Insight into bicyclic thiopeptide biosynthesis benefited from development of a uniform approach for molecular engineering and production improvement†
Heng
Guo
a,
Jiang
Wang
a,
Yeming
Li
a,
Yi
Yu
a,
Qingfei
Zheng
a,
Jiequn
Wu
b and
Wen
Liu
*ab
aState Key Laboratory of Bioorganic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: wliu@mail.sioc.ac.cn; Fax: +86-21-64166128; Tel: +86-21-54925111
bHuzhou Center of Bio-Synthetic Innovation, 1366 Hongfeng Road, Huzhou 313000, China
First published on 26th September 2013
Abstract
The ribosomal origin of thiopeptide antibiotics, a class of sulfur-rich and highly modified poly(thi)azolyl natural products, has recently been uncovered and features complex post-translational modifications (PTMs) of a precursor peptide. Based on molecular engineering and production improvement, we report insight into the biosynthesis of two bicyclic thiopeptide compounds, thiostrepton and nosiheptide. The PTMs of thiostrepton tolerate variations in the first two amino acids of the core peptide part of the precursor peptide: (1) the mutation of Ile1 to Val had no apparent effect on molecular maturation, suggesting that attachment of the quinaldic moiety at position 1 is not residue-dependent for the construction of the side ring system; and (2) the change of Ala2 to Ser led exclusively to the production of an analog that bears a corresponding dehydroamino acid residue, indicating that dehydration at position 2 is site-selective or that the oxazoline formed by cyclodehydration is inaccessible for maturation. For nosiheptide biosynthesis in particular, we provide the first structural evidence that construction of the specific side ring system precedes formation of the common central heterocycle domain and therefore propose that formation of a characteristic thiopeptide framework interweaves both common and specific PTMs that are interdependent. The above efforts benefited from the development of a uniform approach to examine the effectiveness for trans expression of gene encoding precursor peptides and associated PTM capacity. This approach is independent of knowledge regarding organism-specific regulatory mechanisms and potentially applicable to other systems that produce ribosomally synthesized peptide natural products.
Introduction
Thiopeptide antibiotics, having potent activity against many drug-resistant bacterial pathogens, form a peptide natural product family that now comprises nearly 100 distinct entities.1,2Thiopeptides are characterized by a macrocyclic core that contains a six-membered nitrogen heterocycle central to multiple azoles (or azolines) and dehydroamino acids, as shown by the monocyclic member thiocillin and side-ring appending