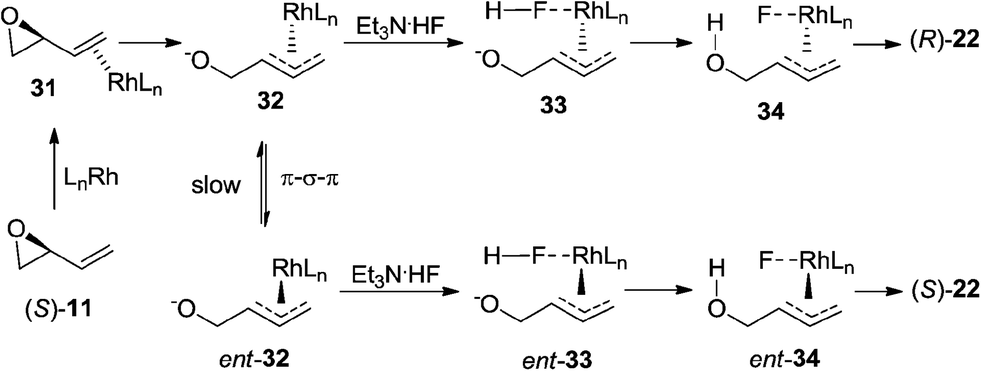 | ||
| Scheme 6 Proposed mechanism for rhodium-catalyzed ring-opening of vinyl epoxides lacking the C(1)-substituent. | ||
Conclusions
We have developed a rhodium-catalyzed methodology that efficiently facilitates the regioselective ring-opening of a variety of vinyl epoxides with Et3N·3HF to provide the corresponding 1,2-addition allylic fluorohydrins in moderate to good yields and with excellent regioselectivity. Mechanistic studies show that the opening of vinyl epoxides occurs with inversion of configuration. The operationally simple, mild, and rapid conditions make this method an attractive strategy for the incorporation of fluorine-18 ion into the allyl moiety of vinyl epoxides. Application of the ring-opening of vinyl epoxide procedure to the preparation of fluorine-18 radiotracers will be reported in due course.Acknowledgements
We thank the University of Iowa for financial support and Dr Dale Swenson for X-ray crystallographic analysis. Q.Z. also thanks the University of Iowa for the Graduate Research Fellowship.Notes and references
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) V. Gouverneur, Science, 2009, 325, 1630 CrossRef CAS PubMed; (c) X.-L. Qui, X.-H. Xu and F.-L. Qing, Tetrahedron, 2010, 66, 789 CrossRef; (d) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (e) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929 RSC.
- Representative examples of non-radioactive aryl–fluorine bonds: (a) D. V. Yandulov and N. T. Tran, J. Am. Chem. Soc., 2007, 129, 1342 CrossRef CAS PubMed; (b) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. García-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661 CrossRef CAS PubMed; (c) N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796 CrossRef CAS PubMed; (d) X. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 7520 CrossRef CAS PubMed; (e) T. Furuya, A. E. Strom and T. Ritter, J. Am. Chem. Soc., 2009, 131, 1662 CrossRef CAS PubMed; (f) P. Tang, T. Furuya and T. Ritter, J. Am. Chem. Soc., 2010, 132, 12150 CrossRef CAS PubMed; (g) P. Tang, W. Wang and T. Ritter, J. Am. Chem. Soc., 2011, 133, 11482 CrossRef CAS PubMed; (h) T. Xu, X. Mu, H. Peng and G. Liu, Angew. Chem., Int. Ed., 2011, 50, 8176 CrossRef CAS PubMed; (i) P. S. Fier and Hartwig, J. Am. Chem. Soc., 2012, 134, 10795 CrossRef CAS PubMed; (j) T. Truong, K. Klimovica and O. Daugulis, J. Am. Chem. Soc., 2013, 135, 9342 CrossRef CAS PubMed; (k) P. S. Fier, J. Luo and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2552 CrossRef CAS PubMed.
