High-purity gold nanocrystal dimers: scalable synthesis and size-dependent plasmonic and Raman enhancement†
Xiaohui
Ji
and
Wensheng
Yang
*
State Key Laboratory for Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: wsyang@jlu.edu.cn; Fax: +86 431-85168868; Tel: +86 431-85168185
First published on 3rd October 2013
Abstract
We report a method for the solution synthesis of directly-fused dimers of gold nanocrystals, including both homo-dimers (two nanocrystals of the same size) and hetero-dimers (two nanocrystals of significantly different sizes) with high purity and high yield. The first key issue in this seeded solution synthesis was the tight integration of the “reduction” of the precursors to form tiny nanocrystals in solution and “random attachment” of these newly formed tiny nanocrystals onto the tips of existing nanocrystals. The second key issue was to decouple the “intra-particle ripening” from the “reduction” and “random attachment”. This new approach was found to be capable of yielding different types of dimers by judiciously tuning the time sequence of “reduction”, “random attachment”, and “intra-particle ripening” in one synthesis. UV-Vis absorption measurements unambiguously demonstrated a two-band feature for the resulting high-purity dimers, indicating strong plasmonic enhancement due to the formation of “hot spots” within the dimers. The two-band absorption spectra of the dimers were further found to be strongly size dependent and intra-particle distance dependent. Consistent with the observation of the formation of “hot spots” by absorption measurements, very strong and size-dependent surface-enhanced Raman scattering was observed for these high-purity, directly fused, and stable dimers of gold nanocrystals.
Introduction
Nanocrystals and nanostructures made of noble metals are promising materials for countless nano-photonic applications due to their strong interaction with light.1–3 Their optical properties are dominated by the localized surface plasmon resonances (LSPR), the collective coherent oscillation of conduction electrons in the metallic nanostructures.4,5 For a given type of noble metal, the resonance energy of the LSPR is highly sensitive to the size, morphology, and configuration of the nanostructures.6,7 Among all types of complex nanostructures, pairs of nanocrystals are the simplest model system to study. The close approach of two nanocrystals leads to interaction of their LSPR, which further results in highly enhanced and localized plasmons or “hot spots” at the junctions between the nanocrystals. Hot spots give rise to large enhancements of the electromagnetic field locally, which enables molecular detection through surface-enhanced Raman scattering (SERS) at or near single molecule sensitivity.8–10Nanocrystal pairs with an intra-particle distance smaller than the diameter of the nanocrystals, known as “dimers”, might generate extremely large plasmonic enhancements in their junctions and have been widely explored as SERS substrates.11–14 Calculations have revealed that, when the spacing between two nanocrystals is below 0.5 nm, especially with slight fusing of the nanocrystals within a dimer to give tiny crevices, the plasmonic electromagnetic enhancements could be 1010 times or greater.15 Importantly, electromagnetic coupling of the plasmon resonance between two nanocrystals follows the principles of molecular hybridization. This means that, when very strong plasmonic enhancement is in place within a dimer, two nanocrystal plasmons hybridize to form a lower energy bonding plasmon mode and a higher energy antibonding plasmon mode.16–19 This offers a convenient tool for characterization of the formation of dimers of noble metal nanocrystals. Thus, if high purity dimers are present in a sample, it should be possible to observe a two-band absorption spectrum for the sample, given that the size of the nanocrystals, the distance between the two nanocrystals, and the degree of fusion between the two nanocrystals are tightly controlled.
The early-stage synthesis of dimers of noble metal nanocrystals has mainly been based on “linking” two nanocrystals by functionalized DNA, organic ligands, and polymer linkers.20–27 These initial efforts typically did not result in a strong plasmonic electromagnetic enhancement, such as a two-band absorption feature, this is likely because of the relatively long intra-particle distance and/or no direct fusion between the two nanocrystals. Recently, this situation has been improved. Xia et al. prepared well-defined silver dimers by etching and dimerization of Ag nanocrystals of various sizes.28,29 Chen et al. showed that Ag growth on Au seeds can form Au–Ag heterodimers by embedding special surface ligands.30 Sun et al. very recently reported a strategy for the synthesis of nanocrystal dimers by the use of colloidal magnetic Fe3O4–SiO2 core–shell particles as the “solid substrates”.31 To obtain nanocrystal dimers and other complex nanostructures of high purity, Chen et al. reported a high-purity separation method for polymer-encapsulated dimers and trimers of noble metal nanocrystals based on an isopycnic density gradient centrifugation method.32–37 Although dimers of noble metal nanocrystals were obtained using these new approaches, several reports have indicated that the yield of the dimers of the nanocrystals was low.38–41
Here we report a method for the solution synthesis of directly-fused dimers of gold nanocrystals, including both homo-dimers (two nanocrystals of the same size) and hetero-dimers (two nanocrystals of significantly different sizes). This seeded solution synthesis produced high-purity and high-yield dimers by judiciously controlling three elementary steps, namely “reduction”, “random attachment”, and “intra-particle ripening”. Optical measurements unambiguously demonstrated very strong, “hot spot” related, and size/morphology dependent plasmonic as well as Raman enhancements for the resulting high-purity dimers.
Experimental
Materials
Tetrachloroauric acid (AR, HAuCl4·4H2O, FW 411.85) was purchased from Shanghai Chemical Reagent Co. Int. Sodium citrate tribasic dihydrate (≧99.0%, Na3C6H5O7·2H2O, FW 294.10) was purchased from Sigma-Aldrich. Sodium hydroxide (>98.5%, NaOH, FW 40.00) was purchased from Bio Basic Inc. 4-Mercaptobenzoic acid (4-MBA) was purchased from Aldrich. All of the chemicals were used without further purification. High-purity water (Pall Purelab Plus) with a resistivity of 18.2 ΩM cm was used for preparation of aqueous solutions in all the experiments. All the glassware used was cleaned in a bath of freshly prepared aqua regia solution (HCl–HNO3, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and then rinsed thoroughly with H2O before use.
:1) and then rinsed thoroughly with H2O before use.
Preparation of gold nanocrystal seeds
Dot-shaped seeds of gold nanocrystals were synthesized by the citrate reduction approach reported in our previous work.42 In a typical procedure, 50 ml aqueous solution of HAuCl4 (0.25 mM) was prepared in a 100 ml flask. After the solution was brought to boiling under stirring, 0.5 ml of 5% sodium citrate solution was added. The reaction was allowed to run until the solution became wine red in color. Transmission electron microscope (TEM) measurements (Fig. S1, ESI†) were carried out to determine the size (∼20 nm in diameter) and size distribution (<10% standard deviation) of the nanocrystals in this typical synthesis.“Reduction” and “random attachment”
In a typical synthesis, the freshly prepared seeds solution (0.2 mM (counted by gold atomic concentration), 25 ml) was mixed with 600 μl of NaOH aqueous solution (23.9 mM) at room temperature for 10 min and in a water bath at 30 °C for 5 min. Subsequently, 600 μl of HAuCl4 aqueous solution (23.9 mM) was added, which resulted in a gold atomic ratio between ions in solution and neutral gold atoms in the seeds (Au3+:Au0 ratio) being 2.8:1. This reaction solution was put in a water bath at 30 °C without stirring. Aliquots were taken to monitor the reaction by UV-Vis absorption. All the experiments were performed under artificial light.
After the reactions with the typical procedure described above were carried for at least one hour, a centrifugation procedure was applied for the isolation of the dimers. This procedure included two steps, with one at 12000 and the other at 8000 rpm. This resulted in the separation of the high-purity dimers as a precipitate from the nanocrystal seeds, reaction side products, and remaining reactants present in the supernatant. After decanting the centrifugation mixture, the dimers in the precipitate were redispersed in a 0.05% sodium citrate solution, this solution was stable for at least a few months.
To obtain different types of dimers in one reaction, a certain volume (1 ml) of reaction mixture was taken at different reaction times, and the samples were immediately centrifuged for isolation. Typically, centrifugation at 12000 rpm was applied for separation of all the nanocrystals from HAuCl4 and the other reaction species. The bottom red precipitate was separated from the top colorless or slightly colored solution by decanting it and then redispersing it in aqueous sodium citrate (0.05%) solution. This solution was centrifuged in two steps. The first step was performed at 4000 rpm to keep the supernatant and the second step at 5000 rpm was used to separate either the dumbbell-shaped or tadpole-shaped nanostructures from the nanocrystal seeds. The precipitate of dumbbells or tadpoles was redispersed in water with sodium citrate (0.05%) and was used for the following “intra-particle ripening” step.
Reactions with variable Au3+:Au0 ratios were performed using the same procedure described above. The gold nanocrystal seeds concentration was the same as that for the typical reaction but the amounts of HAuCl4 and NaOH solutions were varied to obtain a given Au3+:Au0 ratio.
“Intra-particle ripening”
Into the solution of the dumbbell-shaped (or tadpole-shaped) nanostructures obtained above, different amounts of potassium chloride were added to a designated KCl concentration. “Intra-particle ripening” was carried out at room temperature. UV-Vis absorption spectra were measured at different time intervals to monitor the reaction process. To stop the “intra-particle ripening” process, centrifugation at ∼12000 rpm was performed to isolate the nanocrystals from the other chemical reactants, especially chloride ions.
Measurements of AuCl4− concentration in the reaction solution
A procedure published previously42 was adopted to determine the consumption of AuCl4− in the reaction solution. An aliquot of the reaction solution was taken out at a given time interval and then quenched immediately by adding it into a pre-prepared solution of NaCl (0.9 M) and HCl (pH = 1). After being kept for 12 h at room temperature, the absorbance of the supernatant was measured by UV-Vis spectrophotometry to determine the AuCl4− concentration using a standard curve.42Preparation of SERS samples
4-Mercaptobenzoic acid (4-MBA) was used as the probe molecule for SERS measurements. In a typical procedure, 100 μl of the 4-MBA solution (0.01 M) was added into 1.0 ml of the sample solution containing the gold nanostructures. After reacting for 12 h at 4 °C, the 4-MBA-labeled gold nanostructures were separated from the solution by centrifugation at 12000 rpm for 3 min. After decanting the supernatant, the precipitate was re-suspended in 1.0 ml of the citrate solution (0.05%). The centrifugation/decanting process was repeated twice, and the final dispersion was used for SERS measurements.
