 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triaza-macrocyclic complexes of aluminium, gallium and indium halides: fast 18F and 19F incorporation via halide exchange under mild conditions in aqueous solution†
Rajiv
Bhalla
ab,
Christine
Darby
c,
William
Levason
c,
Sajinder K.
Luthra
a,
Graeme
McRobbie
a,
Gillian
Reid
*c,
George
Sanderson
c and
Wenjian
Zhang
c
aGE Healthcare UK, White Lion Road, Amersham, HP7 9LL, UK
bCentre for Advanced Imaging, University of Queensland, Brisbane, Queensland 4072, Australia
cSchool of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk; Tel: +44 (0)23 80593609
First published on 25th October 2013
Abstract
Rapid and complete fluorination of the complexes [MCl3(L)] (L = Me3-tacn, BzMe2-tacn, M = Al, Ga, In) occurs at room temperature via reaction of a MeCN solution of the complex with 3 mol equiv. of KF in water. The Ga and In complexes are also readily fluorinated using R4NF (R = Me or nBu) in MeCN solution, whereas no reaction occurs with the Al species under these conditions. The distorted octahedral fac-trifluoride coordination at M is confirmed in solution by multinuclear (19F, 27Al, 71Ga and 115In) NMR spectroscopic studies, leading to sharp resonances with 19F–71Ga and 19F–115In couplings evident. The [MF3(L)] are extremely stable in aqueous solution and at low pH; they crystallise as tetrahydrates, [MF3(Me3-tacn)]·4H2O, with extended H-bonding networks formed through both F⋯H–O and O⋯H–O contacts. [InF3(BzMe2-tacn)]·1.2H2O also shows intermolecular F⋯H–O hydrogen bonding contacts. The prospects for developing this coordination chemistry further to take advantage of the high metal–fluoride bond energies to enable rapid, late-stage fluorination of large macromolecules under mild conditions for PET imaging applications in nuclear medicine are discussed. This work also demonstrates that F-18 radiolabelling to form [F-18] [GaF3(BzMe2-tacn)] is effected readily at room temperature in aqueous MeCN over 30–60 min on addition of 2.99 mol equiv. of [19F]–KFaq and 0.4 mL [18F]–KFaq (100–500 MBq) to [GaCl3(BzMe2-tacn)] with ca. 30% incorporation.
Introduction
The increased availability of radioisotopes of the main group metals for radiopharmaceutical applications in imaging and therapy (e.g.67Ga, 68Ga, 111In, 113mIn, 117mSn) has driven the development of new coordination chemistry with specific ligand types.1 Fluorine-18 is the radioisotope of choice for medical applications using (non-invasive) PET imaging, owing to its ease of production via cyclotrons that are now widely available, and its short half-life (t1/2 = 109.8 minutes). For radioisotopes with a relative short half-life, there is a drive to introduce the radiolabel in the late stage of the synthesis (and preferably in the final step). Rapid, late-stage fluorination of complex molecules is consequently important for the development of new candidates for PET imaging in nuclear medicine. There has been a significant research effort in this area to provide routes for C–F bond formation reactions, as alternatives to the traditional nucleophilic reactions. Notable successes include the use of electrophilic ‘F+’ and metal-catalysed processes, as reported by Ritter, Groves, Buchwald and others.2–5 There is also a need to develop F-18 labelling methods which permit radiofluorination under mild conditions (neutral pH, room temperature) since this will improve the compatibility of the labelling conditions with a diverse range of biomolecules. Recent efforts towards boron-based agents for F-18 capture include the work of Perrin,6 Gabbaï7 and Tsien.8Recently McBride and others have reported the use of Al–F complexes based upon functionalised bis-carboxylate derivatives of 1,4,7-triazacyclononane for F-18 imaging (Scheme 1). Incorporation of F-18 via formation of M–F bonds with biologically relevant ligand scaffolds provides an exciting alternative to C–F based PET agents.9 McBride et al. have also used the formation of Al–F bonds to label a wide range of biomolecules rapidly in a single step.9 However, the need for elevated temperatures (100 °C) for fluorination in this ‘one-pot’ approach places some limitations on its utility due to the thermal instability of some important high MW biomolecules. In order to further extend the scope of this approach, an increased understanding of the chemistry and properties of fluoride complexes of the Group 13 elements is required.
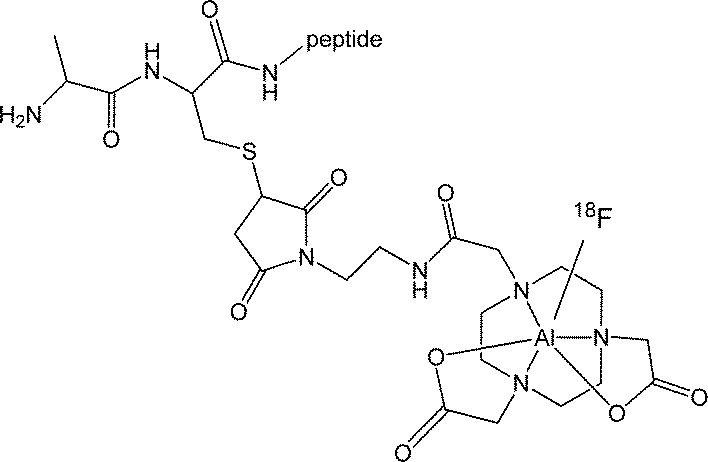 | ||
| Scheme 1 | ||
Metal fluoride coordination complexes are often significantly different from those containing heavier halogens.10 The small, hard and highly electronegative F− significantly influences the electronic environment at the metal centre and hence the binding of other ligands. For example, while ZrX4 (X = Cl, Br, I) readily form complexes with soft ligands such as phosphines and thioethers,11,12 no analogous complexes with ZrF4 are known.13 Further, while GeF414 and WF615 form phosphine adducts, GeX4 and WCl6 (and WBr6) are reduced to lower oxidation states. Few studies have been reported on the coordination chemistry of the Group 13 fluorides.10,16,17
We describe here the chemistry of the Group 13 trihalide complexes [MX3(L)] (L = Me3-tacn, BzMe2-tacn; M = Al, Ga, In; X = F, Cl, Br) (Scheme 2), and demonstrate that using simple neutral triaza macrocyclic frameworks, exchange of Cl− for F−via treatment of the chloro complexes with stoichiometric [R4N]F (R = nBu or Me) in MeCN solution or with aqueous KF in MeCN is rapid and complete under mild conditions (weakly acidic pH) and at room temperature. Further, we demonstrate that treatment of an aqueous MeCN solution of [GaCl3(BzMe2-tacn)] with 2.99 mol equiv. of aqueous [19F]–KF and 0.4 mL of [18F]–KFaq (100–500 MBq) leads to ca. 30% incorporation of the F-18, forming labelled [GaF3(BzMe2-tacn)] at room temperature within 30–60 min.
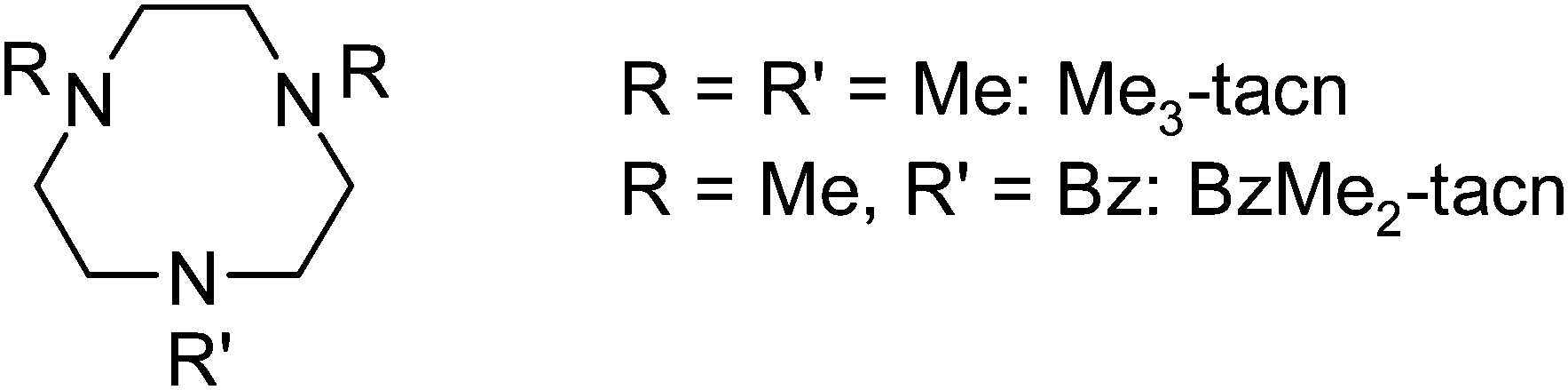 | ||
| Scheme 2 | ||
The ease of fluoride (both F-18 and F-19) incorporation into these preformed complexes at room temperature and in mildly acidic aqueous solution offers potentially significant advantages over McBride's ‘Al–F’ system9 which requires elevated temperature (100 °C) to achieve fluoride uptake, since some biomolecules are unstable at elevated temperatures or under acidic conditions. Hence, the work reported herein provides the very appealing prospect that rapid, late stage F-18 radiolabelling of well-defined, pre-formed metal complexes is possible, and that altering the metal ion to Ga in place of Al may provide further advantages since it facilitates labelling under mild conditions. The pH measured for a freshly prepared solution of [GaCl3(BzMe2-tacn)] in aqueous MeCN was 5.6, while the pH of reaction formulation comprising [GaCl3(BzMe2-tacn)] and KF in aqueous MeCN (unbuffered) was 5.9.