- Representative examples of non-radioactive alkyl–fluorine bonds: (a) D. D. Steiner, N. Mase and C. F. Barbas, Angew. Chem., Int. Ed., 2005, 44, 3706 CrossRef CAS PubMed; (b) M. Marigo, D. Fielenbach, A. Braunton, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 3703 CrossRef CAS PubMed; (c) H. Jiang, A. Falcicchio, K. L. Jensen, M. W. Paixão, S. Bertelsen and K. A. Jørgensen, J. Am. Chem. Soc., 2009, 131, 7153 CrossRef CAS PubMed; (d) T. Wu, J. Yin and G. Liu, J. Am. Chem. Soc., 2009, 131, 16354 CrossRef CAS PubMed; (e) J. Erb, D. H. Paull, T. Dudding, L. Belding and T. Lectka, J. Am. Chem. Soc., 2011, 133, 7536 CrossRef CAS PubMed; (f) P. Kwiatkowski, T. D. Beeson, J. C. Conrad and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 1738 CrossRef CAS PubMed; (g) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681 CrossRef CAS PubMed; (h) R. J. Phillips, K. Hiramatsu and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 8376 CrossRef PubMed; (i) N. A. Cochrane, H. Nguyen and M. R. Gagne, J. Am. Chem. Soc., 2013, 135, 628 CrossRef CAS PubMed; (j) F. Sladojevich, S. I. Arlow, P. Tang and T. Ritter, J. Am. Chem. Soc., 2013, 135, 2470 CrossRef CAS PubMed.
- (a) M. C. Pacheco, S. Purser and V. Gouverneur, Chem. Rev., 2008, 108, 1943 CrossRef CAS PubMed; (b) S. Thibaudeau and V. Gouverneur, Org. Lett., 2003, 5, 4891 CrossRef CAS PubMed; (c) B. Greedy, J. M. Paris, T. Vidal and V. Gouverneur, Angew. Chem., Int. Ed., 2003, 42, 3291 CrossRef CAS PubMed; (d) S. Thibaudeau, R. Fuller and V. Gouverneur, Org. Biomol. Chem., 2004, 2, 1110 RSC; (e) S. Boldon, J. E. Moore and V. Gouverneur, Chem. Commun., 2008, 3622 RSC.
- (a) M. H. Katcher and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 17402 CrossRef CAS PubMed; (b) M. H. Katcher, A. Sha and A. G. Doyle, J. Am. Chem. Soc., 2011, 133, 15902 CrossRef CAS PubMed; (c) C. Hollingworth, A. Hazari, M. N. Hopkinson, M. Tredwell, E. Benedetto, M. Huiban, A. D. Gee, J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 2613 CrossRef CAS PubMed; (d) A. M. Lauer and J. Wu, Org. Lett., 2012, 14, 5138 CrossRef CAS PubMed; (e) E. Benedetto, M. Tredwell, C. Hollingworth, T. Khotavivattana, J. M. Brown and V. Gouverneur, Chem. Sci., 2013, 4, 89 RSC; (f) C. Xue, X. Jiang, C. Fu and S. Ma, Chem. Commun., 2013, 49, 5651 RSC; (g) M.-G. Braun, M. H. Katcher and A. G. Doyle, Chem. Sci., 2013, 4, 1216 RSC.
- J. J. Topczewski, T. J. Tewson and H. M. Nguyen, J. Am. Chem. Soc., 2011, 133, 19318 CrossRef CAS PubMed.
- Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1991, vol. 6 Search PubMed.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed.
- (a) J. Tsuji, H. Kataoka and Y. Kobaysahi, Tetrahedron Lett., 1981, 22, 2575 CrossRef CAS; (b) B. M. Trost and G. A. Molander, J. Am. Chem. Soc., 1981, 103, 5969 CrossRef CAS; (c) B. M. Trost and S. R. Angle, J. Am. Chem. Soc., 1985, 107, 6123 CrossRef CAS.