Determination of SERS enhancement factor
The Raman peak at 1072 cm−1 for 4-MBA was employed for estimation of the enhancement factor (EF) of the hot spot for dimers of gold nanocrystals using a method previously reported in the literature.28 Where the number of bulk molecules (Nbulk) was determined based on the Raman spectrum of a 0.1 M 4-MBA solution in 12 M NaOH (aq.). The number of trapped molecules (Ntrap) in the hot spot region was estimated by assuming that the 4-MBA molecules will be absorbed as a monolayer with a 0.2 nm2 molecular footprint onto a spherical cap having h = r/6.6 located on each gold nanocrystal of the dimer. The nanocrystals were approximated to be a spherical shape with a radius of r and the height of the spherical cap between two nanocrystals was h. The concentration of gold atoms in the dimer solutions for the three types of dimers in Fig. 8 (from bottom to top) was measured by ICP-AES to be 50.2 mg l−1, 90.6 mg l−1 and 127.0 mg l−1, respectively. The obtained Au atom concentration was converted into the nanocrystal concentration by dividing it by the number of Au atoms in one nanocrystal.“Destructive intra-particle ripening”
The sample for “destructive intra-particle ripening” was taken from a typical reaction solution after a very long reaction time interval (24 h). The required nanostructures were isolated by two steps of centrifugation. At 12000 rpm, all the nanostructures were precipitated and the colorless supernatant was decanted. The precipitate was redispersed in 0.05% of sodium citrate and centrifuged once more at 8000 rpm. After decanting the supernatant solution, the nanostructures were redispersed in a 0.05% sodium citrate solution for all measurements as shown in Fig. 9 (right panel).
Measurements
UV-Vis spectra were collected using a SHIMADZU UV-1800 UV-Vis spectrophotometer. Transmission electron microscopy (TEM) observations were carried out with a JEOL JEM-2010 electron microscope with an acceleration voltage of 100 kV. High Resolution TEM (HRTEM) measurements were performed on a JEOL JEM-2100F electron microscope with an acceleration voltage of 200 kV. TEM samples were made by dropping the solutions onto Formvar-coated copper grids. HRTEM samples were made by dropping the solutions onto carbon-coated copper grids. The atomic concentration of gold element in a given dimer solution was analyzed by inductively coupled plasma-atomic emission spectrometry (ICP-AES, Perkin-Elmer 2400 element analyzer). Raman spectra were obtained using a BWTEK MiniRam instrumetn with a 785 nm excitation laser and 30 seconds accumulation time.Results and discussion
Design of synthetic scheme
The synthetic targets of this work were homo- and hetero-dimers of gold nanocrystals with two nanocrystals directly fused together. Our previous work42 revealed that, under certain conditions, newly formed gold nanocrystals with small sizes have a strong tendency to be attached to the surface of existing gold nanocrystals with large sizes in sodium citrate (Na3Ci) reduction. Different from commonly known oriented attachment,43 the attachment observed by us was found to be without crystal structural orientation, and was thus named as “random attachment”. Under the typical reaction conditions studied in the previous report, the small nanocrystals attached were found to rapidly redistribute their atoms onto the existing large nanocrystal through an “intra-particle ripening”44 process. For the synthesis of dot-shaped nanocrystals in a large size range, a tight integration of “random attachment” and “intra-particle ripening” would be ideal to offer synthetic pathways for nearly monodispersed nanocrystals. However, for the synthesis of targeted dimers, breaking this tie would be necessary. In addition, to simplify the optimization of these two processes, the large sized nanocrystals for small ones to attach to were synthesized in advance. This latter design made the synthetic scheme (Scheme 1) similar to that of a seeded growth.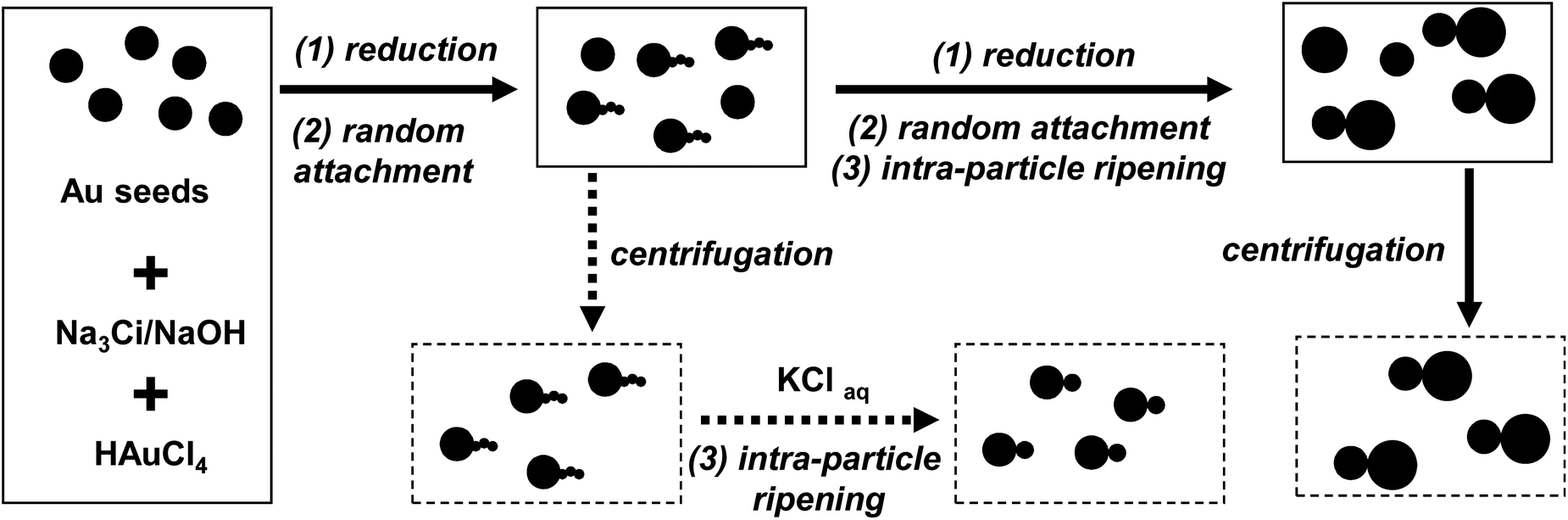 | ||
| Scheme 1 Synthetic scheme for dimers of gold nanocrystals in solution. Dimers of different sizes could be obtained through the dotted pathway at different reaction times (see text for detail). | ||
Gold nanocrystal seeds of a relatively large size and decent size distribution were pre-synthesized using a previously reported procedure.42 Into a solution of these seeds, Na3Ci, NaOH, and HAuCl4 were added in sequence. To initiate the “reduction” of HAuCl4 with Na3Ci, this mixture was brought to 30 °C, instead of 100 °C for synthesis of the seeds. This much reduced temperature was necessary to make the synthesis controllable by slowing down the “reduction” and “intra-particle ripening” steps. Furthermore, under this condition, “reduction” and “random attachment” were tightly integrated. In other words, nearly all the newly formed nanocrystals through “reduction” of the precursors in solution would be allowed to have sufficient time to complete the “random attachment” for realizing a high yield of the targeted dimers.
However, a simple decrease of the temperature did not decouple “intra-particle ripening” from “random attachment” and “reduction”. Early results revealed that the rate of “intra-particle ripening” would strongly depend on solution pH and existence of halide ions.45 The necessary “reduction” process for generation of newly formed small nanocrystals from HAuCl4 would inevitably yield chloride ions. Thus, an increase of the solution pH by the addition of NaOH became a rational choice to further slow down the “intra-particle ripening” during the “reduction” and “random attachment”. Fortunately, this hypothesis worked quite well in terms of prevention of formation of nearly dot-shaped nanocrystals with very large sizes (see next subsection).
The “intra-particle ripening” could be re-initiated by the addition of chloride ions into the reaction solutions taken at different reaction time intervals, this is schematically shown on the bottom row of Scheme 1 (the dotted pathway). In this step, in order to obtain dimers of high purity, it was found that removal of unreacted precursors and possibly some active reaction side products by centrifugation was necessary.
The rationally designed scheme shown in Scheme 1 was verified experimentally and the results are described in the following subsections.