Experimental
Infrared spectra were recorded as Nujol mulls between CsI plates using a Perkin-Elmer Spectrum100 spectrometer over the range 4000–200 cm−1. 1H NMR spectra were recorded in CDCl3 or CD2Cl2 unless otherwise stated, using a Bruker AV300 spectrometer. 19F{1H} NMR spectra used either a Bruker AV300 or Bruker DPX400 (376.5 MHz) spectrometer and are referenced (externally) to CFCl3. 27Al, 71Ga, and 115In NMR spectra were recorded using a Bruker DPX400 spectrometer and are referenced to aqueous [Al(H2O)6]3+ (104.3 MHz), aqueous [Ga(H2O)6]3+ (122.0 MHz) and aqueous [In(H2O)6]3+ at pH = 1 (87.7 MHz) respectively. Microanalyses were undertaken by Medac Ltd. Solvents were dried by distillation prior to use, CH2Cl2 from CaH2, hexane from sodium benzophenone ketyl and MeCN from CaH2. MF3·xH2O, MCl3, MBr3 and [nBu4N]F (1.0 mol dm−3 in thf) (Aldrich) were used as received. Ligands Me3-tacn18 and BzMe2-tacn19 were prepared via the literature methods. [Me4N]F (Aldrich) was dried by azeotropic distillation from toluene. All preparations of chloro and bromo complexes (ESI†) were performed under an atmosphere of dry N2 using Schlenk techniques, and spectroscopic samples were prepared in a dry N2-purged glove box.Preparations
Method 1. AlF3·3H2O (0.100 g, 0.73 mmol) was suspended in freshly distilled water (7 mL). Me3-tacn (0.125 g, 0.73 mmol) was then added and the pale yellow suspension was transferred into a Teflon container and loaded into a stainless steel high pressure vessel (Parr) and heated to 180 °C for 15 h. The vessel was then allowed to cool. A dark yellow-brown solution had formed. A small aliquot of the reaction solution was retained to grow crystals. For the remaining reaction mixture the volatiles were removed in vacuo, giving a light brown solid which was washed with hexane and filtered. The resulting white solid was dried in vacuo. Yield: 0.12 g, 53%. Required for C9H21AlF3N3·3H2O: C, 34.9; H, 8.8; N, 13.6. Found: C, 34.3; H, 8.9; N, 14.7%. 1H NMR (CD3CN, 298 K): δ 2.84–2.76 (m, [6H], tacn-CH2), 2.72–2.65 (m, [6H], tacn-CH2), 2.55 (s, [9H], CH3), 2.19 (s, H2O). IR (Nujol, ν/cm−1): 3438 br (H2O), 1668 (H2O), 633, 614 (Al–F). Slow evaporation of the reaction solvent gave crystals suitable for X-ray diffraction.
Method 2. A solution of KF (0.058 g, 0.99 mmol) in water (2 mL) was added to a suspension of [AlCl3(Me3-tacn)] (0.100 g, 0.33 mmol) in MeCN (5 mL) at room temperature. A white precipitate formed initially which redissolved after a few minutes. NMR spectroscopic data on the solution were as for Method 1.
Method 1. [GaCl3(Me3-tacn)] (0.1 g, 0.28 mmol) was added to CH2Cl2 (8 mL) and stirred for ca. 15 min, the solid mostly dissolved to give a clear solution. [NnBu4]F in thf (1 mol dm−3, 0.84 mL, 0.84 mmol) was added to the mixture via syringe and the reaction was stirred for ca. 10 min, giving a clear, colourless solution. The solution was filtered and the filtrate was taken to dryness in vacuo. The resulting colourless solid was re-dissolved in CH2Cl2, the solution was filtered and the CH2Cl2 was left to evaporate, giving a colourless solid product. Yield: 50%. Required for C9H21F3GaN3·3H2O: C, 30.7; H, 7.7; N, 11.9. Found: C, 30.6; H, 6.9; N, 11.0%. 1H NMR (CD2Cl2, 298 K): δ 2.85–2.94 (br m, [6H], tacn-CH2), 2.67 (s, [9H], Me), 2.55–2.61 (br m, [6H], tacn-CH2, 2.17 (s, H2O)). IR (Nujol, ν/cm−1): 3481, 3429 (H2O), 1648 (H2O), 530, 492 (Ga–F). Colourless crystals were grown from the CH2Cl2 solution of the product upon slow evaporation.
Method 2. [GaCl3(Me3-tacn)] (0.05 g, 0.15 mmol) was suspended in 5 mL anhydrous CH2Cl2. The suspension was treated with [NMe4]F (0.042 g, 0.45 mmol) and stirred at room temperature for 1 h. The [NMe4]Cl by-product was removed by filtration. The resulting colourless filtrate was treated with 5 mL hexane, resulting in a white precipitate which was isolated by filtration and dried in vacuo. Yield: 0.037 g, 74%. Spectroscopic data as for Method 1.
Method 3. [GaCl3(Me3-tacn)] (0.05 g, 0.15 mmol) was suspended in anhydrous MeCN (5 mL). A solution of KF (0.026 g, 0.45 mmol) in water (2 mL) was added drop-wise, leading to rapid dissolution and the formation of a colourless solution. The mixture was stirred for a further 1 h at room temperature. Quantitative by NMR spectroscopic analysis; data as for Method 1.
Method 4. GaF3·3H2O (0.150 g, 0.83 mmol) was suspended in freshly distilled water (7 mL). Me3-tacn (0.142 g, 0.83 mmol) was then added and the pale yellow suspension was transferred into a Teflon container and loaded into a stainless steel high pressure vessel (Parr) and heated to 180 °C for 15 h. The vessel was then allowed to cool. A yellow-brown solution formed. A small aliquot of the reaction solution was retained to grow crystals. For the remaining reaction mixture the volatiles were removed in vacuo, giving a light brown solid which was washed with hexane and filtered. The resulting white solid was dried in vacuo. Yield: 0.21 g, 84%. 1H NMR (CD3CN, 298 K): δ 2.84 (m, [6H], tacn-CH2), 2.72 (m, [6H], tacn-CH2), 2.63 (s, [9H], CH3). IR (Nujol, ν/cm−1): 529, 492 (Ga–F).
Method 1. [GaCl3(BzMe2-tacn)] (0.05 g, 0.10 mmol) was suspended in 5 mL anhydrous CH2Cl2. The suspension was treated with [NMe4]F (0.03 g, 0.30 mmol) and stirred at room temperature for 1 h. The [NMe4]Cl by-product was removed by filtration. The resulting colourless filtrate was treated with 5 mL anhydrous hexane, forming a white precipitate which was isolated by filtration and dried in vacuo. Yield: 0.035 g, 80%. Required for C15H25F3GaN3·4H2O: C, 40.4; H, 8.8; N, 8.6. Found C, 40.9; H, 8.8; N, 8.6%. 1H NMR (D2O, 298 K): δ 7.30 (m, [5H], ArH), 4.73 (s, [2H], Ar-CH2), 3.17 (m, [4H], tacn-CH2), 2.88 (m, [4H], tacn-CH2), 2.73 (s, [6H], CH3), 2.36 (m, [4H], tacn-CH2), 2.25 (s, H2O). IR (Nujol, ν/cm−1): 3390, 1654 (H2O), 526, 515 (Ga–F).
Method 2. As described for [GaF3(Me3-tacn)] Method 3, using [GaCl3(BzMe2-tacn)] (0.05 g, 0.10 mmol) and KF (0.017 g, 0.30 mmol) in water. White solid. 0.035 g, 73%. Spectroscopic data as for Method 1.
