Radical reactivity of the Fe(III)/(II) tetramesitylporphyrin couple: hydrogen atom transfer, oxyl radical dissociation, and catalytic disproportionation of a hydroxylamine†
Thomas R.
Porter
and
James M.
Mayer
*
Department of Chemistry, University of Washington, Seattle, WA 98195-1700, USA. E-mail: mayer@chem.washington.edu; Fax: +1-206-553-2083; Fax: +1-206-553-2083
First published on 16th October 2013
Abstract
The chemistry of low-valent iron porphyrin complexes with oxyl radical reagents has been explored. (meso-Tetramesityl porphyrinato) iron(III) hydroxide, (TMP)FeIII(OH) reacts with the hydroxylamine TEMPO–H (1-hydroxy-2,2,6,6-tetramethylpiperdine) to yield the ferrous porphyrin, (TMP)FeII, together with H2O and TEMPO. This reaction has a second order rate constant k1 = 76 ± 5 M−1 s−1 and likely occurs by concerted e−/H+ transfer. Hydrazines PhNHNHPh and PhNHNH2 similarly yield (TMP)FeII. A subsequent reaction between TEMPO (2,2,6,6-tetramethylpiperdinyl radical) and (TMP)FeII is observed to reversibly form the TEMPO-ligated ferric porphyrin, (TMP)FeIII(TEMPO). A combination of 1H NMR and optical spectroscopies were used to determine the thermodynamic and kinetic parameters for TEMPO binding: K4 (25 °C) = 535 ± 20 M−1, ΔH°4 = −7.0 ± 1.5 kcal mol−1, ΔS°4 = −11 ± 5 cal mol−1 K−1, ΔG‡4(235 K) = 21.3 ± 0.5 kcal mol−1, ΔG‡−4(235 K) = 16.9 ± 0.5 kcal mol−1. The Fe–O bond is remarkably weak. The stable phenoxyl radical 2,4,6-tBu3C6H2O˙ (ArO˙) forms a stronger bond to (TMP)FeII to irreversibly make a similar FeIII(OR) complex. Both (TMP)FeII and (TMP)FeIII(OH) are catalysts for the disproportionation of excess TEMPO–H to TEMPO and TEMP–H (2,2,6,6-tetramethylpiperdine). The lack of reactivity between (TMP)FeII and the alkylated TEMPO–H analogue, TEMPO–CH3, suggests that the disproportionation involves a hydrogen atom transfer step. These results highlight the importance and versatility of the heme FeIII/II couple that is often overshadowed by its higher-valent counterparts.
Introduction
Heme proteins are ubiquitous and have a wide range of functions. In recent years, cytochromes P450 and peroxidases have been a focus of research because of their ability to accomplish difficult oxidative reactions by using high-valent ferryl intermediates (compounds I and II).1 Other heme enzymes, however, accomplish inner-sphere oxidations and reductions using just the FeIII/II couple.2 One example is the autotrophic bacterial multi-heme enzyme hydroxylamine oxidoreductase (HAO),3 which catalyses the oxidation of hydroxylamine to nitrite using just ferric and ferrous oxidation states.4 These low-valent odd-electron transformations tend to be underappreciated relative to their high-valent counterparts.In many cases, the interconversion between heme oxidation states is coupled to proton transfer. Examples range from proton-pumping in cytochrome c oxidase5 to aliphatic hydroxylations by cytochromes P450.1 Reactions involving the transfer of e− and H+ are broadly referred to as proton-coupled electron transfer (PCET) processes, and are ubiquitous in chemistry as well as in biochemistry.6 The hydroxylamine-to-nitrite process mentioned above is one example. The transfer of one proton and one electron in a single kinetic step is hydrogen atom transfer (HAT), or more generally concerted proton–electron transfer (CPET).7 Some enzymes and other catalysts utilize CPET to avoid high energy intermediates, but a variety of pathways are possible for PCET reactions.8 While PCET and CPET are frequently invoked pathways for high-valent heme reactions, low-valent heme reactions of this type have received substantially less attention. The non-heme iron lipoxygenase enzymes use the FeIII/II redox couple to remove H˙ from fatty acid substrates.9
Herein we describe inner sphere redox chemistry of the ferric hydroxide and ferrous states of meso-tetramesitylporphyrinato ligated model complexes. Namely, we address three related topics: PCET reactivity, oxyl radical binding to FeII, and catalytic disproportionation of the hydroxylamine TEMPO–H. Each is presented and discussed in turn below.
meso-Tetramesitylporphyrinato-iron(III) hydroxide, (TMP)FeIII(OH), and related porphyrin complexes have been extensively used as models for heme enzymes by Groves,10 Balch,11 LaMar,11b,c Valentine,12 Nam13 and others. An advantage of this porphyrin ligand is that the steric bulk of the mesityl substituents prevents the formation of μ-oxo dimers, so the ferric hydroxide is readily prepared.11b We first observed that (TMP)FeIII(OH) is readily reduced by the hydroxylamine TEMPO–H to give the ferrous derivative (TMP)FeII, when the reactions are performed and monitored at low concentrations. However, the same reaction gives a different product when performed at higher concentrations, because the TEMPO product reversibly binds to the ferrous heme to form (TMP)FeIII(TEMPO). Reactions with excess TEMPO–H gave yet another set of products, as the iron complexes catalyse the redox disproportionation of TEMPO–H. These results highlight the rich and perhaps underappreciated free-radical chemistry of low-valent heme compounds.
Experimental
Materials
Unless otherwise noted, all chemicals were purchased from Sigma-Aldrich and used without purification. Toluene-d8 and dichloromethane-d2 were purchased from Cambridge Isotope Labs, dried over NaK and CaH2, respectively, and vacuum distilled. Other solvents were purchased from Fischer and dried using a “Grubbs type” Seca Solvent System installed by GlassContour. TEMPO was purified by sublimation. TEMPO–H,14 TEMPO–CH3,15tBu3ArO˙,16 TMPH2,17 (TMP)FeIIICl,18 and P1-phosphazeneH+BF4−,19 were prepared using established literature protocols. All glassware was dried in an oven at 150 °C overnight and pumped into a nitrogen filled glovebox while hot. Celite was dried at 100 °C overnight under vacuum.Instrumentation
All 1H NMR spectra were obtained on Bruker 300 and 500 MHz instruments. Variable temperature spectra were acquired on a Bruker 500 MHz instrument. The chemical shifts reported are referenced to TMS using the residual solvent peak. UV-visible absorption spectra were collected with a Hewlett-Packard 8453 diode array spectrometer equipped with a Unisoku USP-203 cryostat. Kinetic measurements were performed on an OLIS RSM-1000 stopped-flow spectrophotometer.Syntheses
Representative reaction procedure
In a typical NMR-scale reaction, equimolar solutions of (TMP)FeII and TEMPO were prepared in toluene-d8 in the glovebox (4.0 mM). Solutions were added to a J. Young NMR tube and the tube sealed. In a typical optical absorption spectroscopy experiment, equimolar solutions of (TMP)FeII and TEMPO were prepared in toluene in a nitrogen filled glovebox (80 μM). Solutions were added to a 1 cm pathlength quartz cuvette fitted with a Kontes valve and sealed. For both optical and NMR experiments, the sample was inverted several times to ensure that complete mixing had occurred prior to obtaining spectra. 1H NMR data for (TMP)FeIII(TEMPO) and (TMP)FeIII(tBu3ArO) are given in Table 1.| Compound | 1H NMR chemical shifts | Q 1 absorbance | |||
|---|---|---|---|---|---|
| m-H | o-CH3 | p-CH3 | β-pyr | λ max (ε)a | |
| a Values at 25 °C unless otherwise noted. NMR spectra in toluene-d8, with chemical shifts in ppm; n.o. = not observed. Optical spectra: λmax in nm, ε in cm−1 M−1 × 10−3. b At −80 °C. | |||||
| (TMP)FeII | 11.72 | 6.62 | 6.04 | 2.86 | 538 (11.6) |
| (TMP)FeII at −80° C (ref. 10c) | 14.70 | 7.79 | 8.39 | −1.44 | — |
| (TMP)FeIII(TEMPO) | 11.06 | n.o. | 3.26 | 81.0 | — |
| (TMP)FeIII(TEMPO) at −80 °C | 14.24, 13.19 | n.o. | 3.96 | 124 | 565 (8.7)b |
| (TMP)FeIII(tBu3ArO) | 13.76, 12.67 | 5.35 | 3.30 | 89.5 | — |
| (TMP)FeIII(OH) | 12.14, 11.18 | n.o. | 3.29 | 81.5 | 580 (7.5)21 |
Results
I. (TMP)FeIII(OH) as a net hydrogen atom abstractor
The addition of one equivalent of TEMPO–H to a dark green solution of (TMP)FeIII(OH) (80 μM in toluene) forms a bright red solution over approximately 45 minutes. The UV-visible absorption spectrum of the reaction product displayed a Soret band at 418 nm and Q bands at 445 and 538 nm, in good agreement with the spectrum of the known11c 4-coordinate ferrous porphyrin (TMP)FeII and with an independently prepared sample (Table 1).20 These data indicate a net hydrogen atom transfer from TEMPO–H to (TMP)FeIII(OH), as shown in eqn (1).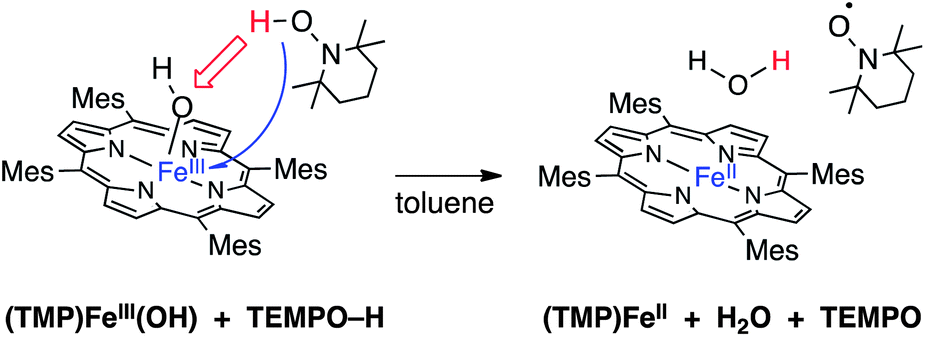 | (1) |
Reaction (1) was monitored by stopped-flow spectrophotometry under pseudo-first order conditions ([TEMPO–H] = 3.6–14.4 mM, Fig. 1 and ESI†). The kinetics are first order in both (TMP)FeIII(OH) and TEMPO–H, with k1 = 76 ± 5 M−1 s−1 at 25 °C (ΔG‡1(25 °C) = 15.0 ± 0.1 kcal mol−1).