Effects of NaOH
Fig. 1(a) illustrates that reduction of HAuCl4 with Na3Ci resulted in rapid appearance of a significant absorbance tail in the wavelength range between 600 and 900 nm. This spectroscopic change in the early stage of the reaction (∼20 min of reaction) was consistent with substantial elongation of the original spherical seeds.42 When the reaction proceeded further, instead of further development of the tail in the 600–900 nm window, the UV-Vis spectra became sharper and sharper. It finally ended up with a pronounced absorption peak in the 520–540 nm range, indicating the nearly spherical shape of the final products.42 For example, the overall spectral features of the 60 min sample in Fig. 1(a)—broadening of the main peak and insignificant absorbance increase between 600 and 900 nm—should be a result of formation of spheroidal-shaped nanocrystals by simultaneous “random attachment” and “intra-particle ripening”42 (see more discussion below). This indicated that reduction of HAuCl4 with Na3Ci in the presence of gold nanocrystal seeds would not sufficiently slow down the “intra-particle ripening” even under the much reduced reaction temperatures.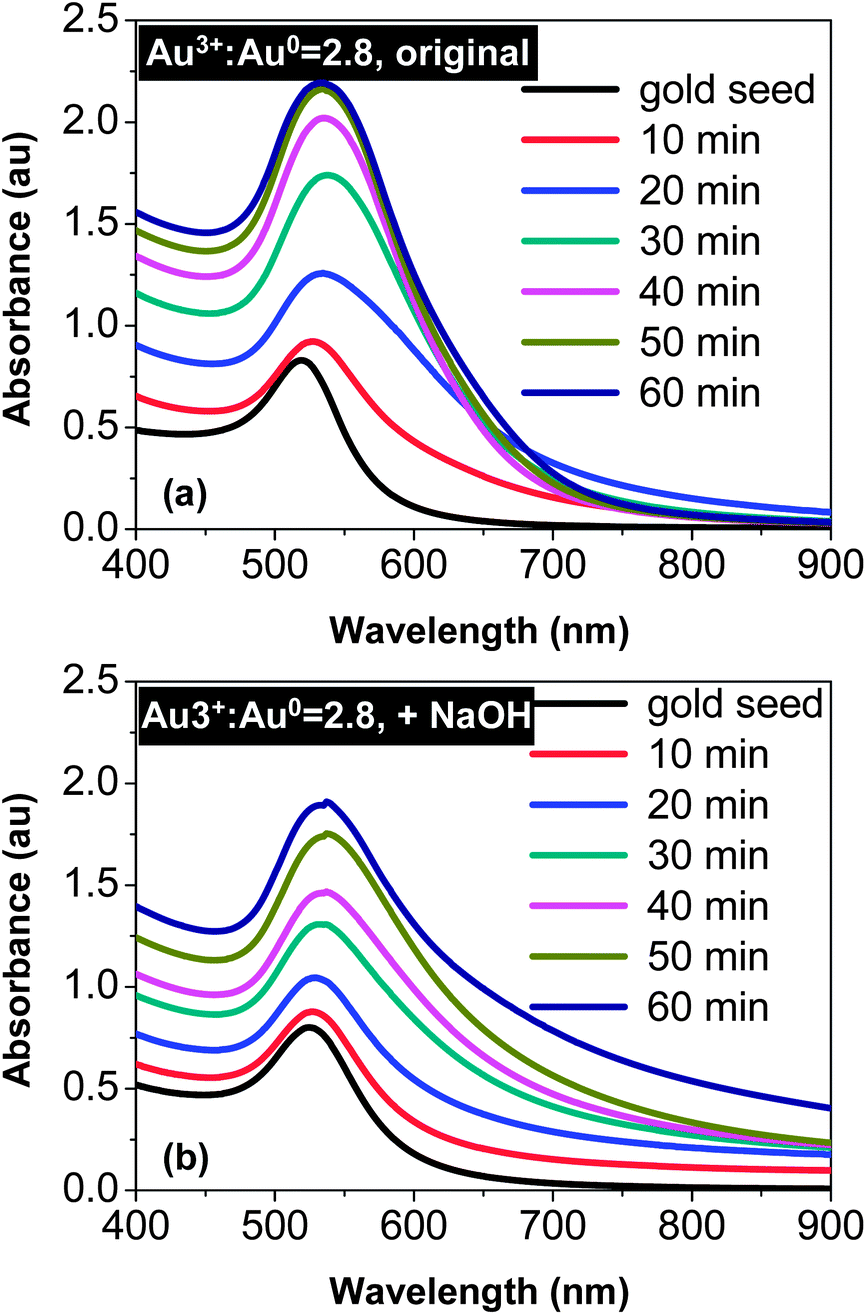 | ||
| Fig. 1 Temporal evolution of UV-Vis spectra of the reaction with a Au3+:Au0 ratio of 2.8:1. (a) Reaction without addition of NaOH. (b) NaOH concentration was 0.6 mM. | ||
Fig. 1(b) illustrates a qualitatively different trend for the comparison reaction with the addition of NaOH. In the early stage (∼20 min of the reaction), the absorbance in the 600–900 nm window gradually increased. However, this increase possessed two distinguishable features in comparison with the corresponding spectra in Fig. 1(a). Firstly, the increase seemed to extend beyond 900 nm (see Fig. 1(b)). Secondly, broadening at the low energy side near the main peak was much smaller in Fig. 1(b) than that in Fig. 1(a). As the reaction proceeded further, the low energy tail in Fig. 1(b) was extended further, in comparison to that illustrated in Fig. 1(a). This indicates a continuous shape-deviation of the nanostructures from the dot-shape observed for the reaction in the presence of NaOH.
The effects of NaOH shown in Fig. 1 (see more results below) were substantial. A plausible explanation is the effects of surface passivation. As a weak acid, the citrate group can exist in solution in different forms, either as citric acid or as citrate ions with different charges. In principle, citrate ions bonded with less hydrogen ions should provide better surface passivation on gold nanocrystals, which should thus slow down the “intra-particle ripening” process. To verify this hypothesis, the temporal evolution of both HAuCl4 concentration and solution pH for the reactions shown in Fig. 1 was studied, and the results (Fig. 2) were found to be supportive.
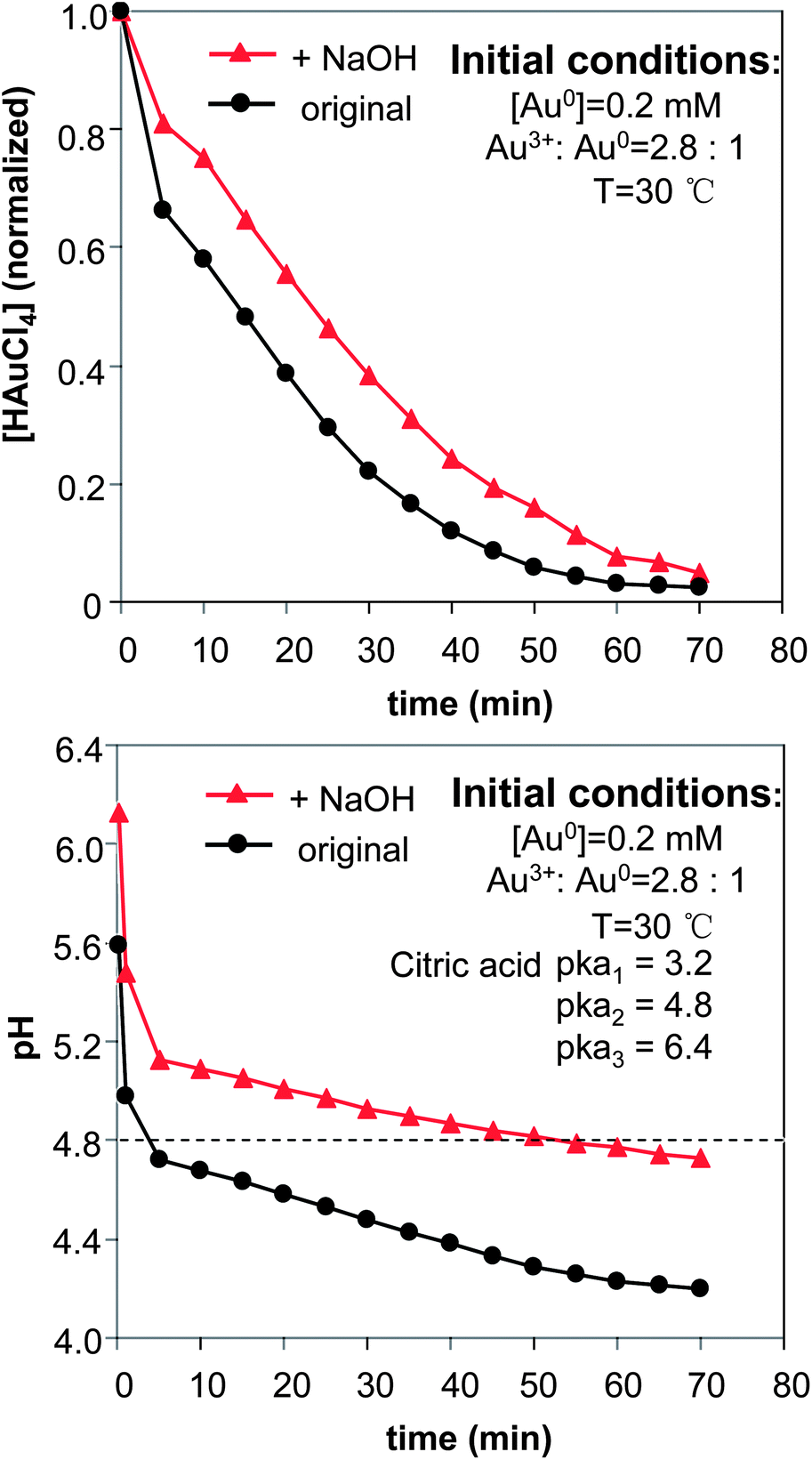 | ||
| Fig. 2 Temporal evolution of the precursor concentration (top) and the pH in the reaction solutions. The concentration of NaOH was 0.6 mM. | ||
The pH of the reaction without addition of NaOH (Fig. 2, bottom) was rapidly dropped to a value below the second ionization constant of citric acid, which is shown as a dashed line in the plot (pKa2 = 4.8). Conversely, the addition of NaOH maintained the pH of the reaction solution above the pKa2 of citric acid most of the time. Only after the reaction had proceeded for a long period of time, did the pH of the reaction solution containing NaOH reduce to below the pKa2 of citric acid. With these results, we tentatively considered that citrate ions with −2 charges—equivalent to HCi2−—would be efficient ligands for the prevention of “intra-particle ripening” in the current system. This consideration was found to be consistent with the results shown in Fig. 3 (see detailed discussion below) as well.
