Effect of the trifluoromethyl group on torquoselectivity in the 4π ring-opening reaction of oxetenes: stereoselective synthesis of tetrasubstituted olefins†
Kohsuke
Aikawa
,
Natsumi
Shimizu
,
Kazuya
Honda
,
Yūta
Hioki
and
Koichi
Mikami
*
Department of Applied Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1-H-113 O-okayama, Meguro-ku, Tokyo 152-8852, Japan. E-mail: mikami.k.ab@m.titech.ac.jp
First published on 9th October 2013
Abstract
A highly stereoselective synthesis of tetrasubstituted olefins bearing a trifluoromethyl group via the thermal 4π electrocyclic ring-opening reaction of oxetenes, simply prepared by the Pd-catalyzed [2+2] cycloaddition reaction of various alkynes with trifluoropyruvate, is achieved. In this reaction process, the trifluoromethyl group prefers inward rotational torquoselectivity because of the orbital interactions between the breaking C–O σ orbital on the oxetene moiety and the C–F σ* orbital in the transition state.
Introduction
The development of reliable synthetic approaches to trifluoromethylated compounds, which are widespread in pharmaceuticals and agrochemicals, has become a hot topic in organic synthesis.1 The trifluoromethylation of (hetero)aromatic compounds has been intensively investigated and excellent progress has been made in recent years.2 A number of synthetic approaches to disubstituted olefins bearing a trifluoromethyl (CF3) group have been reported so far.3 However, stereoselective synthetic methods for tri- and tetrasubstituted olefins bearing a CF3 group are still extremely limited.4,5 These olefins are essential for the development of the catalytic enantioselective construction of trifluoromethylated tertiary and quaternary stereogenic carbon centers using asymmetric catalytic hydrogenations and addition reactions (Scheme 1).6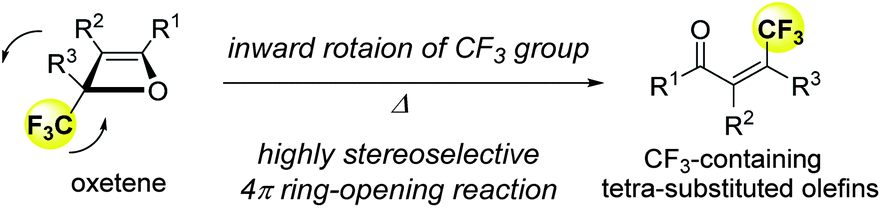 | ||
| Scheme 1 Stereoselective synthesis of tetra-substituted alkenes containing a CF3 group. | ||
On the other hand, the 4π electrocyclic ring-opening reaction of cyclobutene derivatives has received much attention, both synthetically and theoretically. The selectivity of the inward or outward rotation of the substituent in the 2-position is termed “torquoselectivity”. Houk and co-workers have unveiled the stereoelectronic effects of various substituents including CF3, on torquoselectivity in the ring-opening reaction of cyclobutenes.7 Murakami and co-workers also reported the preferences for inward rotation of silyl-, stannyl-, and boryl-substituents in the ring-opening reaction.8 Furthermore, Shindo and co-workers clarified the substituent effects on torquoselectivity in the ring-opening reaction of lithium oxetenoides as the reaction intermediates, which were generated from lithium ynolates and ketones.9,10 In a cyclobutene system, although it has already been demonstrated that the CF3 group of 3-(trifluoromethyl)cyclobutene exhibits a small preference for outward rotation,7a,e,11 the stereoelectronic effects of the CF3 group on torquoselectivity have never been investigated in an oxetene system. We have recently discovered the synthesis of stable oxetenes via the [2+2] cycloaddition reaction of various alkynes with ethyl trifluoropyruvate.12 We proposed that the unprecedented stability of oxetenes containing a CF3 group in the 2-position is attributed to the perfluoroalkyl effect,13 in which a perfluoroalkyl group can increase not only the kinetic but also the thermodynamic stability of a compound. In this communication, we disclose a reliable synthetic approach to tetrasubstituted olefins with the CF3 group through the highly stereoselective 4π electrocyclic ring-opening reaction, using the isolated oxetenes not as reaction intermediates. The stereoselective construction of tetrasubstituted olefins, as well as quaternary stereogenic carbon centers bearing the CF3 group, remains an extremely challenging task due to the unique stereoelectronic effect of the fluorine atom.