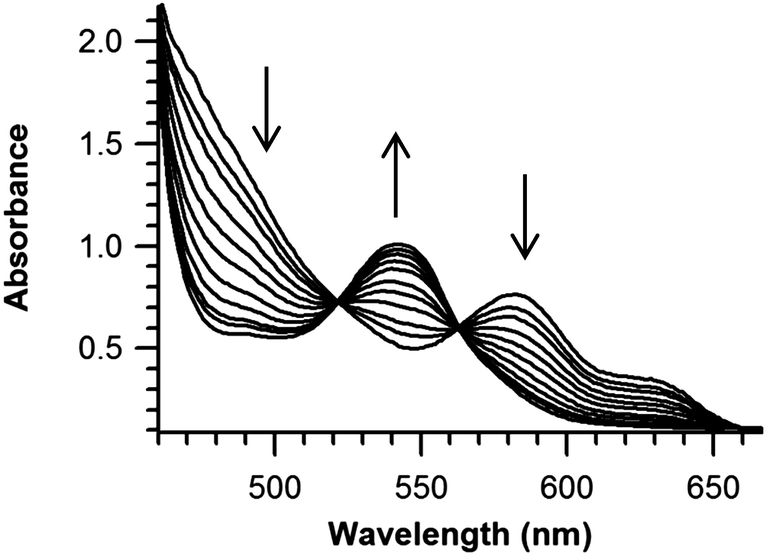 | ||
| Fig. 1 Optical spectral changes for the reaction of (TMP)FeIII(OH) (80 μM) with TEMPO–H (4.0 mM). | ||
The reactivity of (TMP)FeIII(OH) with other potential hydrogen atom donors has been examined in toluene-d8 at 4.0 mM concentrations. TEMPO–H is a strong H-atom donor because its O–H bond dissociation free energy (BDFE) is only 65.2 kcal mol−1.8 The first N–H bond in 1,2-diphenylhydrazine is similar, BDFE = 64.3 ± 1.5 kcal mol−1.8 Adding 0.5 equivalents (1 H˙ equivalent) PhNHNHPh to (TMP)FeIII(OH) resulted in a colour change from dark green to deep red and 1H NMR spectroscopy showed that (TMP)FeII and PhN=NPh had been generated (eqn (2)). The same reaction was carried out at a lower concentrations (80 μM (TMP)FeIII(OH), 40 μM PhNHNHPh) in toluene and monitored optically. Over approximately 8 hours, the characteristic absorption features of (TMP)FeIII(OH) were replaced with those of (TMP)FeII. The weaker H-atom donor PhNHNH2 (BDFE = 67.5 ± 1.5 kcal mol−1)8 also reacted with (TMP)FeIII(OH) but only slowly, requiring approximately 24 hours at RT for 4.0 mM (TMP)FeIII(OH) plus 2.0 mM PhNHNH2 (ref. 22) (eqn (3), and ESI†). 2,4,6-Tri-tert-butyl phenol, with a significantly stronger O–H bond (BDFE = 76.6 kcal mol−1)8 did not react with (TMP)FeIII(OH) over 24 hours.
| 2(TMP)FeIII(OH) + PhNHNHPh → 2 (TMP)FeII + PhN=NPh + 2H2O | (2) |
| 2 (TMP)FeIII(OH) + PhNHNH2 → 2 (TMP)FeII + [PhN=NH] + 2H2O | (3) |
Surprisingly, the reaction between (TMP)FeIII(OH) and TEMPO–H at higher concentrations, 4.0 mM of each reagent, proceeds to a different set of products. Under these conditions, reaction solutions rapidly turn dark brown, rather than the red solution described above for 80 μM solutions. 1H NMR spectra of this reaction mixture showed the complete consumption of TEMPO–H but some (TMP)FeIII(OH) remained. Peaks were also observed for new organic and FeIII products, which were later assigned to 2,2,6,6-tetramethyl piperidine, TEMP–H, and a (TMP)FeIII–TEMPO complex, as described in the next sections.
II. Reactions of (TMP)FeII with oxyl radicals
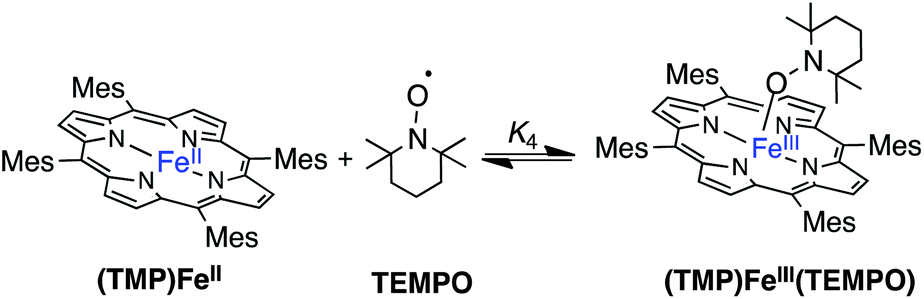 | (4) |
1H NMR spectra of similar reactions with 0.5 or 2 equivalents of TEMPO were surprising because in both cases no (TMP)FeII was identifiable in solution and each reaction appeared to have cleanly generated a different D4h–symmetric [(TMP)FeIII]+ complex. These results suggested an equilibrium reaction in which (TMP)FeII and TEMPO are in rapid equilibrium with a C4v symmetric adduct, (TMP)FeIII(TEMPO) (eqn (4)). In the rapid exchange limit, the observed chemical shifts are the weighted average of all the species present, so different shifts are observed with different reagent ratios.23 This interpretation was confirmed by 1H NMR spectra of (TMP)FeII plus sub-stoichiometric TEMPO at −80 °C, which displayed distinguishable resonances for (TMP)FeII and the C4v-symmetric (TMP)FeIII(TEMPO). Thus at ambient temperatures, TEMPO must be rapidly dissociating from (TMP)FeIII(TEMPO) and re-binding to (TMP)FeII, a process that would give the apparent D4h symmetry observed. The coalescence temperature of ∼235 K and the chemical shift difference of ∼500 Hz, provide estimates of the dissociation rate constant of k−4(235 K) = 1100 ± 200 s−1 and a barrier ΔG‡−4(235 K) = 16.9 ± 0.5 kcal mol−1 (see ESI†).