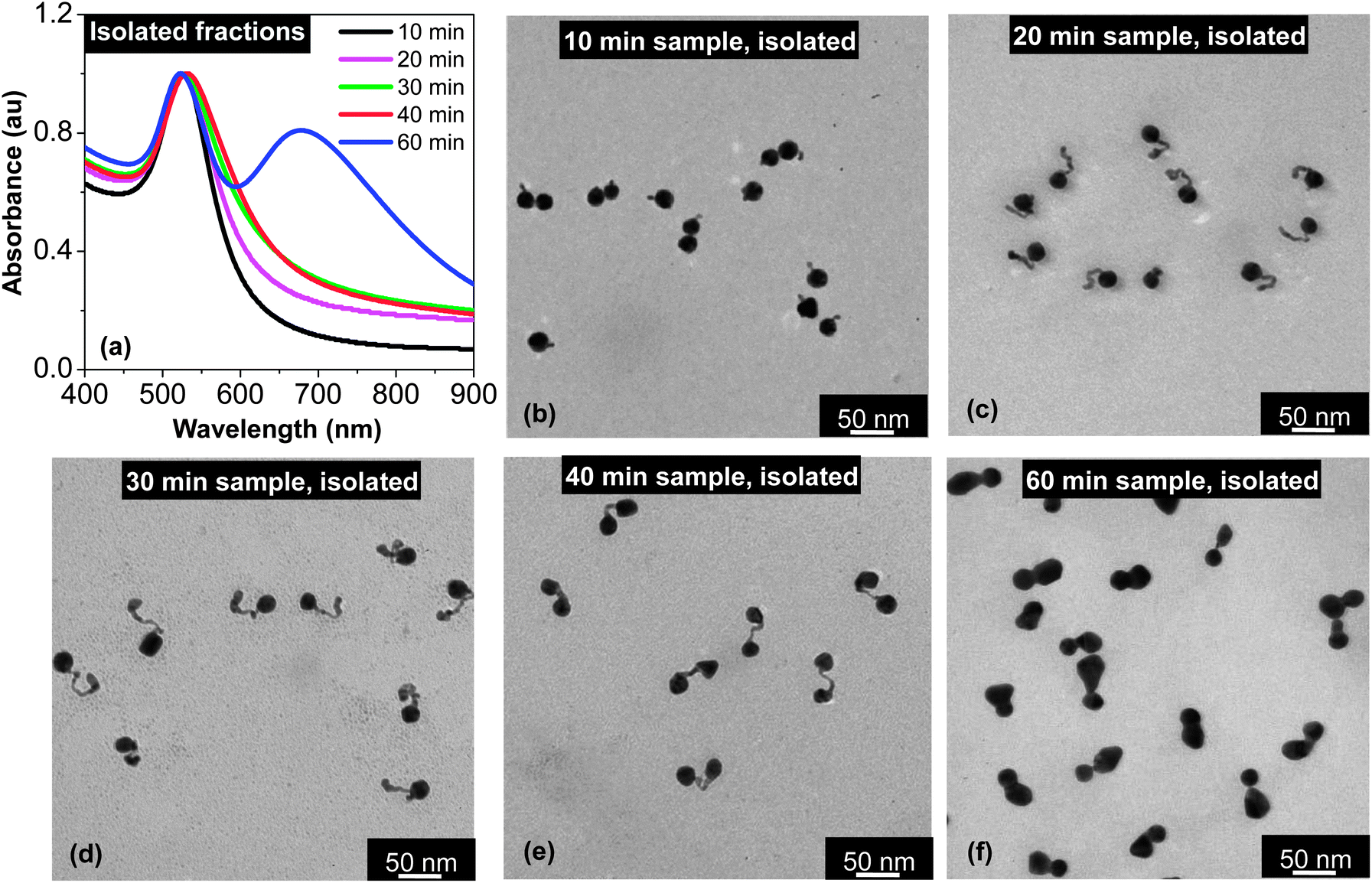 | ||
| Fig. 3 UV-Vis absorption spectra (a) and TEM images (b–e) of the isolated samples taken at different time intervals. For comparison, the absorption spectra of the corresponding samples prior to isolation by centrifugation can be found in Fig. 1(b). Different from Fig. 1(b), the UV-Vis spectra in this plot are normalized at the peak position. | ||
The results in Fig. 2 (top) basically confirmed our expectation that the reduction of the gold ions would be somewhat slowed down with the addition of NaOH.42 It is worth mentioning that the rate difference of reduction between the two reactions was mostly in the initial stage when the pH values of the solutions were high.
The results in this subsection are sufficient to verify the significance of the addition of NaOH into the reaction system. Thus, from this point on, all the results mentioned are related to reactions carried out in the presence of NaOH unless otherwise indicated.
The citrate concentration did not have a major effect on the designated reaction path as long as the reaction pH was fixed and the amount of citrate was sufficient for reduction and stabilization. As revealed by a quantitative study reported previously,42 the formation of a tadpole-shaped (or wirelike) nanostructure by “random-attachment” was possible at a large range of citrate concentration when the pH was low. Conversely, in the presence of the same range of citrate concentration, the system would yield nearly spherical particles through a conventional growth process when the pH was high.
Isolation of products from “reduction” and “random attachment”
In the synthetic approach shown in Scheme 1, isolation of the products from “reduction” and “random attachment” without significant “intra-particle ripening” is essential for synthesis of different types of dimers in one reaction. In addition, successful isolation of the nanostructures should also shed some light on the entire synthetic chemistry.
Fig. 3(a) illustrates the UV-Vis absorption spectra of several samples after isolation by centrifugation using a bench-top centrifuge (see Experimental for details). In comparison to the corresponding spectra in Fig. 1(b), the spectra in Fig. 3(a) are substantially more pronounced in terms of deviation from the spectrum of dot-shaped nanocrystals. The most striking one is the spectrum for the 60 min sample. After the isolation by a simple centrifugation process, this specific sample showed a two-band absorption spectrum (Fig. 3(a)), instead of a barely distinguishable shoulder at ∼700 nm in Fig. 1(b). Another interesting feature for the 60 min sample after isolation was the position and width of its main absorption peak, which was slightly blue-shifted and relatively narrowed after isolation. This latter feature should be a result of removal of the spheroidal nanocrystals formed by over ripening in this final solution (see further discussion below). It should be pointed out that these spectral features for the final solution were found to be universal as long as the Au3+:Au0 ratio in the reaction was not too low (see the results for a reaction with Au3+:Au0 ratio being 1.4:1 in Fig. S2, ESI†).
TEM revealed the surprising purity of all the samples after isolation although the key isolation process for separation of the targeted morphology (Fig. 3(b–f)) from the dot-shaped nanocrystals was carried out by simple centrifugation at ∼4000–5000 rpm using a bench-top centrifuge. Apparently, the volume difference between these nanostructures and the dot-shape seeds could be rather small (see Fig. 3(c) as an example), and the same procedure could not separate the two types of dot-shaped nanocrystals with such a small volume difference. Though the exact mechanism is not known, we hypothesized that this extremely efficient separation was likely associated with the substantially enhanced electromagnetic interactions between polarized and directional surface plasmons for those targeted morphologies shown in Fig. 3.
The morphology of the samples taken at a relatively short reaction time (10–30 min) shown in Fig. 3(b–d) was within the expectation if “intra-particle ripening” was truly stopped in the reaction solution. Evidently, during approximately the first 30 min, the small particles formed by the “reduction” were attached to the seeds in a wirelike manner to form tadpole-shaped aggregates. A subsequent attachment was always occurring at the tip of the tail of a tadpole, which should be a result of the relatively strong electromagnetic field of the tip. According to our previous study,42 such an attachment does not require a lattice orientation.
When the reaction proceeded for about 40 min, the gold precursor in the solution was consumed by about 75% (Fig. 2, top), and the pH value of the reaction approached the pKa2 of citric acid (Fig. 2, bottom). Starting from this point, TEM measurements (Fig. 3(e) and (f)) revealed strong evidence of “intra-particle ripening”. Instead of being tadpole-shaped as in Fig. 3(b)–(d), all the nanostructures in Fig. 3(e) were dumbbell-shaped for the 40 min sample. Compared to the tadpole-shaped nanostructures, the size of both ends of the dumbbell-shaped nanostructure in Fig. 3(e) was similar to the size of the seed nanocrystals. Though two nanocrystals at the ends of a dumbbell-shaped nanostructure were undoubtedly fused together, optical measurements shown in Fig. 3(a) revealed that the plasmonic coupling between the nanocrystals was not strong enough to yield a two-band UV-Vis spectrum. This should be a result of the relatively large distance—approximately 30–40 nm on average—between the two end nanocrystals.
From 40 min to 60 min, “intra-particle ripening” continued and dimers with little spacing between the two end nanocrystals were the dominating products for the 60 min sample (Fig. 3(f)). Consistent with this targeted morphology, a two-band feature was fully developed in the UV-Vis spectrum (Fig. 3(a)).
Although the addition of NaOH slowed down the “intra-particle ripening” in the reaction solution as described above, the results presented in Fig. 3 told us that it did not fully decouple the “intra-particle ripening” from “reduction” and “random attachment”. When the reaction proceeded for about 40 min, “intra-particle ripening” already showed a strong influence on the morphology of the nanostructures as described above. At this moment, there were still about 25% of the precursors left in the solution. Even at one hour, the precursors were not consumed completely (Fig. 2, top) although the targeted dimers were obtained without additional ripening treatment (Fig. 3(f)). However, the results shown in Fig. 3 and those to be described later revealed that, though the decoupling of “intra-particle ripening” from “reduction” and “random attachment” was incomplete, it was sufficient for synthesis of the targeted products. Therefore, no further optimization of the reaction conditions for the “reduction” and “random attachment” steps was pursued.
Isolation of the intermediate and final products of the original reaction confirmed that, without addition of NaOH, “intra-particle ripening” was initiated too early to yield such a variety of shape-controlled nanostructures as shown in Fig. 3. For instance, the 30 min sample for the reaction without NaOH (Fig. S3, ESI†) gave a mixture of extremely tightly fused dimers and short rods, instead of the tadpoles shown in Fig. 3(d). The 60 min sample for the reaction without the addition of NaOH (Fig. S3, ESI†) was dominated by deeply intra-particle ripened nanostructures, i.e., nearly spherical dots with large sizes. These results were found to be consistent with the UV-Vis measurements shown in Fig. 1 (top).
Initiation of “intra-particle ripening” by chloride ions after isolation
In Scheme 1, one reaction could yield many different types of dimers including both hetero-dimers and homo-dimers. The key idea was to take the nanostructures shown in Fig. 3 as the intermediates and reinitiate “intra-particle ripening” for these isolated nanostructures. To confirm this design principle, a systematic study of chloride ion concentration versus rate of “intra-particle ripening” was carried out.Fig. 4 illustrates the temporal evolution of UV-Vis absorption of the 40 min sample with isolation (the nanostructures in Fig. 3(e)) under different chloride ion concentrations. For the reaction with [KCl] = 1.5 mM, a slight increase of absorbance at around 700 nm slowly occured within 30 min and no noticeable changes were observed for the main absorption peak (Fig. 4(a)). As the KCl concentration increased to 3.0 mM, increase of the absorbance at around 700 nm was accelerated and gradually developed into a shoulder. When the KCl concentration further increased to 6.0 mM, the shoulder at 700 nm was developed very rapidly, and at the same time, the main peak slightly shifted to blue and became narrow. After this initial stage, the 700 nm shoulder shifted to blue readily, indicating that the intra-particle distance between the two nanocrystals within a dimer reduced too rapidly.