Results and discussion
Initially, oxetene derivatives were synthesized employing the procedure illustrated in Scheme 2. The catalytic [2+2] cycloaddition of ethyl trifluoropyruvate 2 with alkyne 1a in the presence of the dicationic rac-BINAP–Pd complex (5 mol%) provided the corresponding oxetene 3a in 95% yield. Subsequent reduction of the ester group by LiAlH4, followed by protections (Nbz, Bz, and TBS) of the alcohol group led to oxetenes 5 in good yields. All the oxetenes could be easily purified by silica-gel column chromatography, without undergoing the 4π ring-opening reaction.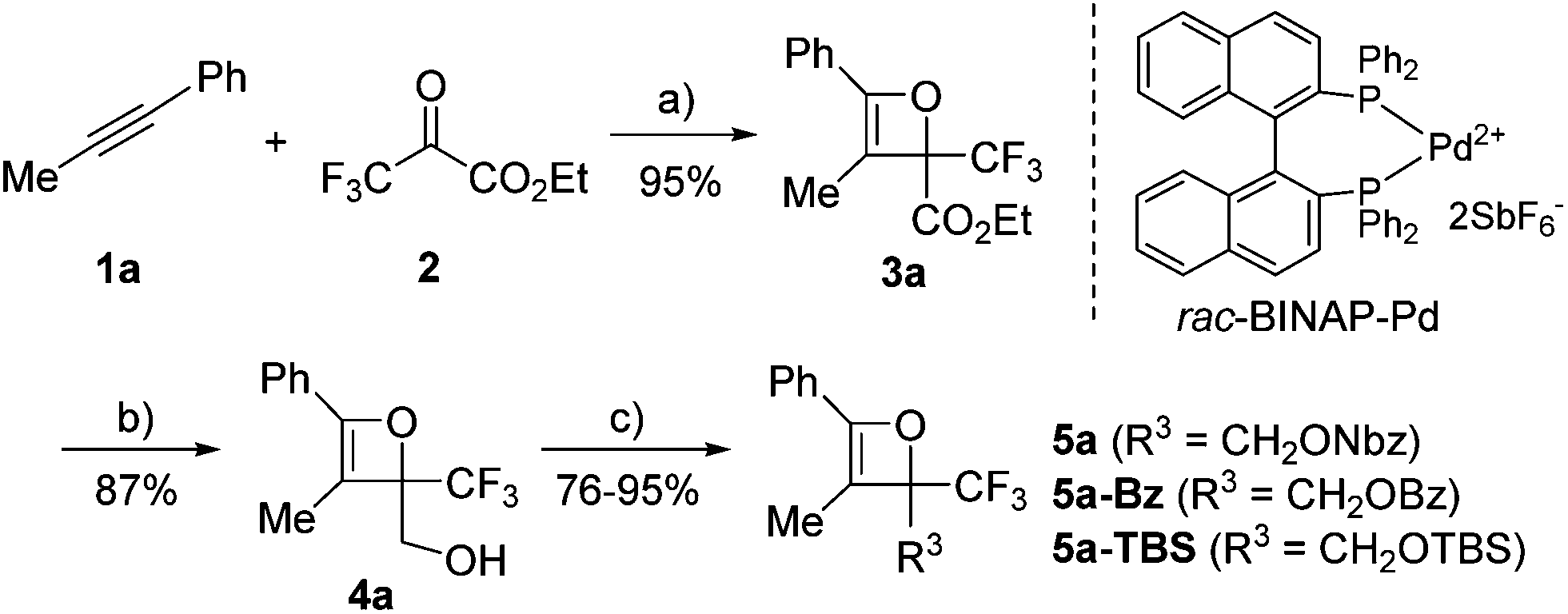 | ||
| Scheme 2 Synthesis of oxetene derivatives: (a) rac-BINAP–Pd (5 mol%), CH2Cl2, −20 °C, 12 h; (b) LiAlH4, THF, 0 °C, 30 min; (c) 4-NO2C6H4COCl (NbzCl), NEt3, CH2Cl2, RT, 2 h (95%); BzCl, NEt3, CH2Cl2, RT, 2 h (89%); TBSCl, imid., DMF, RT, 6 h (76%). | ||
When heating in toluene-d8, the electrocyclic ring-opening reaction of oxetene 3a proceeded to give a mixture of the corresponding Z- and E-isomers, with the Z-isomer predominating in a ratio of 81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 (Table 1, entry 1). The half-life (t1/2) of the oxetene at 70 °C was 138 hours, demonstrating the extremely stable nature of the CF3-oxetenes at elevated temperatures, compared to the oxetenes obtained previously.14 Of particular note, it was found that the use of oxetene 5a, with a methylene group, dramatically increased the stereoselectivity (entry 2). The structures of 5a and (Z)-6a were unequivocally determined by single crystal X-ray analysis (Fig. 1). Replacing 5a by 5a-Bz and 5a-TBS gave similar stereoselectivities (entries 3 and 4), but the more electron-donating R3-substituent decreased the thermal stability of the oxetenes. While the reaction of the unprotected oxetene 4a provided the corresponding olefin 6a-OH with complete stereoselectivity, many by-products were also obtained (entry 5). Increased temperature (100 °C) accelerated the reaction, but the stereoselectivity of the olefins remained almost the same regardless of temperature. In addition, the ratio of Z/E was constant during the course of the reaction, even after heating for a longer period of time. These results indicate that the ratio of Z/E was kinetically determined. The reactions gave almost quantitative yields, except for entry 5.