Method 1. [InCl3(Me3-tacn)] (0.214 g, 0.54 mmol) was added to CH2Cl2 (8 mL) and stirred for ca. 15 min, this gave a cloudy suspension. [NnBu4]F in thf (1 mol dm−3, 1.63 mL, 1.63 mmol) was added to the mixture via a syringe and stirred for ca. 2 h. The solution was filtered and the white precipitate collected, washed with hexane and dried in vacuo. Yield: 0.150 g, 70%. Required for C9H21F3InN3·H2O: C, 29.9; H, 6.4; N, 11.6. Found: C, 29.9; H, 6.1; N, 11.5%. 1H NMR (CD2Cl2, 298 K): δ 3.09–3.15 (br m, [6H], tacn-CH2), 2.93 (m, [9H], Me), 2.72–2.82 (br m, [6H], tacn-CH2), 2.19 (s, H2O). IR (Nujol, ν/cm−1): 3392 br (H2O), 1669 (H2O), 479, 462, 443 (In–F). Colourless crystals were grown from the CH2Cl2 solution of the product upon slow evaporation.
Method 2. [InCl3(Me3-tacn)] (0.060 g, 0.17 mmol) was suspended in 5 mL anhydrous CH2Cl2. The suspension was treated with [NMe4]F (0.047 g, 0.51 mmol) and stirred at room temperature for 1 h. The [NMe4]Cl by-product was removed by filtration. The resulting colourless filtrate was treated with 5 mL anhydrous hexane, forming a white precipitate which was isolated by filtration and dried in vacuo. Yield: 0.044 g, 76%. Spectroscopic data as for Method 1.
Method 3. A Teflon reactor vessel was charged with freshly distilled water (7 mL), InF3·3H2O (0.200 g, 0.90 mmol) and Me3-tacn (0.154 g, 0.90 mmol). The Teflon container was loaded into a stainless steel high pressure vessel (Parr) and heated to 180 °C for 15 h. The vessel was then allowed to cool. A dark yellow-brown solution had formed. A small aliquot of the reaction solution was retained to grow crystals. For the remaining reaction mixture the volatiles were removed in vacuo, giving a light brown gum which was washed with hexane. The hexane was decanted and the remaining volatiles removed in vacuo to give a light brown solid. Yield 0.246 g, 0.62 mmol, 69%. Spectroscopic data as for Method 1.
F-18 radiolabelling of [GaCl3(BzMe2-tacn)]
Experiments were analysed on a Gilson 322 HPLC system with a Gilson 156 UV detector. Dionex Chromeleon 6.8 Chromatography data recording software was used to integrate the UV and radiochemical peak areas. Preparative HPLC: Luna 5μ C18(2) 100 × 10 mm (mobile phase A = 100% water; B = 100% MeCN). Flow rate 3 mL min−1. Gradient 0 to 5 min (10% B), 5–20 min (10–90% B), 20–25 min (90% B), 25–26 min (10% B).Analytical HPLC: Luna 5μ C18(2) 250 × 4.6 mm (mobile phase A = 10 mM ammonium acetate, B = 100% MeCN). Flow rate 1 mL min−1. Gradient 0–15 min (10–90% B), 15–20 min (90% B), 20–21 min (90–10% B), 21–26.5 min (10% B).
ESI+ mass spectra were recorded from direct injection of the products onto a Thermo Finnigan mass spectrometer fitted with an LCQ advantage MS pump.
Procedure: In a typical experiment [GaCl3(BzMe2-tacn)] (0.001 g, 2.36 μmol) was dissolved in a mixture of MeCN (0.5 mL) and H2O (0.1 mL). This solution was added to 0.4 mL of an aqueous solution containing [18F]–KF (100 to 500 MBq) and [19F]–KF (7.05 μmol) and the vial was allowed to stand at room temperature for 30 to 60 min. The crude reaction solution was diluted with water so that approximately 10% of the solvent composition was organic. Preparative HPLC on the diluted crude reaction solution confirmed ca. 30% incorporation of F-18 into the metal complex (based upon integration of the radio peaks) after one hour. Peak 1: Rt = 2.2 min (18F−). Peak 2: Rt = 9.0 min (complex). The MeCN was then removed in vacuo and the product was made up with PBS and ethanol to give a ca. 10% ethanol final product (pH 7.2).
Peak 2 was run through an analytical HPLC system giving a single radio and UV peak at Rt 6.2 min (RCP 99%).
Peak 2: ESI+ MS (MeCN/NH4OAc): found m/z = 354 [GaF2(BzMe2-tacn)]+; 391 [GaF3(BzMe2-tacn) + NH4]+. The product is stable to chemical and radiochemical degradation for at least two hours in phosphate buffered saline and ethanol – see ESI.†
X-ray crystallography
Details of the crystallographic data collection and refinement parameters are given in the ESI.† Crystals were obtained as described. Data collection used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum rotating anode generator with VHF Varimax optics (100 μm focus) with the crystal held at 120 K (N2 cryostream) or a Bruker-Nonius FR591 rotating anode diffractometer fitted with confocal mirrors and with the crystal held at 120 K (N2 cryostream). Mo-Kα X-radiation (λ = 0.71073 Å). Structure solution and refinement were generally routine,20,21 except as described below, with hydrogen atoms on C added to the model in calculated positions and using the default C–H distance. For [AlCl3(Me3-tacn)] the data were collected as orthorhombic, but during the structure solution it became clear that in fact the crystal was monoclinic. The data were therefore reprocessed as monoclinic, giving 96% completeness. The refinement used TWIN/BASF commands to model disorder. For [InCl3(BzMe2-tacn)] the crystal quality was rather poor, hence the final residuals are higher than normal, although the coordination environment is not in doubt.Results and discussion
The Group 13 fluorides, MF3·3H2O, are poorly soluble in organic solvents, and hence there are rather few examples where these have been used directly to form metal fluoro-complexes with neutral ligands under conventional conditions.10 Notable exceptions include mer-[GaF3(pyridine)3] (prepared by prolonged refluxing of the constituents in thf),22 [GaF3{1,4,7-tris(2-amino-3,5-di-butylbenzyl)-1,4,7-triazacyclononane}],23 and direct reaction of InF3·3H2O with 2,2′-bipy or 1,10-phen (L–L) forms the distorted octahedral species [InF3(L–L)(H2O)].24 Diimine and amine complexes of MF3 (M = Al, Ga, In) have been obtained using hydrothermal syntheses at elevated temperature (∼180 °C),25 while reaction of AlN or InN and NH4F in supercritical ammonia (400 °C) forms [AlF3(NH3)2] and [InF2(NH2)(NH3)] respectively.26Direct reaction of AlF3·3H2O and Me3-tacn under hydrothermal conditions (180 °C, 15 h) led to formation of [AlF3(Me3-tacn)]·4H2O as a white solid in good yield. The IR spectrum shows two bands in the far-IR region attributed to the a1 and e stretching modes from the facial MF3 unit of a distorted octahedron (C3v). There is also clear evidence for H-bonded H2O, which turn out to be an important feature of these complexes and is described in more detail below. The 1H NMR spectrum (CD3CN) is characteristic of facially coordinated Me3-tacn. NMR spectroscopic measurements in D2O also confirm the stability of the trifluoro species in aqueous solution. The 27Al [I = 5/2, 100% abundance, Q = 0.149 × 10−28 m2, RC = 1170] and 19F{1H} NMR spectra (Table 1) each show a singlet.
| Complex | δ 27Al/71Ga/115In/ppm; (w1/2/Hz) | δ 19F{1H} (ppm) | Solvent |
|---|---|---|---|
| a n.o. = not observed. | |||
| [AlF3(Me3-tacn)] | 19.0 (60); 18.5 (52) | −176.1; −169.9 | MeCN; D2O |
| [AlCl3(Me3-tacn)] | 34.5 (30) | — | CH2Cl2 |
| [AlBr3(Me3-tacn)] | 18.9 (80) | — | CH2Cl2 |
| [GaF3(Me3-tacn)] | 42.0 (q, 1JGaF ∼ 490 Hz); 44.6 (br q) | −180.9 (two br q); −173 (br) | CH2Cl2; D2O |
| [GaCl3(Me3-tacn)] | 93.9 (60) | — | CH2Cl2 |
| [GaBr3(Me3-tacn)] | −29.3 (180) | — | MeCN |
| [InF3(Me3-tacn)] | 64 (q, 1JInF ∼ 600 Hz); n.o. | −215 (br); −192.5 (br) | MeCN; D2O |
| [InCl3(Me3-tacn)] | 268 (750) | — | CH2Cl2 |
| [InBr3(Me3-tacn)] | n.o.a | — | MeCN |
| [AlF3(BzMe2-tacn)] | 19.8 (100) | −161.5 (F), −161.7 (2F) | MeCN |
| [AlCl3(BzMe2-tacn)] | 36.5 (45) | — | CH2Cl2 |
| [AlBr3(BzMe2-tacn)] | 20.1 (35) | — | CH2Cl2 |
| [GaF3(BzMe2-tacn)] | 44.9 (q 1JGaF ∼ 445Hz) | −172.8 (br) | D2O |
| [GaCl3(BzMe2-tacn)] | 92.8 (360) | — | MeCN |
| [InF3(BzMe2-tacn)] | n.o.a | −220 (br) | MeCN |
| [InCl3(BzMe2-tacn)] | 265 (2200) | — | MeCN |
The crystal structure is consistent with the spectroscopic data, confirming the distorted octahedral coordination at Al via a tridentate Me3-tacn ligand and three fac F− ligands (Fig. 1(a)).