bicyclic members thiostrepton and nosiheptide (Fig. 1). In the past several years, we and others have established the paradigm of thiopeptide biosynthesis—a ribosomally synthesized precursor peptide associated with complex post-translational modifications (PTMs), thereby setting the stage for sequence engineering of the precursor peptide to diversify the peptidyl skeleton.3–5 This diversification, which is challenging current chemical synthesis-based approaches due to the structural complexity of thiopeptides, is of significant interest in the search for new analogs suited to fight progressively emerging bacterial resistance in the clinic and for increase of our understanding in thiopeptide biosynthesis by evaluating the effect of precursor peptide variation on PTM capacity.
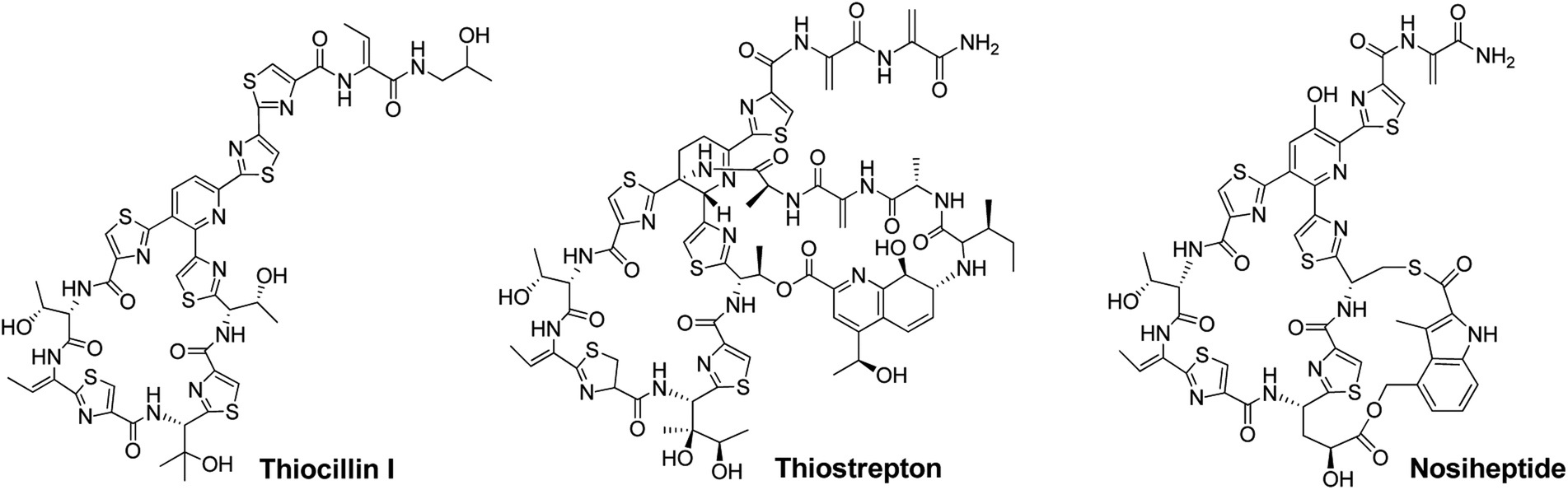 | ||
| Fig. 1 Structures of representative thiopeptides, including the monocyclic member thiocillin I and bicyclic members thiostrepton and nosiheptide. | ||
To put sequence mutagenesis into practice, there are two major prerequisites that should be considered: a robust system for thiopeptide production and a carrier platform facilitating genetic manipulation of the precursor peptide-encoding gene (termed prep). For example, Walsh et al. effectively produced GE37468 in S. coelicolor M1152 and berninamycin in S. lividans TK24 by testing various Streptomyces strains as heterologous hosts.6,7 This group developed a plasmid-based system for the trans expression of prep alone; therefore, site-directed mutagenesis of the precursor peptide of GE37468 or thiocillin can be easily performed.8–10 These efforts generated hundreds of thiopeptide variants and provided structural evidence for the key intramolecular cyclization step to form the central heterocycle.6–10 However, trans expression of prep may not always rescue thiopeptide production in the mutant strain in which even prep may be deleted in-frame, as exemplified for thiostrepton in S. laurentii.11,12 There are two possibilities regarding the failure: (1) the prep sequence participates in the transcriptional control of PTM-encoding genes; and (2) trans expression of prep is not achieved in an effective way. To solve this problem, Kelly et al. performed prep mutations in situ on a fosmid that harbors the entire biosynthetic gene cluster.11,12 Although introduction of the resulting fosmid variants into the prep-deleted mutant strain proved effective for analog production, the complexity arising from genetic manipulation of a large construct may argue against systematic screening of thiopeptide variants by codon randomization of the precursor peptide. Because the regulatory mechanism for producing each thiopeptide is