 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The acid test: the chemistry of carboxylic acid functionalised {Cr7Ni} rings†
George F. S.
Whitehead
,
Jesùs
Ferrando-Soria
,
Lorna G.
Christie
,
Nicholas F.
Chilton
,
Grigore A.
Timco
,
Fabrizio
Moro
and
Richard E. P.
Winpenny
*
School of Chemistry and Photon Science Institute, The University of Manchester, Oxford Road, M13 9PL, Manchester, UK. E-mail: richard.winpenny@manchester.ac.uk; Fax: +44 (0)161-275-4616
First published on 22nd October 2013
Abstract
The synthesis, structures and magnetism of a new nano-scale constructs using carboxylic acid functionalised {Cr7Ni} rings are reported. Well-established carboxylate chemistry is used to synthesise of spectacular assemblies including a {Zn4O} tetrahedron surrounded by an octahedron of {Cr7Ni} rings.
Until recently the nuclearity of cage complexes produced by coordination chemistry has lagged behind the vast polymetallic cages made by the polyoxometallate (POMs) community. Compounds such as the {Mo368} nano-hedgehog and the {Mo176} and {Mo154} wheels reported by Műller and co-workers remain a benchmark that coordination chemists strive to match.1–3 Coordination chemists are catching up, beginning with the {Mn84} ring reported by Tasiopoulos et al.4 and arriving recently at an {Fe168} cage.5 Xu et al., while reporting a {Pd84} ring,6 noted the remarkable frequency with which certain factors appear, e.g. most of the gigantic structures are multiples of fourteen, and the few that aren't, e.g. the {Mo176} wheel and the {Fe64} reported by the Gao group,7 contain multiples of eight. This curious observation perhaps suggests that such structures arise from the agglomeration of building blocks. However such individual building blocks themselves are not isolable for any of these species, although the {Fe64} cage contains {Fe8} units that can be related to isolable {Fe8} cages.7
By contrast, extended lattices are often constructed from building blocks where the individual building blocks are isolable; the vast literature on metal–organic frameworks relies heavily on lattices arising from polycarboxylate links between copper dimers or oxo-centred zinc tetramers.8 Some such lattices show interesting magnetic properties,9 while others have been proposed as molecular rheostats and switches10 and still others are luminescent.11
Making large 0d-cage complexes by linking together isolable units is rare – which seems a curious lacuna. POMs are an exception, with POMs used as ligands for metal cages, although invariably the 3d-cages is grown in the presence of the POM, rather than being isolated separately.12 Where dimers of cages have been reported, interesting physics, e.g. the biasing of quantum effects between identical clusters,13 and there are proposals that linked cages could be used in quantum information processing.14 Therefore our aim was to design strategies where different 3d-cages can be brought together to make very large molecules, which could be an elegant and simple way to make nanoscale assemblies with control.
We have begun studies where we isolate cage complexes, in our case heterometallic rings,15 and use these as building blocks to make nanoscale assemblies of cages. Previously we have shown that we can introduce a pyridine functionality onto such cages, and use this pyridine to bind to copper dimers,16a,b oxo-centred metal triangles and much larger cages, producing compounds containing up to sixty metal centres.16c To take this concept further we wished to introduce a second type of binding functionality, and we chose a carboxylate. Here we report our initial studies using this approach.
The compound studied here is [nPr2NH2][Cr7NiF8(O2CtBu)16] 1, which is a heterometallic ring containing seven chromium(III) and a single nickel(II) arranged in an octagon, bridged on the outer edge by sixteen carboxylates (in this case dimethylpropanoic acid), eight in the plane of the metals and eight alternating above and below the plane, and on the inner edge by eight fluorides. The ring is templated around a central ammonium cation.15
To introduce a carboxylic acid functional group, a di-carboxylic acid was reacted with 1 using methods similar to those used previously.16 The acid chosen was iso-phthalic acid as it has a delocalised π-system between the carboxylic acid groups that can facilitate through-bond spin propagation and so allow spin communication. It is also rigid, which should aid crystallisation. The monosubstituted species was successfully synthesised by reacting unsubstituted {Cr7Ni} with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 excess of iso-phthalic acid in 1,2-dichlorobenzene and toluene. The resultant mixture was then purified by column chromatography to give [nPr2NH2][Cr7NiF8(O2CtBu)15(O2CC6H4CO2H)] 2 in a 28.6% yield. The compound crystallises as a hydrogen-bonded dimer (Fig. 1) with O⋯O distances of 2.74(3) Å, close to the mean found in carboxylic acid dimers on the Cambridge structural database (CSD) of 2.681(15) Å.17
:1 excess of iso-phthalic acid in 1,2-dichlorobenzene and toluene. The resultant mixture was then purified by column chromatography to give [nPr2NH2][Cr7NiF8(O2CtBu)15(O2CC6H4CO2H)] 2 in a 28.6% yield. The compound crystallises as a hydrogen-bonded dimer (Fig. 1) with O⋯O distances of 2.74(3) Å, close to the mean found in carboxylic acid dimers on the Cambridge structural database (CSD) of 2.681(15) Å.17
 | ||
| Fig. 1 Structure of 2 in the crystal, showing the formation of hydrogen bonded dimers in the crystal structure. Colours: Cr = green, Ni = turquoise, O = red, F = yellow, C = grey. H-atoms, central ammonium cation and Me-groups omitted for clarity. | ||
The {Cr7Ni} ring substitutes on a Cr⋯Ni edge as these edges are approximately 1010 times more reactive than Cr⋯Cr edges, based on simple exchange rates.18 The iso-phthalate group occupies an axial position on the ring, perpendicular to the plane of the metal octagon. The axial position is preferably substituted over the equatorial carboxylates as these are stabilised by the fluorides in the trans position to them.