An oximato-iron(III) adduct, R=N–O–FeIII(TMP), closely related to (TMP)FeIII(TEMPO), has been similarly prepared from the fluorenone oxime and (TMP)FeIIIOH (eqn (5)).10j This product is stable, and was structurally characterized. In addition, slow dissociation of the fluorenyl oxyimyl radical was indicated and radical formation was facilitated by the addition of CO.
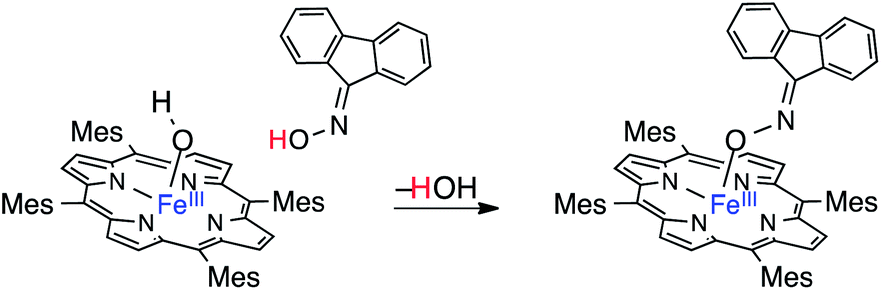 | (5) |
To study the TEMPO dissociation equilibrium (eqn (4)), multiple 4.0 mM samples of (TMP)FeII in toluene-d8 were prepared with various concentration of TEMPO. Their 1H NMR spectra at 25 °C showed peaks with positions that changed as a function of TEMPO concentration (Fig. 2). Plotting the porphyrin chemical shifts vs. [TEMPO] showed smooth curves that were extrapolated to high [TEMPO] to give the chemical shifts of pure (TMP)FeIII(TEMPO) (Fig. S4†). Reasonable estimates of these chemical shifts could be made for m-H, p-CH3, and β-pyrrolic peaks (Table 1) but was not possible for the o-CH3 peak because significant broadening of this signal was observed even at low concentrations of TEMPO. Taking the observed chemical shifts of each sample as the weighted average of the shifts of (TMP)FeIII(TEMPO) and (TMP)FeII in solution and assuming mass balance, a plot of the [(TMP)FeIII(TEMPO)]/[(TMP)FeII] ratio vs. the concentration of free TEMPO in solution gives the equilibrium constant. K4 was calculated using the β-pyrrolic, m-aryl, and p-CH3 resonances to give a value of 520 ± 25 M−1 (see ESI†).
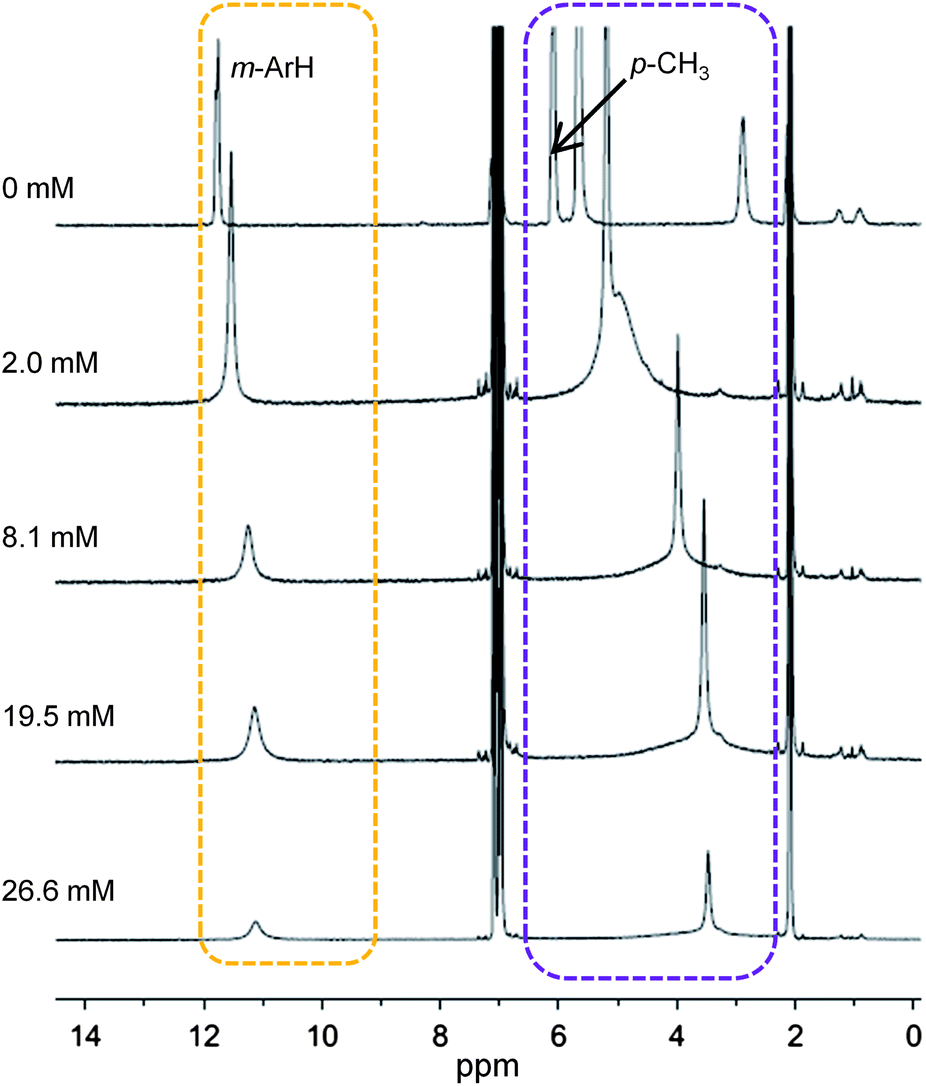 | ||
| Fig. 2 1H NMR spectra of 4.0 mM (TMP)FeII with 0 to 26.6 mM of TEMPO in toluene-d8 on a 500 MHz spectrometer at 25 °C. The dotted box at left shows the shift of the meta-aryl protons, and the box at the right shows the shift of the p-CH3 groups, indicating the equilibrium formation of (TMP)FeIII(TEMPO). The broad, downfield β-pyrrolic peaks are not shown. | ||
The TEMPO binding equilibrium was also studied by optical spectroscopy, using 80 μM (TMP)FeII and one equivalent of TEMPO in toluene, at temperatures from 25 °C to −90 °C. As the temperature decreased, the Q band associated with (TMP)FeII at 538 nm began to disappear and a new Q band assigned to (TMP)FeIII(TEMPO) at 565 nm grew in. Equilibrium constants were obtained at each temperature from the absorbance at three different wavelengths (487, 538, and 565 nm), assuming mass balance for the iron species and that the extinction coefficients in Table 1 are independent of temperature (and ignoring the very small absorbance due to free TEMPO). The value obtained at 25 °C, K4(25 °C)optical = 550 ± 25 M−1, is in good agreement with that obtained by NMR measurements, and together those give a consensus value of 535 ± 20 M−1. This K4 is consistent with the differences observed in the above experiments, that the TEMPO adduct is mostly formed at NMR concentrations but mostly dissociated at the lower concentrations used for optical studies.
A van't Hoff analysis of TEMPO binding gives ΔH°4 = −7.0 ± 1.5 kcal mol−1 and ΔS°4 = −11 ± 5 cal K−1 mol−1 (Fig. 3 and ESI†). The Fe–O bond in (TMP)FeIII(TEMPO) is remarkably weak, with a bond dissociation enthalpy (BDE) of only 7 kcal mol−1 (a BDFE of only 4 kcal mol−1). Combining K4(235 K) = 1.3 × 104 M−1 with the dissociation rate constant (k−4(235 K)) gives the rate constant for TEMPO addition k4(235 K) = K4k−4 = (1.4 ± 0.5) × 107 M−1 s−1 and ΔG‡4 (235 K) = 21.3 ± 0.5 kcal mol−1.