system-dependent and has not been characterized thus far, it is necessary to develop a uniform approach to evaluate the effectiveness of both PTM capacities in the host and trans expression of prep in an easily manageable platform. By addressing this common query, we herein report the generation of new analogs and improvement in production of thiostrepton and nosiheptide, thereby yielding insight into the biosynthesis of bicyclic thiopeptide members.
Results and discussion
We first reported this idea in the thiostrepton-producing system S. laurentii (Fig. 2A). Upon sequence analysis of tsrH (termed tsrA in ref. 13),13,14 the prep of thiostrepton, we chose the codons GGA and GAG, corresponding to Gly-30 and Glu-7 within the leader peptide part, for in situ mutations into the stop codons TGA and TAG, respectively. These single “base”-based mutations are outside the regions predicted to be involved in forming hairpin structures for transcription, thereby minimizing potential polar effects on the expression of PTM-encoding genes in both directions. Furthermore, the products of mutant preps would be truncated at the leader peptide by translation, preventing an impact on the catalytic efficiency of thiopeptide formation (truncated peptides may lack the interaction with PTM enzymes at the protein level). Consequently, the mutant strains SL2057 (mutation of GGA to TGA) and SL2051 (GAG to TAG) were constructed (Fig. 2A) and their product profiles were examined by fermentation, demonstrating that thiostrepton production was completely abolished as anticipated (Fig. 3A and Table S6†). We then constructed various pSET152-based recombinant plasmids (Fig. S5†), in which a single copy of tsrH is under the control of the constitutive promoter for the erythromycin resistance gene ermE15,16 or flanks its original upstream and downstream sequence regions that vary in length (mimic to the native regulation in S. laurentii, as that for restoring GE37468 production by trans complementation).7 These plasmids were individually introduced into the prep-mutant strain SL2051, by integration into the chromosome via the pSET152 vector-mediated, site-specific recombination.17,18 The resulting recombinants were fermented and compared for thiostrepton production (Table S6†), leading to identification of SL2052 (Fig. 3A, III), which produced thiostrepton with the highest yield at 41.0 ± 2.4 mg L−1, 54.2% of that of the wild type strain (74.0 ± 3.4 mg L−1). This strain was derived from SL2051 in which the GAG codon for Glu-7 of TsrH was mutated to the stop codon TAG (Fig. 2A and 4A). SL2052 contains the recombinant plasmid pSL2050 (Fig. S5†), in which tsrH flanks a 530 bp upstream sequence and a 288 bp downstream