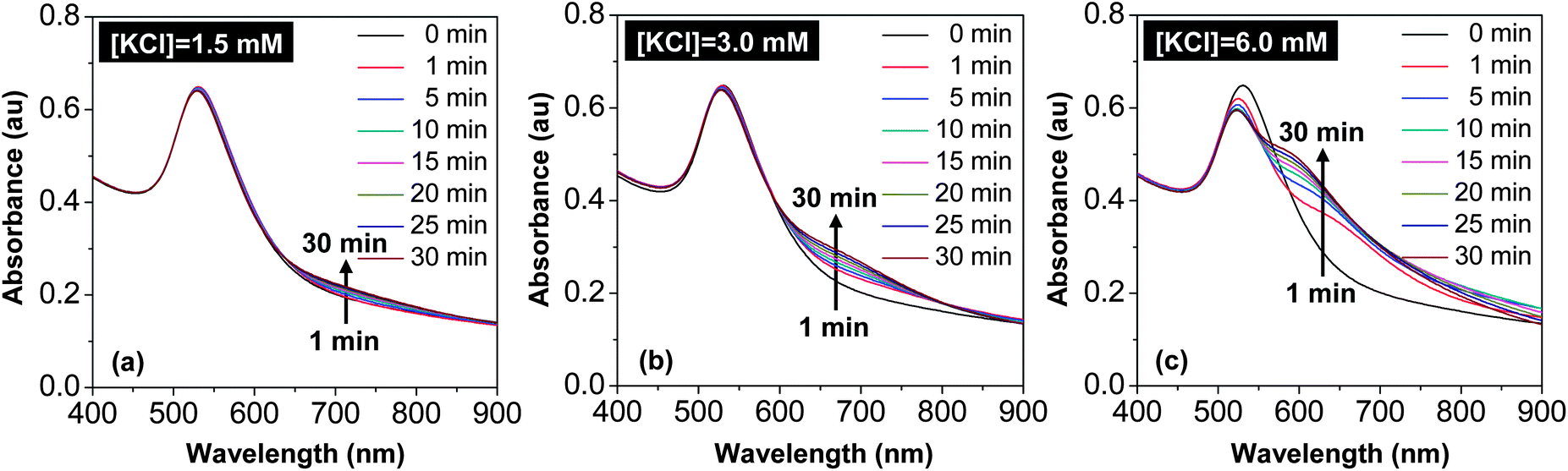 | ||
| Fig. 4 Temporal evolution of UV-Vis spectra of three reactions with different KCl concentrations as labeled. Note: the spectrum labeled “0 min” was measured using the 40 min sample after isolation by centrifugation. | ||
Overall, the trend of changes upon increase of chloride ion concentration shown in Fig. 4 was found to be consistent with the original design. Furthermore, this trend was found to be semi-quantitatively reproducible. As shown in Fig. 4, [KCl] being 3 mM was found to be a suitable concentration, and the 1.5 mM (6.0 mM) reaction was too slow (fast). For a specific sample after isolation by centrifugation from the “reduction” solution, it was always possible to optimize the ideal concentration of the chloride ions and the suitable reaction time for accomplishing “intra-particle ripening”. For a specific sample after isolation, however, the suitable reaction time at a given concentration of KCl was found to differ somewhat from one reaction to another possibly because the degree of removal of chloride ions by centrifugation varied. Generally, the later the sample was taken out of the reaction solution, the shorter the “intra-particle ripening” time would be.
For the samples after isolation by centrifugation (Fig. 3), their corresponding dimers formed by separate “intra-particle ripening” using KCl treatment were generally of high-purity. Fig. 5(a) illustrates a large field TEM image of the dimers by “intra-particle ripening” using KCl for the 40 min sample after isolation (Fig. 2(e)). A higher resolution TEM image (Fig. 5(b)) clearly revealed the homo-dimer structure, which is substantially different from the nanostructures before the “intra-particle ripening” treatment (Fig. 3(e)). Statistically, this sample was found to be dominated by homo-dimers, with 366 (∼72.5%) dimers out of 505 nanostructures in a randomly selected image. As shown in Fig. 5(c), the majority (92 counts, 18.2%) of the non-dimer nanostructures were particle aggregates that were induced by the relatively high concentration of nanostructures on the TEM grids needed for statistics. The single dot fraction (47 counts, 9.3%) included those over-ripened nanostructures as indicated in Fig. 5(a). In repeated experiments, the dimer fraction was found to be in the range between 70 and 80% using the same classification catagories.
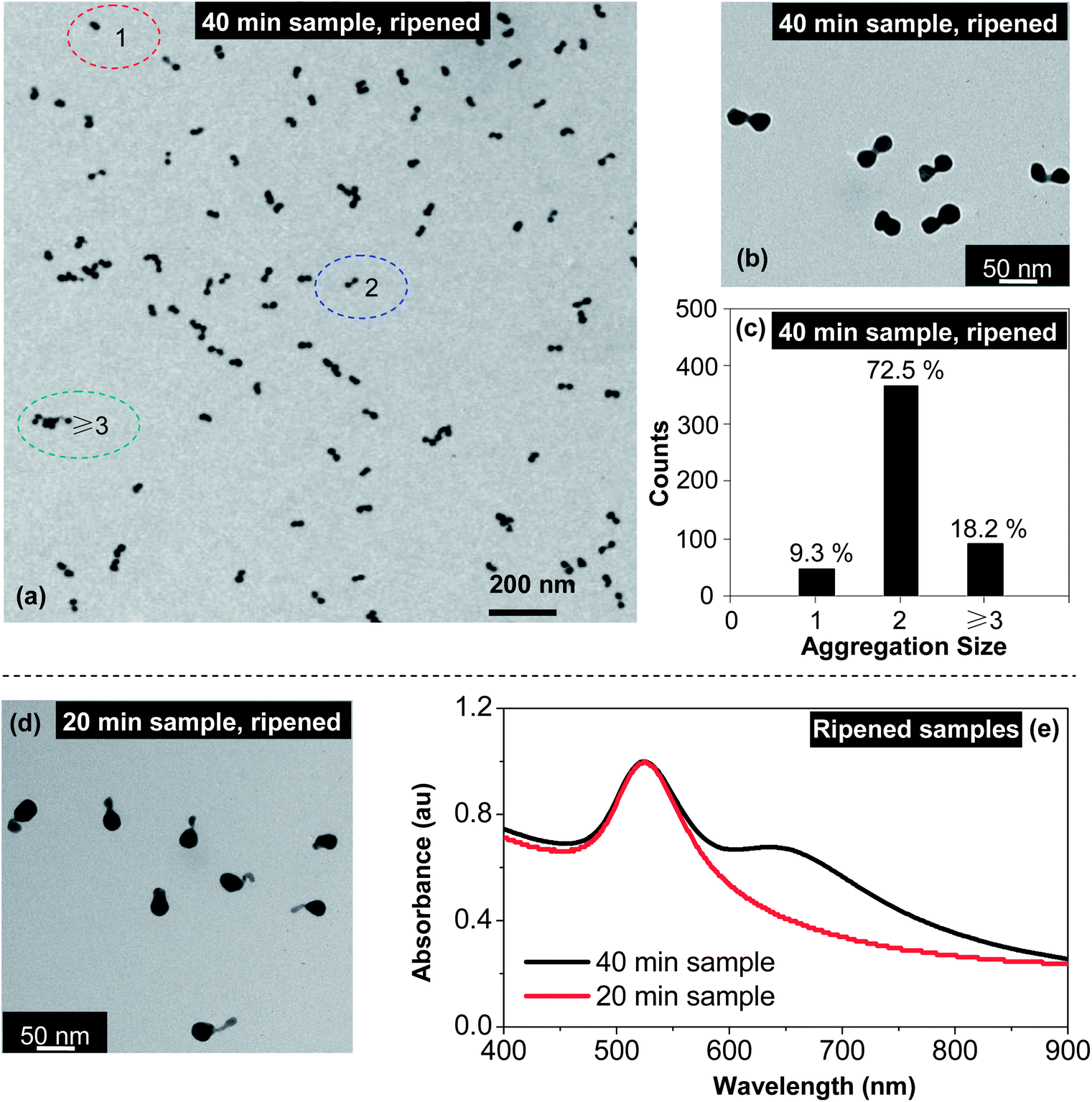 | ||
| Fig. 5 (a) A low magnification TEM image of a ripened sample (the sample was taken out of the “reduction” reaction at 40 min). (b) A relatively high magnification TEM image of the same sample in (a). (c) Statistical results for the same sample in (a) with ∼505 nanostructures. (d) TEM image of another ripened sample (the sample was taken out of the “reduction” reaction at 20 min. (e) UV-Vis spectra of two ripened samples shown in (a) and (d). | ||
Fig. 5(d) illustrates a hetero-dimer sample obtained by “intra-particle ripening” with KCl treatment, the original sample before the ripening process was the one associated with Fig. 3(c) (the 20 min sample with isolation through centrifugation). Evidently, the long tail of the tadpole-shaped nanostructures in Fig. 3(c) was mostly gone. It should be pointed out that there was room for further optimization for this type of hetero-dimers, namely a large seed with a small nanocrystal. As shown in Fig. 5(d), the degree of “intra-particle ripening” did not occur at the same rate for different nanostructures under the current conditions, resulting in hetero-dimers with a different fusing distance.
Fig. 5(e) shows the UV-Vis spectra for two samples associated with Fig. 5(b) and (d). For the 20 min sample, the most apparent change was the significant increase of the absorbance between 600 and 900 nm. Probably because one of the nanocrystals in this hetero-dimer sample was too small, the spectrum did not develop a clear two-band feature. Conversely, a two-band feature for the 40 min sample after ripening (Fig. 5(e)) was fully developed, and the main peak became somewhat blue shifted and narrowed in comparison to the corresponding spectrum before ripening (Fig. 3(a)).