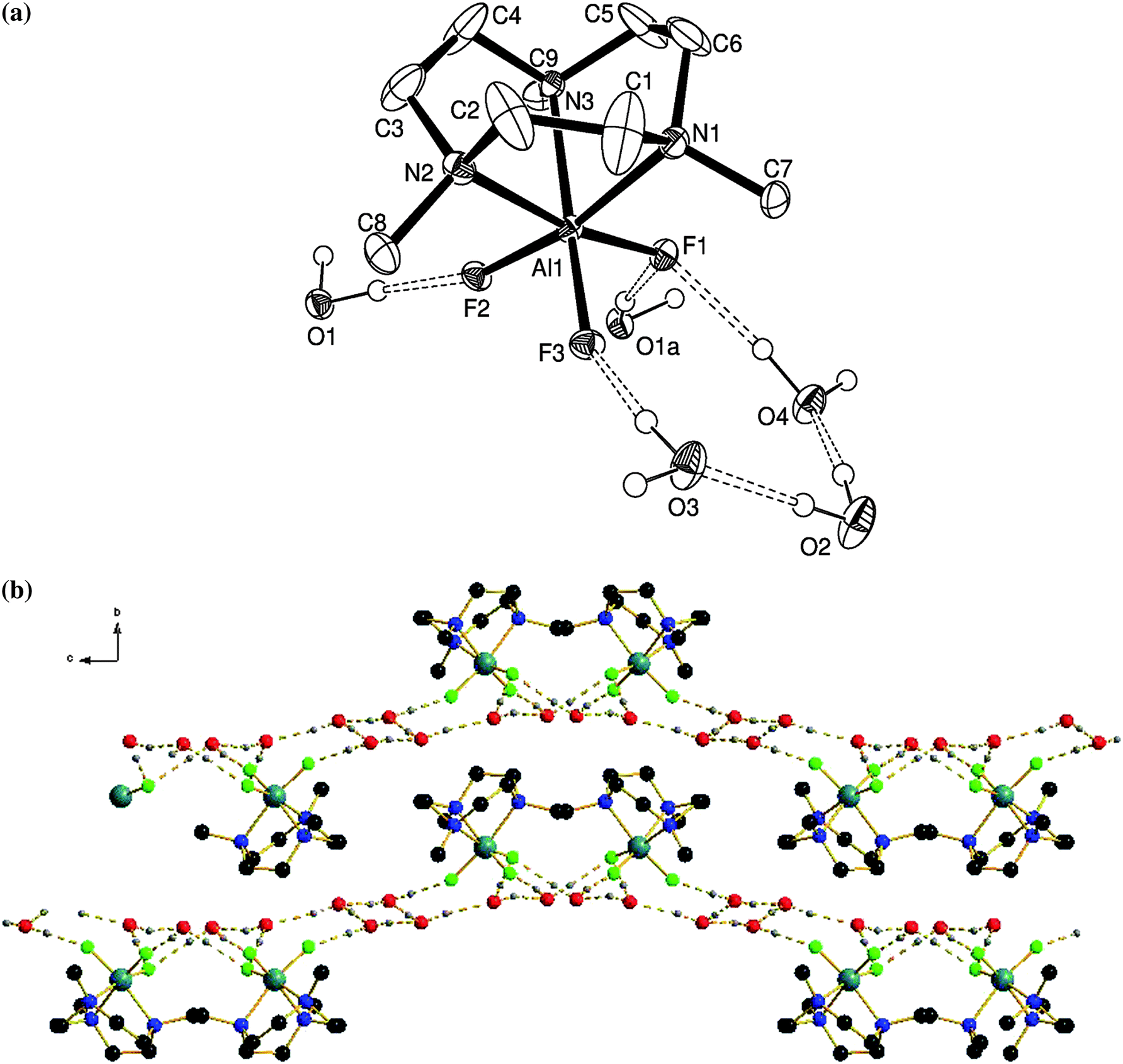 | ||
| Fig. 1 (a) Structure of [AlF3(Me3-tacn)]·4H2O showing the atom labelling scheme. Ellipsoids are drawn at the 40% probability level and H atoms bonded to C are omitted for clarity. (b) View along the a-axis of the extended structure assembled through intermolecular H-bonding (colour key: teal = Al, green = F, blue = N, red = O, black = C). | ||
The asymmetric unit contains four H2O molecules as well as one [AlF3(Me3-tacn)] molecule. The water molecules are involved in an array of H-bonding interactions both with the F atoms in the [AlF3(Me3-tacn)], as well as between the H2O molecules themselves, giving H2O⋯F1 = 2.806(3), H2O⋯F2 = 2.701(3), H2O⋯F3 = 2.688(3), H2O⋯F1 = 2.802(4) Å. This gives rise to the extensively H-bonded array illustrated in Fig. 1(b).
The GaF3 analogue, [GaF3(Me3-tacn)]·4H2O, obtained and characterised similarly, also crystallises as a tetrahydrate (below). These [MF3(Me3-tacn)]·4H2O species are remarkably stable in the solid and also in solution in H2O, CH2Cl2, MeCN etc. This led us to consider the possibility of that introduction of F−via exchange reactions with the heavier halide analogues (Cl− or Br−) may be synthetically viable and serve as an alternative route to the [MF3(Me3-tacn)] (M = Al, Ga or In), by virtue of the M–F bonds formed being stronger than those involving the heavier halides.10
In order to test this idea, the complexes, fac-[MX3(Me3-tacn)] (M = Al, Ga, In; X = Cl, Br) and [MX3(BzMe2-tacn)] (M = Al, X = Cl, Br; M = Ga, X = Cl; M = In; X = Cl or Br) were obtained in high yield from direct reaction of MX3 with the triaza macrocycle in anhydrous CH2Cl2 or MeCN. Confirmation of their identities follows from microanalyses, IR and 1H NMR spectroscopic data and from crystal structures of representative examples.
Multinuclear solution 27Al, 71Ga and 115In NMR studies support the formulations – Table 1. For the Al and Ga complexes, the spectra show a single resonance despite the moderate quadrupole moments associated with the 27Al and 71Ga [I = 3/2, 39.6% abundance, Q = 0.112 × 10−28 m2, RC = 322] nuclei. This is consistent with the proposed distorted six-coordinate geometry with local C3v symmetry, leading to the electric field gradient (EFG) being close to zero, and hence relatively sharp lines.27 The 115In NMR spectra were less informative due to the much larger quadrupole moment, which sometimes led to the resonance not being observed for [InX3(R3-tacn)] (X = Cl or Br) [115In: I = 9/2, 95.7% abundance, Q = 1.16 × 10−28 m2, RC = 1920]. The multinuclear NMR studies show that the chloro complexes are more resistant to hydrolysis/solvolysis in solution than the bromo species, and that while the complexes are stable in anhydrous CH2Cl2 or MeCN, stronger donor solvents such as dmf or dmso lead to decomposition. The powdered solids may be stored under N2 for many months without degradation.
Trace hydrolysis of [GaX3(Me3-tacn)] (X = Cl or Br) produces the face-sharing bioctahedral dimers [{(Me3-tacn)Ga}2(μ-OH)3]X3·3CH2Cl2, and the crystal structure of the Br derivative was also determined (ESI†). Wieghardt and co-workers have described the hydrolysis of [InCl3(Me3-tacn)] to form dinuclear μ-hydroxy and tetranuclear μ-oxo derivatives.28
While the bromo complexes are readily hydrolysed, the chloro species are much more stable, and therefore considered excellent candidates for the fluorination studies.