region. Introduction of pSL2050 into SL2057 (Fig. 2A, generated by mutation of the GGA codon for Gly-30 to the stop codon TGA) was also capable of restoring the production; however, the yield of the resulting strain SL2060 was lower than that of SL2052 (22.9 ± 1.0 mg L−1, Table S6†). Thus, the stop codon-based mutation of prep at Glu-7 maintains the thiostrepton biosynthetic machinery; and SL2052 serves as an ideal host to examine TsrH production via trans expression, for which pSL2050, with an approximately 1 kb inserted DNA fragment containing tsrH, is a suitable platform for genetic manipulation.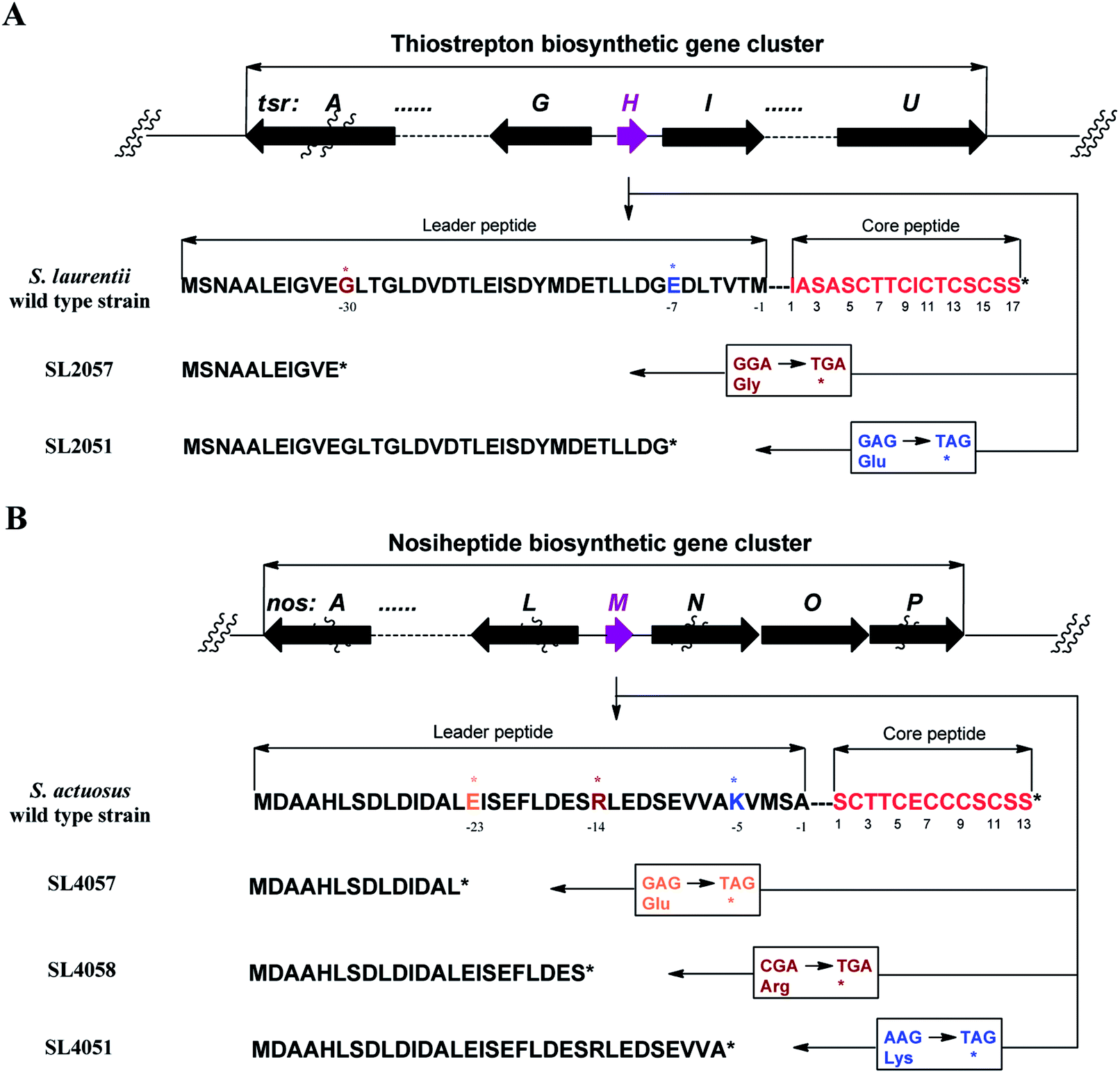 | ||
| Fig. 2 Single “base”-based mutation into a stop codon. (A) Genotypes of the S. laurentii wild type strain, SL2057 (by mutation of GGA for Gly-30 into TGA, ochre) and SL2051 (by mutation of GAG for Glu-7 into the stop codon TAG, blue), and corresponding encoded precursor peptides. (B) Genotypes of the S. actuosus wild type strain, SL4057 (by mutation of GAG for Glu-23 into TAG, yellow), SL4058 (by mutation of CGA for Arg-14 into TGA, ochre) and SL2051 (by mutation of AAG for Lys-5 into TAG, blue), and corresponding encoded precursor peptides. | ||
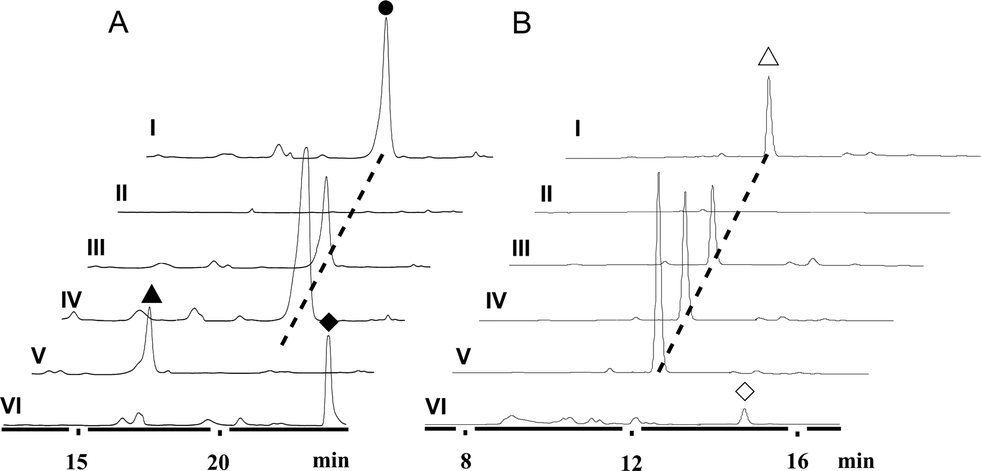 | ||
| Fig. 3 HPLC analysis of thiopeptide production. (A) Thiostrepton (●) and variants 1 (▲) and 2 (◆). I, wild type S. laurentii; II, SL2051 (with mutation of GAG for Glu-7 of TsrH into TAG); III, SL2052 (SL2051 derivative, containing pSL2050 for expressing tsrH in trans); IV, SL2055 (containing pSL2050 for tsrH duplication); V, SL2053 (SL2051 derivative, containing pSL2051 for producing TsrH–Ile1Val); and VI, SL2054 (SL2051 derivative, containing pSL2052 for producing TsrH–Ala2Ser). (B) Nosiheptide (△) and truncated intermediate 3 (◇). I, wild type S. actuosus; II, SL4051 (with mutation of AAG for Lys-5 of NosM to TAG); III, SL4052 (SL4051 derivative, containing pSL4050 for expressing nosM in trans); IV, SL4053 (containing pSL4050 for nosM duplication); V, SL4054 (containing pSL4051 for nosM triplication); and VI, SL4056 (SL4055 derivative, containing pSL4051 for nosM triplication). | ||
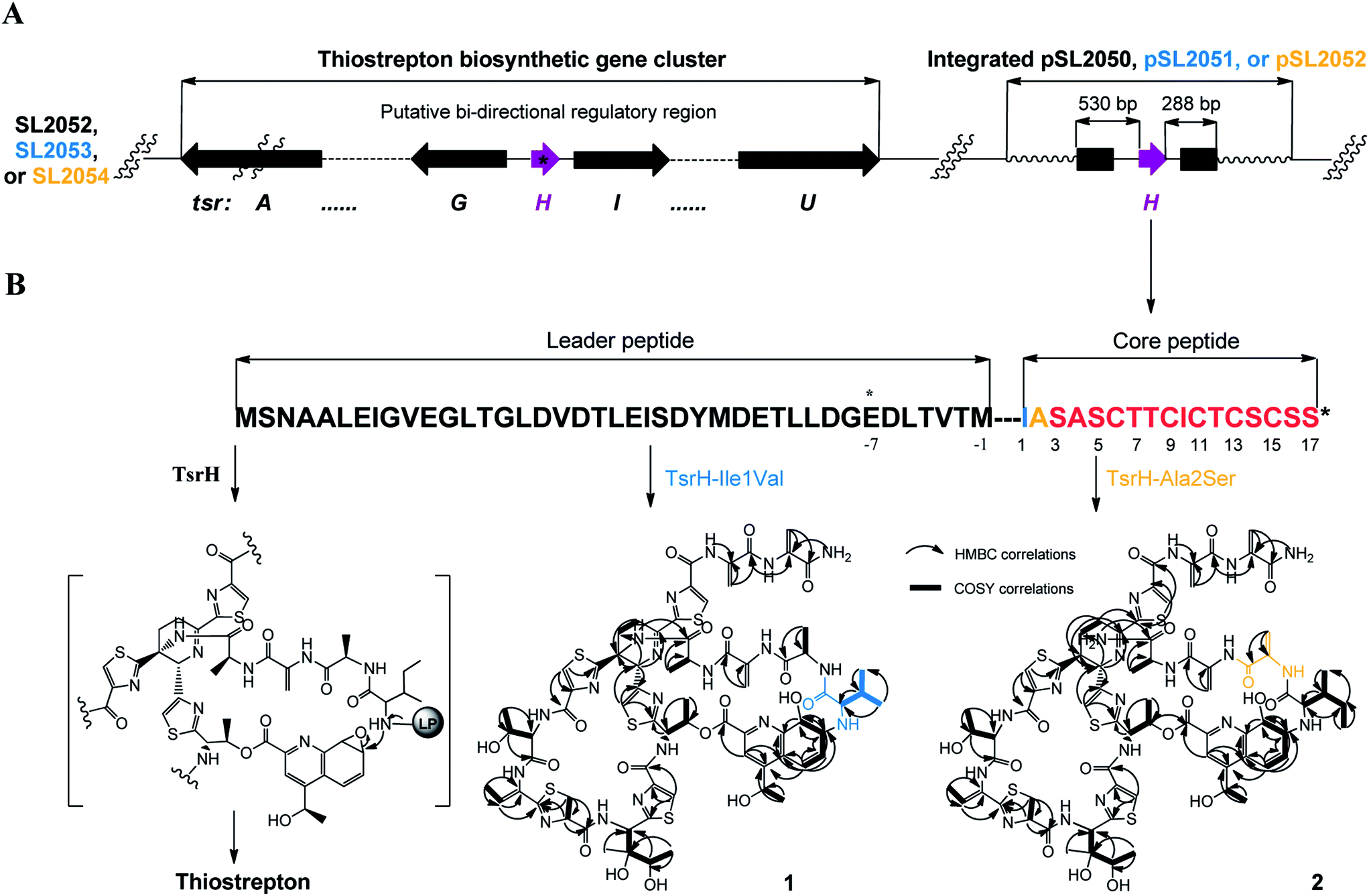 | ||
| Fig. 4 Thiostrepton engineering. (A) Genotypes of the recombinant strains SL2052 (producing thiostrepton), SL2053 (producing 1), and SL2054 (producing 2). The gene prep and PTM-encoding genes are shown in purple and black, respectively. The asterisk indicates the prep that bears a stop codon-based mutation. (B) Precursor peptide TsrH (black) and its variants, TsrH–Ile1-Val (blue) and TsrH–Ala2-Ser (yellow), and corresponding thiostrepton variants 1 and 2 (1H–1H COSY and HMBC correlations are labeled). LP, leader peptide. | ||