:19 (Table 1, entry 1). The half-life (t1/2) of the oxetene at 70 °C was 138 hours, demonstrating the extremely stable nature of the CF3-oxetenes at elevated temperatures, compared to the oxetenes obtained previously.14 Of particular note, it was found that the use of oxetene 5a, with a methylene group, dramatically increased the stereoselectivity (entry 2). The structures of 5a and (Z)-6a were unequivocally determined by single crystal X-ray analysis (Fig. 1). Replacing 5a by 5a-Bz and 5a-TBS gave similar stereoselectivities (entries 3 and 4), but the more electron-donating R3-substituent decreased the thermal stability of the oxetenes. While the reaction of the unprotected oxetene 4a provided the corresponding olefin 6a-OH with complete stereoselectivity, many by-products were also obtained (entry 5). Increased temperature (100 °C) accelerated the reaction, but the stereoselectivity of the olefins remained almost the same regardless of temperature. In addition, the ratio of Z/E was constant during the course of the reaction, even after heating for a longer period of time. These results indicate that the ratio of Z/E was kinetically determined. The reactions gave almost quantitative yields, except for entry 5.
|
|
|||||
|---|---|---|---|---|---|
| Entity | Oxetene | R3 (olefin) | t 1/2 [h] | k [h−1] |
Z:Ed,e |
| a Toluene-d8 (1.0 M) was used as a solvent. b Half-life of oxetenes at 70 °C. c At 70 °C. d Z/E stereoselectivity at 70 °C, and at 100 °C in parentheses. e Determined by 19F NMR analysis. f Many by-products were obtained. g Purification by silica-gel chromatography gave only (Z)-6a-OH in 36% yield. | |||||