Following the successful synthesis of the desired functionalised ring, the piperidinium and tetramethylpiperidinium salts were prepared to increase the reactivity of the carboxylate groups and allow the ring to undergo salt metathesis reactions. These salts were prepared by adding piperidine to 2 in acetone in a 1:1 ratio.
The solvent was removed and recrystallized from a minimal amount of hot acetone yielding the products {[C5H10NH2][nPr2NH2][Cr7NiF8(O2CtBu)15(O2CC6H4CO2)]} 3 and {[C9H18NH2][nPr2NH2][Cr7NiF8(O2CtBu)15(O2CC6H4CO2)]} 4 respectively. In each case the protonated piperidinium cation is found hydrogen-bonding to the carboxylate-functionalised ring (Fig. 2 and S1†) with N⋯O distances of 2.61(3), 2.673(18) and 2.686(18), 2.697(17) Å respectively, which are similar to the mean distance for these systems found on the CSD, 2.765(5) Å.17 The hydrogen-bonding in the two species is markedly different. In 3 the H-bonding adopts a chair conformation, whereas in 4 the bonding is planar. This difference in conformation is likely due to steric requirements of the cation; the bulky methyl groups of 4 force the planar conformation.
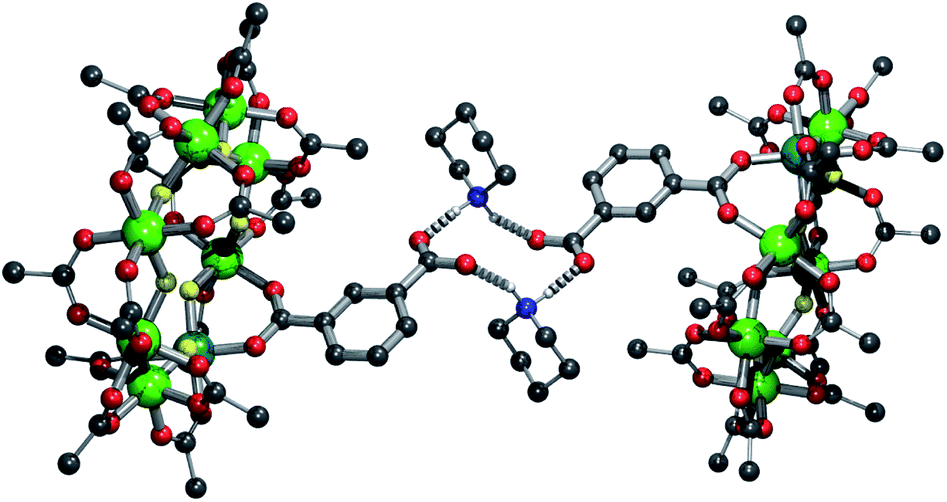 | ||
| Fig. 2 Structure of 3 in the crystal, showing the hydrogen bonding present. Colours and omissions as Fig. 1 plus N = blue. | ||
Salt metathesis reactions of 3 or 4 with copper(II) perchlorate in acetone leads to a thirty-six metal complex {{[nPr2NH2][Cr7NiF8(O2CtBu)15(O2CC6H4CO2)]}4[Cu4(OH)4(O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CMe2)]} 5 consisting of four rings and a four copper(II) square (Fig. 3). In the square four five-coordinate copper(II) sites are bridged on each edge by a hydroxide and a ring-carboxylate. The Jahn–Teller axis in each case is directed toward a central coordinating acetone.