We applied this optimized SL2051–pSL2050 trans complementary system to an examination of the variability of the first two amino acids residing in the peptidyl skeleton of thiostrepton (Fig. 4). The pSL2050 derivatives, pSL2051 (encoding the precursor peptide variant TsrH–Ile1Val) and pSL2052 (encoding TsrH–Ala2Ser), were constructed by site-specific mutagenesis and individually introduced into the host SL2052, to provide the peptide substrate analogs in situ by trans expression. Each of the resulting recombinant strains produced one distinct thiopeptide (Fig. 3A, V and VI), with a robust yield of 36.4 ± 1.7 mg L−1 for compound 1 in SL2053 (carrying pSL2051) or 52.3 ± 2.2 mg L−1 for compound 2 in SL2054 (carrying pSL2052), validating that the biosynthetic machinery of thiostrepton tolerates these substrates for PTMs and molecular maturation. The fermentation cultures were then prepared on a large scale, allowing for purification and elucidation of their structures by spectral analysis (ESI Supplementary Results†). Consistent with the variation in the TsrH sequence, compound 1 ([M + Na]+m/z: calcd 1672.4659 for C71H83N19O18S5, found 1672.4696) bears Val1 (Fig. 4B), distinct from Ile1 in the parent compound. Position 1 is of importance to thiostrepton formation and serves as one of the linkages to the quinaldic moiety, epoxidation of which has previously been proposed to facilitate nucleophilic addition of the amino group of the residue for closure of the side ring system and release of the leader peptide.19,20 Clearly, the Ile1Val mutation has no apparent effect on thiopeptide maturation, suggesting that the downstream PTMs are not residue-dependent at this position. The Ala2Ser mutation produced compound 2 ([M + Na]+m/z: calcd 1684.4659 for C71H83N19O18S5, found 1684.4642), which contains a dehydroalanine (Dha) at position 2 (Fig. 4B). In theory, Ser residues in known thiopeptides have two possible fates by modification to either oxazoline (via cyclodehydration) or Dha (via dehydration). The fact that the Ala2Ser mutation exclusively produced compound 2 featuring Dha at position 2 indicates that these PTMs are site-selective or that in this case, oxazoline formed by cyclodehydration is inaccessible for thiopeptide maturation. Furthermore, we evaluated the effects of the Ile1Val and Ala2Ser variations on the biological activities of the resulting new thiopeptides 1 and 2. As shown in in vitro assays against the test strains Bacillus substilis and Staphylococcus aureus, whereas compound 1 exhibited minimum inhibitory concentrations (MICs) 1-fold lower than those of thiostrepton, compound 2 produced MICs 1–3-fold higher than those of the parent compound (Table S5†).
