1,4-Disilacyclohexa-2,5-diene: a molecular building block that allows for remarkably strong neutral cyclic cross-hyperconjugation†
Julius
Tibbelin
a,
Andreas
Wallner
af,
Rikard
Emanuelsson
a,
Filip
Heijkenskjöld
b,
Martin
Rosenberg
ac,
Kaoru
Yamazaki
ad,
Djawed
Nauroozi
e,
Leif
Karlsson
b,
Raimund
Feifel
*b,
Roland
Pettersson
a,
Judith
Baumgartner
f,
Sascha
Ott
e and
Henrik
Ottosson
*a
aDepartment of Chemistry–BMC, Uppsala University, Box 576, 751 23 Uppsala, Sweden. E-mail: Henrik.Ottosson@kemi.uu.se
bDepartment of Physics and Astronomy, Uppsala University, Box 516, 751 20 Uppsala, Sweden. E-mail: Raimund.Feifel@physics.uu.se
cDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark
dDepartment of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan
eDepartment of Chemistry – Ångström laboratory, Uppsala University, Box 523, 751 20 Uppsala, Sweden
fInstitut für Anorganische Chemie, Technische Universität Graz, Stremayrgasse 9, A-8010 Graz, Austria
First published on 26th September 2013
Abstract
2,3,5,6-Tetraethyl-1,4-disilacyclohexa-2,5-dienes with either four chloro (1a), methyl (1b), or trimethylsilyl (TMS) (1c) substituents at the two silicon atoms were examined in an effort to design rigid compounds with strong neutral cross-hyperconjugation between π- and σ-bonded molecular segments arranged into a cycle. Remarkable variations in the lowest electronic excitation energies, lowest ionization energies, and the first oxidation potentials were observed upon change of substituents, as determined by gas phase ultraviolet (UV) absorption spectroscopy, ultraviolet photoelectron spectroscopy (UPS), and cyclic voltammetry. A particularly strong neutral cyclic cross-hyperconjugation was observed in 1c. Its lowest electron binding energy (7.1 eV) is distinctly different from that of 1b (8.5 eV). Molecular orbital analysis reveals a stronger interaction between filled π(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and π(SiR2) group orbitals in 1c than in 1a and 1b. The energy shift in the highest occupied molecular orbital is also reflected in the first oxidation potentials as observed in the cyclic voltammograms of the respective compounds (1.47, 0.88, and 0.46 V for 1a, 1b and 1c, respectively). Furthermore, 1,4-disilacyclohexadiene 1c absorbs strongly at 273 nm (4.55 eV), whereas 1a and 1b have no symmetry allowed excitations above 215 nm (below 5.77 eV). Thus, suitably substituted 1,4-disilacyclohexa-2,5-dienes could represent novel building blocks for the design of larger cross-hyperconjugated molecules as alternatives to traditional purely cross-π-conjugated analogues, and could allow for design of molecules with properties that are not accessible to those that are exclusively π-conjugated.
C) and π(SiR2) group orbitals in 1c than in 1a and 1b. The energy shift in the highest occupied molecular orbital is also reflected in the first oxidation potentials as observed in the cyclic voltammograms of the respective compounds (1.47, 0.88, and 0.46 V for 1a, 1b and 1c, respectively). Furthermore, 1,4-disilacyclohexadiene 1c absorbs strongly at 273 nm (4.55 eV), whereas 1a and 1b have no symmetry allowed excitations above 215 nm (below 5.77 eV). Thus, suitably substituted 1,4-disilacyclohexa-2,5-dienes could represent novel building blocks for the design of larger cross-hyperconjugated molecules as alternatives to traditional purely cross-π-conjugated analogues, and could allow for design of molecules with properties that are not accessible to those that are exclusively π-conjugated.
Introduction
π-Conjugation and σ-conjugation are well-established concepts in chemistry since many decades and compounds containing these conjugation topologies have been investigated for purely fundamental reasons as well as in the course of development in various applied areas.1–3 Conjugation in the classical sense is the stabilizing interaction between local orbitals of π-symmetry and occurs in molecules with alternating single and multiple bonds.4 π-Conjugation can further be divided into linear- or cross-conjugation depending on the connectivity in the bond alternating path. While a linearly conjugated compound provides strong delocalization across the entire π-bonded path, a cross-conjugated system only provides strong delocalization up until the crossing point.5 However, stabilizing conjugative interactions between adjacent orbitals are not restricted to π-bonded compounds, and σ-conjugation is observed in heavy alkanes, such as oligo- and polysilanes.2 Indeed, Mulliken and co-workers early emphasized that “the differences in conjugating power” between unsaturated and saturated groups are “quantitative rather than qualitative”.6 Yet, the combination of σ- and π-conjugation topologies into a strong σ/π-conjugation, or rather hyperconjugation, in neutral compounds is less explored,7 and even less exploited in applications. Herein, we report on a joint theoretical and experimental study of a compound class, the 1,4-disilacyclohexa-2,5-dienes (1, Fig. 1), in which π- and σ-bonded molecular segments are joined into a rigid cyclic structure and where the segments interact in a cross-hyperconjugated manner.8 These species could complement 1-silacyclopenta-2,4-dienes (siloles) which are extensively investigated organosilicon compounds of which the oligomers and polymers are used in organic electronics.9,10 | ||
| Fig. 1 The 1,4-disilacyclohexa-2,5-dienes (1) examined herein, and an example (2) of the previously investigated neutral hydrocarbons of Sekiguchi, Lee and co-workers which display significant neutral hyperconjugative σ/π-interaction (ref. 11). | ||
Hyperconjugation is exceptionally rare in neutral pure hydrocarbons, even though such a conjugation is observed in the recent perfluoroaryltetrahedranes of Sekiguchi, Lee and co-workers (2, Fig. 1) composed of one or two strained tetrahedranes which interact with an adjacent phenyl group.11 Strong interaction between σ- and π-bonded molecular segments is also observed in poly-1,1-siloles in which the monomers are connected via the Si atoms (3; Fig. 2) as these display significant coupling between the localized π*-orbitals of the diene segments and the delocalized σ*-orbitals (cf., linear combinations of local π(Si(SiR3)2 orbitals)) of the Si backbone, respectively.12 However, oligomeric and polymeric compounds composed of different segments which are either σ- or π-bonded, such as the linearly linked silanylene–thiophene oligomers 4, have drawbacks because the rotational flexibility about the connecting single bonds affects the σ- and π-orbital overlap and, thus, the conjugation strength.13–19 It should therefore be important to design neutral molecules and repeat units in oligomers/polymers with rigid structures that internally display strong hyperconjugative interactions.
 | ||
| Fig. 2 Earlier investigated oligomers and polymers with interacting σ- and π-bonded segments; the oligo/polysilole (3) and poly(silanylene-thiophene) (4), respectively (ref. 9–19). | ||
Recently, we examined the electronic structure of bis(phenylethynyl)methanes and silanes in dependence of the substituents at the central C or Si atom, and found that when the substituents at the central atom are σ-electron donor groups such as the trimethylsilyl group the electronic structures of these compounds resemble that of a regular cross-π-conjugated hydrocarbon.20,21 Such molecules have two hyperconjugated paths which are united by the π-symmetric group orbitals of the central ER2 moiety having the same function as the π-orbitals in a geminally connected CC double bond. These compounds can therefore be labelled as cross-hyperconjugated. Indeed, hyperconjugative interaction between ethylene fragments bonded to the silicon atom in dimethyldivinylsilane has been shown earlier with photoelectron spectroscopy,22 however, a SiMe2 segment only provides for very weak interaction.20 We now utilize these findings and investigate 1, a rigid cyclic structure composed of two ER2 moieties connected via two ethenylene groups and first described in 1963 by West and Bailey,23 as a molecule with potentially optimal hyperconjugative interaction. The all-carbon analogue, cyclohexa-1,4-diene, has previously been studied theoretically as well as experimentally, both in the ground and the excited states. A weak hyperconjugation has been demonstrated in these earlier studies and attributed to π(CH2) group orbitals composed of the C–H σ- and σ*-bond orbitals interacting with the π-orbitals of the CC double bond.24 Moreover, from UPS studies of the tetrakis(TMS) substituted cyclohexa-1,4-diene Bock and Kaim found a significant shift of the HOMO to a higher electron binding energy when compared to the parent cyclo-1,4-hexadiene.25
The silole ring has six electrons in orbitals of π-character; four in the π-orbitals of the 1,3-butadiene segment and two in the π(SiR2) group orbital. Yet, despite the 6π-electron systems, the geometries of various siloles indicate that cyclic conjugation is modest.26,27 In this context it is particularly noteworthy that the electronic structure of siloles is valence isolobal to that of pentafulvenes (Fig. 3), cross-π-conjugated hydrocarbons in which the optical properties and degree of aromaticity can be varied extensively through substitution.28–31
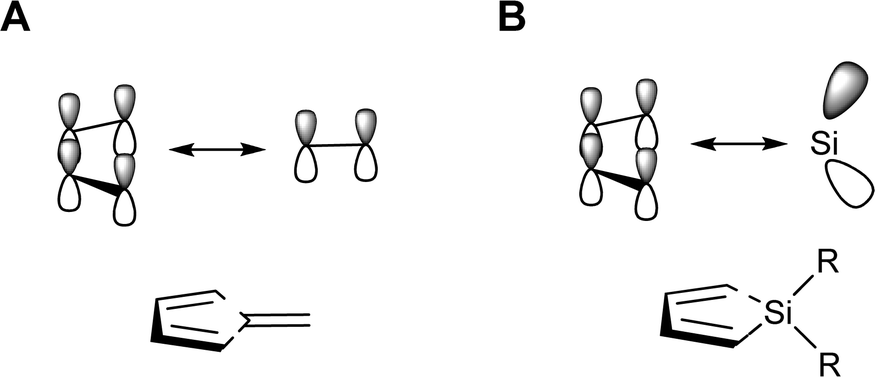 | ||
| Fig. 3 Schematic drawings of the valence isolobal analogy of the interactions between the π-symmetric basis orbitals of (A) pentafulvene and (B) silole, respectively. | ||
Our hypothesis is now that 1 is a good template for the design of neutral cyclic cross-hyperconjugated molecules. In a similar way that the π-orbital interaction in siloles is valence isolobal with that of pentafulvenes, the π-orbital interaction in 1 is isolobal to that between the π-orbitals of the endocyclic and the exocyclic CC double bonds in para-xylylene (5, Fig. 4). Para-xylylene is a cyclic cross-π-conjugated compound,24 and by analogy, properly substituted derivatives of 1 are potentially cyclic cross-hyperconjugated. The question thus arises whether 1,4-disilacyclohexa-2,5-diene can provide a template for the design of optically and electronically useful compounds.