CMe2)]} 5 consisting of four rings and a four copper(II) square (Fig. 3). In the square four five-coordinate copper(II) sites are bridged on each edge by a hydroxide and a ring-carboxylate. The Jahn–Teller axis in each case is directed toward a central coordinating acetone.
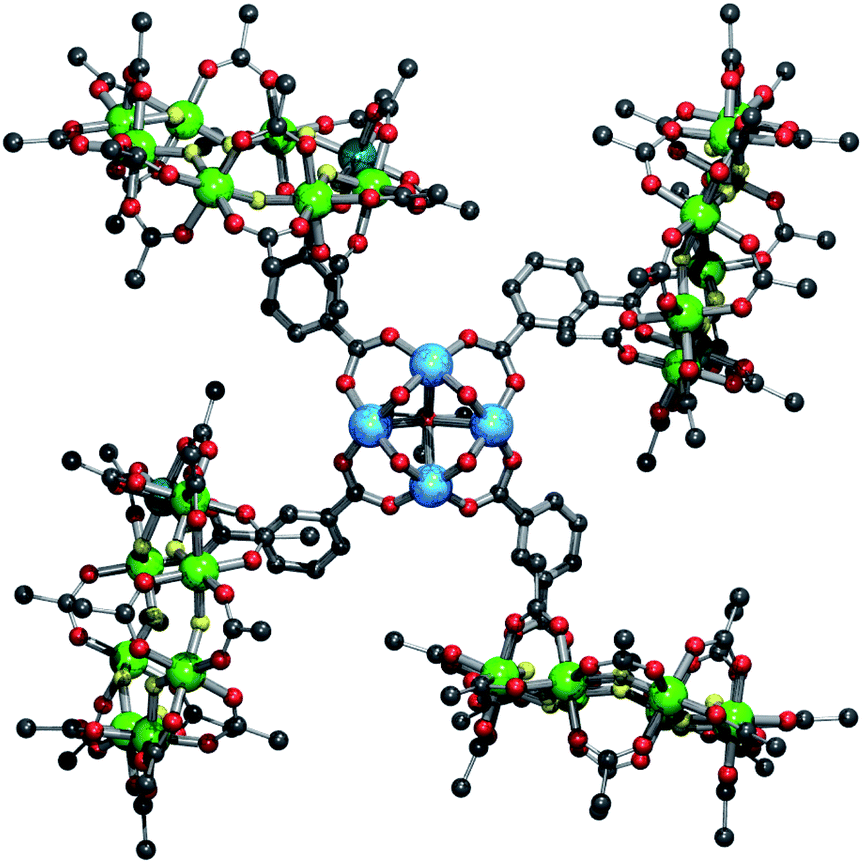 | ||
| Fig. 3 Structure of 5 in the crystal. Colours and omissions as Fig. 1 plus Cu = light blue. | ||
A similar moiety has been observed where copper bridged by bis(2-carboxyethyl)isocyanurate with a gadolinium bound to the bridging hydroxides.19 Complexes with cobalt, manganese and nickel and a tetra anionic p-tert-butylsulfonylcalix-[4]-arene ligand have also shown a similar motif.20 However, the metal centres in these previous cases are all 6-coordinate and the complexes contain a central bridging anion, either hydroxide or nitrate. In the case of the tetra-copper square the central site is occupied by a weakly-coordinating acetone, Cu–O(acetone) = 2.695(18) Å.
To facilitate packing around the copper square the rings lie perpendicular to the plane of the structure and steric requirements force the phenyl group of the iso-phthalic acid group to be twisted slightly out-of-plane with the bridging carboxylates, with a twist angle of 21.78(2)° with respect to copper square carboxylates. This slight mis-alignment of the π-system might result in disruption of any spin communication between the rings and the copper square. The use of less bulky substituents on the ring i.e. propionate groups instead of pivalate groups, may result in better packing.
The same reaction with zinc(II) perchlorate yields an oxo-centred zinc tetrahedron, analogous to zinc oxyacetate [Zn4O(CH3COO)6],21 with an iso-phthalate group attached to a ring bridging each edge of the tetrahedron giving {{[nPr2NH2][Cr7NiF8(O2CtBu)15(O2CC6H4CO2)]}6[Zn4O]} 6 (Fig. 4). As the mid-point of each edge of a tetrahedron describes an octahedron, this leads to a slightly distorted octahedral arrangement of six heterometallic rings containing fifty-two metal centres (Fig. 4 and 5).