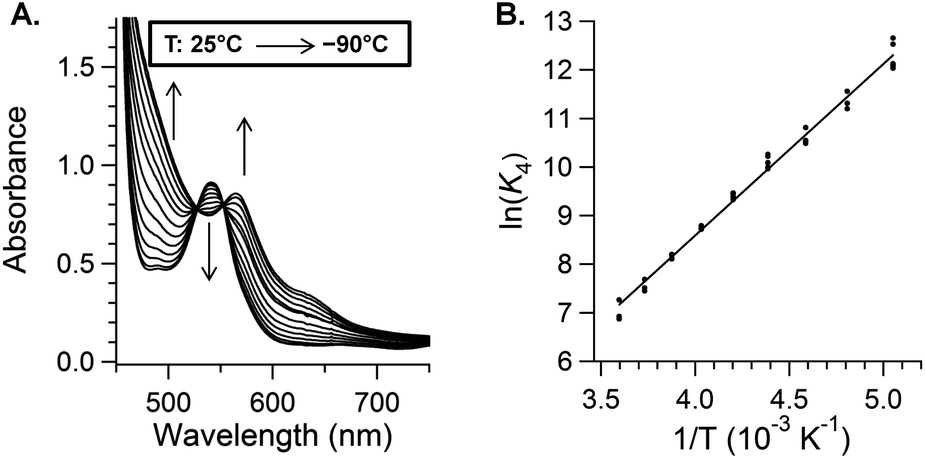 | ||
| Fig. 3 (A) Optical spectra of (TMP)FeII (80 μM) and one equivalent of TEMPO in toluene at temperatures ranging from 25 °C to −90 °C. (B) van't Hoff plot of ln(K4) vs. 1/T. | ||
1H NMR spectra of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of (TMP)FeII plus tBu3ArO˙ in toluene-d8 have a broad downfield β-pyrrolic resonance at 89 ppm (Table 1), indicating the oxidation of iron(II) to iron(III).11a Interestingly, two resonances are observed for the m-aryl protons, as is typical for (TMP)FeIIIX species with C4v symmetry. These data indicate that tBu3ArO˙ is bound to the iron centre: (TMP)FeIII(tBu3ArO). The pair of m-aryl resonances shows that dissociation of the aryloxyl radical is slow on the NMR timescale, so that the two sides of the porphyrin ring are inequivalent. This slow exchange is also indicated by 1H NMR spectra of solutions with (TMP)FeII with 0.5 equivalents of tBu3ArO˙, which show separated signals for (TMP)FeIII(tBu3ArO) and (TMP)FeII at 25 °C.
:1 mixtures of (TMP)FeII plus tBu3ArO˙ in toluene-d8 have a broad downfield β-pyrrolic resonance at 89 ppm (Table 1), indicating the oxidation of iron(II) to iron(III).11a Interestingly, two resonances are observed for the m-aryl protons, as is typical for (TMP)FeIIIX species with C4v symmetry. These data indicate that tBu3ArO˙ is bound to the iron centre: (TMP)FeIII(tBu3ArO). The pair of m-aryl resonances shows that dissociation of the aryloxyl radical is slow on the NMR timescale, so that the two sides of the porphyrin ring are inequivalent. This slow exchange is also indicated by 1H NMR spectra of solutions with (TMP)FeII with 0.5 equivalents of tBu3ArO˙, which show separated signals for (TMP)FeIII(tBu3ArO) and (TMP)FeII at 25 °C.
(TMP)FeIII(tBu3ArO) reacts with excess distilled, degassed water to cleanly form (TMP)FeIII(OH) and tBu3ArOH, by 1H NMR spectroscopy (eqn (6)). This reaction is consistent with the inability of (TMP)FeIII(OH) to abstract a hydrogen atom from tBu3ArOH that was mentioned above. This reaction is also essentially the reverse of reactions (1 + 4) and of reaction (5).
| (TMP)FeIII(tBu3ArO) + H2O → (TMP)FeIII(OH) + tBu3ArOH | (6) |
III. The catalytic disproportionation of TEMPO–H by (TMP)FeII
In a reaction originally formulated as a control experiment, one equivalent of TEMPO–H was added to 4.0 mM toluene-d8 solution of (TMP)FeII. No reaction was expected as both of these reagents typically act as reductants. To our surprise, the solution slowly turned from deep red to green. After 24 hours, the 1H NMR spectrum indicated the presence of (TMP)FeIII(OH), a (TMP)FeII/(TMP)FeIII(TEMPO) equilibrium mixture, and a set of resonances for 2,2,6,6-tetramethylpiperidine (TEMP–H), in a 1:1:1 ratio (eqn (7), Fig. S9†). The piperidine product was identified by GC/MS and by comparison of its 1H NMR signals to an authentic sample. The chemical shifts for (TMP)FeII + TEMPO/(TMP)FeIII(TEMPO) are sensitive to the position of the equilibrium, so these shifts indicate the concentration of TEMPO. The concentration of TEMPO was determined to be half of the initial concentration of TEMPO–H, indicating the overall product stoichiometry of 1:1:1:1 as indicated in eqn (7). When 2 equivalents of TEMPO–H were used, (TMP)FeII was completely converted to (TMP)FeIII(OH), one equivalent of TEMP–H and presumably one equivalent of NMR silent TEMPO (eqn (8)).| (TMP)FeII + TEMPO–H → ½ (TMP)FeIII(OH) + ½ (TMP)FeII + ½ TEMP–H + ½ TEMPO | (7) |
| (TMP)FeII + 2TEMPO–H → (TMP)FeIII(OH) + TEMP–H + TEMPO | (8) |
Since the (TMP)FeIII(OH) product could be reduced by additional TEMPO–H back to (TMP)FeII (eqn (1) above), reactions (8) and (1) indicate that the disproportionation of TEMPO–H should be catalytic in iron, using either (TMP)FeII or (TMP)FeIII(OH). To test this, 12 equivalents of TEMPO–H were added to a toluene-d8 solution of (TMP)FeIII(OH). After 24 hours, 1H NMR spectra showed the presence of 4 equivalents of TEMP–H and a broad upfield water resonance, and the (TMP)FeIII(OH) was unchanged, consistent with the expected stoichiometry (eqn (9)).
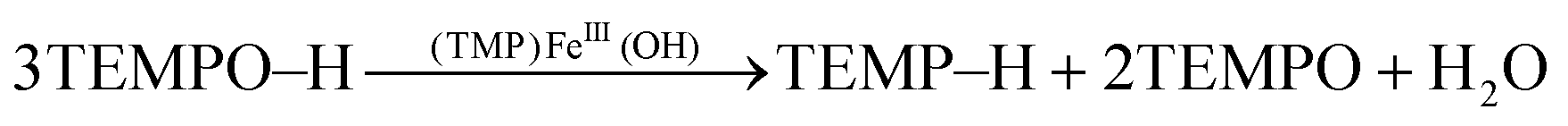 | (9) |
Over 100 turnovers of TEMPO–H disproportionation was observed in benzene-d6 over 9 hours with 0.08 mM (TMP)FeII loading and 33 mM TEMPO–H. The disappearance of TEMPO–H resonances matches the growth of TEMP–H resonances in a 3:1 ratio with nearly quantitative yield (∼96% mass balance), determined by 1H NMR with a hexamethylbenzene internal standard (Fig. 4A). The reaction is substantially inhibited by the addition of excess TEMPO (Fig. 4B), presumably due to the equilibrium reaction of TEMPO with (TMP)FeII previously described (eqn (4)).