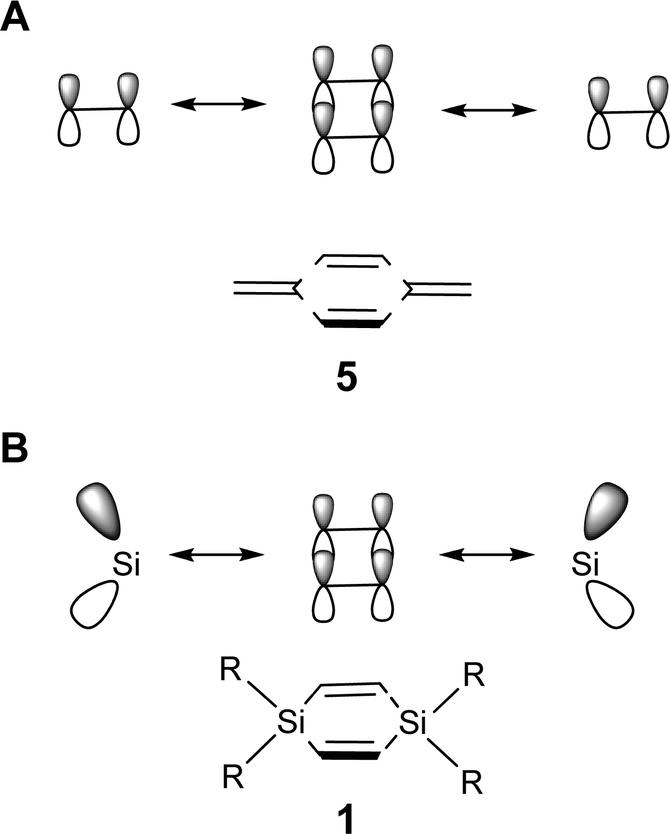 | ||
| Fig. 4 Schematic drawings of the valence isolobal analogy of the interactions between the π-symmetric basis orbitals of (A) para-xylylene (5) and (B) 1,4-disilacyclohexa-2,5-diene (1), respectively. | ||
The earlier studies on the parent 1,4-disilacyclohexa-2,5-diene (1) have focused on synthesis rather than spectroscopy.32–35 In the present study, 1,4-disilacyclohexa-2,5-dienes with either Cl (1a), Me (1b), or SiMe3 (1c, TMS) substituents at the Si atoms and ethyl substituents at the sp2 hybridized C atoms (Fig. 1) were synthesized and examined by X-ray crystallography, UV photoelectron spectroscopy (UPS), cyclic voltammetry and gas phase UV absorption spectroscopy. As a reference for an essentially non-cross-hyperconjugated compound, the all-carbon analogue, i.e., cyclohexa-1,4-diene (6), was also examined by gas phase UV absorption spectroscopy. The computational part of our investigation was also extended to a larger set of 1,4-disilacyclohex-2-enes and related compounds (Fig. 5) so as to reveal the effect of unifying two cross-hyperconjugated moieties into a cycle as compared to ordinary linear hyperconjugation and acyclic cross-hyperconjugation, the latter exemplified by the bis(ethynyl)silanes.20,21 Properties, such as electronic transitions, ionization energies, and how these are coupled to the substitution pattern of the different compounds, are analysed and discussed. Our aim is to deduce a new monomer unit which in an optimal manner combines σ- and π-bonded segments into a rigid and strongly cross-hyperconjugated cyclic framework. This unit could represent a novel structural motif to be used in oligomers and polymers for various optical and/or electronic applications, similar to the silole unit.
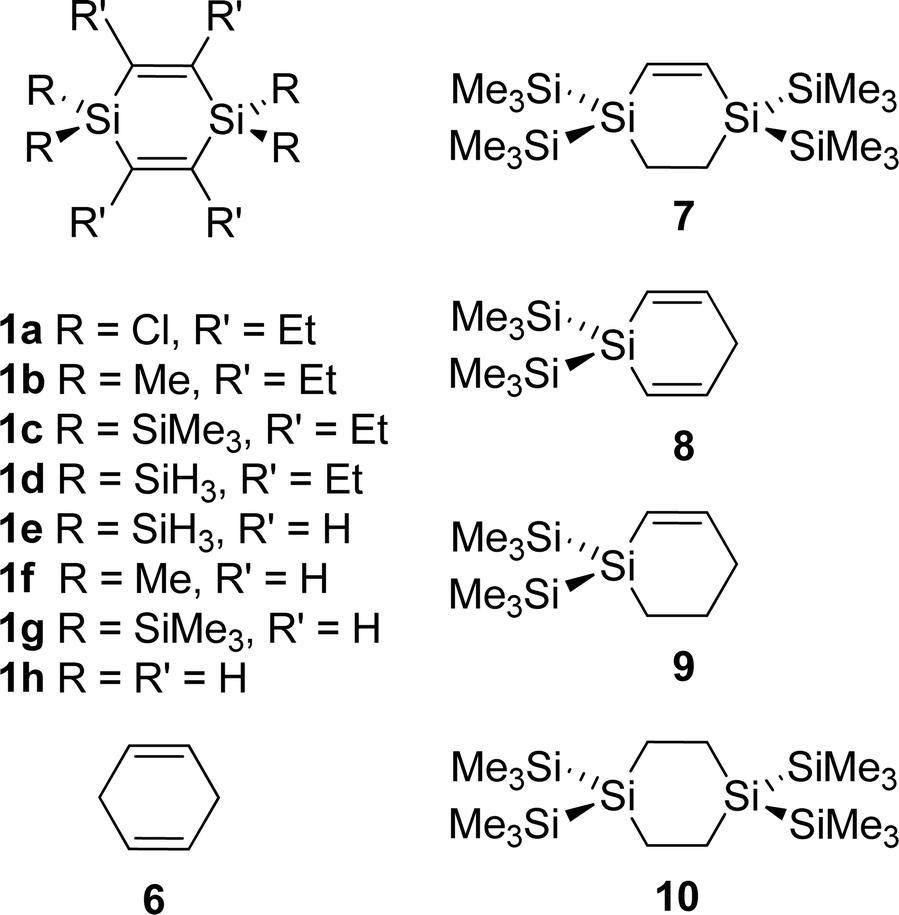 | ||
| Fig. 5 The substituted 1,4-disilacyclohexa-2,5-dienes (1a–h), the all-carbon reference cyclohexa-1,4-diene (6), and model compounds (7–10) explored computationally to examine the extent of cyclic cross-hyperconjugation. | ||
Results and discussion
The goal of the presently reported investigation is to find ways to maximize the neutral cyclic cross-hyperconjugative interaction between the local π(CC) and π(ER2) orbitals in 1,4-disilacyclohexa-2,5-dienes 1a–c. For that reason we first discuss the qualitative molecular orbital (MO) diagram and the calculated MOs before progressing to the experimental data. At the end we compare, through quantum chemical calculations, the strongly cross-hyperconjugated 1c with several model compounds (7–10) which allows for an evaluation of the cyclic cross-hyperconjugation in 1c as compared to hyperconjugation and acyclic cross-hyperconjugation. We also discuss what effect the rigidification in 1, when compared to the previously studied bis(phenylethynyl)methanes and silanes,20,21 has on the electronic structure and how this could be exploited in future molecular design.