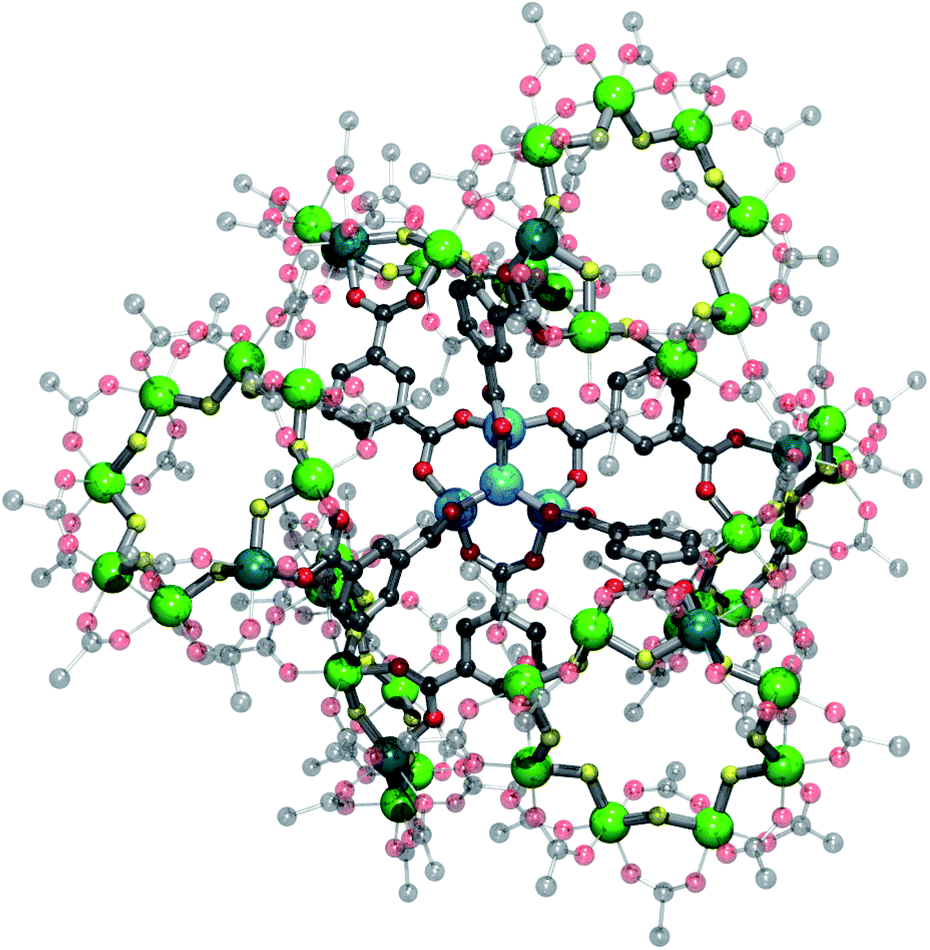 | ||
| Fig. 4 Structure 6 in the crystal viewed down a pseudo three-fold axis, i.e. down a Zn–O bond of the central tetrahedron. The front three {Cr7Ni} rings shown as full lines, the back three rings shown as lines. Colours as Fig. 1 plus Zn = light blue. | ||
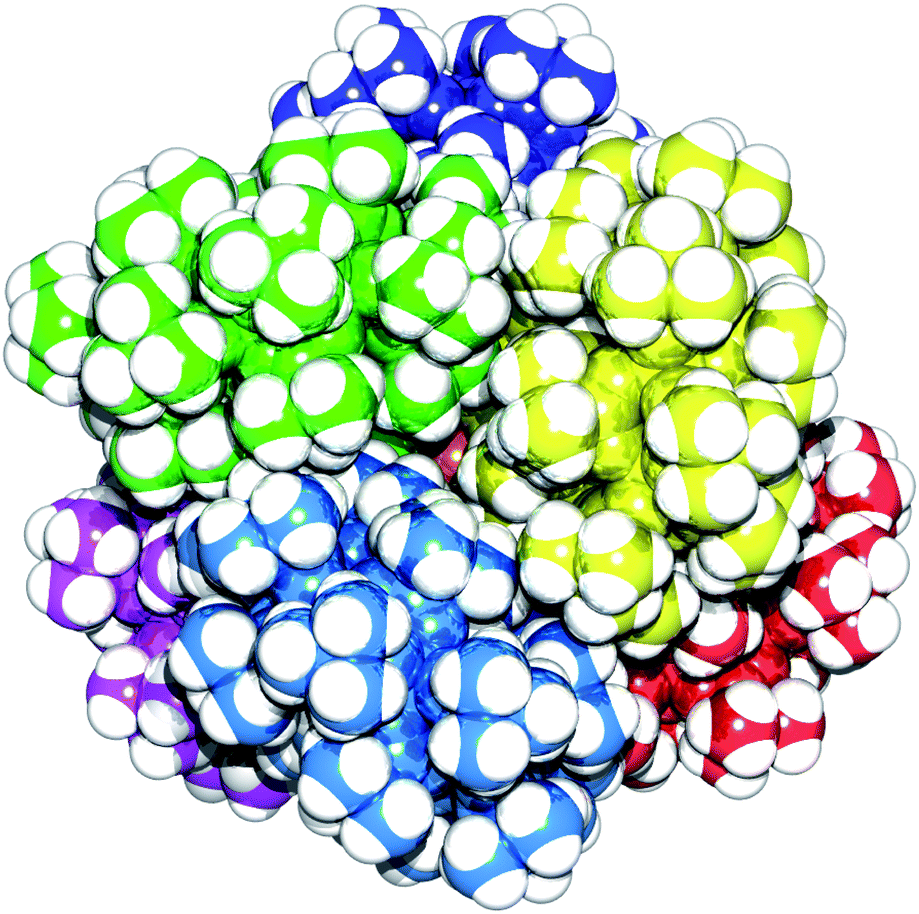 | ||
| Fig. 5 Space filling representation of 6 viewed down a pseudo threefold axis, highlighting the octahedral arrangement of the rings around the Zn4O core. The non-hydrogen atoms of each of the six rings in the structure are shown in a different colour. | ||
The fact this structure forms in the first place is quite extraordinary considering the steric requirements involved, but the molecule crystallises well. Each ring is crystallographically unique in the structure and the geometry of the zinc tetrahedron is distorted with the central oxygen slightly offset from the centre of the tetrahedron with Zn–O bond distances of 1.916(12), 1.956(10), 1.965(13) and 1.948(13) Å. There is also a varying degree of twist of the phenyl rings out-of-plane of the bridging carboxylate groups, with the twist angles ranging from 2.49(2)° to 31.60(2)° with respect to the zinc tetrahedron carboxylates. Examination of a space-filling representation of the structure (Fig. 5) shows how well the heterometallic rings lock together about the central {Zn4O} core.
The crystallography for the complexes is not trivial. The sheer size of the complexes (in 6, the entire molecule resides in the asymmetric unit), the inherent disorder of the t-butyl moieties of the pivalate groups and the presences of very large solvent accessible voids results in crystals that are very weakly diffracting, even with the use of a synchrotron source. This is reflected in high R-factors and a generally poor data to parameter ratio for some of the refined crystal structures (see ESI†). Despite this, we are confident of the models obtained as the metal sites, fluorides, oxygens and most importantly the iso-phthalate groups are all well-defined.