| 1 | 3a | CO2Et (6a-CO2Et) | 138 | 5.0 × 10−3 | 81:19 (77:23) |
| 2 | 5a | CH2ONbz (6a) | 134 | 5.2 × 10−3 | 97:3 (96:4) |
| 3 | 5a-Bz | CH2OBz (6a-Bz) | 80 | 8.7 × 10−3 | 97:3 (97:3) |
| 4 | 5a-TBS | CH2OTBS (6a-TBS) | 43 | 1.6 × 10−2 | 98:2 (98:2) |
| 5f | 4a | CH2OH (6a-OH) | — | — | 100:0g |
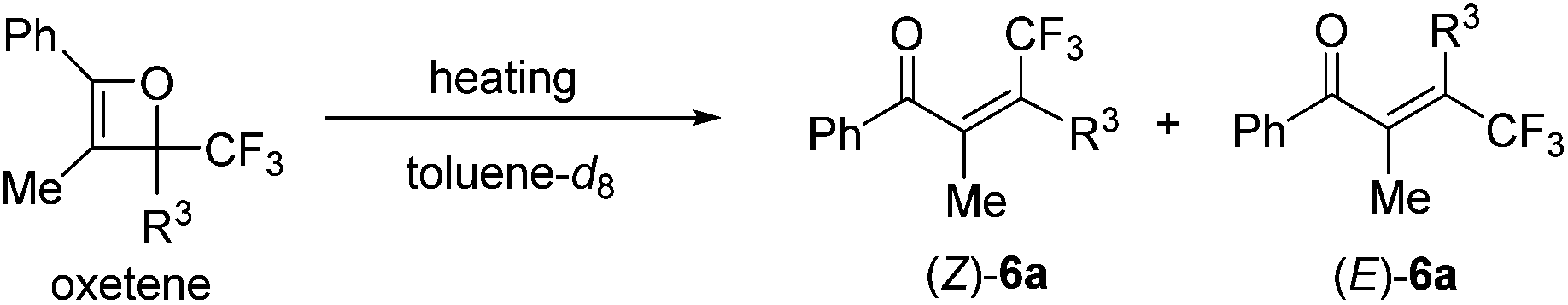
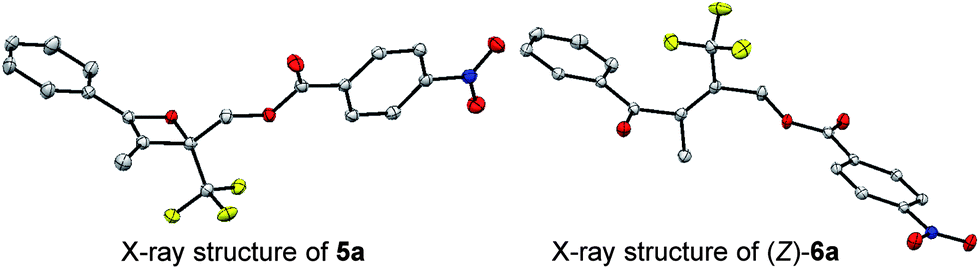 | ||
| Fig. 1 X-Ray structures of oxetene and olefin. | ||
In contrast to oxetene 4a, the reaction of unprotected oxetene 7, easily prepared from ethynylbenzene and ethyl trifluoropyruvate in two steps, was executed to give the trisubstituted olefin (Z)-8 quantitatively and with complete stereoselectivity (Scheme 3, eqn (1)). This Z-isomer bearing a CF3 group cannot be synthesized through a Wittig reaction, which leads to the E-isomer.4b,c Unprotected oxetene 9 also reacted to provide the desired cyclic product 10 in quantitative yield (Scheme 3, eqn (2)).
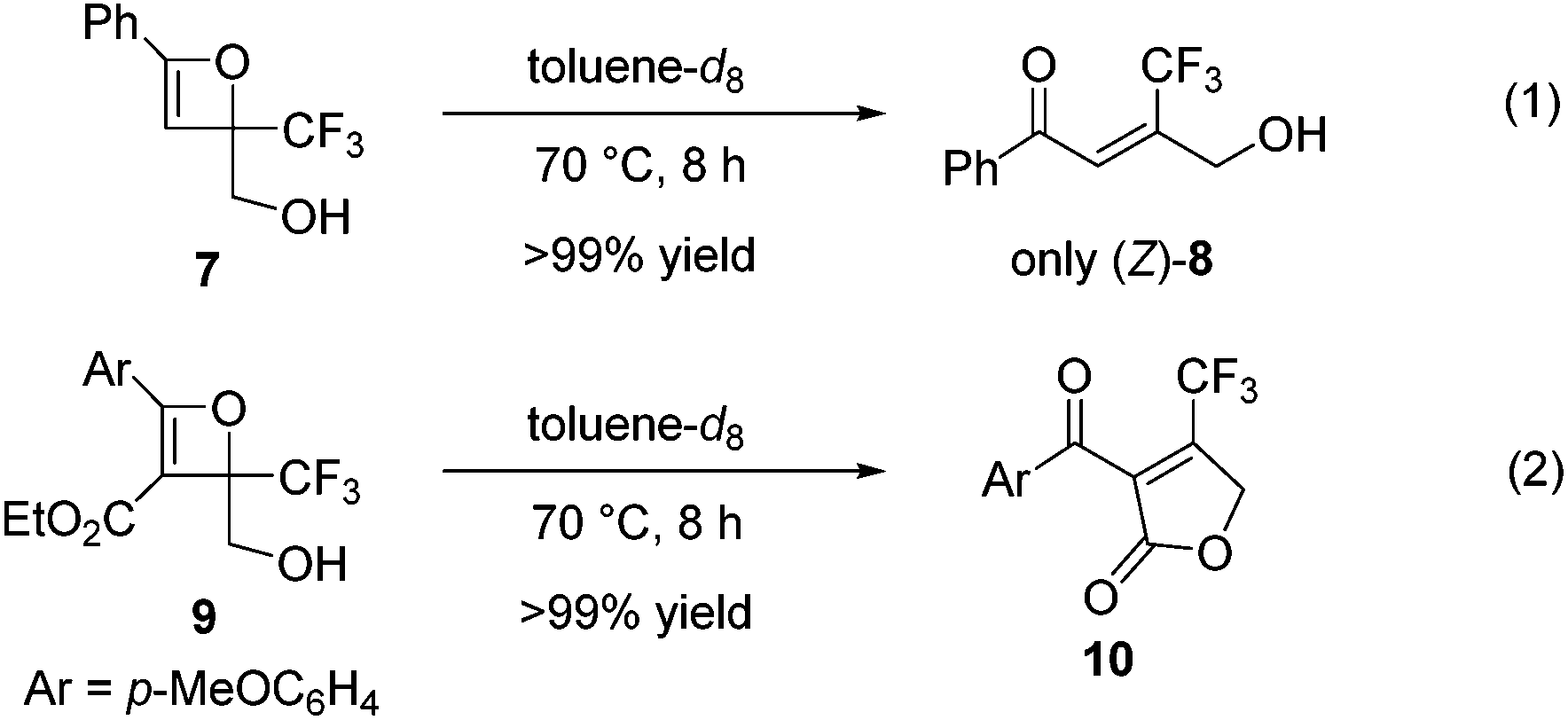 | ||
| Scheme 3 Reactions of unprotected oxetenes. | ||
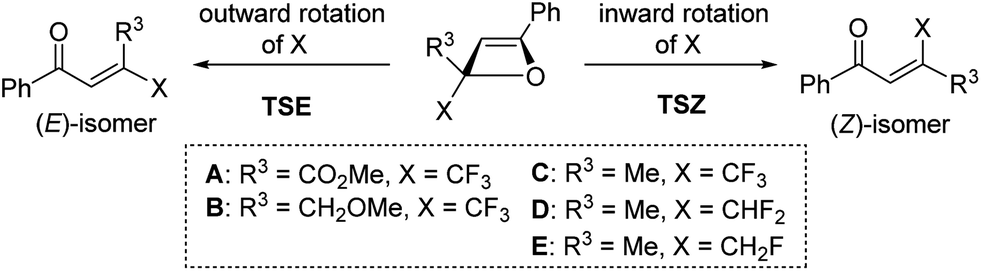
In a cyclobutene system, Houk and co-workers showed that the CF3 group of 3-(trifluoromethyl)cyclobutene exhibits a slight preference for outward rotation because of electrostatic repulsion between the CF3 group and the π-system of cyclobutene.7e Therefore, we carried out computational studies (B3LYP/6-31G(d) level calculations) to clarify the counterintuitive inward rotation of the CF3 group in the oxetene system (Table 2). The activation