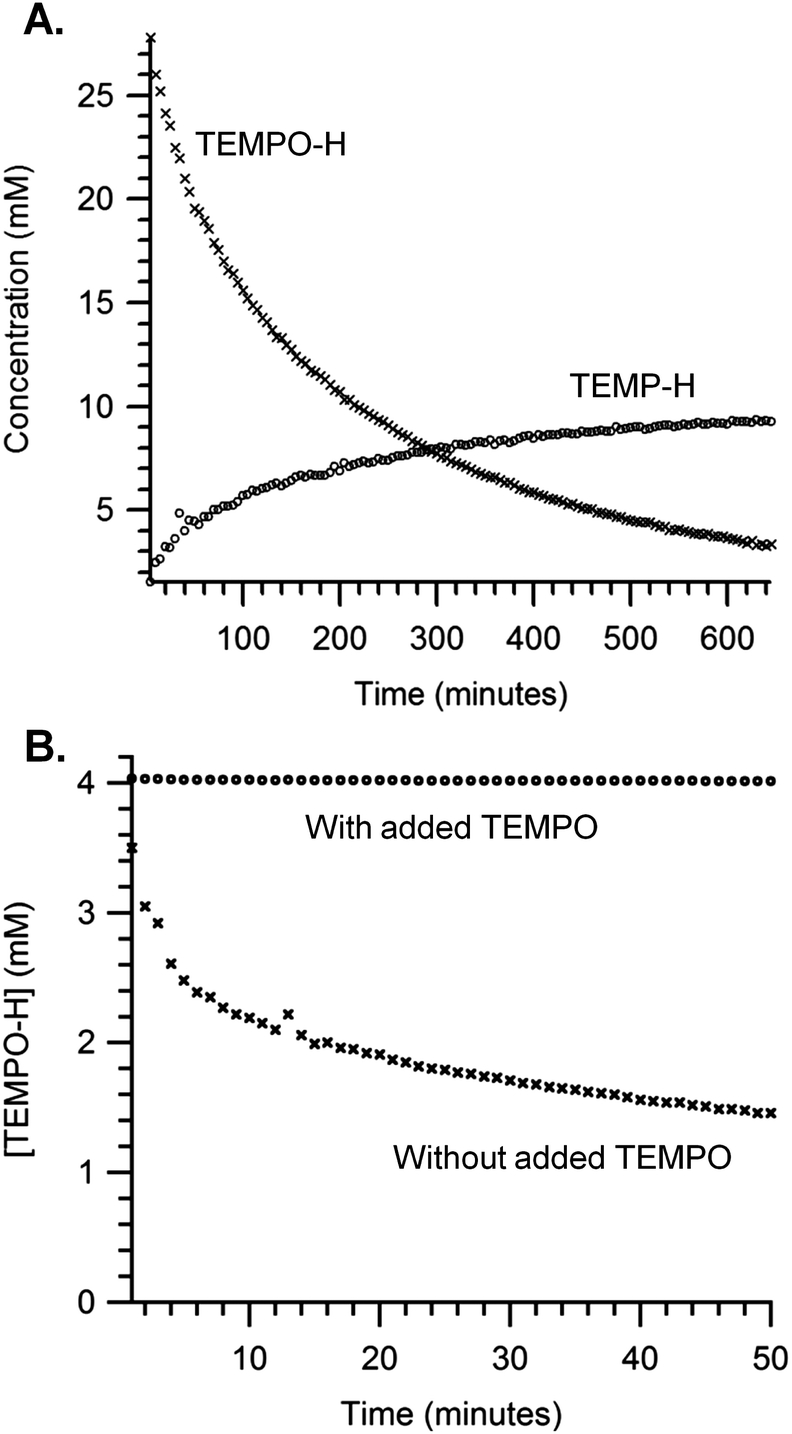 | ||
| Fig. 4 Kinetic traces for the disproportionation of TEMPO–H catalysed by (TMP)FeII in toluene-d8. Concentrations were determined by 1H NMR using a hexamethylbenzene internal standard. (A) The disappearance of TEMPO–H (×) (initial concentration = 33 mM) and the appearance of TEMP–H (○) vs. time in the presence of 0.08 mM (TMP)FeII. (B) The disappearance of TEMPO–H (initial concentration = 4 mM) vs. time with 0.04 mM (TMP)FeII in the absence (×) or presence (●) of 20 mM TEMPO added at t = 0. | ||
To probe the mechanism of the catalytic disproportionation, the reactivity of the methoxy-amine TEMPO-CH3 was explored. No reaction was observed between TEMPO–CH3 and (TMP)FeII or (TMP)FeIII(OH), even after heating at 75 °C for 48 h. Since TEMPO–H reduces itself in reaction (8), we also tested whether TEMPO-CH3 could be reduced by TEMPO–H, PhNHNHPh, or tBu3ArOH in the presence of (TMP)FeII. However, no products derived from TEMPO-CH3 were observed.
Discussion
I. Net H-atom transfer to (TMP)FeIII(OH); the “effective BDFE” of (TMP)FeII + H2O
(TMP)FeIII(OH) reacts with TEMPO–H, PhNHNHPh and PhNHNH2 by net hydrogen atom transfer to give (TMP)FeII, water and the oxidized donor (TEMPO, PhN=NPh or [PhN=NH]). The TEMPO–H reaction (eqn (1), repeated here) is quantitative and therefore provides thermochemical information (independent of the mechanism of the reaction, which is discussed below).| (TMP)FeIII(OH) + TEMPO–H → (TMP)FeII + TEMPO + H2O | (1) |
Reaction (1) shows that the effective O–H BDFE of the (TMP)FeII + H2O combination should be comparable to or greater than the O–H BDFE of TEMPO–H (65.2 kcal mol−1).7 The effective BDFE is defined in 10; the term effective is used because eqn (10) is not a simple bond homolysis. It encompasses both the (unfavourable) binding of H2O to (TMP)FeII and the cleavage of the (TMP)FeII(HO–H) bond. Because the reduced product, (TMP)FeII + H2O, is two molecules, the thermochemical analysis of eqn (1) is not as simple as a typical H-atom transfer reaction X + HY → XH + Y, for which Keq has no units. The experimental execution of eqn (1) was done at 80 μM concentrations, and these low concentrations will entropically favour the products (3 species vs. 2), as compared with 1 M standard state. This provides an extra driving force under the reaction conditions, so the effective BDFE of (TMP)FeII + H2O could be slightly lower than the BDFE of TEMPO–H and reaction would still proceed to completion. We conclude that the BDFE of [(TMP)FeII + H2O] ≥ 64 kcal mol−1.
To complement this lower limit, a firm upper limit is provided by the downhill reaction of (TMP)FeIII(tBu3ArO) with H2O to form (TMP)FeIII(OH) + tBu3ArOH (eqn (6)). Eqn 10–12 sum to eqn (6), as shown below. Reaction (12), the dissociation of aryloxyl from FeII, was shown above to be very unfavourable. Therefore, the effective BDFE of (TMP)FeII + H2O must be substantially less than the BDFE of 2,4,6-tri-tert-butyl phenol, BDFE = 76.6 kcal mol−1.8
| (TMP)FeII + H2O → (TMP)FeIII(OH) + H˙, ΔG°9 = effective BDFE | (10) |
| H˙ + tBu3ArO˙ → tBu3ArOH, ΔG°10 = −BDFE(tBu3ArOH) | (11) |
| (TMP)FeIII(tBu3ArO) → (TMP)FeII + tBu3ArO˙, ΔG°11 ≫ 0 | (12) |
| (TMP)FeIII(tBu3ArO) + H2O → (TMP)FeIII(OH) + tBu3ArOH, ΔG°6 = ΔG°9 + ΔG°10 + ΔG°11 | (13) |
The conclusion that 64 ≤ BDFE[(TMP)FeII + H2O] ≪ 76.6 is consistent with the reactions of (TMP)FeIII(OH) with hydrazines. 1,2-Diphenylhydrazine, in which the first N–H bond (BDFE = 64.3 ± 1.5 kcal mol−1) is likely even weaker than that in TEMPO–H and phenylhydrazine, in which the first N–H bond is slightly stronger (BDFE = 67.5 ± 1.5 kcal mol−1).8 There are clearly kinetic as well as thermodynamic effects in play, because PhNHNHPh reacts more slowly than TEMPO–H even though its XH bond is likely weaker. These data suggest an effective BDFE of 67 ± 3 kcal mol−1.
The mechanism of H+ and e− transfer from TEMPO–H to (TMP)FeIII(OH) could occur by (i) concerted proton–electron transfer (called CPET or H-atom transfer), (ii) initial proton transfer then electron transfer (PT/ET), (iii) initial electron transfer then proton transfer (ET/PT), or (iv) an initial protolytic ligand-exchange mechanism (see below).
The stepwise ET/PT and PT/ET paths are unlikely based on thermochemical arguments. TEMPO–H is a poor one-electron reductant, E = 0.71 V in MeCN,8 while (TMP)FeIII(OH) is reported to be quite difficult to reduce, E1/2 = −1.23 V in dichloromethane (all potentials vs. Cp2Fe+/0).21 While the differences in solvent preclude a completely quantitative comparison, the rough estimate is that ET is nearly 2 V or 45 kcal mol−1 endoergic. This value is substantially higher than the observed kinetic barrier for reaction (1), ΔG‡ = 15.0 ± 0.1 kcal mol−1, even considering the uncertainties involved. Thus it is unlikely the reaction of (TMP)FeIII(OH) with TEMPO–H proceeds through an ET/PT mechanism. In a similar vein, initial PT is unlikely because TEMPO–H is a very weak acid (pKa in MeCN of 41 (ref. 8)), and (TMP)FeIII(OH) is not very basic. (TMP)FeIII(OH) is not protonated in CD2Cl2 by 20 equivalents of the weak acid tert-butylimino-tris(pyrolidino)phosphorane hydrofluoroborate, P1-phosphazene H+BF4−, which has a pKa in MeCN of 28.42 (ref. 24) (see ESI†). The ET/PT and PT/ET mechanisms are particularly unlikely in toluene solution because the formation of charged intermediates is even more difficult in this low-polarity solvent. The results are thus consistent with a mechanism of H-atom transfer (concerted H+ and e− transfer), which is very common for TEMPO–H.