To investigate whether there are any interactions between the cluster components, the compounds were studied by magnetometry. We would expect any interaction between the rings and the bound clusters to be weak, so only observable at low temperature. The major difficulty in the study is the potential presence of unidentified solvent molecules in the lattice (e.g. in 6 there are solvent cavities present in the X-ray structure that could potentially contain eighty-two molecules of MeCN), hence determination of molecular weight and a diamagnetic correction for the compounds is challenging. This introduces a significant source of error to all magnetic measurements. All plots are shown with molecular weight and diamagnetic corrections calculated without solvent, consistent with elemental analysis on dried samples. The number of spin centres present in 5 and 6 (thirty-six and forty-eight respectively) make modelling the data challenging.
The variable temperature behaviour of the product χmT (where χm is the molar magnetic susceptibility) (Fig. 6 and S7†) for compounds 5 and 6 is in each case similar, showing a gradual decline with temperature; the values found are close to but not identical with the sum of four and six {Cr7Ni} rings, respectively.22 Similarly, the low temperature magnetisation for 5 and 6 are also very similar to the sum of four and six {Cr7Ni} rings (Fig. 7 and S8†). While it is unsurprising that the magnetic measurements of compound 6, containing a diamagnetic tetra-zinc core, closely resemble the sum of six non-interacting rings, it was not expected that compound 5 should appear to behave as four non-interacting {Cr7Ni} rings.