energy for inward rotation in TSZa of oxetene A, with CO2Me as an R3-substituent, was slightly lower than that in TSEa for outward rotation, because the two electron-accepting groups would be simultaneously substituted in the 2-positions (entries 1 and 2). In the case of oxetenes B and C with methoxymethyl and methyl substituents, the activation energies in TSZb and TSZc were 2.7 and 4.1 kcal mol−1 lower than those for outward rotation, respectively (entries 3–6). These theoretical stereoselectivities agreed well with our experimental results (Table 1). On the other hand, oxetenes D and E bearing CHF2 and CH2F groups instead of a CF3 group also indicated a preference for the inward rotation of these groups, although this was only demonstrated by the theoretical results (entries 7–10). Interestingly, it was found the preference for inward rotation follows the order: CHF2 (ΔΔE≠ = 5.8 kcal mol−1), CF3 (4.1 kcal mol−1), CH2F (1.0 kcal mol−1).15 As shown in Fig. 2a, the C1–F1 and C3–H1 bonds, which are almost anti-periplanar to the breaking C–O bond, were found to be slightly longer than the other bonds in TSZ and TSE of oxetenes C, D, and E. In sharp contrast to the same bond lengths of C3–H1 (1.101 Å) in TSE, the C1–F1 bonds of TSZ were longer in the order of CH2F (1.404 Å), CHF2 (1.381 Å), CF3 (1.366 Å), because the positive charge developed on the C1 carbon would be better stabilized by C1–H4 and C1–H5 bonds compared with C1–F2 and C1–F3 bonds. In the case of TSZ, it was also found that the C3–H3 bond is almost syn-periplanar to the breaking C–O bond.