However, a protolytic ligand-exchange mechanism is also possible. This path would involve TEMPO–H substituting for the OH ligand on (TMP)FeIII(OH) with proton transfer to form water and (TMP)FeIII(TEMPO), which subsequently dissociates TEMPO. Such a protolytic pathway is likely followed by the closely related fluorene oxime reaction10j shown in eqn (5) above. The displacement of ArOH from (TMP)FeIII(OAr) by water (eqn (13)) has to be hydrolytic because the HAT pathway is precluded by the high water O–H BDFE. This mechanism has been studied for exchange of phenolate ligands in (TMP)Fe(OAr) with carboxylic acids and alcohols.25 The alcohol exchange reactions are quite slow, with rate constants ∼10−2 M−1 s−1 at 60 °C, and are slower for less nucleophilic and more crowded alcohols.
Based on these precedents, we tentatively favour an HAT mechanism for the TEMPO–H reaction rather than a hydrolytic one. TEMPO–H is a crowded molecule, yet it reacts with (TMP)Fe(OH) many orders of magnitude faster than alcohols react with (TMP)Fe(OPh). TEMPO–H has a very low acidity (pKa 31 (ref. 8a) in DMSO, vs. 16.2 for fluorenone oxime26) making it less likely to participate in protolytic reactions. Finally, the qualitative rate comparison observed here, k(TEMPOH) > k(PhNHNHPh) ≫ k(PhNHNH2), is more consistent with a hydrogen atom transfer mechanism based on the X–H bond strengths, but clearly do not parallel the pKa values: TEMPO–H, 31 > PhNHNH2, 28.8 > PhNHNHPh, 26.2 (in DMSO8a). The very weak O–H bond in TEMPO–H (BDE ∼70 kcal mol−1) biases its reactivity toward HAT as opposed to the fluorene oxime (BDEs ∼70 vs. ∼82 kcal mol−1 (ref. 27)). These mechanistic arguments are, however, suggestive rather than definitive.
II. Binding of stable oxyl radicals to (TMP)FeII
The binding of the oxyl radicals TEMPO (2,2,6,6-tetramethylpiperdine-N-oxyl radical) and tBu3ArO˙ (2,4,6-tri-tert-butyl phenoxyl radical) to (TMP)FeII is reminiscent of the much-studied reversible binding of O2 to ferrous hemes.28 The product (TMP)FeIII(OR) complexes are well described as high-spin FeIII species based on their 1H NMR spectra.11a The binding of TEMPO is reversible, with K4 = 535 ± 20 M−1 from both NMR and optical studies. A van't Hoff analysis gives the TEMPO-iron bond dissociation enthalpy as only –ΔH°4 = 7.0 ± 1.5 kcal mol−1. This is a very weak bond (the Fe–O homolytic bond dissociation free energy (BDFE) is 3.7 ± 0.1 kcal mol−1). To our knowledge, these are the first measurements of such Fe–O homolytic bond energies, other than for the binding of O2.The (1.4 ± 0.5) × 107 M−1 s−1 and 1100 s−1 rate constants for TEMPO association and dissociation are near the middle of the ranges of related values for O2 binding to natural and synthetic hemes. These vary from 104 to almost 109 M−1 s−1 for kon, and from 2 to 105 s−1 for koff.28
Related redox coordination chemistry of TEMPO with transition metal complexes has received significant attention, for instance in the area of living radical polymerizations.29 Waymouth and co-workers have shown that TEMPO reversibly binds Ti(III) sandwich complexes in an η1 fashion through the TEMPO oxygen.30 With Cp2TiIVCl(TEMPO), for instance, Ti–O bond homolysis occurs at 60 °C in the presence of CCl4 to afford Cp2TiIVCl2, TEMPO, and ˙CCl3. The Ti–O BDE of Cp2TiIVCl(TEMPO) was determined to be approximately 27 kcal mol−1.30c
The reaction between (TMP)FeII and TEMPO bears a particularly close resemblance to the reactivity of (TMP)RhII with TEMPO reported by de Bruin and co-workers.31 The Rh complex makes a somewhat stronger bond, ΔH°Rh-TEMPO = 14.7 ± 1.4 kcal mol−1. The higher bond energy for RhIII–TEMPO is reasonable considering that second row transition metals typically form stronger bonds than first row metals, and that RhII is generally a less favourable oxidation state than FeII.
The entropy for the association of TEMPO to (TMP)RhII was reported as ΔSo = −30.6 ± 4.6 cal mol−1 K−1, much more negative than the analogous value reported here for (TMP)FeIII(TEMPO), –11 ± 5 cal mol−1 K−1. Only a small part of this difference is due to the entropy of the spin multiplicity change. The entropy of the electron spin states is given by S° = Rln(2S + 1), where R is the ideal gas constant and S is the spin multiplicity of the system. So in the binding of (TMP)RhII and TEMPO, both S = 1/2, to give the diamagnetic, S = 0 (TMP)RhIII(TEMPO) complex, spin accounts for a positive entropy change of ∼3 cal mol−1 K−1. The reaction of intermediate spin S = 1 (TMP)FeII (ref. 11c) with S = 1/2 TEMPO results in the formation of a high-spin S = 5/2 (TMP)FeIII(TEMPO) complex has an associated entropy change of only ∼ −1 cal mol−1 K−1. Thus the differences in the spin entropy of the two systems accounts for only ∼4 cal mol−1 K−1 of the 20 cal mol−1 K−1 measured difference, suggesting that there must be significant difference in the structural and vibrational change upon TEMPO coordination between the rhodium and iron compounds.
The tBu3ArO˙ radical binds significantly more strongly to (TMP)FeII than TEMPO. From one perspective this is surprising because of the high degree of steric bulk on both the iron porphyrin as well as the phenoxide. tBu3ArO˙ would seem to be slightly more bulky than TEMPO, having t-butyl groups bound to the carbons β to the oxyl centre while TEMPO has two methyl groups in those positions. The stronger bond in (TMP)FeIII(tBu3ArO) is likely due primarily to tBu3ArO˙ being a less stabilized radical than TEMPO, as evidenced by the O–H bond being 11.5 kcal mol−1 stronger in tBu3ArO–H vs. TEMPO–H. If the tBu3ArO and TEMPO ligands were rigidly held between the mesityl substituents, the (TMP)FeIII(OR) compounds would have C2v symmetry with inequivalent β-pyrrolic signals, but these resonances are broad and cannot be resolved.