Cl−/19F− exchange reactions
Reagents such as Me3SiF and Me3SnF are often convenient fluoride sources in synthetic chemistry, e.g. [AlCl3(py)n] (n = 1 to 3) and Me3SiF in pyridine affords [AlF2(py)4]Cl.29 However, for F-18 applications, the radio-fluorine is produced as F− ions, and hence it is more desirable to be able to use the fluoride directly as Na18F (or K18F or R4N18F). Therefore, in this work we have investigated both tetraalkylammonium fluorides (in organic solvents) and aqueous KF as the fluoride source.Initial studies were performed by addition of three mol equiv. of a 1 mol dm−3 thf solution of [NnBu4]F to a suspension of [MCl3(R3-tacn)] in MeCN. For the Ga systems this led to rapid and complete dissolution at room temperature over ca. 5 min, and in situ71Ga and 19F{1H} NMR studies (Table 2) show complete conversion to [GaF3(R3-tacn)]. For the more symmetrical Me3-tacn system, a quartet is observed in the 71Ga NMR spectrum (MeCN) due to coupling to three F− ligands (δ71Ga = 42.0, 1JGaF ∼ 490 Hz – Fig. 2), and although not fully resolved, the 19F{1H} NMR spectrum shows a single resonance (−180.0 ppm) with coupling to 69/71Ga, providing unequivocal evidence for formation of [GaF3(Me3-tacn)].
| Complex | d(M–X)/Å | d(M–N)/Å | ∠(X–M–X)/° | ∠(N–M–N)/° |
|---|---|---|---|---|
| [AlF3(Me3-tacn)]·4H2O | 1.740(2), 1.744(2), 1.757(2) | 2.098(3), 2.106(3), 2.115(3) | 96.65(12), 96.00(11), 95.81(12) | 82.20(13), 82.13(13), 82.37(13) |
| [AlCl3(Me3-tacn)] | 2.267(2), 2.274(2), 2.276(2) | 2.111(3), 2.126(4), 2.141(4) | 94.03(7), 93.74(6), 94.21(7) | 83.03(14), 81.64(14), 81.97(14) |
| [GaF3(Me3-tacn)]·4H2O | 1.851(3), 1.858(3), 1.881(3) | 2.126(4), 2.126(4), 2.140(4) | 95.81(12), 94.87(13), 94.23(12) | 82.2(1), 82.4(2), 81.6(2) |
| [GaCl3(Me3-tacn)] | 2.3087(5), 2.3177(9), 2.3217(5) | 2.1644(13), 2.1755(13), 2.1960(14) | 94.38(2), 93.98(2), 94.49(2) | 81.90(5), 80.80(5), 80.83(5) |
| [InF3(Me3-tacn)]·4H2O | 2.0318(11), 2.0391(12), 2.0570(11) | 2.287(2), 2.289(2), 2.291(2) | 96.33(5), 95.04(5), 94.79(5) | 78.68(6), 78.91(6), 78.44(6) |
| [InBr3(Me3-tacn)]·CH2Cl2 | 2.5987(8), 2.6006(8), 2.6046(8) | 2.344(4), 2.345(5), 2.347(4) | 96.66(3), 95.06(2), 96.15(3) | 76.4(2), 76.8(2), 76.2(2) |
| [InF3(BzMe2-tacn)]·1.2H2O | 2.4443(13), 2.4518(14), 2.4581(14) | 2.312(4), 2.342(4), 2.371(4) | 96.72(5), 96.72(5), 98.24(5) | 77.96(6), 77.14(6), 77.80(6) |
| [InCl3(BzMe2-tacn)] | 2.4443(13), 2.4518(14), 2.4581(14) | 2.312(4), 2.342(4), 2.371(4) | 95.32(5), 96.26(5), 98.31(5) | 77.1(2), 77.4(2), 76.3(2) |
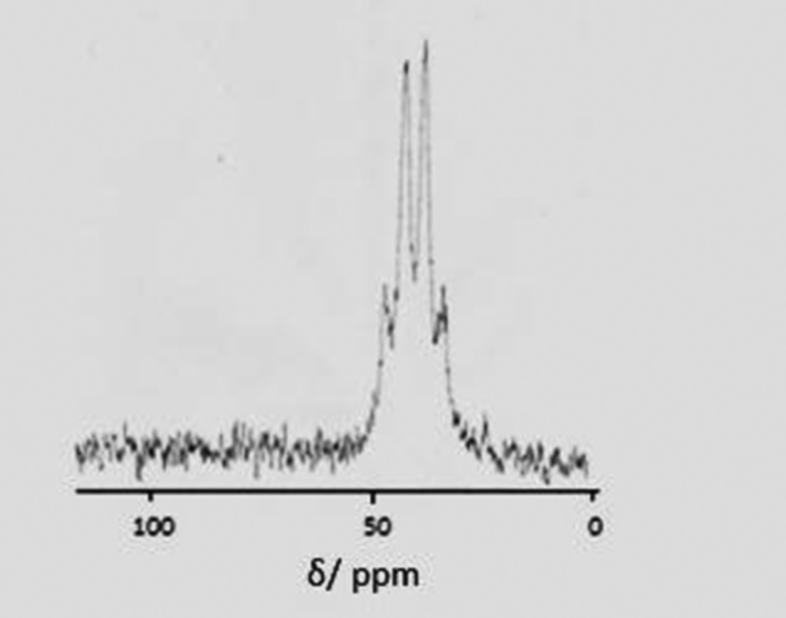 | ||
| Fig. 2 71Ga NMR spectrum of [GaF3(Me3-tacn)] (CH2Cl2) showing the quartet coupling (1JGaF ∼ 490 Hz). | ||
Notably, like [AlF3(Me3-tacn)], [GaF3(Me3-tacn)] also crystallises as a tetrahydrate, [GaF3(Me3-tacn)]·4H2O (Fig. 3), with an extensive H-bonding array involving intermolecular O⋯F and O⋯O interactions.
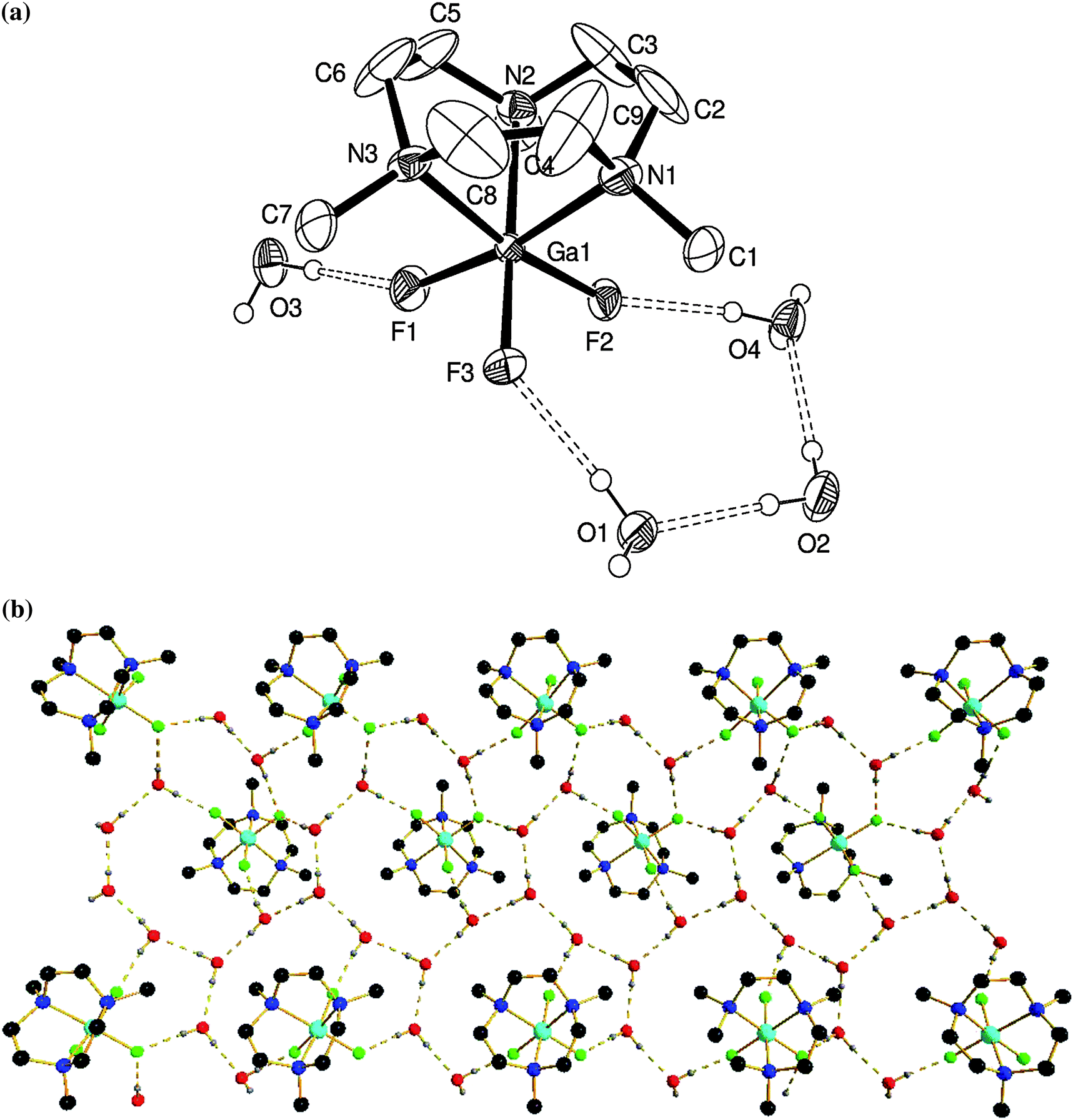 | ||
| Fig. 3 (a) View of the structure of [GaF3(Me3-tacn)]·4H2O with numbering scheme adopted. Ellipsoids are shown at the 40% probability level and H atoms (except those on the H2O molecules) are omitted for clarity. (b) View down the c-axis of the extended structure showing the H-bonding interactions (colour key: turquoise = Ga, green = F, blue = N, red = O, black = C). | ||
The [GaF3(R3-tacn)] complexes were subjected to a range of experimental conditions that showed the trifluoro-complexes are unaffected by (i) prolonged heating (2 h at 40–50 °C) in MeCN, (ii) the presence of a 10-fold excess of Cl− in MeCN, (iii) standing for several days in aqueous solution, (iv) the presence of acid (aqueous HBF4) and (v) the presence of excess F− either in MeCN or H2O.