|
|
||||
|---|---|---|---|---|
| Entity | Oxetene | Transition state | ΔE≠ (kcal mol−1) | ΔΔE≠ (kcal mol−1) |
| 1 | A | TSEa | 28.1 | +1.0 |
| 2 | A | TSZa | 27.1 | 0 |
| 3 | B | TSEb | 29.0 | +2.7 |
| 4 | B | TSZb | 26.3 | 0 |
| 5 | C | TSEc | 30.3 | +4.1 |
| 6 | C | TSZc | 26.2 | 0 |
| 7 | D | TSEd | 30.0 | +5.8 |
| 8 | D | TSZd | 24.2 | 0 |
| 9 | E | TSEe | 24.3 | +1.0 |
| 10 | E | TSZe | 23.3 | 0 |
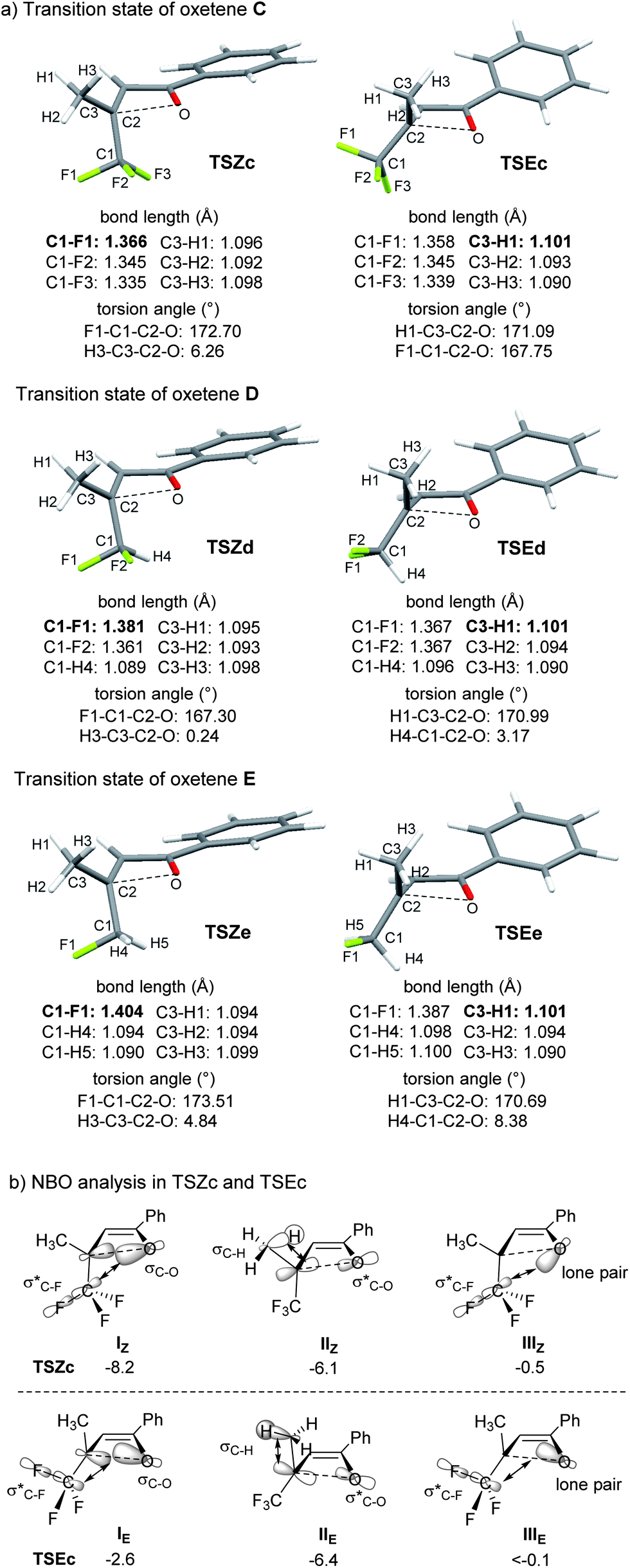 | ||
| Fig. 2 Effect of fluoromethyl groups on torquoselectivity. (a) Selected bond lengths and torsion angle of TSZ and TSE of oxetenes C, D, and E; (b) representative secondary interactions (I–III) between the NBOs of TSZc and TSEc, and the energies of these interactions in kcal mol−1. | ||
Next, NBO (natural bond orbital) analysis was carried out for TSZc and TSEc of oxetene C, referring to the approach reported by Shindo and Mori9 (Fig. 2b).16 The NBO analysis disclosed the secondary orbital interactions between the breaking C–O σ orbital and the C–F σ* orbital (I).17 The orbital interaction IZ in TSZc was much larger in energy than IE in TSEc, by 5.6 kcal mol−1. The interactions IIIZ and IIIE between the nonbonding orbital of oxygen on the oxetene moiety and the C–F σ* orbital were much smaller.9d In contrast, the interactions IIZ and IIE between the breaking C–O σ* orbital and the C–H σ orbital indicated favorability towards E-selectivity, i.e. outward rotation of the CF3 group, but the difference was much smaller. These results show that the orbital interaction IZ (8.2 kcal mol−1) is the dominant factor in determining the torquoselectivity for inward rotation of the CF3 group, with the assistance of the orbital interaction IIZ (6.1 kcal mol−1).18
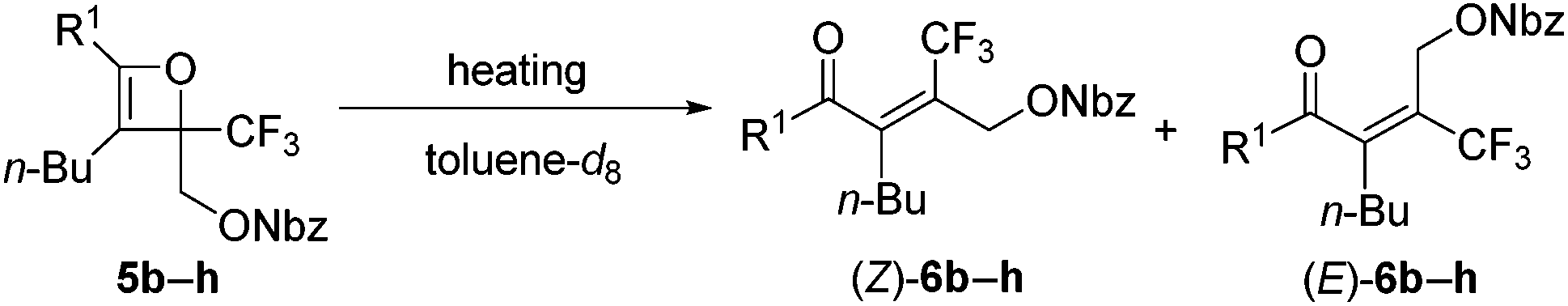