We next expanded this screening strategy to S. actuosus, the nosiheptide-producing system.21 The codons GAG, CGA and AAG, corresponding to Glu-23, Arg-14 and Lys-5 within the leader peptide-encoding region were chosen for mutation to the stop codons TAG, TGA and TAG, respectively (Fig. 2B). The resulting mutant strains, SL4057, SL4058 and SL4051, were confirmed to be unable to produce nosiheptide and then screened by introduction of the pSET152-based constructs that share the prep nosM but differ in length of flanking upstream and downstream sequences or in promoter (Fig. 3B and S5, and Table S6†). This screening resulted in a recombinant SL4052 in which the production of nosiheptide was effectively restored (Fig. 3B, III), with a yield of 6.6 ± 0.5 mg L−1, 88.9% of that of the wild type strain (7.4 ± 0.6 mg L−1). SL4052, derived from SL4051 by mutation of AAG for Lys-5 (of the precursor peptide NosM) to the stop codon TAG (Fig. 2B), harbors an integrated construct pSL4050 that contains nosM along with 972 bp upstream and 69 bp downstream original sequences (Fig. S5†), facilitating nosiheptide engineering. To enhance the production of nosiheptide, we introduced the validated platform pSL4050 into the wild type S. actuosus strain, producing SL4053. This strain, carrying two nosMs, one in trans and one in cis (within the gene cluster), yielded 12.5 ± 0.9 mg L−1 of nosiheptide (Fig. 3B, IV), demonstrating a positive prep-dose effect on nosiheptide production. Duplication of nosM led to generation of the construct pSL4051 (Fig. S5†) and subsequent introduction of this plasmid into the wild type strain produced the resulting recombinant SL4054, which has three nosMs (two in trans and one in cis), with a further improved yield (17.6 ± 1.2 mg L−1) (Fig. 3B, V). The fact that thiopeptide production apparently depends on the supply of the precursor peptide substrate was also found in S. laurentii, given the fact that SL2055, in which tsrH is duplicated by introduction of pSL2050 into the wild type strain, yielded 97.5 ± 3.6 mg L−1 of thiostrepton (Fig. 3A, IV).
We subsequently exploited the production improvement by the pSL4051 construct to characterize the low-yield intermediate in nosiheptide biosynthesis arising from gene inactivation. Initially, we deleted nosO, the counterpart of tclM in thiocillin biosynthesis shown to be involved in the formation of the six-membered central heterocycle.22 The mutant strain, SL4055, only produced a trace of the new compound ([M + H]+m/z: calcd 1296.4356 for C54H49O14N13S6, found 1296.4324). We integrated pSL4051 into the chromosome of SL4055, yielding SL4056 in which nosM was triplicated (Fig. 5A). SL4056 produced an apparently improved yield at 3.4 ± 0.2 mg L−1 (Fig. 3B, VI), which facilitated the purification of this labile compound. The obtained product, compound 3 (Fig. 5B), was analyzed and eventually characterized to be a peptide with fully-installed thiazoles and Dhas but apparently without the central heterocycle domain (ESI Supplementary Results†), a key hallmark of all thiopeptide members that is absent in other ribosomally synthesized and post-translationally modified peptide natural products.22 Compound 3 is likely a shunt product generated by spontaneous hydrolysis of the leader peptide: tautomerization of Dha1 to methyl imine facilitates nucleophilic attack by H2O to produce a hydrated intermediate, and a Cα–N bond cleavage then releases 3. This finding is consistent with that of Walsh et al. in the tclM-inactivated strain and provides significant insight into the timing of PTMs in nosiheptide biosynthesis.23