For the gallium systems, clean fluorination is also effected using [NMe4]F in MeCN (the [NMe4]Cl by-product is more readily separated than [NnBu4]Cl). Addition of aqueous KF to a suspension of [GaCl3(R3-tacn)] in MeCN also leads to rapid and complete fluorination at room temperature. This confirms that Cl−/F− exchange is faster than any competing hydrolysis reactions under these conditions.
The [InCl3(R3-tacn)] behave similarly with both [NR4]F in MeCN, although the Cl−/F− exchange reaction is slower (ca. 30–45 min) to reach completion at room temperature compared to the Ga systems. The 115In spectrum (MeCN) of [InF3(Me3-tacn)] shows a well-resolved 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3:1 quartet (Fig. 4) at 64 ppm (1JInF ∼ 600 Hz), confirming the complete exchange of Cl− for F−.
:3:3:1 quartet (Fig. 4) at 64 ppm (1JInF ∼ 600 Hz), confirming the complete exchange of Cl− for F−.
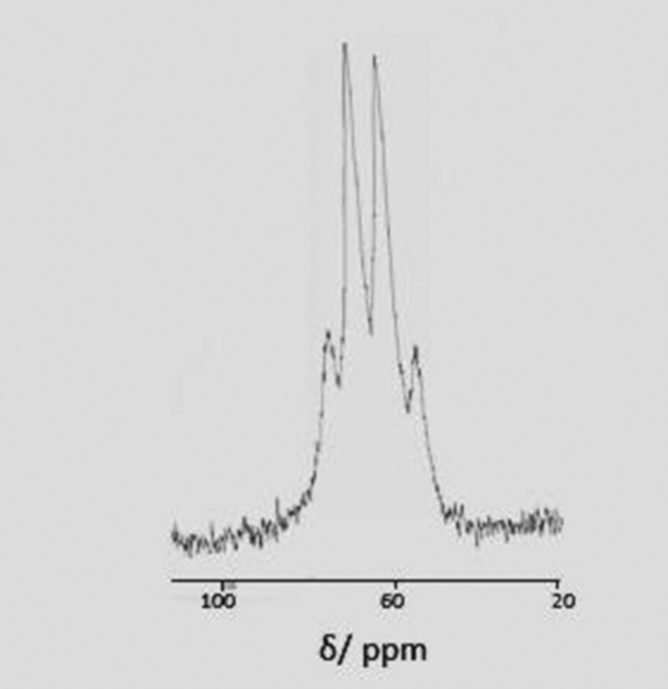 | ||
| Fig. 4 115In NMR spectrum of [InF3(Me3-tacn)] (MeCN). The splitting pattern arises through the coupling of three equivalent fluorides to the indium centre (1JInF ∼ 600 Hz). | ||
Both [InF3(Me3-tacn)]·4H2O (Fig. 5) and [InF3(BzMe2-tacn)]·1.2H2O (Fig. 6) were also characterised crystallographically. Although none of the [MF3(Me3-tacn)] complexes in this study are isomorphous, they all adopt very similar structures and crystallise as tetrahydrates, showing a very strong tendency for the F ligands to engage in extensive F⋯H–OH hydrogen-bonding, while [InF3(BzMe2-tacn)]·1.2H2O, shows the H2O molecules form significant interactions with F1 and F2, F⋯H–OH ∼ 2.8 Å.
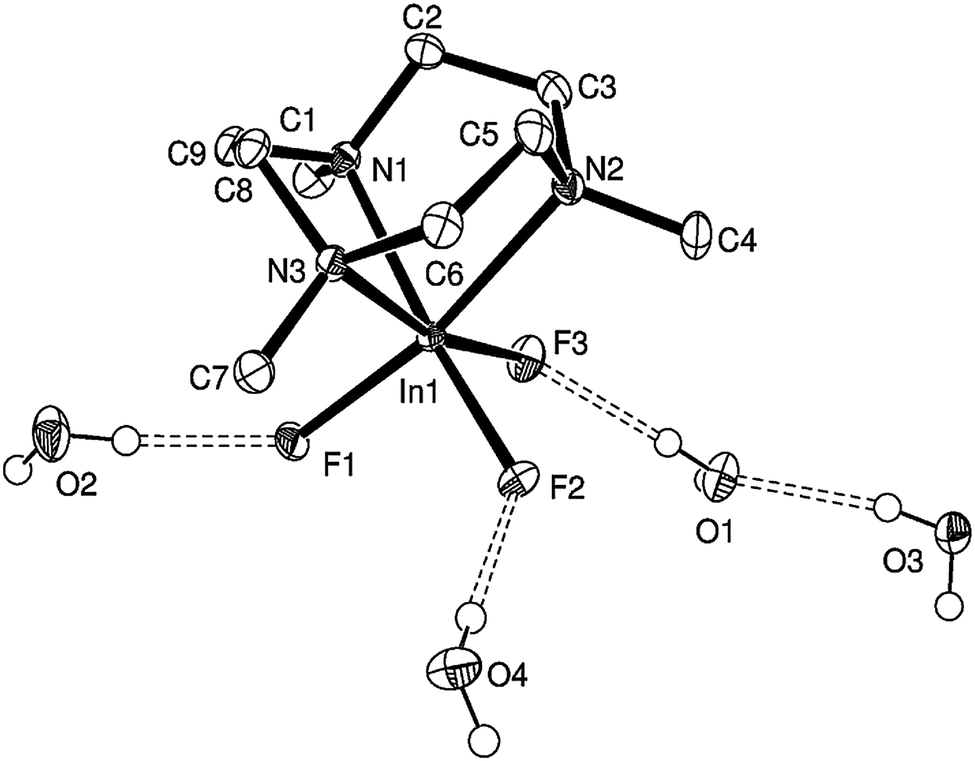 | ||
| Fig. 5 View of the structure of [InF3(Me3-tacn)]·4H2O with numbering scheme adopted and showing the F⋯H–O interactions. Ellipsoids are shown at the 40% probability level and H atoms (except those on the H2O molecules) are omitted for clarity. | ||
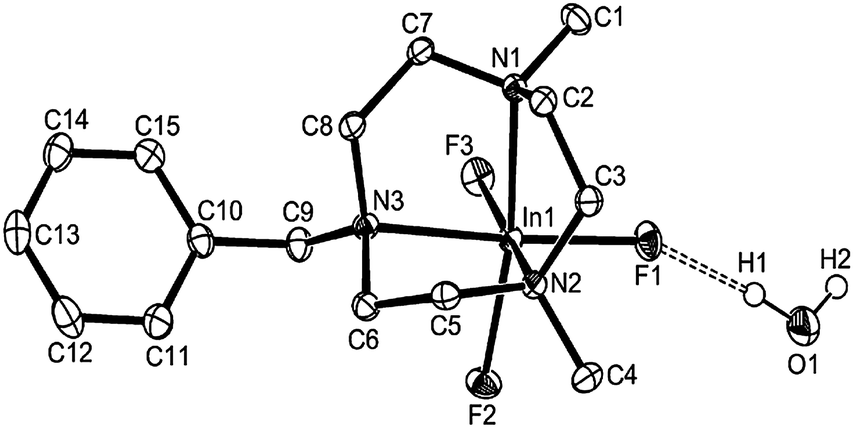 | ||
| Fig. 6 Structure of [InF3(BzMe2-tacn)]·H2O with atom numbering scheme. Ellipsoids are shown at the 50% probability level and H atoms (except those on O1) are omitted for clarity. | ||
Unlike the Ga and In analogues, [AlCl3(Me3-tacn)] does not react with either [NnBu4]F or [NMe4]F in neat MeCN at room temperature, even over several hours. Heating the reaction mixture causes partial decomposition, forming [AlF4]− and releasing the R3-tacn, but there is no evidence in the 19F{1H} and 27Al NMR spectra for formation of [AlF3(Me3-tacn)] under these conditions. This was unexpected, and the reason for the failure is not entirely clear, however, it may be a result of the smaller ionic radius of Al3+, which would disfavour an associative (A) or associative interchange (Ia) ligand substitution mechanism. However, we were able to demonstrate that addition of aqueous KF to a MeCN suspension of [AlCl3(Me3-tacn)] does lead to clean conversion to form [AlF3(Me3-tacn)] at room temperature, the spectroscopic signature of the product matching that formed via hydrothermal synthesis from AlF3·3H2O (above). This suggests that the more polar (cf. MeCN) H2O solvent is involved in a solvent assisted substitution mechanism.30
F-18 radiolabelling
Based upon the results from the Cl−/19F− exchange reactions the gallium(III) systems were identified as the most promising candidate for the F-18 radiolabelling experiments. Furthermore, inclusion of the benzyl chromophore in [GaCl3(BzMe2-tacn)] allows the fate of the complex to be monitored in parallel with the radio-trace by using UV-visible spectroscopy. Radiolabelling was carried out on a 1 mg scale by dissolving [GaCl3(BzMe2-tacn)] in aqueous MeCN, adding 2.99 mol equiv. of aqueous [19F]–KF and 0.4 mL of [18F]–KFaq (100–500 MBq) and allowing the solution to stand at room temperature for between 30 and 60 min. The crude reaction solution was purified by preparative HPLC using a water–MeCN mobile phase. This gave a single product peak at Rt = 9.0 min (ca. 30% incorporation after one hour). The purified species was eluted through an analytical HPLC system using a 10 mM aq. NH4OAc–MeCN mobile phase, giving a single peak in the radio-chromatograph at Rt = 6.1 min. ESI+ mass spectrometric analysis of this species post elution gave an m/z and isotope pattern consistent with the species [GaF3(BzMe2-tacn) + NH4]+ (m/z = 391; 100%) – see ESI.