Molecular orbitals
Fig. 6 shows the MOs of π-symmetry and considers a D2h symmetric 1,4-disilacyclohexa-2,5-diene with substituents R and R′ at the silicon and carbon atoms, respectively. These MOs are derived through combinations of suitable local π-orbitals of the two CC bonds combined into a 2 × (CC) fragment (marked blue in Fig. 6) and the two SiR2 moieties of a 2 × (SiR2) fragment (marked red). The interaction includes the symmetry-adapted in-phase and out-of-phase combinations of the four πCC and π*CC orbitals of 2 × (CC) with the symmetry-adapted in-phase and out-of-phase combinations of the four π(SiR2) and π*(SiR2) orbitals of the 2 × (SiR2) fragment. For further details on how this diagram was constructed and discussion about the unoccupied orbitals, see the ESI.†
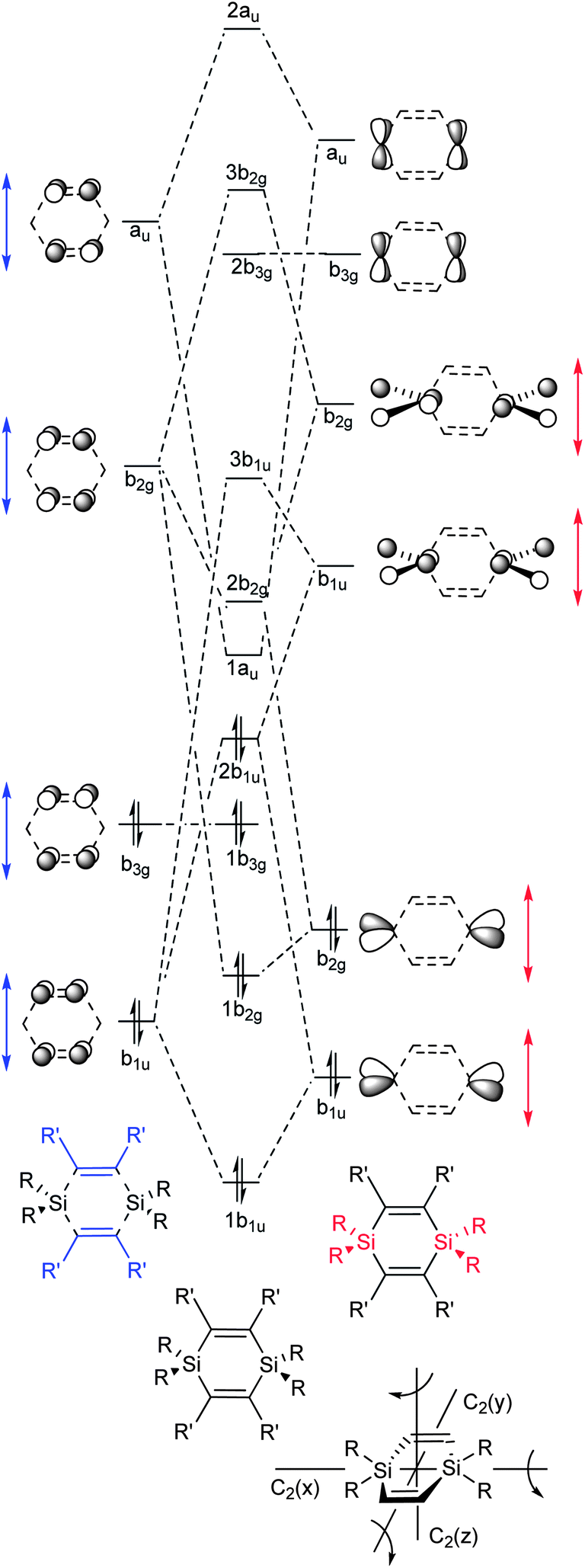 | ||
| Fig. 6 Qualitative molecular orbital (MO) diagram of D2h symmetric 1,4-disilacyclohexa-2,5-diene with the lowest few occupied and unoccupied MOs of π-character constructed from suitable fragment orbitals. Red arrows indicate changes in fragment orbital energies in dependence of substituents R and blue arrows indicate changes of substituents R′. The orbitals are labelled in accordance with the irreducible representations of the D2h point group. The three C2 rotational axes are arranged as shown in the structure in the lower right corner. | ||
From Fig. 6 it is clear that the energy of MO 2b1u will vary depending on the energy of the b1u group orbital of 2 × (SiR2) relative to that of the b1u group orbital of 2 × (CC). Thus, the less electronegative R becomes, the higher the energy of this MO will become. The variation in orbital interaction strength can be recognized from the suitable π-symmetry orbitals of 3-hexene and H2SiR2. At the B3LYP/6-31G(d) level the π(CC) of 3-hexene is located at −6.40 eV whereas the π(SiR2) of H2SiR2 is found at −11.45 (R = Cl), −8.38 (R = Me), and −6.53 eV (R = TMS) (see ESI† for orbital plots). With R = TMS, 2b1u can thus be expected to be of particularly high energy, and indeed, this orbital becomes HOMO for 1c (orbital 2b1, Fig. 7). The 1b3g MO, on the other hand, is not equally affected by the choice of the substituents at Si as it is primarily localized to the CC bonds. This orbital should instead vary in energy upon a change of substituents R′.
 | ||
| Fig. 7 Energy variations of the two occupied frontier molecular orbitals 1b3 and 2b1 and the two unoccupied frontier orbitals 1a and 2b2 of 1a–c calculated at the B3LYP/6-31G(d) level. | ||
For 1a–c, the HOMO − 1, HOMO, LUMO, and LUMO + 1 correspond to either of the 1b3g, 2b1u, 1au and 2b2g orbitals of Fig. 6, and as the symmetry is reduced from D2h to D2 the four MOs become 1b3, 2b1, 1a, and 2b2. These four MOs of 1c are displayed in Fig. 8, and which orbital is HOMO (1b3 or 2b1) and which one is LUMO (1a or 2b2) depends on R (Fig. 7). It is noteworthy that no occupied MO with σ-symmetry is found between the 2b1 and 1b3 orbitals for any of the three compounds. Furthermore, there is a striking similarity of the calculated MOs of 1c with those of para-xylylene (5, Fig. 8) as only LUMO and LUMO + 1 change place between the two compounds. Two further items can be noted in particular. First, HOMO of 1c (−5.24 eV) is nearly isoenergetic to the HOMO of 5 (−5.35 eV) at the B3LYP/6-31G(d) level. Secondly, the a-symmetric orbital which is LUMO + 1 in 5 can interact with the a-symmetric group orbital of 2 × (SiR2) composed of the two 3d(Si) AOs of 1c. As a consequence, this MO is lowered in energy so that it becomes the LUMO of 1c.
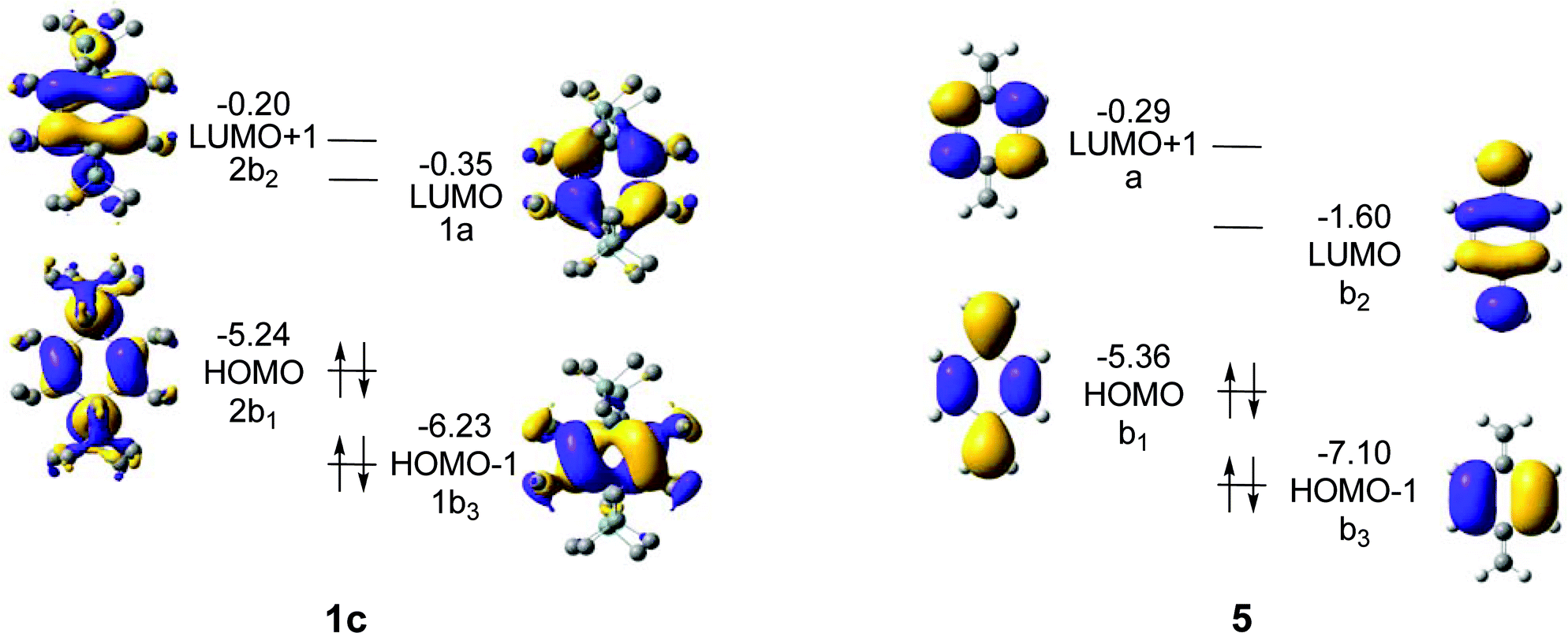 | ||
| Fig. 8 Frontier orbitals, orbital energies in eV, and orbital symmetries of 1c and para-xylylene (5), as given for the D2 point group, calculated at the B3LYP/6-31G(d) level. The hydrogen atoms in 1c are omitted for clarity. | ||
When going from 1a to 1b, all four MOs move up in energy, but the energy change is slightly larger for the occupied 2b1 and the unoccupied 2b2 than for 1b3 and 1a, reflecting the fact that the first two MOs directly involve the substituents R. However, the most significant change occurs when going from 1b to 1c because a large split in the orbital energies of 1b3 and 2b1 is observed; the latter is raised from −6.24 to −5.24 eV while 1b3 merely changes from −6.31 to −6.23 eV. It can furthermore be noted that 1b3 has no 3d(Si) AO contribution from 2 × (SiR2), in line with the qualitative MO-diagram of Fig. 6.