- Several representative examples, see: (a) B. M. Trost and A. Tenaglia, Tetrahedron Lett., 1988, 29, 2931 CrossRef CAS; (b) B. M. Trost, E. J. McEachern and F. D. Toste, J. Am. Chem. Soc., 1998, 120, 12702–12703 CrossRef CAS; (c) B. M. Trost and E. J. McEachern, J. Am. Chem. Soc., 1999, 121, 8649–8650 CrossRef CAS; (d) X.-Q. Yu, F. Yoshimura, F. Ito, M. Sasaki, A. Hirai, K. Tanino and M. Miyashita, Angew. Chem., Int. Ed., 2008, 47, 750–754 CrossRef CAS PubMed.
- K. Fagnou and M. Lautens, Org. Lett., 2000, 2, 2319 CrossRef CAS PubMed.
- (a) S. Bruns and G. Haufe, J. Fluorine Chem., 2000, 104, 247 CrossRef CAS; (b) G. Haufe, S. Bruns and M. Runge, J. Fluorine Chem., 2001, 112, 55 CrossRef CAS; (c) G. Hauge and S. Bruns, Adv. Synth. Catal., 2002, 344, 165 CrossRef; (d) J. A. Kalow and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 3268 CrossRef CAS PubMed; (e) J. A. Kalow and A. G. Doyle, J. Am. Chem. Soc., 2011, 133, 16001 CrossRef CAS PubMed.
- J. Zhu, G. C. Tsui and M. Lautens, Angew. Chem., Int. Ed., 2012, 51, 12353 CrossRef CAS PubMed.
- There are two examples of regioselective opening of vinyl epoxides with triethylamine trihydrofluoride in the absence of the metal catalyst, see: (a) A. Hedhli and A. Baklouti, J. Fluorine Chem., 1995, 70, 141 CrossRef CAS; (b) L. Hunter, D. O'Hagan and A. M. Z. Sawin, J. Am. Chem. Soc., 2006, 128, 16422–16423 CrossRef CAS PubMed.
- To confirm the relative stereochemistry of 2A, it was converted into allylic benzoate, which underwent hydrogenation to afford the trans-1,2-fluoride whose relative stereochemistry was confirmed by 1H NMR and COSY analyses. See ESI† for details.
- Heating the reaction to 40 °C and 60 °C in THF resulted in 37% and 30% NMR yield, respectively.
- Although THF provides fluorohydrin 2A in higher NMR yield than Et2O, use of Et2O as a solvent in rhodium-catalyzed ring-opening of vinyl epoxide 1 resulted in a biphasic mixture. This phase separation eliminates the need for an aqueous workup, and the crude product 2A was extracted from a biphasic mixture and subsequently treated with 4-flurobenzoyl chloride to form isolable allylic fluoride 2.
- (a) R. Bittman, N. M. Witzke, T.-C. Lee, M. L. Blank and F. Snyder, J. Lipid Res., 1987, 28, 733–738 CAS; (b) G. Haufe and A. Burchardt, Eur. J. Org. Chem., 2001, 4501–4507 CrossRef CAS; (c) A. Burchardt, T. Takahashi, Y. Takeuchi and G. Haufe, J. Org. Chem., 2001, 66, 2078–2084 CrossRef CAS PubMed; (d) R. Bittman, H.-S. Byun, K. C. Reddy, P. Samadder and G. Arthur, J. Med. Chem., 1997, 40, 1391–1395 CrossRef CAS PubMed.
- It is known that regioselective opening of enantiopure vinyl epoxide 11 provided allylic fluorohydrin with opposite stereochemistry.14a.
- See the ESI† for details.
- This result is consistent with what has been reported for the rhodium-catalyzed regioselective opening of vinyl epoxides with alcohols and anilines.11.
- Based on the result obtained with epoxide R-(8) in Scheme 4b, we hypothesized that vinyl epoxides bearing the C(1)-substituted group are likely to occur via SN1 mechanism.
- For an alternative enylrhodium intermediate, see: P. A. Evans and J. D. Nelson, J. Am. Chem. Soc., 1998, 120, 5581 CrossRef CAS.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427 RSC.
- A. Hazari, V. Gouverneur and J. M. Brown, Angew. Chem., Int. Ed., 2009, 48, 1296 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for all new compounds. CCDC 964273. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc51949j |
| This journal is © The Royal Society of Chemistry 2014 |