† The presence of associated [NH4]+ in these species was also confirmed from independent mass spectrometry experiments on the preformed [GaF3(BzMe2-tacn)] complex with and without added cation. Thus, introduction of one mol equiv. of [NH4][PF6] leads to the appearance of a strong peak at m/z = 391.This behaviour is attributed to the presence of the highly electronegative facial GaF3 fragment which can form electrostatic and/or H-bonding interactions with the hard [NH4]+ cation introduced during the labelling experiments and HPLC analysis – similar to the strong F⋯H–OH interactions to the associated water molecules observed crystallographically (vide supra). There is also literature precedent for this behaviour in alkaline earth or lanthanide complexes of AsF3 such as [Ca(AsF3)(AsF6)2], in which the pyramidal AsF3 molecule behaves as a Lewis base, bonding to the metal cation via bridging fluorides (with further interactions between Ca2+ and the [AsF6]− anions).31
The purified species was dried under vacuum and treated with phosphate buffered saline (PBS) and ethanol, giving a formulation of 10% ethanol with pH 7.2. Subsequent analysis by analytical HPLC confirmed that the species was stable under these conditions for at least 2 hours. The radiochemical purity (RCP) of the purified product remained high over this period (98−99% RCP) – ESI.†
X-ray structural comparisons
In view of the very different stabilities of the [MX3(R3-tacn)] complexes and the differing reactivities towards F−/Cl− exchange observed across the series, it was of interest to compare the structural properties of the species to attempt to ascertain any significant structural trends which might provide some insights. For comparison with the trifluoro complexes already described, crystal structures of [MX3(R3-tacn)] were therefore determined for a range of M with X = Cl or Br, specifically for [MX3(Me3-tacn)] (M = Al, X = Cl; M = Ga, X = Cl; M = In, X = Br) – Fig. 7 and ESI, and for [InCl3(BzMe2-tacn)] (ESI†). Each structure shows the expected distorted octahedral coordination environment at M, comprising a tridentate triamine macrocycle and three mutually facial X ligands.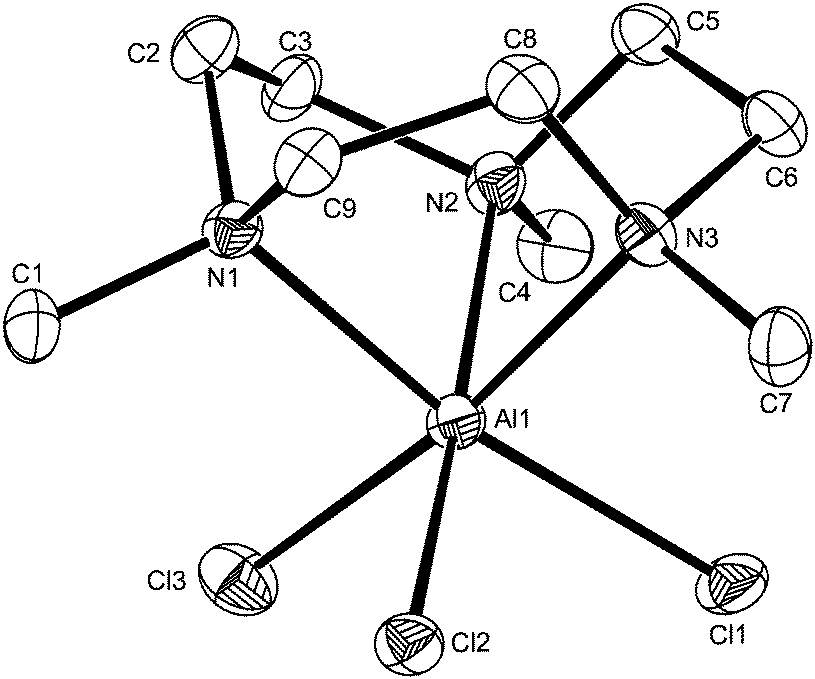 | ||
| Fig. 7 Structure of [AlCl3(Me3-tacn)] with atom numbering scheme. Ellipsoids are shown at the 50% probability level and H atoms are omitted for clarity. | ||
In contrast to the fluorides, the chloro- and bromo-complexes are discrete molecular entities, and show no incorporation of solvent molecules in the crystal lattice.
Table 2 summarises the key geometric parameters for the series of complexes. Comparing the M–N and M–F distances within the series [MF3(Me3-tacn)]·4H2O (M = Al, Ga, In) reveals that upon going from Al to Ga the M–F bond distances increase by ∼0.12 Å, and from Ga to In the increase is ∼0.19 Å. These changes are almost exactly in line with expectation based on the increasing ionic radii for the six-coordinate trivalent metal ions down Group 13 (Al3+ = 0.535 Å; Ga3+ = 0.62 Å; In3+ = 0.80 Å).32
In contrast however, the M–N bond distances for Ga complex are only ∼0.02 Å longer than for the Al complex, whereas from Ga to In the M–N bonds increase by ∼0.17 Å. Also, comparing the Al–N bond distances in [AlF3(Me3-tacn)]·4H2O with those in [AlCl3(Me3-tacn)] reveals a very small increase of only ∼0.02 Å, whereas in the Ga systems [GaF3(Me3-tacn)]·4H2O the Ga–N distances are ca. 0.04–0.05 Å shorter than in [GaCl3(Me3-tacn)]. These observations suggest that the 9-membered triaza macrocycle may be too large for optimal facial coordination to the smallest Al3+ ion, whereas the larger Ga3+ and In3+ fit rather better. This may also account for the differences observed in the Cl−/F− exchange reactions in MeCN solution; i.e. the Al3+ centre is sterically less accessible to the F− entering group in MeCN, whereas the halide exchange in aqueous MeCN probably undergoes a solvent (H2O) assisted substitution.
The trend in X–M–X and N–M–N angles across the series correlates with the trends in bond distances. In all cases the angles involving halide ligands are ∼94–98°, with no obvious trend with changing X or M. In contrast, the N–M–N angles in the Al3+ and Ga3+ complexes are essentially invariant (∼82°), while those involving In3+ are rather more acute, ∼77°, reflecting the elongated In–N bond distances.