For LUMO, with a nodal plane coinciding with the SiR2 plane, the variation in energy of this MO with R is the smallest among the frontier orbitals of Fig. 7.
Thus, the qualitative MO diagram agrees with the quantitative computations because the 2b1u orbital (HOMO of 1b and 1c at the B3LYP level) changes in energy by a change of substituents R. In contrast, the LUMOs do not vary as extensively with R, and for this reason there are large differences in the HOMO-LUMO energy gap (ΔεH-L) between the three compounds, with values of 5.47 (1a), 6.03 (1b), and 4.89 eV (1c) at B3LYP/6-31G(d) level. Taking into account the earlier reported ΔεH-L of 6.43 eV for the parent species at the same level of computation,36 one may argue that the 1,4-disilacyclohexa-2,5-diene can be a highly valuable template for extensive variations in electronic and optical properties.
Synthesis
The synthesis of 1a is based on Jung's procedure,37 and subsequent addition of MeLi or TMSCl and lithium yielded 1b and 1c, respectively (Scheme 1).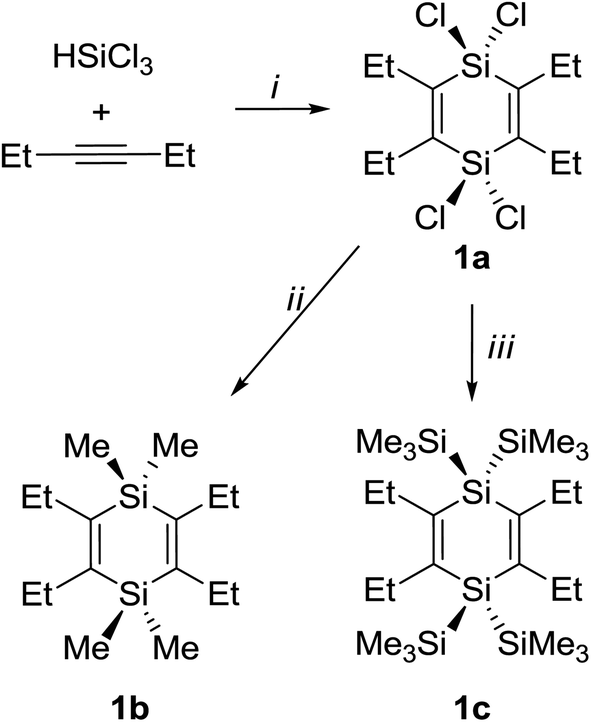 | ||
| Scheme 1 Reagents and conditions: (i) Bu4PCl, 180 °C (sealed tube reaction), 10 h, 75%; (ii) MeLi, Et2O, −78 °C to r.t., 12 h, 68%; (iii) TMSCl, Li, THF, −78 °C to r.t., 13 h, 71%. | ||
Geometric structure
Here we primarily discuss the geometrical parameters of 1c. The geometries of 1a and 1b are discussed in the ESI.†The crystal structure of 1c reveals that this 1,4-disilacyclohexa-2,5-diene adopts a slight twist-chair conformer (1c-I, Fig. 9) with Ci symmetry and C–Si–CC dihedral angles within the ring of approximately 13°. B3LYP/6-31G(d) calculations show that the non-planar structure is a result of the steric bulk as replacement of the Et groups by smaller Me groups leads from a D2 to a D2h symmetric molecule with a planar ring. These calculations reveal that a second conformer with the Et groups arranged in an up-down-up-down fashion (conformer 1c-II, Fig. 9) is essentially isoenergetic with 1c-I when based on the calculated free energy at 298 K. However, M06-2X/6-311G(d), a dispersion corrected DFT method suitable to handle sterically congested molecules, indicates that conformer 1c-II is 2.0 kcal mol−1 higher in energy than a C1 symmetric conformer of 1c-I (the Ci symmetric conformation of 1c-I is a second-order saddle point at 0.6 kcal mol−1 higher energy). Yet, both conformers should be populated at the elevated temperatures of the gas phase UV absorption and UPS spectral measurements (up to 350 °C).
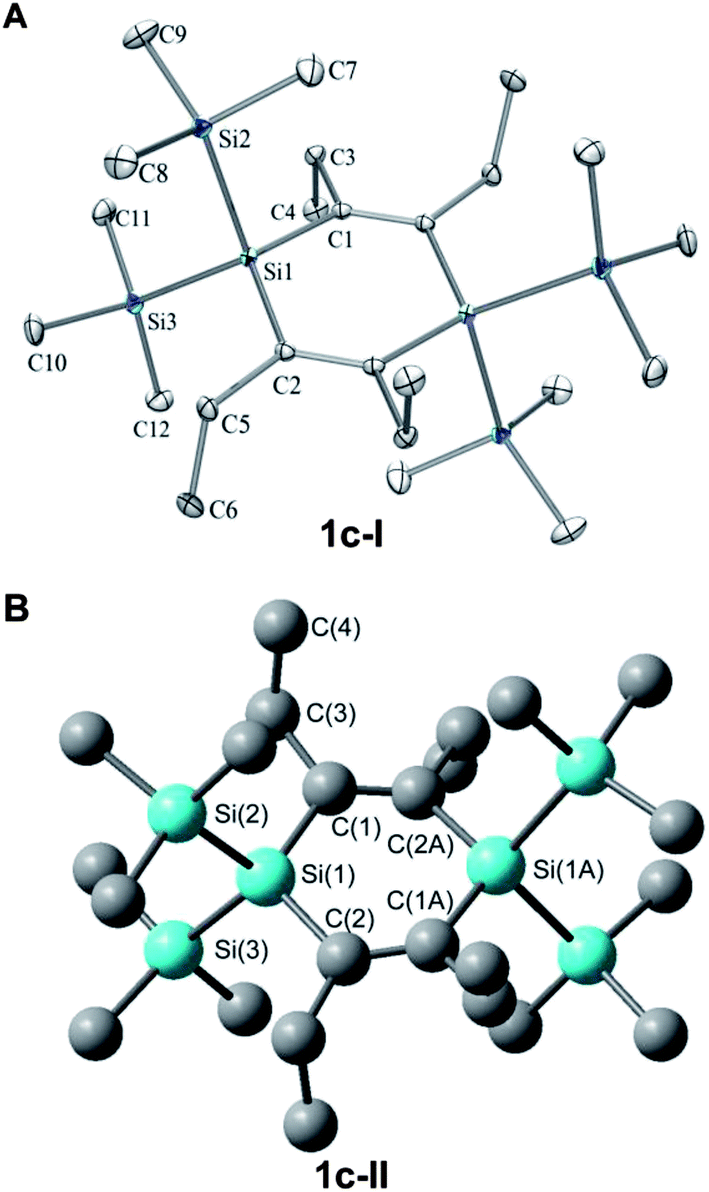 | ||
| Fig. 9 (A) Crystal structure of 1c (conformer 1c-I) with the measured data (normal print), and the B3LYP/6-31G(d) calculated data (in parentheses normal print), and (B) the calculated geometry of the conformer 1c-II (lower structure, data in parentheses italics). Selected bond lengths (Å) and angles (deg), hydrogens omitted for clarity: Si(1)-C(1) 1.8845(13) (1.907, 1.905), Si(1)-Si(2) 2.3685(6) (2.410, 2.402), Si(1)-Si(3) 2.3690(6) (2.400, 2.402), C(1)-C(2A) 1.3456(18) (1.361, 1.361), C(1)-C(3) 1.5297(16) (1.533, 1.534), C(3)-C(4) 1.5307(19) (1.5421, 1.542), Si(2)-Si(1)-Si(3) 109.33(3) (110.04, 110.36), Si(1A)-Si(1)-Si(2) 140.33 (137.06, 124.82), Si(1A)-Si(1)-Si(3) 110.33 (112.90, 124.82), Si(2)-Si(1)-C(1) 113.66(4) (112.51, 111.28). | ||
Overall, there is good agreement between the crystal structure and the calculated structure of 1c (selected bond distances and angles are given in the caption of Fig. 9). The only notable differences are the B3LYP/6-31G(d) bond lengths, which are slightly longer than the experimental ones, in line with earlier observations.38
The important geometrical parameters for the evaluation of the potential cross-hyperconjugation are the CC and Si–C bond lengths. A discussion of these bond lengths should be carried out in comparison with those of cross-π-conjugated para-xylylene 5. In the X-ray crystal structure of 1c-I the CC double bonds (1.346 Å) are slightly longer than ordinary CC double bonds (1.33 Å),39 and the Si–C bond lengths (1.885 Å) of the ring are moderately elongated as compared to regular Si–C single bonds (1.87 Å).40 The elongation of the CC bonds partially stems from steric congestion between the substituents as revealed through comparisons between the 1,4-disilacyclohexa-2,5-dienes 1d–h (see ESI†). There is also a hyperconjugative component that impacts the geometry, and this is revealed through a comparison with 5. At the B3LYP/6-31G(d) level the CC bond lengths of 1c are longer than those calculated for the cross-π-conjugated 5 (1.361 vs. 1.349 Å, respectively). Replacement of the ethyl groups (1g, Fig. 5) with hydrogen atoms leads to a shortening of the CC bonds to 1.351 Å, i.e., essentially identical lengths as found for 5. In contrast, the two isolated CC bonds in cyclohexa-1,4-diene 6 (1.335 Å) are significantly shorter than in 1g. The calculated CC bonds in 1g are also longer than those of the parent 1,4-disilacyclohexa-2,5-diene (1h), which are 1.344 Å. Thus, the CC bond elongations observed in 1c to some extent stem from cross-hyperconjugation. This observation is also true at higher levels of computations because CCSD/6-311G(d) calculations show that 6 has CC bond lengths of 1.339 Å compared to 1,1,4,4-tetrasilyl substituted 1e at 1.355 Å and cross-π-conjugated 5 at 1.349 Å.