Synthesis of dimers by variation of Au3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Au0 ratio
:Au0 ratio
Gold nanocrystal dimers with different morphology/size could also be made by variation of the initial Au3+:Au0 ratio. Fig. 6 illustrates four types of dimers synthesized with the same type of seeds and with similar reaction conditions except HAuCl4 concentration, i.e., Au3+:Au0 ratio. Different from most dimers described in the above subsections, the dimers in Fig. 6 were all directly isolated from the final “reduction” solution by a centrifugation procedure at ∼12000 and 8000 rpm using a benchtop centrifuge, and no separate “intra-particle ripening” was performed. This corresponds to the top pathway in Scheme 1.
 | ||
| Fig. 6 TEM images of gold nanocrystal dimers prepared under different Au3+:Au0 ratios. | ||
Within a dimer in Fig. 6, the size of the newly formed nanocrystal increased as HAuCl4 concentration increased, given that the size of the seeds was approximately constant. However, this size increase of the second nanocrystal within a dimer was not proportional to the amount of HAuCl4 added into each reaction. This implies that some of the monomers accumulated onto the seeds, instead of being completely applied for growth of the second nanocrystal in a dimer.
Structural analysis of the dimers
The structure of the nanostrucutres in this report was studied systematically using HRTEM. The seed nanocrystals were evidently multi-twined gold nanocrystals (Fig. S1, ESI†). The crystal domain orientation in tadpole-shaped nanostructures was found to be random along the long tail (data not shown), which is the same as that reported in our previous report.42Fig. 7 shows HRTEM images of a typical dumbbell-shaped nanostructure and a representative dimer. Fig. 7(a) and (b) revealed that both types of nanostructures were polycrystalline in nature, which is consistent with the “random-attachment” growth model. Fig. 7(a′) and (b′) are magnified images for the narrow connection regime between two ends of either dumbbell-shaped nanostructure or dimer. In Fig. 7(a′), the (200) lattice fringes were labeled at the boundary between the narrow connection and each of the ends. Clearly, the (200) lattice fringes at adjacent crystal domains were not perpendicular to each other (Fig. 7(a′)). This means that neither of the end nanocrystals was oriented-attached to the narrow connection between the two ends.
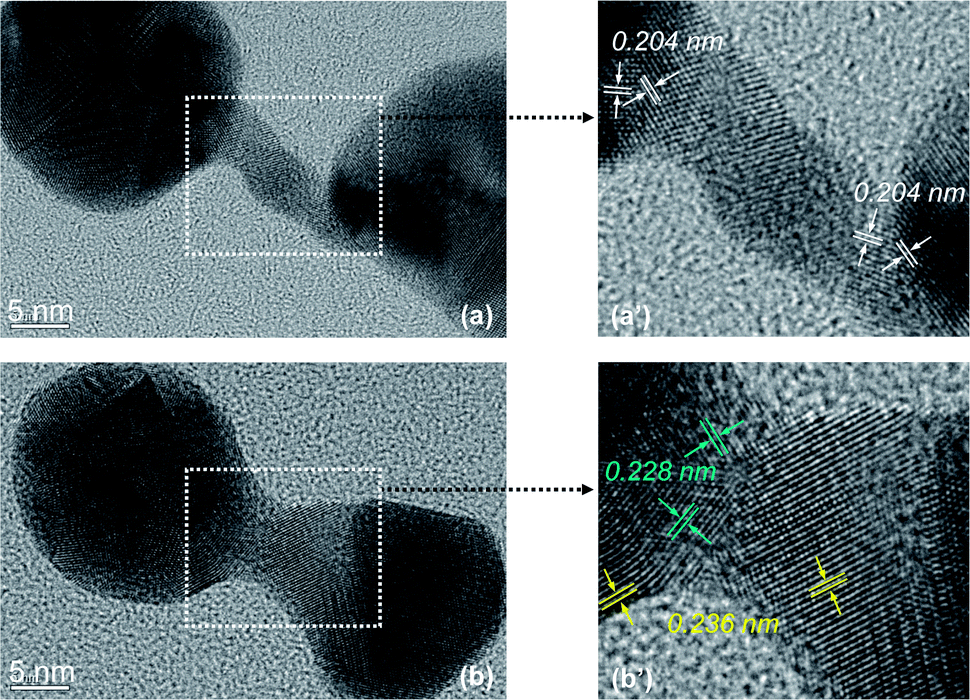 | ||
| Fig. 7 HRTEM images of gold nanocrystal dimers with specific morphologies. | ||
In addition to the non-oriented nature of the boundary between the end nanocrystal and the narrow yet short connection in the dimer (Fig. 7(b′)), two sets of lattice spacing were identified. One was 0.236 nm, which is consistent with the regular spacing between (111) lattice planes. The other was 0.228 nm, which should also be the spacing between (111) lattice planes. This slightly smaller spacing for the latter might be a result of significant lattice strain in such a complex nanostructure.
Overall, the HRTEM measurements presented in Fig. 7 revealed different types of lattice imperfection in the nanostructures disclosed in this report. Such imperfections were thought to be consistent with the “random-attachment” involved in the formation of the corresponding nanostructures.
Size dependent UV-Vis absorption and SERS of the dimers
Here, the size dependence shall be discussed with the size of one nanocrystal in the dimer, namely the seed nanocrystal, being fixed. In Fig. 8, three types of dimers were all grown with the seed nanocrystals being 22 nm as the final average size, and the size of the newly formed nanocrystal attached to the seed in a dimer was 28 nm, 22 nm, and 6 nm (from top to bottom, Fig. 8), respectively. For simplicity, these three types of dimers from top to bottom in Fig. 8 shall be described as “22–28 dimer”, “22–22 dimer”, and “22–6 dimer”, respectively. Among them, the “22–22 dimer” was a homo-dimer and the other two types were hetero-dimers.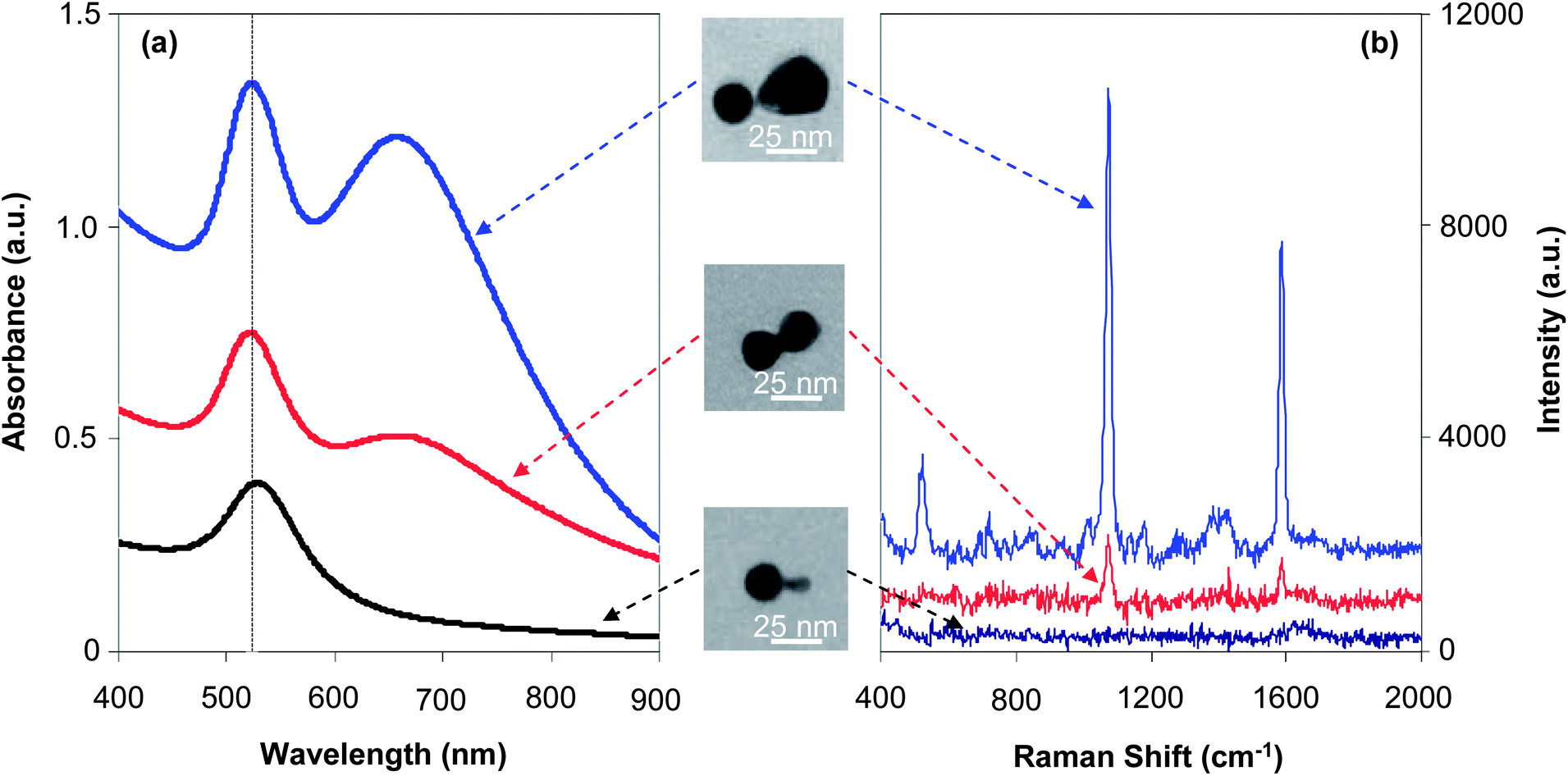 | ||
| Fig. 8 UV-Vis absorption spectra (a) and SERS spectra (b) recorded for three types of dimers with the same size of seeds. | ||
Fig. 8(a) illustrates the UV-Vis spectra of three types of dimers, which qualitatively reveals an expected evolution in terms of two-band absorption due to formation of hot spots. The two-band feature in the absorption spectrum of “22–28 dimers”—the largest one in the series—was well developed, with two peaks having similar peak heights, indicating a very strong plasmonic enhancement. For the medium sized dimer sample, i.e., “22–22 dimers”, the two-band feature was reasonably developed but the peak height of the low energy one was significantly lower than that of the main peak. The smallest dimers, namely “22–6 dimers”, only showed an absorbance increase in the 600–900 nm window without a clear indication of the low energy peak (also see Fig. S4, ESI†).