III. Catalytic disproportionation of TEMPO–H
(TMP)FeIII(OH) catalyses the disproportionation of TEMPO–H to TEMPO, TEMP–H and water (eqn (9)). Over 100 turnovers have been observed with no sign of catalyst degradation. Catalysis is fast initially, but slows over time. The chemistry reported above suggests that the catalytic cycle likely proceeds in a ping-pong fashion, as depicted in Scheme 1. The substrate oxidation part presumably proceeds by PCET from TEMPO–H to (TMP)FeIII(OH). Then the slowing of the catalysis is due to the reversible binding of the TEMPO product to (TMP)FeII, removing it from the catalytic cycle.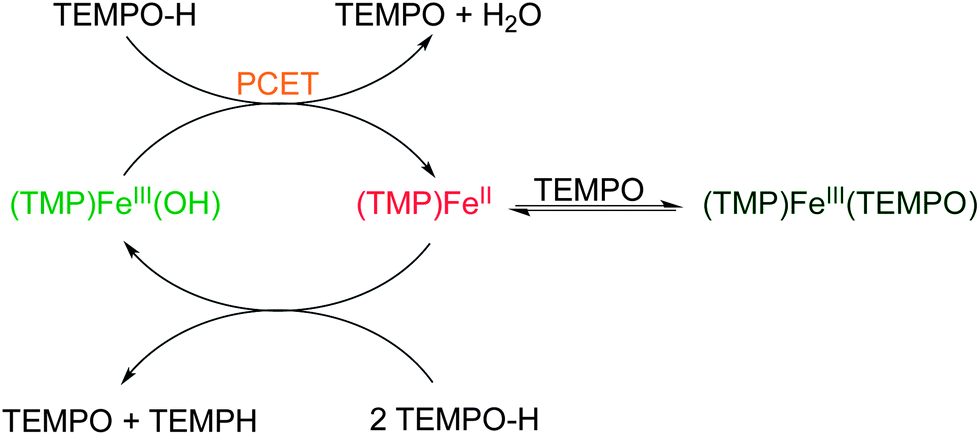 | ||
| Scheme 1 Proposed reaction cycle for the catalytic disproportionation of TEMPO–H by (TMP)FeIII(OH). | ||
The substrate reduction side, (TMP)FeII + 2 TEMPO–H → (TMP)FeIII(OH) + TEMPO + TEMP–H, has also been observed in stoichiometric reactions. The mechanism, however, remains obscure, especially how the N–O bond is cleaved. The lack of reactivity between (TMP)FeII and TEMPO–CH3 argues against a pathway in which TEMPO–R transfers OR (R = H or CH3) to (TMP)FeII to form the tetramethylpiperdinyl radical. The N–O homolytic bond strength of TEMPO–CH3 should be weaker than that of TEMPO–H (because CH3 is more electron donating than H), so OR transfer should be more facile for TEMPO–CH3 than for TEMPO–H. Although sterics could conceivably play a role, the strong binding of the bulky phenoxyl radical tBu3ArO to (TMP)FeII suggests that attack of TEMPO–CH3 should not be precluded. Reduction of TEMPO–H does not occur by outer-sphere electron transfer from (TMP)FeII, because TEMPO–H is unreactive with the much stronger reductant cobaltocene (over 24 h in toluene-d8).
The disproportionation of TEMPO–H by (TMP)FeIII(OH) is potentially related to the biochemical conversion of hydroxylamine to nitrite by the multi-heme enzyme hydroxylamine oxidoreductase (HAO).3 This complicated enzyme has inspired a number of different studies into the reactivity of hydroxylamine with both heme32 and non-heme33 transition metal complexes. For instance, two equivalents hydroxylamine react with (TPP)FeIIICl in MeOH/CHCl3 to give NH4Cl, water, and (TPP)FeNO.32a,b Water soluble iron porphyrin complexes catalyse the disproportionation of hydroxylamine to high yields of NH3 (the reduced product) and a mixture of N2 and N2O as the oxidized products.32c The mechanism is not well understood in either case, but there is some evidence for a pre-equilibrium formation of (porphyrin)FeIII(NH2OH)2+, and the possibility of radical pathways has been discussed.32a,b NH2OMe is catalytically decomposed to ⅓ NH3, ⅓ N2 and MeOH by aquapentacyanoferrate in aqueous buffer.33 A mechanism of ˙NH2 abstraction by iron(II) to yield a ˙OCH3 radical was suggested based on studies with the radical trap DMPO, although such an N–O cleavage is not indicated by the chemistry reported here (and spin adducts of DMPO can also be generated via non-radical mechanisms in the presence of ferric ions34).
TEMPO–H may be considered a simpler surrogate for hydroxylamine. Its oxidation stops at the stable TEMPO radical, while removal of H˙ form hydroxylamine starts a cascade to ½ N2 and H2O.35 The bond strength of the NH2O–H bond in water is only 7.4 kcal mol−1 stronger than that of TEMPO–H.8 So the results reported here indicate that hydrogen atom transfer from NH2OH to (heme)FeIII(OH) is a reasonable step in the catalytic cycle of HAO and related enzymes.
Conclusions
Described here are a number of inner-sphere reactions of the (TMP)FeIII/II redox couple (TMP = meso-tetramesitylporphyrinato). (TMP)FeIII(OH) reacts with compounds with very weak OH and NH bonds, TEMPO–H, PhNHNHPh and PhNHNH2, with transfer of a proton to the hydroxide ligand and an electron to the iron centre. This is a convenient way to generate (TMP)FeII. The data are consistent with either a pathway involving hydrogen atom transfer (concerted H+ + e− transfer), or by initial hydrolytic conversion of (TMP)FeIII(OH) + TEMPO–H to (TMP)FeIII(TEMPO) + H2O followed by TEMPO dissociation. The thermochemical data rule out pathways involving initial electron or proton transfer. Analysis of the reactivity with different hydrogen atom transfer reagents gives an estimate of 67 ± 3 kcal mol−1 for the effective bond dissociation free energy (BDFE) of (TMP)FeII + H2O → (TMP)FeIII(OH) + H˙.The ferrous porphyrin (TMP)FeII homolytically binds oxyl radicals to generate (TMP)FeIII–OR complexes. With TEMPO, this reaction is rapid and reversible on the NMR timescale at room temperature but is slower at low temperatures. This is, to our knowledge, the first reported case of a (porphyrin)FeIII–TEMPO complex. Equilibrium studies show that the Fe–O bond is remarkably weak, with a bond dissociation enthalpy is only 7.0 ± 1.5 kcal mol−1 (the BDFE is 4 kcal mol−1).
Both (TMP)FeII and (TMP)FeIII(OH) catalyse the disproportionation of the hydroxylamine TEMPO–H to ⅔ TEMPO + ⅓ TEMP–H (2,2,6,6-tetramethylpiperidine). This type of catalytic disproportionation has been observed with hydroxylamine and metal porphyrins but no radical intermediates have been directly observed. The results reported here suggest that the previously reported reactions of hydroxylamine with metal porphyrins may involve hydrogen atom transfer.
In discussions of substrate transformations by heme cofactors, the conversation is often dominated by high-valent intermediates, and the chemistry of lower valent heme species can be overlooked. The results reported here serve to highlight both the rich inner-sphere chemistry of the lesser-explored FeIII/II couple as well as the critical role that PCET can play in these systems.
We gratefully acknowledge financial support from the U.S. National Institute of Health (GM50422) and the University of Washington Department of Chemistry. We thank Dr Alexander Fox for useful discussions.
Notes and references
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 9, 3947 CrossRef PubMed.
- F. P. Guengerich, Chem. Res. Toxicol., 2000, 14, 611 CrossRef.
- N. Igarashi, H. Moriyama, T. Fujiwara, Y. Fukumori and N. Tanaka, Nat. Struct. Biol., 1997, 4, 276 CrossRef CAS PubMed.
- M. L. Fernandez, D. A. Estrin and S. E. Bari, J. Inorg. Biochem., 2008, 102, 1523 CrossRef CAS PubMed.
- V. R. I. Kaila, M. I. Verkhovsky and M. Wilkström, Chem. Rev., 2010, 110, 7062 CrossRef CAS PubMed.
- J. M. Mayer, Annu. Rev. Phys. Chem., 2004, 55, 363 CrossRef CAS PubMed.
- C. Constentin, D. H. Evans, M. Robert, J.-M. Saveant and P. S. Singh, J. Am. Chem. Soc., 2005, 127, 12490 CrossRef PubMed.
- (a) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961 CrossRef CAS PubMed; (b) BDFE values in toluene are assumed to be the same as those reported in benzene; (c) BDFE values in DMSO were taken from reference 8a and converted to BDFE values in toluene using Abraham's model. See ESI for more details†.
- (a) J. Z. Haeggström and C. D. Funk, Chem. Rev., 2011, 111, 5866 CrossRef PubMed; (b) M. P. Meyer and J. P. Klinman, J. Am. Chem. Soc., 2011, 133, 430 CrossRef CAS PubMed; (c) S. Fukuzumi, Helv. Chim. Acta, 2006, 89, 2425 CrossRef CAS.