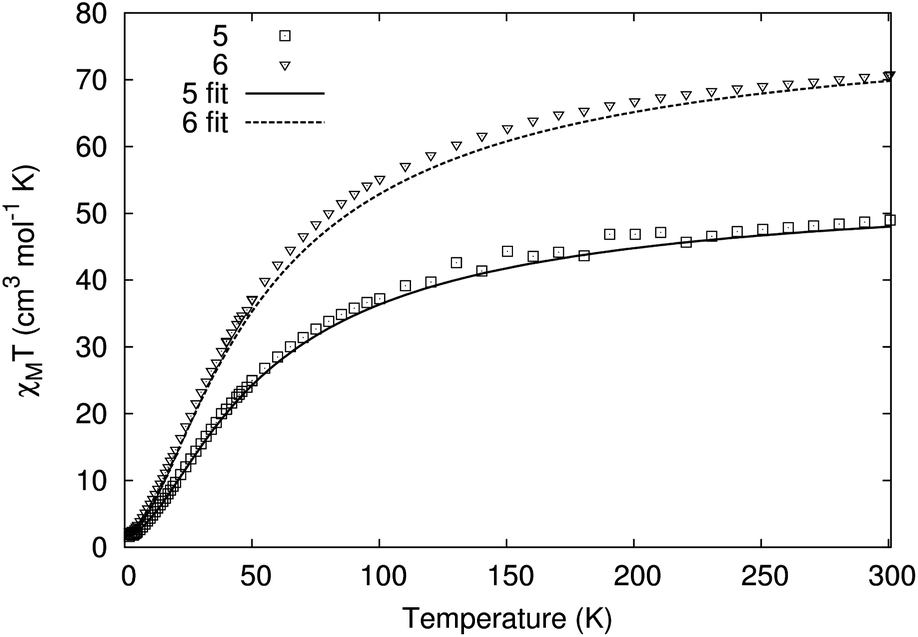 | ||
| Fig. 6 The product of magnetic susceptibility and temperature (χmT) against temperature (T) for 5 (open squares) and, 6 (open triangles measured on powder samples in an external field of 0.1 T). The fits of the data, as described in the text, are shown as full lines for 5 and dashed lines for 6. | ||
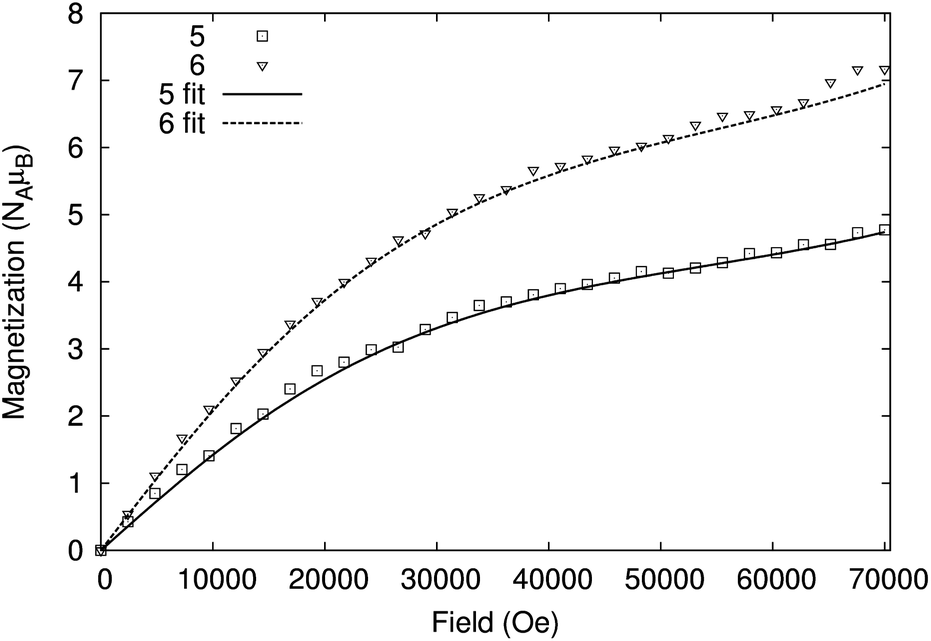 | ||
| Fig. 7 Magnetisation against field for compounds 5 (open squares) and 6 (open triangles) measured on powder samples at 2 K. The best fit of the data are shown as full and dashed lines respectively, using parameters described in the text. | ||
The copper square at the centre of 5 has not previously been reported and therefore we have no precedent for its behaviour. The Cu–O–Cu angles involving the bridging hydroxides are 102.6(5)°; magnetostructural correlations beginning with Hatfield24 for hydroxide bridged dimers would predict a strong anti-ferromagnetic coupling (JCu–Cu/kB = 565 K) for such an angle. This corroborates the observation that, even at room temperature, the central copper cluster appears to be diamagnetic. While the magnetic behaviour of 5 and 6 are close to simple sums of non-interacting rings, the data show a non-negligible discrepancy (Fig. S7 and S8†), which led to further investigation.
Firstly, we examined the effect of a small interaction between the rings, treating each ring as an effective S = 1/2 centre. However all attempts resulted in low temperature magnetization profiles that did not match the experiment, indicating that any interaction between the rings is very small in magnitude and not detectable by thermodynamic magnetometry measurements. Secondly, returning to sums of non-interacting rings, we investigated whether the exchange interactions involving the substituted nickel(II) site could have been altered. Using a microscopic exchange Hamiltonian we examined this possibility, however no parameter set that reproduced the data was obtainable.