The main peak for all three types of dimers was nearly at the same position, which was almost the same as that of the seed nanocrystals (see one comparative example in Fig. S4, ESI†). Comparatively, the “22–28 dimers” and “22–22 dimers” had a quite similar peak width for the main peak, which was found to be narrower than that of the “22–6 dimer” sample (Fig. S4, ESI†). These results along with the much less developed two-band feature discussed in the above paragraph implied that “hot spots” would not be significant for the smallest dimers in the series.
Fig. 8(b) shows typical Raman spectra for the same series of dimers. Two strong peaks at 1072 and 1583 cm−1 are known characteristic Raman peaks for 4-mercaptobenzoic acid (4-MBA). The peak at 1072 cm−1 is a combination of the phenyl ring-breathing mode, C–H in-plane bending, and C–S stretching. The peak at 1583 cm−1 can be assigned to the phenyl ring-stretching vibration.28
The SERS measurements (Fig. 8(b)) were found to be consistent with the absorption properties discussed above (Fig. 8(a)) for the same series of dimers. The “22–28 dimer”—the largest in the series—showed very strong Raman peaks. Conversely, the Raman peaks were barely visible for the smallest dimer in the series, namely the “22–6 dimer”. Under the same conditions, the medium sized dimer (“22–22 dimer”) sample showed medium Raman signals in Fig. 8(b). This trend could be semi-quantitatively evaluated using a method reported in the literature.28 The Raman EF of the “hot spot” for different dimers were calculated as 8.2 × 103, 6.4 × 104, and 4.5 × 105 for “22–6 dimer”, “22–21 dimer”, and “22–28 dimer”, respectively. These EF values were found to be consistent with some values reported in the literature. For example, it was reported that the ensemble-averaged EFs at the hot spots of the dimers and trimers of silver-coated gold nanocrystals (∼20 nm for each nanocrystal) should be in the range of 105 to 107.35
It should be pointed out that the EF calculation based on the peak at 1072 cm−1 described above was quite rough (see details in Experimental). Generally, because of the non-spherical shape of both nanocrystals in a dimer and a certain degree of non-uniformity revealed by TEM measurements, the EF of the “hottest” molecule under optimal conditions should greatly exceed the estimated average values. In addition, it is well-known that the SERS effects could be sensitive to the wavelength of the excitation. Specifically, the “22–6 dimers” might become more effective using a short wavelength excitation that matches the plasmonic resonance. Judged by the UV-Vis spectra, however, the wavelength dependence of “22–6 dimers” and spherical seed nanocrystals would be similar. If this was the case, based on literature reports,46 the wavelength dependence should be insignificant in comparison to the EF differences found for three types of dimers.
Morphology dependent UV-Vis absorption and SERS of gold nanostructures
The morphology dependence of the optical properties of the dimers was briefly illustrated in Fig. 3 and 5. TEM measurements revealed that, for the 40 min sample obtained by isolation from the reaction solution without further “intra-particle ripening” by KCl, the morphology was dumbbell-shaped, and could also be considered to be homo-dimers with a large intra-particle distance (Fig. 3(e)). After the KCl treatment, the morphology of the nanostructures became that of a standard homo-dimer with two nanocrystals closely fused together (Fig. 5(b)). Comparing the UV-Vis absorption spectra of these two samples (Fig. 3(a) for before and Fig. 5(e) for after the KCl treatment), one could see dramatic changes, including the appearance of a two-band absorption feature, a blue-shifted main peak, and narrowing of the main peak. To further illustrate these dramatic changes of the absorption properties for these different dimers, a direct comparison was plotted in Fig. S5 (ESI†). We believe that the dramatic spectral differences shown in Fig. S5 (ESI†) could be a result of the spacing between the two nanocrystals, which is in good agreement with theoretical predictions.8,13,14,16 With both samples being homo-dimers of similar sizes, the results in Fig. S5 (ESI†) could be considered as a good example of the morphology-dependence of the optical properties of the dimers.Up to this point, “intra-particle ripening” has been described as being a key step for the construction of dimers. In fact, it could also be used for the destruction of the dimers. For clarity, destruction of existing dimers through “intra-particle ripening” shall be referred to as “destructive intra-particle ripening” in this report. Although “destructive intra-particle ripening” should be avoided for the synthesis of dimers, it could be employed for studying the morphology-dependence of the optical properties of gold nanostructures (Fig. 9).
The TEM pictures in Fig. 9 revealed that, upon “destructive intra-particle ripening”, a high-purity sample of dimers was converted to a mixture of short rods and spheroids. Simultaneously, the UV-Vis spectrum after the “destructive intra-particle ripening” changed dramatically (Comparing Fig. 9(a) and (d)). The two-band feature disappeared and the main peak became very broad. In addition, the absorbance beyond 700 nm almost dropped to baseline in Fig. 9(d). The spectral features in UV-Vis shown in Fig. 9(d) were thought to be reasonably consistent with gold rods with a small aspect ratio,47 and they imply that the plasmonic enhancements were largely reduced by the destruction of the dimer morphology.
Raman scattering measurements were in good agreement with removal of “hot spots” through the “destructive intra-particle ripening”. As shown in Fig. 9(c) and (f), the Raman peaks became barely visible after the “destructive intra-particle ripening”. For the relatively smaller dimers, though reduction of Raman signals was observed (Fig. S6, ESI†), the change was less significant than that shown in Fig. 9. This is consistent with the dramatic reduction of the effects of hot spot as the size of the dimers decreased as shown in Fig. 8.
Stability and yield
It is worth mentioning that, although “destructive intra-particle ripening” was demonstrated in Fig. 9, the dimers discussed in this report could be stored stably with their native surface passivation, which should be mainly citrates as suggested by Fig. 2. Neither polymer nor another coating was needed to retain their morphology as long as the nanostructures were isolated from the reaction solutions and redispersed in Na3Ci solution (Fig. 10). Given the relatively weak bonding of citrate ions on the surface of the dimers, the high purity dimers synthesized using the method reported here could be employed as stable SERS substrates for various molecules.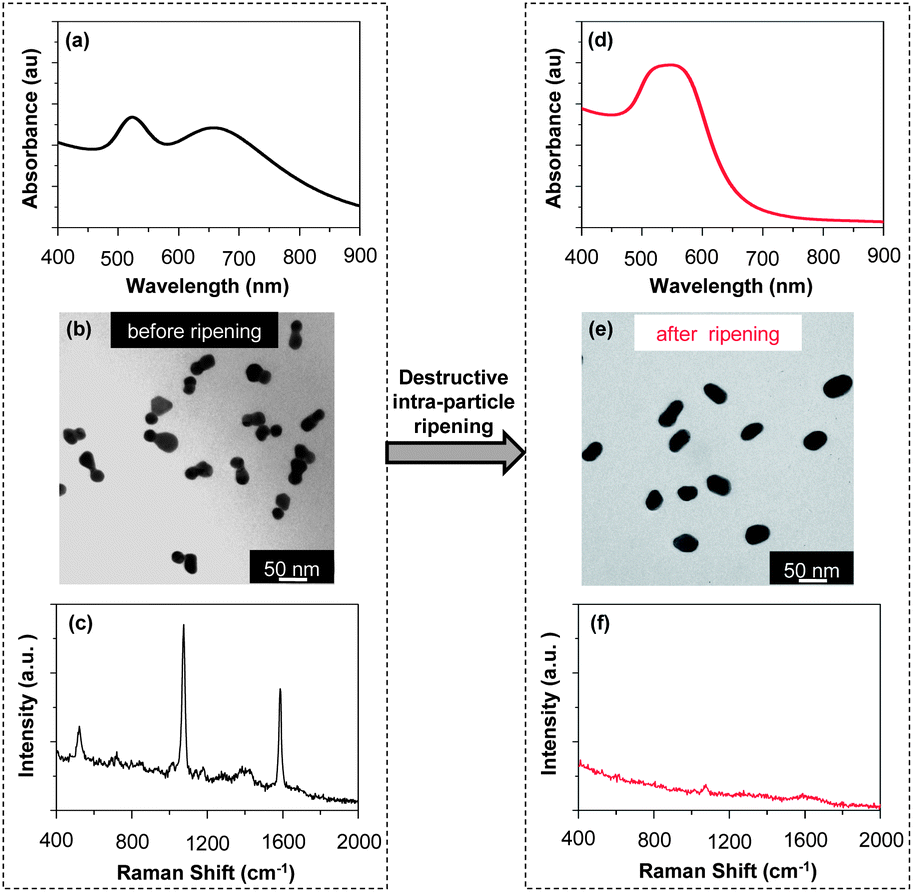 | ||
| Fig. 9 Morphology-dependent optical properties of gold nanostructures. | ||
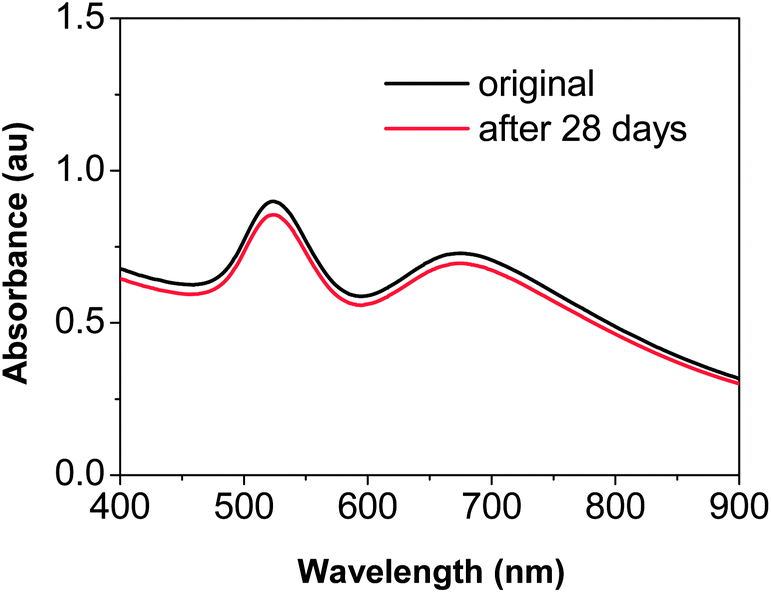 | ||
| Fig. 10 UV-Vis absorption spectra of the “22–28 dimers” before and after being stored at 4 °C for 28 days in 0.05% Na3Ci solution. | ||
As potential SERS substrates, the yield of the dimers in a specific reaction would be an important measure. For the dimers with a very small newly formed nanocrystal attached to the seeds, for example the 10 min sample in Fig. 3, the yield was relatively low. As long as the reaction proceeded to a sufficient degree, the resulting dimers were found to have a quite high yield. For instance, for the hetero-dimers giving the largest SERS in Fig. 8 (the “22–28 dimer”), the final yield of high-purity dimers was around 71%. This yield was estimated on the basis of nanocrystal seeds. It means that 71% of the nanocrystal seeds were found to form dimers after isolation, with the others being either individual dot-shaped or spheroid-shaped nanocrystals were removed by simple centrifugation treatments.