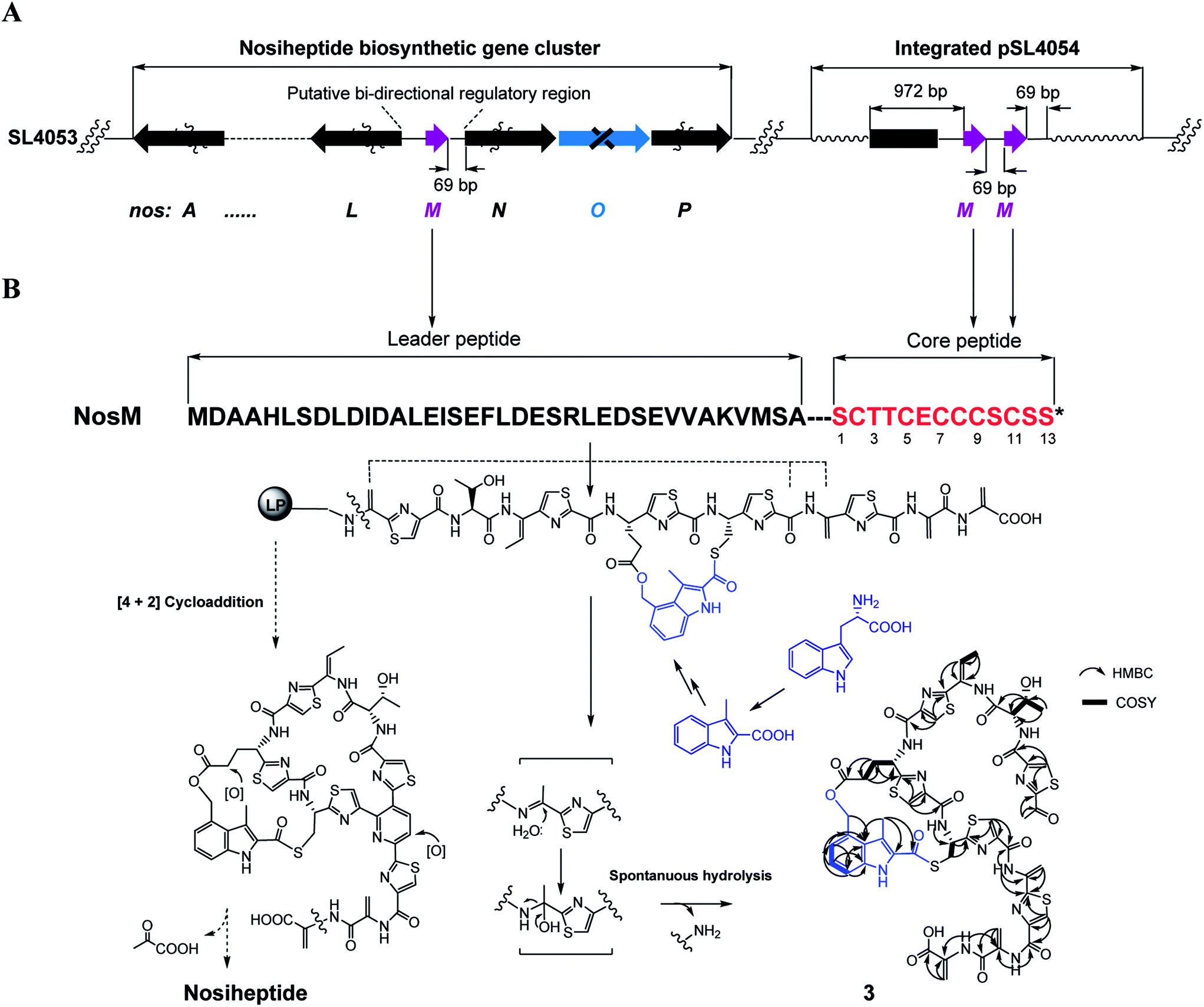 | ||
| Fig. 5 Production of compound 3. (A) Genotypes of the recombinant strains SL4056. The gene prep and PTM-encoding genes are shown in purple and black, respectively. (B) Precursor peptide of nosiheptide, linear intermediate, and shunt product 3 (1H–1H COSY and HMBC correlations are labeled). While dashed arrows indicate the proposed biosynthetic pathway of nosiheptide, solid arrows show the predicted degradation process to generate 3. | ||
Remarkably, the indolic side ring system of nosiheptide is completely formed in compound 3 (Fig. 5B). To our knowledge, this is the first structural evidence for bicyclic thiopeptide members that building of the side ring precedes formation of the central heterocycle domain and the associated construction of a characteristic thiopeptide framework. The indole moiety of nosiheptide is derived from L-tryptophan,24 which is independent of the precursor peptide, requiring an unusual radical-mediated fragmentation–recombination to process the carbon side chain (Fig. 5B). Closure of the side ring via an ester linkage at Glu6 of the precursor peptide appears dispensable to form the thiopeptide framework, as evidenced by previous characterization of an indole-containing, side ring-opening analog of nosiheptide;21 however, the indole moiety addition to the unmodified Cys8 residue of the precursor peptide is considered necessary, as indicated by the fact that deconstruction of the indole moiety failed to produce any monocyclic thiopeptide intermediate.21,25 Thiopeptide biosynthesis is known to share the common PTMs including cyclodehydration/dehydrogenation for polyazol(in)es, dehydration for dehydroamino acids and cycloaddition for the central heterocycle.3–5 This process has not been reproduced in vitro, due in part to the poorly understood biosynthetic logic that may involve certain specific PTMs. In fact, many specific PTMs, including methylation, oxidoreduction, C-terminal decarboxylation and cyclic residue formation, have been found to precede cycloaddition in the biosynthesis of thiocillin or GE37468.7,22 Except for indole ring attachment, compound 3 bears no further specific PTMs, including regioselective oxidation of Glu6 at the γ-position and dealkylation of the C-terminal Dha to form the amide moiety,26 indicating that these PTMs, as well as hydroxylation of the central pyridine domain, may take place after cycloaddition. Therefore, attachment of the indole moiety by S-acylation of the Cys8 residue may be the only specific but indispensible PTM prior to cycloaddition. Although the timing of this reaction with the generation of thiazoles or Dhas remains unclear, the above observations suggest that formation of the characteristic thiopeptide framework of nosiheptides interweaves both the common and specific PTMs that are interdependent. In comparison to nosiheptide, the potent parent compound, compound 3 was nearly inactive against the test strains B. substilis and S. aureus (Table S5†), demonstrating the necessity of the central domain-containing macrocyclic scaffold to the activity of the thiopeptide.