Photoelectron spectroscopy
The experimental verification of the substituent effect on the electronic structures of 1,4-disilacyclohexa-2,5-dienes was obtained through photoelectron and UV absorption spectroscopies, as well as through cyclic voltammetry. The cationic valence electronic states of 1b and 1c were studied by conventional photoelectron spectroscopy using HeIα radiation at hν = 21.22 eV for ionization. Interpretation of the spectra were made on the basis of outer valence Green's function (OVGF) calculations at the OVGF/6-311+G(d)//B3LYP/6-31G(d) level. The D2 symmetric conformers 1b-II and 1c-II were used to facilitate rationalization in terms of qualitative MO-theory. The ionization energies of the Ci symmetric conformers I differ minutely, and tables which enable comparisons between conformers I and II are given in the ESI.†With regard to 1b, the valence photoelectron spectrum shows a well-defined onset at 7.9 eV followed by broad rounded bands (Fig. 10). The computed energies group around the spectral features so that a rather good understanding is achievable despite the strong overlap of bands. The experimental and calculated energies for the lower states are summarized in Table 1 along with our interpretations (for a complete table see the ESI†). The first band with a peak maximum at 8.5 eV represents ionization from three different orbitals that are close in energy according to the computations. The two lowest of these are π-type orbitals with b1 and b3 symmetries, in line with the qualitative MO-diagram, while the third is a σ-orbital. The second band (structural feature 3) at 9.4 eV represents a single σ-type orbital of b3 symmetry, whereas the third band with a peak maximum at 10.3 eV corresponds to ionization from four different orbitals, pair-wise of π- and σ-symmetry, respectively. The former two can be characterized as in-phase (b1) and out-of-phase (b2) combinations of the two π(SiMe2) group orbitals, where the first also has contributions from π(CC) (cf.Fig. 6).
 | ||
| Fig. 10 The photoelectron spectrum of 1b excited using HeIα radiation at 21.22 eV. The bands are numbered as in Table 1. Ionization energies calculated at the OVGF/6-311+G(d)//B3LYP/6-31G(d) level are included as bars on the energy axis. | ||
| Structure number | Binding energy (exp) | Binding energy (calc) | Assignment (orbital type) | Comment |
|---|---|---|---|---|
| a Calculated at ROVGF/6-311+G(d)//B3LYP/6-31G(d) level. | ||||
| 1 | 7.9 | Onset | ||
| 1 | 8.5 | 8.20 | b1 (π) | Peak max |
| 8.27 | b3 (π) | Peak max | ||
| 2 | 8.9 | 8.38 | b1 (σ) | Shoulder |
| 3 | 9.4 | 9.24 | b3 (σ) | Peak max |
| 4 | 10.3 | 10.11 | a (σ) | Peak max |
| 10.26 | b2 (π) | |||
| 10.32 | b2 (σ) | |||
| 5 | 10.7 | 10.60 | b1 (π) | Shoulder |
The pressure that could be obtained in the ionization chamber for 1c was much lower than for 1b. Due to these experimental constraints we recorded the spectral region between 5.5 eV and 9.5 eV several times and summed the individual spectra (Fig. 11). However, the region above 9.5 eV was included in only one of the recordings. The statistics are therefore not as good as for 1b, in particular in the higher binding energy region where signals from remaining H2O and N2 in the spectrometer are present. Yet, in the lower energy range the spectrum seems to be free of such influences. Some broad structures can be identified in this part, and their energies are given along with orbital characters in Table 2.
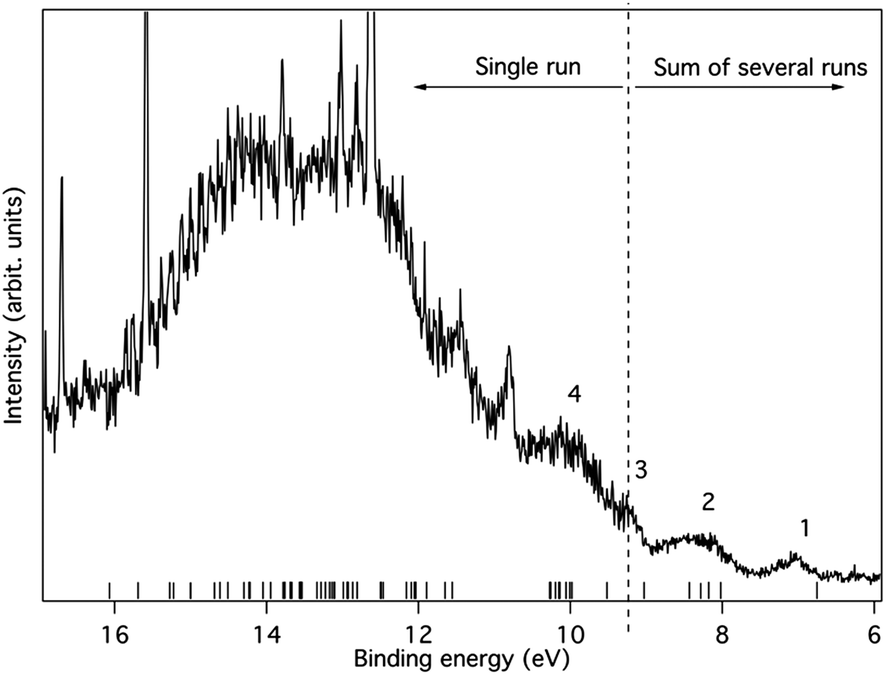 | ||
| Fig. 11 The photoelectron spectrum of 1c excited using HeIα radiation at 21.22 eV. The bands are numbered in accordance with Table 2. Ionisation energies calculated at the OVGF/6-311+G(d)//B3LYP/6-31G(d) level are included as bars on the energy axis. | ||
| Structure number | Binding energy (exp) | Binding energy (OVGF) | Assignment (orbital type) | Comment |
|---|---|---|---|---|
| a Calculated at the ROVGF/6-311+G(d)//B3LYP/6-31G(d) level. | ||||
| 1 | 6.7 | Onset | ||
| 7.1 | 6.76 | b1 (π) | Peak max | |
| 2 | 7.6 | Onset | ||
| 8.3 | 8.02 | b3 (π) | ||
| 8.18 | b2 (π) | |||
| 8.28 | b1 (σ) | |||
| 8.43 | b3 (σ) | |||
| 3 | 9.3 | 9.03 | a (σ) | |
| 9.52 | b1 (π) | |||
| 9.98 | b2 (σ) | |||
The first band is located at 7.1 eV (peak maximum), and according to the calculations it corresponds to the ionization from a single π-orbital of b1 symmetry. It should particularly be noted that the energy is substantially lower than for any orbital of 1b, suggesting that the influence of the SiMe3 substituent character is significant. Thus, in line with the MO-theoretical description of Fig. 6, the Si(SiMe3)2 segment provides local π(ER2) orbitals which are matched energetically with the π-orbitals of the 2 × (CC) fragment, pushing up the MO which is the out-of-phase combination of the b1 symmetric group orbitals of the 2 × (CC) and 2 × (SiR2) segments when compared to 1b. This confirms experimentally the large energy difference between HOMO and HOMO − 1 of 1c, as compared to 1a and 1b, observed in the B3LYP calculations (cf.Fig. 7).