Conclusions and outlook
This work has shown that direct fluorination of the distorted octahedral Group 13 tacn-based complexes, [MCl3(R3-tacn)] (M = Al, Ga, In) occurs cleanly and rapidly at room temperature in aqueous MeCN using KF (as well as in MeCN solution via [NR4]F) to produce very stable [MF3(R3-tacn)] complexes, all of which are isolated as hydrates. We have also shown that radiolabelling an aqueous solution of [GaF3(BzMe2-tacn)] with F-18 doped [19F]–KF leads to ca. 30% incorporation of F-18, forming [GaF3(BzMe2-tacn)] rapidly at room temperature under mildly acidic conditions which is stable over at least 2 hours in PBS buffered solution. The rapidly growing interest in metal complexes as high affinity binders for F-18 in PET imaging applications in nuclear medicine, together with the remarkably high stability of the Group 13 metal trifluoro complexes described herein, lead to an enticing prospect for producing new generations of metal-fluoride based PET imaging agents. It is notable that McBride's ‘Al–F’ system9 requires elevated temperature (100 °C) to achieve fluoride uptake, whereas in the chemistry described here, Cl−/F− exchange is achieved rapidly using KF at room temperature in aqueous solution both on a preparative and radio-tracer scale. This suggests that rapid, late stage F-18 radiolabelling of pre-formed metal complexes may be an attractive alternative strategy, and provides evidence that introducing metal ions such as Ga in place of Al may offer further advantages. It should be noted that in order to demonstrate the radiolabelling of the Ga compound in the present work at room temperature, we have used 1 mg (2.36 μmol) of the pre-formed [GaCl3(Me2Bz-tacn)] complex, whereas McBride et al. in their systems6 were able to label at lower quantity, but require heating to achieve good labelling. Further work in our laboratories is aimed at modifying the ligands to enable room temperature radiolabeling of the complexes using less material.Acknowledgements
We thank GE Healthcare and the EPSRC for a CASE studentship (G. S.). We also thank Dr M. Webster for assistance with the crystallographic analyses, J. Ali (GE Healthcare) for support on the mass spectrometry studies and Dr G. J. Langley (Southampton) for helpful discussions.Notes and references
- G. Bandoli, A. Dolmella, F. Tisato, M. Porchia and F. Refosco, Coord. Chem. Rev., 2009, 253, 56 CrossRef CAS PubMed; D. E. Reichert, J. S. Lewis and C. J. Anderson, Coord. Chem. Rev., 1999, 184, 3 CrossRef; C. J. Anderson and M. J. Webb, Chem. Rev., 1999, 99, 2219 CrossRef PubMed.
- K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS PubMed; E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639 CrossRef PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; P. P. Tang, T. Furuya and T. Ritter, J. Am. Chem. Soc., 2010, 132, 12150 CrossRef PubMed.
- W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322 CrossRef CAS PubMed.
- D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. García-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661 CrossRef CAS PubMed.
- R. Ting, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Am. Chem. Soc., 2005, 127, 13094 CrossRef CAS PubMed; R. Ting, C. Harwig, U. auf dem Keller, S. McCormick, P. Austin, C. M. Overall, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Am. Chem. Soc., 2008, 130, 12045 CrossRef PubMed.
- Z. Li, K. Chansaenpak, S. Liu, C. R. Wade, P. S. Conti and F. P. Gabbaï, Med. Chem. Commun., 2012, 3, 1305 RSC.
- R. Ting, T. A. Aguilera, J. L. Crisp, D. J. Hall, W. C. Eckelman, D. R. Vera and R. Y. Tsien, Bioconjugate Chem., 2010, 21, 1811 CrossRef CAS PubMed.
- P. Laverman, W. McBride, R. Sharkey, A. Eek, L. Joosten, W. Oyen, D. Goldenberg and O. Boerman, J. Nucl. Med., 2010, 51, 454 CrossRef CAS PubMed; W. McBride, C. D'Souza, R. Sharkey, H. Karacay, E. Rossi, C. Chang and D. Goldenberg, Bioconjugate Chem., 2010, 21, 1331 CrossRef PubMed; W. McBride, R. Sharkey, H. Karacay, C. D'Souza, E. Rosso, P. Laverman, C. Chang, O. Boerman and D. Goldenberg, J. Nucl. Med., 2009, 50, 991 CrossRef PubMed; W. McBride, C. D'Souza, H. Karacay, R. Sharkey and D. Goldenberg, Bioconjugate Chem., 2012, 23, 538 CrossRef PubMed; D. Shetty, S. Y. Choi, J. M. Jeong, J. Y. Lee, L. Hoigebazar, Y.-S. Lee, D. S. Lee, J. K. Chung, M. C. Lee and Y. K. Chung, Chem. Commun., 2011, 47, 9732 RSC.
- S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1460 RSC.
- W. Levason, M. L. Matthews, B. Patel, G. Reid and M. Webster, Dalton Trans., 2004, 3305 RSC.
- R. Hart, W. Levason, B. Patel and G. Reid, J. Chem. Soc., Dalton Trans., 2002, 3153 RSC.
- S. L. Benjamin, W. Levason, D. Pugh, G. Reid and W. Zhang, Dalton Trans., 2012, 41, 12548 RSC.
- M. F. Davis, W. Levason, G. Reid and M. Webster, Dalton Trans., 2008, 2261 RSC.
- S. El-Kurdi, A.-A. Al-Terkawi, B. M. Schmidt, A. Dimitrov and K. Seppelt, Chem.–Eur. J., 2010, 16, 505 CrossRef PubMed.
- The Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities, ed. S. Aldridge and A. J. Downs, Wiley, NY, 2011, ch. 1 Search PubMed.
- The Chemistry of Aluminium, Gallium, Indium and Thallium, ed. A. J. Downs, Blackie, London, UK, 1st edn, 1993 RSC; W. Levason and G. Reid, J. Chem. Soc., Dalton Trans., 2001, 2953 RSC; W. Levason, G. Reid and W. Zhang, Dalton Trans., 2011, 40, 8491 RSC.
- K. Wieghardt, P. Chaudhuri, B. Nuber and J. Weiss, Inorg. Chem., 1982, 21, 3086 CrossRef CAS.
- M. J. Belousoff, M. B. Duriska, B. Graham, S. R. Batten, B. Moubaraki, K. S. Murray and L. Spiccia, Inorg. Chem., 2006, 45, 3746 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXS-97, Program for crystal structure solution, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for crystal structure refinement, University of Göttingen, Germany, 1997 Search PubMed.
- S. Petricek, A. Demsar, P. Bukovec, L. Golic and J. V. Brencic, Acta Chim. Slov., 1997, 44, 317 CAS.
- F. N. Penkert, T. Weyhermüller and K. Wieghardt, Chem. Commun., 1998, 557 CAS.
- A. B. Ilyukhin and M. A. Malyarik, Zh. Neorg. Khim., 1999, 44, 1511 CAS; M. A. Malyarik, S. P. Petrsyants, A. B. Ilyukhin and Y. A. Busalaev, Zh. Neorg. Khim., 1991, 36, 2816 Search PubMed.
- P. Petrosyants and A. B. Ilyukhin, Zh. Neorg. Khim., 2010, 55, 33 Search PubMed.
- D. R. Ketchum, G. L. Schimek, W. T. Pennington and J. W. Kolis, Inorg. Chim. Acta, 1999, 294, 200 CrossRef CAS.
- J. Mason, Multinuclear NMR, Plenum, NY, 1987, ch. 5 Search PubMed.
- K. Wieghardt, M. Kleine-Boymann, B. Nuber and J. Weiss, Inorg. Chem., 1986, 25, 1654 CrossRef CAS.
- A. Dimitrov, D. Heidemann and E. Kemnitz, Inorg. Chem., 2006, 45, 10807 CrossRef CAS PubMed.
- A. Bodor, I. Tóth, I. Bányai, Z. Szabó and G. T. Hefter, Inorg. Chem., 2000, 39, 2530 CrossRef CAS.
- M. Tramšek and B. Žemva, J. Fluorine Chem., 2004, 125, 1919 CrossRef PubMed; M. Tranšek, P. Benkič, A. Turičnik, G. Tavčar and B. Žemva, J. Fluorine Chem., 2002, 114, 143 CrossRef; M. Tramšek and B. Žemva, J. Fluorine Chem., 2006, 127, 1275 CrossRef PubMed.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Preparative and spectroscopic details for the corresponding bromo complexes, HPLC traces and crystal structures of [GaCl3(Me3-tacn)], [InBr3(Me3-tacn)], [InCl3(BzMe2-tacn)], [{(Me3-tacn)Ga}2(μ-OH)3]Br3·3CH2Cl2 and [NMe4]3[Al2F9] and cif files for all of the crystal structures. CCDC 926503–926512. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc52104d |
| This journal is © The Royal Society of Chemistry 2014 |