Conclusions
We developed a uniform approach to evaluate the effectiveness of trans expression of genes encoding precursor peptides of thiopeptides. The platforms that carry prep are easily manageable, mutagenizable, and able to complement the hosts that harbor the stop codon-based prep mutant while maintaining PTM capacity. This approach is independent of knowledge regarding the regulatory mechanisms of thiopeptide production. We first practised this strategy in the thiostrepton-producing system of S. laurentii and then validated its generality in the nosiheptide-producing system of S. actuosus. Utilization of the approach-based outputs facilitated examination of peptidyl skeleton variability by sequence permutation and improvement in thiopeptide production, and ultimately yielding of insight into the biosynthesis of bicyclic thiopeptide compounds.Acknowledgements
We thank Prof. Heinz G. Floss, University of Washington, for providing of the strains S. laurentii ATCC 31255 and S. actuosus ATCC 25421. This work was supported in part by grants from NNSF (91213303 and 31000033), STCSM (13XD1404500), “973 program” (2010CB833200 and 2012CB721100), and MST (2012AA02A706) of China.Notes and references
- M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685–714 CrossRef CAS PubMed.
- J. Li, X. Qu, X. He, L. Duan, G. Wu, D. Bi, Z. Deng, W. Liu and H. Y. Ou, PLoS One, 2012, 7, e45878 CAS.
- C. Li and W. L. Kelly, Nat. Prod. Rep., 2010, 27, 153–164 RSC.
- C. T. Walsh, S. J. Malcolmson and T. S. Young, ACS Chem. Biol., 2012, 7, 429–442 CrossRef CAS PubMed.
- Q. Zhang and W. Liu, Nat. Prod. Rep., 2013, 30, 218–226 RSC.
- S. J. Malcolmson, T. S. Young, J. G. Ruby, P. Skewes-Cox and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8483–8488 CrossRef CAS PubMed.
- T. S. Young, P. C. Dorrestein and C. T. Walsh, Chem. Biol., 2012, 19, 1600–1610 CrossRef CAS PubMed.
- M. G. Acker, A. A. Bowers and C. T. Walsh, J. Am. Chem. Soc., 2009, 131, 17563–17565 CrossRef CAS PubMed.
- A. A. Bowers, M. G. Acker, A. Koglin and C. T. Walsh, J. Am. Chem. Soc., 2010, 132, 7519–7527 CrossRef CAS PubMed.
- A. A. Bowers, M. G. Acker, T. S. Young and C. T. Walsh, J. Am. Chem. Soc., 2012, 134, 10313–10316 CrossRef CAS PubMed.
- C. Li, F. Zhang and W. L. Kelly, Mol. Biosyst., 2011, 7, 82–90 RSC.
- C. Li, F. Zhang and W. L. Kelly, Chem. Commun., 2012, 48, 558–560 RSC.
- W. L. Kelly, L. Pan and C. Li, J. Am. Chem. Soc., 2009, 131, 4327–4334 CrossRef CAS PubMed.
- R. Liao, L. Duan, C. Lei, H. Pan, Y. Ding, Q. Zhang, D. Chen, B. Shen, Y. Yu and W. Liu, Chem. Biol., 2009, 16, 141–147 CrossRef CAS PubMed.
- J. Wu, Q. Zhang, W. Deng, J. Qian, S. Zhang and W. Liu, Appl. Environ. Microbiol., 2011, 77, 7508–7516 CrossRef CAS PubMed.
- Y. Chen, W. Deng, J. Qian, J. Chu, Y. Zhuang, S. Zhang and W. Liu, Appl. Environ. Microbiol., 2008, 74, 1820–1828 CrossRef CAS PubMed.
- M. Bierman, R. Logan, K. O'Brien, E. T. Seno, R. N. Rao and B. E. Schoner, Gene, 1992, 116, 43–49 CrossRef CAS.
- H. M. Thorpe and M. C. Smith, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5505–5510 CrossRef CAS.
- L. Duan, S. Wang, R. Liao and W. Liu, Chem. Biol., 2012, 19, 443–448 CrossRef CAS PubMed.
- Q. Zhang, Y. Li, D. Chen, Y. Yu, L. Duan, B. Shen and W. Liu, Nat. Chem. Biol., 2011, 7, 154–160 CrossRef CAS PubMed.
- Y. Yu, L. Duan, Q. Zhang, R. Liao, Y. Ding, H. Pan, E. Wendt-Pienkowski, G. Tang, B. Shen and W. Liu, ACS Chem. Biol., 2009, 4, 855–864 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- A. A. Bowers, C. T. Walsh and M. G. Acker, J. Am. Chem. Soc., 2010, 132, 12182–12184 CrossRef CAS PubMed.
- U. Mocek, A. R. Knaggs, R. Tsuchiya, T. Nguyen, J. M. Beale and H. G. Floss, J. Am. Chem. Soc., 1993, 115, 7557–7568 CrossRef CAS.
- Y. Ding, Y. Yu, H. Pan, H. Guo, Y. Li and W. Liu, Mol. Biosyst., 2010, 6, 1180–1185 RSC.
- Y. Yu, H. Guo, Q. Zhang, L. Duan, Y. Ding, R. Liao, C. Lei, B. Shen and W. Liu, J. Am. Chem. Soc., 2010, 132, 16324–16326 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, bacterial strains and plasmids, and compound characterization. See DOI: 10.1039/c3sc52015c |
| This journal is © The Royal Society of Chemistry 2014 |