The second band is broader and stronger, indicating ionization from more than one orbital. The peak maximum is observed at approximately 8.3 eV, and the calculations put four orbitals in this energy range. These orbitals are mostly of π-character and primarily located on the ring. The photoelectron band is therefore expected to resemble the outermost band of 1b, both in energy and general shape, due to the mutual similarity between the orbitals. This similarity is indeed observed as seen in Fig. 12 where the lower binding energy regions of the photoelectron spectra of 1b and 1c are shown in comparison to the numerical results. The only notable difference between the two compounds is found in the energies needed for ionization from the HOMOs.
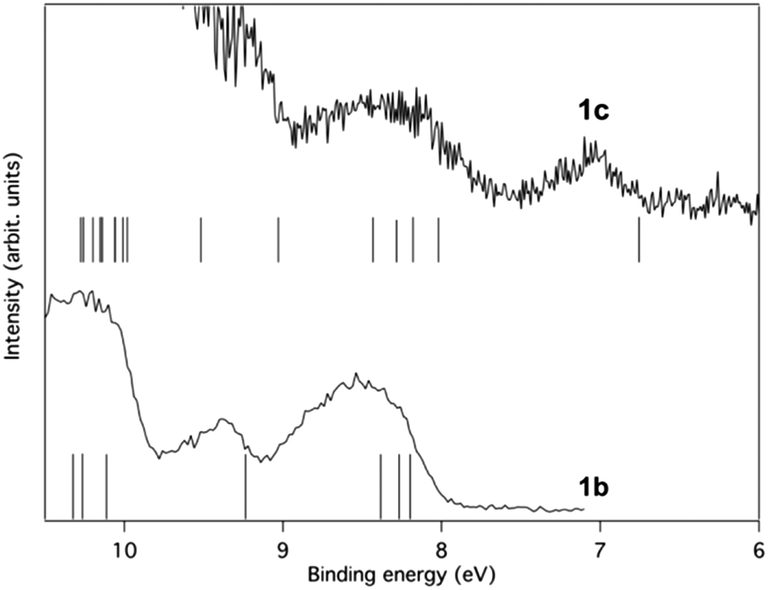 | ||
| Fig. 12 A comparison of the lower binding energy regions of the photoelectron spectra of 1b and 1c excited using HeIα radiation at 21.22 eV. Ionisation energies calculated at the OVGF/6-311+G(d)//B3LYP/6-31G(d) level are included as bars on the energy axis. | ||
Cyclic voltammetry
To provide further evidence for the role of the substituents on the frontier orbital energies, compounds 1a–1c were investigated by cyclic voltammetry. Fig. 13 displays the anodic scan of compounds 1a (black), 1b (blue) and 1c (red). All three compounds show electrochemically irreversible oxidation processes at 1.47, 0.88 and 0.46 V vs. Fc/Fc+, respectively. Thus, the oxidative peak potential of 1c is cathodically shifted by 400 mV compared to that of 1b and by 1 V relative to 1a. These findings strongly support the results obtained in the valence photoelectron spectroscopy as well as in the computational studies. The findings also hint to extensive variations in redox properties that could be useful for future applications.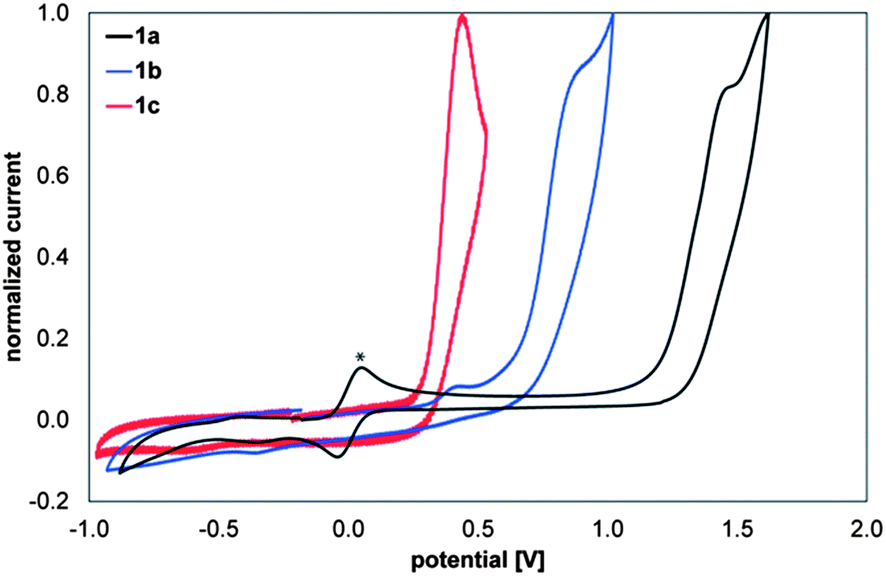 | ||
| Fig. 13 Cyclic voltammograms (anodic scans) of 1a (black), 1b (blue), 1c (red) (1 mM solutions in CH2Cl2) containing 0.1 M NBu4PF6vs. Fc+/0, v = 200 mV s−1 * Fc+/Fc0 couple as internal standard. | ||
UV absorption spectroscopy
The excitation energies of the three 1,4-disilacyclohexa-2,5-dienes 1a–c and cyclohexa-1,4-diene 6 were recorded by gas phase UV absorption spectroscopy (Fig. 14). The wavelengths and extinction coefficients at the absorption peaks are summarized in the ESI.† The UV absorption spectra of 1a and 1b were only recorded in the gas phase as solvent interferences when recorded in cyclohexane were so strong that no reliable spectral information was obtained. For 1c, both gas phase and solution spectra were recorded, and the gas phase measurement clearly reveals a thermal redistribution of the curve as compared to the measurement at room temperature in cyclohexane, a result of vibrational broadening. | ||
| Fig. 14 UV absorption spectra of 1a (black), 1b (blue), 1c (red), and 6 (green) recorded in gas phase, and for 1c also the spectrum recorded at room temperature in cyclohexane solution (black dashed curve). | ||
Compounds 1a and 1b have similar characteristics as their spectra are composed of two overlapping bands; one centered at 210–220 nm and the second in the range 190–200 nm. The strongest absorptions are found in the lower wavelength range (198 and 196 nm for 1a and 1b, respectively). 1,4-Disilacyclohexa-2,5-diene 1c, on the other hand, shows notably different spectral features, with the most intriguing feature being the strong absorption at 273 nm (4.55 eV). A comparison with the UV absorption spectrum of cyclohexa-1,4-diene (6) further reveals the difference between the SiR2 and CH2 units as mediators between the two CC bonds because 6 does not absorb above 220 nm. Moreover, the molecular extinction coefficients of the excitations of 6 are much smaller than for the excitations of any of the three 1,4-disilacyclohexa-2,5-dienes.
In 1c, two weakly chromophoric trisilane segments have been incorporated, and it is apparent from the UV spectrum that these couple strongly with the two CC bonds because the first absorption of permethylated trisilane, Si3Me8, has been reported at 216 nm (5.75 eV).41 Hence, the chromophore of 1c constitutes the complete cross-hyperconjugated cycle involving the two CC bonds and the two trisilane segments.
In order to analyze the experimental UV absorption spectra, excitation energies and oscillator strengths were calculated using time-dependent DFT (TD-DFT) at the TD-PBE0/6-31+G(2d)//B3LYP/6-31G(d) level. To allow for a facile comparison with the D2h symmetric 5 we discuss the type-II conformers for all three compounds, although both conformers I and II will be nearly equally populated at the elevated temperatures used. The calculated excitation energies and oscillator strengths of conformer I closely resemble those of conformer II (for a full comparison between I and II, see the ESI†). In general, the calculated excitation wavelengths (energies) of the three compounds 1a-II to 1c-II agree with the recorded values of Fig. 14.
The most interesting spectrum among the 1,4-disilacyclohexa-2,5-dienes studied is displayed by 1c. Its first calculated excitation is a dark transition at 311 nm (3.98 eV) of B1 symmetry and it has significant character of an excitation from HOMO to LUMO (cf.Fig. 8). However, the second excitation calculated at 276 nm (4.50 eV) is strongly allowed and of B3 symmetry. This transition should correspond to the strong peak observed experimentally at 273 nm and it has a significant contribution of the HOMO to LUMO + 1 excitation, but also of the HOMO − 1 to LUMO excitation. As the transition has HOMO to LUMO + 1 excitation character, i.e., a transition between the two MOs which have clear cross-hyperconjugative character involving both trisilane and olefin fragments, this transition can only be observed in 1c and closely related 1,4-disilacyclohexadiene derivatives.