- (a) R. C. Haushalter, M. Nakamura, T. E. Nemo, B. J. Evans and J. T. Groves, J. Am. Chem. Soc., 1981, 103, 2884 CrossRef; (b) T. E. Nemo and J. T. Groves, J. Am. Chem. Soc., 1983, 105, 5786 CrossRef; (c) T. E. Nemo and J. T. Groves, J. Am. Chem. Soc., 1983, 105, 6243 CrossRef; (d) R. Quinn, J. T. McMurry, M. Nakamura, G. Lang, B. Boso and J. T. Groves, J. Am. Chem. Soc., 1985, 107, 354 CrossRef; (e) J. A. Gilbert and J. T. Groves, Inorg. Chem., 1986, 25, 123 CrossRef; (f) Y. Watanabe and J. T. Groves, J. Am. Chem. Soc., 1986, 108, 7834 CrossRef PubMed; (g) Y. Watanabe and J. T. Groves, J. Am. Chem. Soc., 1988, 110, 8443 CrossRef; (h) Z. Gross, M. K. Stern and J. T. Groves, Inorg. Chem., 1994, 33, 5065 CrossRef; (i) J. T. Groves, Y. Z. Han, Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montello, Plenium, New York, 3rd edn, 2004, pp 1–34 Search PubMed; (j) C. C.-Y. Wang, D. M. Ho and J. T. Groves, J. Am. Chem. Soc., 1999, 121, 12094 CrossRef CAS.
- (a) R.-J. Chen, L. Latos-Grażyński and A. L. Balch, Inorg. Chem., 1982, 21, 2412 CrossRef; (b) L. Latos-Grażyński, R.-J. Chen, G. N. La Mar and A. L. Balch, J. Am. Chem. Soc., 1982, 104, 5992 CrossRef; (c) Y.-W. Chan, R.-J. Chen, G. N. La Mar, L. Latos-Grażyński, M. W. Renner and A. L. Balch, J. Am. Chem. Soc., 1984, 106, 7779 CrossRef; (d) L. Latos-Grażyński, M. W. Renner and A. L. Balch, J. Am. Chem. Soc., 1985, 107, 2983 CrossRef; (e) R. D. Arasasingham, C. R. Cornman and A. L. Balch, J. Am. Chem. Soc., 1989, 111, 7800 CrossRef CAS.
- (a) M. F. Sisemore, M. Selke, J. N. Burstyn and J. S. Valentine, Inorg. Chem., 1997, 36, 979 CrossRef CAS PubMed; (b) M. Selke and J. S. Valentine, J. Am. Chem. Soc., 1998, 120, 2652 CrossRef CAS; (c) D. L. Wertz and J. S. Valentine, Struct. Bonding, 2000, 97, 37 CrossRef CAS; (d) D. L. Wertz, M. F. Sisemore, J. Driscoll and J. S. Valentine, J. Am. Chem. Soc., 1998, 120, 5331 CrossRef CAS.
- (a) M. H. Lim, S.-Y. Oh and W. Nam, Inorg. Chem., 2000, 39, 5572 CrossRef; (b) T. Kamachi, T. Kuono, W. Nam and K. Yoshikawa, J. Inorg. Biochem., 2006, 100, 751 CrossRef CAS PubMed; (c) A.-R. Han, Y. J. Jeong, Y. Kang, J. Y. Lee, M. S. Seo and W. Nam, Chem. Commun., 2008, 1076 RSC; (d) Y. Kang, H. Chen, Y. J. Jeong, W. Lai, E. H. Bae, S. Shaik and W. Nam, Chem.–Eur. J., 2009, 15, 10039 CrossRef CAS PubMed; (e) W. Nam, Acc. Chem. Res., 2007, 40, 522 CrossRef CAS PubMed.
- E. A. Mader, E. R. Davidson and J. M. Mayer, J. Am. Chem. Soc., 2007, 129, 5153 CrossRef CAS PubMed.
- G. M. Whitesides and T. L. Newirth, J. Org. Chem., 1975, 40, 3448 CrossRef CAS.
- V. W. Manner, T. F. Markle, J. H. Freudenthal, J. P. Roth and J. M. Mayer, Chem. Commun., 2008, 256 RSC.
- R. W. Wagner and J. S. Lindsey, J. Org. Chem., 1989, 54, 828 CrossRef.
- J. Jiao, I. Schmidt, M. Taniguchi, J. S. Lindsey and D. F. Bocian, Langmuir, 2008, 24, 12047 CrossRef CAS PubMed.
- J. Willaredt, H. Schlemper, M. Keller, D. Schmitt, H. Fritz and R. Schwesinger, Chem. Ber., 1994, 127, 2435 CrossRef.
- The conclusion that the product is 4-coordinate (TMP)FeII rather than a 5-coordinate aquo complex is based on 1H NMR spectra. We observe the same spectrum for (TMP)FeII generated from (TMP)FeIIICl under rigorously moisture free conditions with cobaltocene and for (TMP)FeII + H2O generated from (TMP)FeIII(OH) + 0.5 eq PhNHNHPh (eqn 2). Furthermore, alcohols, ethers, and H2O are known to be poor ligands for ferrous porphyrins (i.e. Keq for THF binding to (TPP)FeII in C6H6 = ∼5 M−1): T. Mashiko and D. Dolphin, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, Pergamon Press, New York, 1st edn, 1987, ch. 21, p. 813 Search PubMed.
- (a) C. Swistak, X. H. Mu and K. M. Kadish, Inorg. Chem., 1987, 26, 4360 CrossRef CAS; (b) Converted from the reported E1/2 = –0.86 V vs. SCE in CH2Cl2.
- PhN
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH is bracketed since it is unstable to decomposition and is not observed by 1H NMR. See P.-K. Huang and E. M. Kosower, J. Am. Chem. Soc., 1968, 90, 2367 CrossRef CAS.
NH is bracketed since it is unstable to decomposition and is not observed by 1H NMR. See P.-K. Huang and E. M. Kosower, J. Am. Chem. Soc., 1968, 90, 2367 CrossRef CAS. - J. Sandström, Dynamic NMR Spectroscopy, Academic Press, New York, 1st edn, 1982 Search PubMed.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019 CrossRef CAS PubMed.
- M. W. Nee and J. R. L. Smith, J. Chem. Soc., Dalton Trans., 1999, 3373 RSC.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, Fl, 2007 Search PubMed.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561 CrossRef CAS PubMed.
- D. Benoit, V. Chaplinski, R. Bradslau and C. Hawker, J. Am. Chem. Soc., 1999, 121, 3904 CrossRef CAS.
- (a) M. K. Mahanthappa, K. Huang, A. P. Cole and R. M. Waymouth, Chem. Commun., 2002, 502 RSC; (b) K. Huang and R. M. Waymouth, J. Am. Chem. Soc., 2002, 124, 8200 CrossRef CAS PubMed; (c) K. Huang, J. H. Han, A. P. Cole, C. B. Musgrave and R. M. Waymouth, J. Am. Chem. Soc., 2005, 127, 3807 CrossRef CAS PubMed.
- K. S. Chan, X. Z. Li, W. I. Dzik and B. de Bruin, J. Am. Chem. Soc., 2008, 130, 2051 CrossRef CAS PubMed.
- (a) I. K. Choi, Y. Liu, Z. Wei and M. D. Ryan, Inorg. Chem., 1997, 36, 3113 CrossRef CAS PubMed; (b) S. E. Bari, V. T. Amorebieta, M. M. Gutiérrez, J. A. Olabe and F. Doctorovich, J. Inorg. Biochem., 2010, 104, 30 CrossRef CAS PubMed; (c) G. E. Alluisetti, A. E. Almaraz, V. T. Amorebieta, F. Doctorovich and J. Olabe, J. Am. Chem. Soc., 2004, 126, 13432 CrossRef CAS PubMed.
- M. M. Gutiérrez, J. A. Olabe and V. T. Amorebieta, Inorg. Chem., 2011, 50, 8817 CrossRef PubMed.
- (a) K. Ranguelova and R. P. Mason, Magn. Reson. Chem., 2011, 49, 152 CrossRef CAS PubMed; (b) L. Eberson, J. Chem. Soc., Perkin Trans. 2, 1994, 171 RSC; (c) K. Makino, T. Hagiwara, A. Hagi, M. Nishi and A. Murakami, Biochem. Biophys. Res. Commun., 1990, 172, 1073 CrossRef CAS PubMed; (d) F. Leinisch, K. Ranguelova, E. F. DeRose, J. Jiang and R. P. Mason, Chem. Res. Toxicol., 2011, 24, 2217 CrossRef CAS PubMed.
- J. Lind and G. Merényi, J. Phys. Chem. A, 2006, 110, 192 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: BDFE value interconversion calculations, Keq and van't Hoff calculations, stopped flow kinetic data, TEMPO–H disproportionation mass balance plot, and additional NMR data. See DOI: 10.1039/c3sc52055b |
| This journal is © The Royal Society of Chemistry 2014 |