Conclusions
In summary, high-purity, high-yield, directly fused, and stable homo-dimers and hetero-dimers of gold nanocrystals were synthesized by judiciously controlling three elementary steps, namely “reduction”, “random attachment”, and “intra-particle ripening”. The UV-Vis absorption measurements unambiguously demonstrated a two-band feature for the dimers, indicating the existence of “hot spots” within those dimers. The two-band absorption spectra of dimers were further found to strongly depend on the size of both nanocrystals in a dimer and the intra-particle distance within a dimer. Consistent with the observation of the formation of “hot spots” by absorption measurements, very strong and size-dependent surface-enhanced Raman scattering was observed. This simple yet high-performance synthetic scheme should further open the door towards the synthetic control of the optical properties of noble metal nanostructures. For example, it might be possible to synthesize different types of “hot spot” nanostructures by the use of nanocrystals with rods, prisms, and other shapes as seeds. Such nanostructures should be of interest for both the fundamental understanding of plasmon coupling as well as for technical applications.Acknowledgements
This work was supported by the Natural Science Foundation of China (no. 20803029, 51072064).Notes and references
- S. Lal, S. Link and N. J. Halas, Nat. Photonics, 2007, 1, 641–648 CrossRef CAS.
- N. Berkovitch and M. Orenstein, Nano Lett., 2011, 11, 2079–3689 CrossRef CAS PubMed.
- J. Li, X. Tian, S. Li, J. R. Anema, Z. Yang, Y. Ding, Y. Wu, Y. Zeng, Q. Chen, B. Ren, Z. Wang and Z. Tian, Nat. Protoc., 2012, 8, 52–65 CrossRef PubMed.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- S. Link and M. A. El-Sayed, Annu. Rev. Phys. Chem., 2003, 54, 331–366 CrossRef CAS PubMed.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- E. Hao and G. C. Schatz, J. Chem. Phys., 2004, 120, 357–366 CrossRef CAS PubMed.
- J. P. Camden, J. A. Dieringer, Y. Wang, D. J. Masiello, L. D. Marks, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2008, 130, 12616–12617 CrossRef CAS PubMed.
- S. L. Kleinman, E. Ringe, N. Valley, K. L. Wustholz, E. Phillips, K. A. Scheidt, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2011, 133, 4115–4122 CrossRef CAS PubMed.
- S. L. Kleinman, B. Sharma, M. G. Blaber, A. I. Henry, N. Valley, R. G. Freeman, M. J. Natan, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2013, 135, 301–308 CrossRef CAS PubMed.
- A. Lombardi, M. P. Grzelczak, A. Crut, P. Maioli, I. Pastoriza-Santos, L. M. Liz-Marzán, N. D. Fatti and F. Vallée, ACS Nano, 2013, 7, 2522–2531 CrossRef CAS PubMed.
- F. Huang and J. J. Baumberg, Nano Lett., 2010, 10, 1787–1792 CrossRef CAS PubMed.
- J. A. Scholl, A. García-Etxarri, A. L. Koh and J. A. Dionne, Nano Lett., 2013, 13, 564–569 CrossRef CAS PubMed.
- J. M. Mcmahon, A. I. Henry, K. L. Wustholz, M. J. Natan, R. G. Freeman, R. P. Van Duyne and G. C. Schatz, Anal. Bioanal. Chem., 2009, 394, 1819–1825 CrossRef CAS PubMed.
- J. B. Lassiter, J. Aizpurua, L. I. Hernandez, D. W. Brandl, I. Romero, S. Lal, J. H. Hafner, P. Nordlander and N. J. Halas, Nano Lett., 2008, 8, 1212–1218 CrossRef CAS PubMed.
- A. M. Funston, C. Novo, T. J. Davis and P. Mulvaney, Nano Lett., 2009, 9, 1651–1658 CrossRef CAS PubMed.
- C. E. Talley, J. B. Jackson, C. Oubre, N. K. Grady, C. W. Hollars, S. M. Lane, T. R. Huser, P. Nordlander and N. J. Halas, Nano Lett., 2005, 5, 1569–1574 CrossRef CAS PubMed.
- S. Sheikholeslami, Y. W. Jun, P. K. Jain and A. P. Alivisatos, Nano Lett., 2010, 10, 2655–2660 CrossRef CAS PubMed.
- M. Ringler, T. A. Klar, A. Schwemer, A. S. Susha, J. Stehr, G. Raschke, S. Funk, M. Borowski, A. Nichtl, K. Kürzinger, R. T. Phillips and J. Feldmann, Nano Lett., 2007, 7, 2753–2757 CrossRef CAS PubMed.
- R. Sardar, T. B. Heap and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2007, 129, 5356–5357 CrossRef CAS PubMed.
- S. Bidault, F. J. G. de Abajo and A. Polman, J. Am. Chem. Soc., 2008, 130, 2750–2751 CrossRef CAS PubMed.
- T. J. Yim, Y. Wang and X. Zhang, Nanotechnology, 2008, 19, 435605–6 CrossRef PubMed.
- M. P. Busson, B. Rolly, B. Stout, N. Bonod, E. Larquet, A. Polman and S. Bidault, Nano Lett., 2011, 11, 5060–5065 CrossRef CAS PubMed.
- Y. Cheng, M. Wang, G. Borghs and H. Chen, Langmuir, 2011, 27, 7884–7891 CrossRef CAS PubMed.
- X. Wang, G. Li, T. Chen, M. Yang, Z. Zhang, T. Wu and H. Chen, Nano Lett., 2008, 8, 2643–2647 CrossRef CAS PubMed.
- L. Zhu, H. Wang, X. Shen, L. Chen, Y. Wang and H. Chen, Small, 2012, 8, 1857–1862 CrossRef CAS PubMed.
- W. Li, P. H. C. Camargo, X. Lu and Y. Xia, Nano Lett., 2009, 9, 485–490 CrossRef CAS PubMed.
- W. Li, P. H. C. Camargo, L. Au, Q. Zhang, M. Rycenga and Y. Xia, Angew. Chem., Int. Ed., 2010, 49, 164–168 CrossRef CAS PubMed.
- Y. Feng, J. He, H. Wang, Y. Y. Tay, H. Sun, L. Zhu and Y. Chen, J. Am. Chem. Soc., 2012, 134, 2004–2007 CrossRef CAS PubMed.
- Y. Hu and Y. Sun, J. Am. Chem. Soc., 2013, 135, 2213–2221 CrossRef CAS PubMed.
- X. Sun, S. M. Tabakman, W. S. Seo, L. Zhang, G. Zhang, S. Sherlock, L. Bai and H. Dai, Angew. Chem., Int. Ed., 2009, 48, 939–942 CrossRef CAS PubMed.
- S. Li, Z. Chang, J. Liu, L. Bai, L. Luo and X. Sun, Nano Res., 2011, 4, 723–728 CrossRef CAS.
- G. Chen, Y. Wang, L. H. Tan, M. Yang, L. S. Tan, Y. Chen and H. Chen, J. Am. Chem. Soc., 2009, 131, 4218–4219 CrossRef CAS PubMed.
- G. Chen, Y. Wang, M. Yang, J. Xu, S. J. Goh, M. Pan and H. Chen, J. Am. Chem. Soc., 2010, 132, 3644–3645 CrossRef CAS PubMed.
- L. Bai, X. Ma, J. Liu, X. Sun, D. Zhao and D. G. Evans, J. Am. Chem. Soc., 2010, 132, 2333–2337 CrossRef CAS PubMed.
- O. Akbulut, C. R. Mace, R. V. Martinez, A. A. Kumar, Z. Nie, M. R. Patton and G. M. Whitesides, Nano Lett., 2012, 12, 4060–4064 CrossRef CAS PubMed.
- P. H. C. Camargo, Y. Xiong, L. Ji, J. M. Zuo and Y. Xia, J. Am. Chem. Soc., 2007, 129, 15452–15453 CrossRef CAS PubMed.
- B. Lim, H. Kobayashi, T. Yu, J. Wang, M. J. Kim, Z. Y. Li, M. Rycenga and Y. Xia, J. Am. Chem. Soc., 2010, 132, 2506–2507 CrossRef CAS PubMed.
- G. Krylova, L. J. Giovanetti, F. G. Requejo, N. M. Dimitrijevic, A. Prakapenka and E. V. Shevchenko, J. Am. Chem. Soc., 2012, 134, 4384–4392 CrossRef CAS PubMed.
- J. H. Lee, G. H. Kim and J. M. Nam, J. Am. Chem. Soc., 2012, 134, 5456–5459 CrossRef CAS PubMed.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939–13948 CrossRef CAS PubMed.
- R. L. Penn and J. F. Banfield, Science, 1998, 281, 969–971 CrossRef CAS.
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2001, 123, 1389–1395 CrossRef CAS.
- L. Zhao, X. Ji, X. Sun, J. Li, W. Yang and X. Peng, J. Phys. Chem. C, 2009, 113, 16645–16651 CAS.
- J. Fang, S. Du, S. Lebedkin, Z. Li, R. Kruk, M. Kappes and H. Hahn, Nano Lett., 2010, 10, 5006–5013 CrossRef CAS PubMed.
- H. Wu, X. Ji, L. Zhao, S. Yang, R. Xie and W. Yang, Colloids Surf., A, 2012, 415, 174–179 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6. See DOI: 10.1039/c3sc52135d |
| This journal is © The Royal Society of Chemistry 2014 |