We also examined the emissive properties of 1c but no emission could be detected upon excitation at 273 nm. However, 1c also does not photodecompose when irradiated with λ > 220 nm light, indicating that non-radiative pathways bring it back to the electronic ground state. In contrast, a rapid photodegradation, possibly due to silylene extrusion from the trisilane segments, is observed upon irradiation at shorter wavelengths than 220 nm.
On the impact of cyclic cross-hyperconjugation in 1c
To determine the importance of the cyclic nature of the cross-hyperconjugation in 1cvs. linear hyperconjugation and acyclic cross-hyperconjugation we calculated the UV absorption and electron binding energies for a series of additional compounds (7–10, Fig. 5) at TD-PBE0/6-31+G(2d)//B3LYP/6-31G(d) and OVGF/6-311+G(d)//B3LYP/6-31G(d) levels (results from additional TD-M06-2X/6-31+G(2d)//B3LYP/6-31G(d) calculations are found in the ESI†). The tetraethyl analogue of one of these compounds, 7, could at first glance be generated experimentally through hydrogenation of 1c. However, despite numerous attempts using different conditions we were unable to reduce any of the double bonds in 1c, presumably due to the extensive steric bulk of the four ethyl and four TMS substituents.Computations of 1g, the derivative of strongly cross-hyperconjugated 1c with hydrogens instead of ethyl substituents, reveal that the ethyl groups have a small effect on the electronic structure of 1c (first excitations at 299 and 311 nm (4.16 and 3.98 eV) for 1g and 1c, respectively, and a moderate effect on the first binding energy (7.14 and 6.76 eV, respectively)). The replacement of one of the CC double bonds by a saturated C–C single bond to yield 7, rendering this compound linearly hyperconjugated, results in a significant change in the calculated spectral properties with the first electronic excitation at 263 nm (4.71 eV) and a first binding energy of 7.76 eV. Breakage of the cyclic nature of the cross-hyperconjugation by replacement of one of the Si(SiMe3)2 groups with a CH2 group, yielding the acyclic cross-hyperconjugated 8, has a similar large effect leading to a first transition at 261 nm (4.75 eV) and a first binding energy of 7.53 eV. Replacing one of the CC double bonds of 8 with a saturated C–C bond leading to 9 with a short linearly hyperconjugated path, gives a compound which is less hyperconjugated according to the TD-DFT and OVGF results (first transition at 236 nm (5.25 eV) and binding energy at 7.77 eV). Finally, replacing both the double bonds of 1g with saturated C–C bonds results in compound 10 with two isolated Si(SiMe3)2 segments. This compound has experimentally been found to not absorb above 220 nm in accordance with computations.42 Taken together, this shows on the effect of having two cross-hyperconjugated moieties unified in a cycle.
On the orientational effect of cross-hyperconjugation
In comparison to our previously examined bis(phenylethynyl)methanes and silanes20,21 the two SiR2 segments of the disilacyclohexa-1,4-dienes 1 are not connected to ethynylene (–C≡C–) fragments but to ethenylene (–CC–) fragments. In the earlier studies we used the ethynylenes as spacers so as to reduce the steric congestion between two phenyl groups. However, ethenylene fragments are more attractive targets for two reasons; (i) they can be substituted so as to further influence the electronic and optical properties of the system, and (ii) the local π(CC) orbitals are of higher energy than local π(CC) orbitals, providing a better orbital energy match with the π(SiR2) orbitals.
In 1, the geometry is constrained, however to probe the orientational effect of the ethenylene moieties several model compounds, 11–14 (Fig. 15A) were investigated computationally using the same levels as above. For these compounds we gradually drove the two CC–C–C dihedral angles ω from 0° to 180°, keeping C2 symmetry. The planar structures should provide for optimal cross-hyperconjugation while the remaining structures have the ethenyl groups out-of-plane. From the calculations the ω-dependences of the first excitation energies and first binding energies are obvious for 11 and 12; when 11 and 12 are planar (0 and 180°) their first binding energies are at the lowest values whereas for the first excitation energies the lowest energies are found at 180° while slightly higher values are observed at 0°. This raise in excitation energy should stem from steric congestion between the terminal hydrogen atoms of the ethenyl groups (in 11, α = 126° at ω = 0° and α = 114° at ω = 180°) which influences the excitation energy (Fig. 15). On the contrary, if the two double bonds are connected as in 8, avoiding the steric congestion and reducing the angle α, the excitation energy is again lowered (see above). Hence, the rotation of the double bonds out-of-plane displays the importance of the rigid structure for optimal cross-hyperconjugation. Neither the methyl substituted 13 nor the chloro substituted 14 show the same dependence on ω, but instead display more modest variations as is expected for two nearly isolated double bonds. For discussions and comparisons between the different orbitals, see the ESI.†
 | ||
| Fig. 15 (A) The model compounds 11–14, the CC–C–C dihedral angles ω (one marked in red), and the C–C–C valence angle α. (B) The calculated first excitation energies of the 21A and 11B states of 11–14 calculated at TD-PBE0/6-31+G(2d)//B3LYP/6-31G(d) level. (C) The calculated first binding energies of 11–14 calculated at OVGF/6-311+G(d)//B3LYP/6-31G(d) level. | ||
Comparison with previous work and future synthetic targets
When compared with our previously reported bis(phenylethynyl)methanes and silanes20,21 the ethenylene fragments can interact more strongly with the central ER2 segment than can ethynylene fragments because bis(ethynyl)silanes with SiMe3 and Me substituents, when compared to 12 and 13, have higher first excitation and electron binding energies (for R = SiMe3: 237 nm (5.23 eV) and 8.1 eV, and for R = Me: 209 nm (5.93 eV) and 10.0 eV, respectively). Indeed, these values are similar or slightly above the values found for 12 and 13 at ω = 90° (Fig. 15), where the conjugation is the weakest. Attachment of additional ethyl substituents at the ends of the triple bonds, so as to allow for comparison with the ethyl substituted 1, marginally lowers the first excitation and binding energies. However, for the double bonds it is essential to lock the conformation into a planar structure for optimal conjugation. Taken together, it is apparent that the ethenylene fragments are attractive as they provide for both further substitution opportunities and for stronger interaction with the ER2 group than the ethynylene fragments. One structure with (near) optimal and fixated neutral hyperconjugation is our disilacyclohexa-1,4-dienes, yet, the computed results on 12 indicate that even stronger cyclic cross-hyperconjugation can be achieved through a “cis”-conformation with ω = 180°, provided a suitable rigid structure can be tailored. Overall, it is likely that rigid planar structures with properly substituted ER2 fragments provide for substantial σ/π-interactions and represent new synthetic targets for research on optically and electronically active compounds. Oligomers of such compounds with potential connecting points from both the π-system and the ER2 segment should be attractive targets and investigations into these and related compounds are on-going in our laboratories.Conclusions
Herein we have shown that 1,4-disilacyclohexa-2,5-dienes are a compound class that allows for remarkable variations in the electronic and optical characteristics through the choice of the substituents. The large tunability arises from the fact that two SiR2 segments as parts of the cyclic 1,4-disilacyclohexa-1,4-diene allow for neutral cyclic cross-hyperconjugation to various degrees. A strong cyclic cross-hyperconjugation is found with R = SiMe3 and much weaker when R = Cl and Me as manifested in the valence photoionization energies, the first oxidation potentials, and the lowest UV absorption energies. One can predict that a change of the substituents at the CC double bonds provides for further tuning of the electronic and optical properties. The neutral cross-hyperconjugation strengths in the 1,4-disilacyclohexadienes were clarified with quantum chemical calculations combined with a MO-theoretical analysis. Interestingly, the orbital pattern and the electronic structure of 1c has similarities with that of the cross-π-conjugated para-xylylene. The 1,4-disilacyclohexa-2,5-diene is therefore a new monomer unit which in an optimal way combines σ- and π-bonded segments into a rigid neutral and cyclic cross-hyperconjugated framework. It should represent an interesting structural motif for new molecules with applications in the molecular and organic electronics areas, and it could allow for design of strongly hyperconjugated compounds with properties that are not available to purely π-conjugated compounds.
Acknowledgements
We thank Dr. Carla Puglia for valuable discussions. We are grateful for financial support from the Swedish Research Council, from Uppsala University for the U3MEC KoF07 Initiative, from the Wenner-Gren Foundations in the form of a postdoctoral fellowship to A.W., and from the Göran Gustafsson Foundation (S.O and D.N.). K. Y. is grateful for the fellowship and Grant-in-Aid for JSPS Fellows Number 251672 from Japan Society for the Promotion of Science (JSPS). The National Supercomputer Center (NSC) in Linköping, Sweden is acknowledged for a generous allotment of computer time.Notes and references
- See complete volume, Chem. Rev., 2005, 105, 3433–3947 Search PubMed.
- (a) R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS; (b) J. Michl and R. West, Acc. Chem. Res., 2000, 33, 821–823 CrossRef CAS PubMed.
- T. A. Skotheim and J. R. Reynolds, Handbook of Conducting Polymers, CRC Press LLC, Bocata Raton, 2007 Search PubMed.
- R. Hoffmann, C. Janiak and C. Kollmar, Macromolecules, 1991, 24, 3725–3746 CrossRef CAS.