Finally, we found that the data could be well described by the sums of non-interacting {Cr7Ni} rings with a small paramagnetic impurity (chosen as CrIII, S = 3/2, g = 2; Fig. 6 and 7). The magnetization and magnetic susceptibility data were fitted simultaneously using the program PHI,23 with the microscopic Hamiltonian:
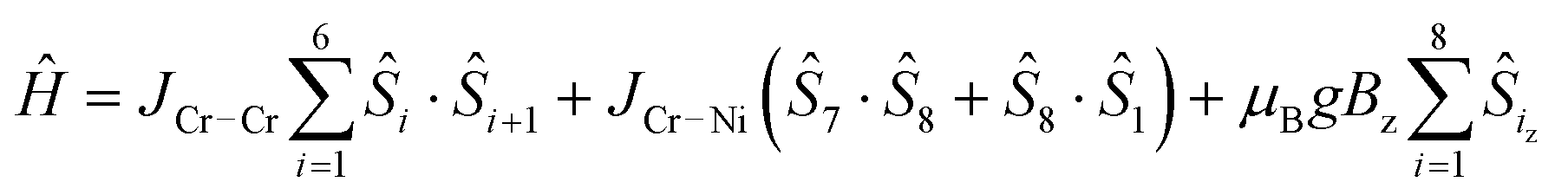
The experimentally determined exchange parameters for the {Cr7Ni} ring were fixed,22 however a single g value for the ring was allowed to vary from an initial value of 2.0, along with the impurity concentration. Extremely good agreement with the experimental data was obtained with g = 1.97 and 1.94, for 5 and 6 respectively, both displaying paramagnetic impurities of 2.6%.
Conclusions
We have demonstrated that new large nanoscale objects can be constructed using similar principles to those we have previously used with a different functionality. However there is a major difference here: for rings with pyridine functionality we could react them with pre-formed polymetallic cages.16 This is straightforward Lewis-base Lewis-acid chemistry. With the carboxylate functionality, the functionalised rings drive the formation of the central clusters from simple metal salts in situ. This resembles the chemistry using POMs as ligands.12 Thus the results extend the versatility of our approach in using simple coordination chemistry principles to make large complex structures. In compound 6 the core is very well-known, but in 5 we are able to support a [Cu4] square that has no close precedent. Compound 6 contains fifty-two metal centres and is one of the larger 3d-metal cages reported, and certainly one of the most structurally sophisticated involving three different metal ions within a hierarchal “assembly of cages” structure.Acknowledgements
This research was funded by the EPSRC (UK) and R.E.P.W. holds a Royal Society Wolfson Research Merit Award. N.F.C. thanks The University of Manchester for a President's Doctoral Scholarship. We also thank Dr David Allan and his team for help in use of X-ray at the synchrotron at Diamond Light Source, and we thank DLS for beam-time. We thank Prof David Collison for helpful discussions.Notes and references
- A. Műller, E. Beckmann, H. Bögge, M. Schmidtmann and A. Dress, Angew. Chem., Int. Ed., 2002, 41, 1162–1167 CrossRef.
- A. Műller, E. Krickemeyer, H. Bögge, M. Schmidtmann, C. Beugholt, P. Kögerler and C. Lu, Angew. Chem., Int. Ed., 1998, 37, 1220–1223 CrossRef.
- D. Zhong, F. L. Sousa, A. Műller, L. Chi and H. Fuchs, Angew. Chem., Int. Ed., 2011, 50, 7018–7021 CrossRef CAS PubMed.
- A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef CAS PubMed.
- Z. M. Zhang, S. Yao, Y.-G. Li, R. Clérac, Y. Lu, Z.-M. Su and E.-B. Wang, J. Am. Chem. Soc., 2009, 131, 14600–14601 CrossRef CAS PubMed.
- F. Xu, H. N. Miras, R. A. Scullion, D.-L. Long, J. Thiel and L. Cronin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11609–11612 CrossRef CAS PubMed.
- T. Liu, Y.-J. Zhang, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2008, 130, 10500–10501 CrossRef CAS PubMed.
- For example: (a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. L. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed; (b) L. F. Jones, A. Prescimone, M. Evangelisti and E. K. Brechin, Chem. Commun., 2009, 2023–2025 RSC; (c) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed; (d) G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC; (e) X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schroder, J. Am. Chem. Soc., 2009, 131, 2159–2171 CrossRef CAS PubMed.