- (a) H. Wang, R. Helgeson, B. Ma and F. Wudl, J. Org. Chem., 2000, 65, 5862–5867 CrossRef CAS; (b) M. Bruschi, M. G. Giuffreda and H. P. Lüthi, Chem.–Eur. J., 2002, 8, 4216–4227 CrossRef CAS; (c) N. F. Phelan and M. Orchin, J. Chem. Educ., 1968, 45, 633–637 CrossRef CAS.
- R. S. Mulliken, C. A. Rieke and W. G. Brown, J. Am. Chem. Soc., 1941, 63, 41–56 CrossRef CAS.
- (a) R. S. Mulliken, J. Chem. Phys., 1939, 7, 339–352 CrossRef CAS; (b) I. V. Alabugin and T. A. Zeidan, J. Am. Chem. Soc., 2002, 124, 3175–3185 CrossRef CAS PubMed; (c) J. B. Lambert and R. A. Singer, J. Am. Chem. Soc., 1992, 114, 10246 CrossRef CAS; (d) I. V. Alabugin, K. M. Gimore and P. W. Peterson, WIREs Comput. Mol. Sci., 2011, 1, 109–141 CrossRef CAS.
- We use the term cross-hyperconjugation instead of hypercross-conjugation to stress that the geminal connectivity of the conjugated paths should have higher priority than the specific conjugation type.
- For recent reviews in silole-containing oligomers and polymers see, e.g.: (a) K. Tamao and S. Yamaguchi, J. Organomet. Chem., 2000, 611, 5–11 CrossRef CAS; (b) S. Yamaguchi and K. Tamao, J. Organomet. Chem., 2002, 653, 223–228 CrossRef CAS; (c) S. Yamaguchi and K. Tamao, Chem. Lett., 2005, 2 CrossRef CAS; (d) J. Chen and Y. Cao, Macromol. Rapid Commun., 2007, 28, 1714–1742 CrossRef CAS; (e) J. Ohshita, Macromol. Chem. Phys., 2009, 210, 1360–1370 CrossRef CAS.
- M. Ishikawa and J. Ohshita, in Organic Conductive Molecules and Polymers, ed. H. S. Nalwa, Wiley, New York, 1997, ch. 30 Search PubMed.
- (a) M. Nakamoto, Y. Inagaki, M. Nishina and A. Sekiguchi, J. Am. Chem. Soc., 2009, 131, 3172–3173 CrossRef CAS PubMed; (b) T. Ochiai, M. Nakamoto, Y. Inagaki and A. Sekiguchi, J. Am. Chem. Soc., 2011, 133, 11504–11507 CrossRef CAS PubMed; (c) Y. Inagaki, M. Nakamoto and A. Sekiguchi, J. Am. Chem. Soc., 2011, 133, 16436–16439 CrossRef CAS PubMed; (d) A. Chrostowska, A. Dargelos, P. Baylère, A. Graciaa, Y. Inagaki, M. Nakamoto, V. Ya. Lee and A. Sekiguchi, ChemPlusChem, 2013, 78, 398–401 CrossRef CAS.
- Y. Yamaguchi, Synth. Met., 1996, 82, 149–153 CrossRef CAS.
- J. Ohshita and A. Kunai, Acta Polym., 1998, 49, 379–403 CrossRef CAS.
- J. Ohshita, H. Takata, H. Kai, A. Kunai, K. Komaguchi, M. Shiotani, A. Adachi, K. Sakamaki, K. Okita, Y. Harima, Y. Kunugi, K. Yamashita and M. Ischikawa, Organometallics, 2000, 19, 4492–4498 CrossRef CAS.
- J. Ohshita, D.-H. Kim, Y. Kunugi and A. Kunai, Organometallics, 2005, 24, 4494–4496 CrossRef CAS.
- D.-H. Kim, J. Ohshita, T. Kosuge, Y. Kunugi and A. Kunai, Chem. Lett., 2006, 266–267 CrossRef CAS.
- J. Ohshita, Y. Izumi, D.-H. Kim, A. Kunai, T. Kosuge, Y. Kunugi, A. Naka and T. Ishikawa, Organometallics, 2007, 26, 6150–6154 CrossRef CAS.
- J. Ohshita, Y. Hatanaka, S. Matsui, T. Mizumo, Y. Kunugi, Y. Honsho, A. Saeki, S. Seki, J. Tibbelin, H. Ottosson and T. Takeuchi, Dalton Trans., 2010, 39, 9314–9320 RSC.
- J. Ohshita, Y. Hatanaka, S. Matsui, Y. Ooyama, Y. Harima and Y. Kunugi, Appl. Organomet. Chem., 2010, 8, 540–544 Search PubMed.
- R. Emanuelsson, A. Wallner, E. A. M. Ng, J. R. Smith, D. Nauroozi, S. Ott and H. Ottosson, Angew. Chem., Int. Ed., 2013, 52, 983–987 CrossRef CAS PubMed.
- E. Göransson, R. Emanuelsson, K. Jorner, L. Hammarström and H. Ottosson, Chem. Sci., 2013, 4, 3522–3532 RSC.
- U. Weidner and A. Schweig, Angew. Chem., Int. Ed. Engl., 1972, 11, 536–537 CrossRef CAS.
- R. West and R. E. Bailey, J. Am. Chem. Soc., 1963, 85, 2871–2872 CrossRef CAS.
- (a) R. Hoffmann, Acc. Chem. Res., 1971, 4, 1–9 CrossRef CAS; (b) R. Srinivasan, L. S. White, A. R. Rossi and G. A. Epling, J. Am. Chem. Soc., 1981, 103, 7299–7304 CrossRef CAS.
- H. Bock and W. Kaim, J. Am. Chem. Soc., 1980, 102, 4429–4438 CrossRef CAS.
- R. West, H. Sohn, U. Bankwitz, J. Calabrese, Y. Apeloig and T. Müller, J. Am. Chem. Soc., 1995, 117, 11608–11609 CrossRef CAS.
- R. West, H. Sohn, D. R. Powell, T. Müller and Y. Apeloig, Angew. Chem., Int. Ed. Engl., 1996, 35, 1002–1004 CrossRef CAS.
- B. T. Stępień, T. M. Krygowski and M. K. Cyrański, J. Org. Chem., 2002, 67, 5987–5992 CrossRef PubMed.
- H. Ottosson, K. Kilså, K. Chajara, M. C. Piqueras, R. Crespo, H. Kato and D. Muthas, Chem.–Eur. J., 2007, 13, 6998–7005 CrossRef CAS PubMed.
- A. P. Scott, I. Agranat, P. U. Biedermann, N. V. Riggs and L. Radom, J. Org. Chem., 1997, 62, 2026–2038 CrossRef CAS PubMed.
- A. Stanger, J. Org. Chem., 2006, 71, 883–893 CrossRef CAS PubMed.
- O. M. Nefedov and M. N. Manakov, Angew. Chem., Int. Ed. Engl., 1966, 5, 1021–1038 CrossRef CAS.
- W. H. Atwell and D. R. Weyenberg, Angew. Chem., Int. Ed. Engl., 1969, 8, 469–477 CrossRef CAS.
- P. P. Gaspar and R. West, in The Chemistry of Organosilicon Compounds, Wiley, New York, 1998, vol. 2, pp. 2463–2568 Search PubMed.
- F. Wudl, R. D. Allendoerfer, J. Demirgian and J. M. Robbins, J. Am. Chem. Soc., 1971, 93, 3160–3162 CrossRef CAS.
- A. Kunai, J. Ohshita, T. Iida, K. Kanehara, A. Adachi and K. Okita, Synth. Met., 2003, 37, 1007–1008 CrossRef.
- S.-H. Kang, J. S. Han, M. E. Lee, B. R. Yoo and N. Jung, Organometallics, 2003, 22, 2551–2553 CrossRef CAS.
- B. Ma, J.-H. Lii, H. F. Schaefer III and N. L. Allinger, J. Phys. Chem., 1996, 100, 8763–8769 CrossRef CAS.
- (a) L. S. Bartell and R. A. Bonham, J. Chem. Phys., 1959, 31, 400–404 CrossRef CAS; (b) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orphen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- M. Kaftory, M. Kapon and M. Botoshansky, in The Chemistry of Organic Silicon Compound, ed. Z. Rapport and Y. Apeloig, Wiley, Chichester, UK, vol. 2, ch. 5, p. 181 Search PubMed.
- C. G. Pitt, J. Am. Chem. Soc., 1969, 91, 6613–6622 CrossRef CAS.
- A. Wallner, R. Emanuelsson, J. Baumgartner, C. Marschner and H. Ottosson, Organometallics, 2013, 32, 396–405 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full computational and experimental procedures as well as details on the gas phase UV absorption spectroscopy, analysis of the impact of steric and electronic factors on the geometries of 1,4-disilacyclohexa-2,5-dienes 1a–h, orbital plots, absolute electronic energies, Cartesian coordinates, tables with TD-DFT data, tables with complete photoelectron spectroscopic data and X-ray crystallographic information for compound 1c. CCDC 775978. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc52389f |
| This journal is © The Royal Society of Chemistry 2014 |