- For example: H. Miyasaka, K. Nakata, L. Lecren, C. Coulon, Y. Nakazawa, T. Fujisaki, K.-I. Sugiura, M. Yamashita and R. Clérac, J. Am. Chem. Soc., 2006, 128, 3770–3783 CrossRef CAS PubMed.
- M. H. Chisholm, F. Feil, C. M. Hadad and N. J. Patmore, J. Am. Chem. Soc., 2005, 127, 18150–18158 CrossRef CAS PubMed.
- L. Zhang, Y.-Y. Qin, Z.-J. Li, Q.-P. Lin, J.-K. Cheng, J. Zhang and Y.-G. Yao, Inorg. Chem., 2008, 47, 8286–8293 CrossRef CAS PubMed.
- For example: (a) J. M. Clemente-Juan, H. Andres, J. J. Borras-Almenar, E. Coronado, H.-U. Gűdel, M. Aebersold, G. Kearly, H. Buttner and M. Zolliker, J. Am. Chem. Soc., 1999, 121, 10021–10027 CrossRef CAS; (b) S. G. Mitchell, P. I. Molina, S. Khanra, H. N. Miras, A. Prescimone, G. J. T. Cooper, R. S. Winter, E. K. Brechin, D. L. Long, R. J. Cogdell and L. Cronin, Angew. Chem., Int. Ed., 2011, 50, 9154–9157 CrossRef CAS PubMed; (c) X. Feng, P. Kögerler, Y. Furukawa, M. Speldrich and M. Luban, Angew. Chem., Int. Ed., 2011, 50, 5212–5216 CrossRef PubMed.
- W. Wernsdorfer, N. Aliaga-Alcalde, D. N. Hendrickson and G. Christou, Nature, 2002, 416, 406–409 CrossRef PubMed.
- (a) F. Meier, J. Levy and D. Loss, Phys. Rev. Lett., 2003, 90, 047901 CrossRef; (b) F. Troiani, A. Ghirri, M. Affronte, S. Carretta, P. Santini, G. Amoretti, S. Piligkos, G. Timco and R. E. P. Winpenny, Phys. Rev. Lett., 2005, 94, 207208 CrossRef CAS; (c) G. Aromí, D. Aguila, P. Gamez, F. Luis and O. Roubeau, Chem. Soc. Rev., 2012, 41, 537–546 RSC.
- (a) G. A. Timco, T. B. Faust, F. Tuna and R. E. P. Winpenny, Chem. Soc. Rev., 2011, 40, 3067–3075 RSC; (b) G. A. Timco, E. J. L. McInnes and R. E. P. Winpenny, Chem. Soc. Rev., 2013, 42, 1796–1806 RSC.
- (a) G. A. Timco, S. Carretta, F. Troiani, F. Tuna, R. J. Pritchard, C. A. Muryn, E. J. L. McInnes, A. Ghirri, A. Candini, P. Santini, G. Amoretti, M. Affronte and R. E. P. Winpenny, Nat. Nanotechnol., 2009, 4, 173–178 CrossRef CAS PubMed; (b) G. F. S. Whitehead, B. Cross, L. Carthy, V. A. Milway, H. Rath, A. Fernandez, S. L. Heath, C. A. Muryn, R. G. Pritchard, S. J. Teat, G. A. Timco and R. E. P. Winpenny, Chem. Commun., 2013, 49, 7195–7197 RSC; (c) G. F. S. Whitehead, F. Moro, G. A. Timco, W. Wernsdorfer, S. J. Teat and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2013, 52, 9932–9935 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- S. F. Lincoln, Helv. Chim. Acta, 2005, 88, 523–545 CrossRef CAS.
- Q. Zhu, S. Xiang, T. Sheng, D. Yuan, C. Shen, C. Tan, S. Hu and X. Wu, Chem. Commun., 2012, 48, 10736–10738 RSC.
- T. Kajiwara, T. Kobashi, R. Shinagawa, T. Ito, S. Takaishi, M. Yamashita and N. Iki, Eur. J. Inorg. Chem., 2006, 1765–1770 CrossRef CAS.
- H. Koyama and Y. Saito, Bull. Chem. Soc. Jpn., 1954, 27, 112–114 CrossRef CAS.
- R. Caciuffo, T. Guidi, S. Carretta, P. Santini, G. Amoretti, C. Mondelli, G. A. Timco and R. E. P. Winpenny, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 174407 CrossRef.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- V. H. Crawford, H. W. Richardson, J. R. Wasson, D. J. Hodgson and W. E. Hatfield, Inorg. Chem., 1976, 15, 2107–2110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of synthesis and structural studies are available. CCDC 952420, 952422–952425. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc52095a |
| This journal is © The Royal Society of Chemistry 2014 |