The effect of the R1-substituent on the torquoselectivity and the thermal stability of oxetenes 5b–h, with n-Bu in the 3-position, were investigated under the same reaction conditions (Table 3). Oxetenes 5b–e with electron-withdrawing and -donating aromatic substituents, gave excellent stereoselectivities (entries 1–4). Importantly, it was clarified that the R1-substituent in the 4-position does not electronically affect torquoselectivity in the 4π ring-opening reaction, due to the same stereoselectivities being observed regardless of temperature. In contrast, the R1-substituent electronically influences the thermal stability of oxetenes; more electron-donating aromatic substituents facilitated the 4π ring-opening reaction (entry 4), while more electron-withdrawing substituents suppressed the reaction (entry 1). Interestingly, in spite of there being no change in stereoselectivity, the thermal stability of oxetenes 5f and 5g, with electron-donating mesityl substituents, was dramatically enhanced (entries 5 and 6). The stability of oxetenes would be increased when the conjugation between the π-system of oxetene and the R1-substituent was not extended; as can be seen from the X-ray structures (Fig. 3), the torsion angle (C1–C2–C3–C4) of oxetene 5l was found to be significantly more acute than that of 5g-CO2Et (ref. 12) (3.2° vs. 79.7°) (also 5a, 1.5°). The distortion of 5g-CO2Et from planar geometry obviously stems from the steric repulsion between methyl groups in the 2,6-positions of the mesityl and tert-butyl substituents. The C2–C3 bond lengths of 5g-CO2Et (1.468 Å) were also found to be somewhat longer than those of 5l (also 5a) (1.446 (1.453) Å). On the other hand, not only aromatic but also alkenyl substituents gave the corresponding tetrasubstituted olefin 6h in excellent stereoselectivity (entry 7).
|
|
|||||
|---|---|---|---|---|---|
| Entity | R1 | t 1/2 [h] | k [h−1] |
Z:Ed,e |
|
| a Toluene-d8 (1.0 M) was used as a solvent. b Half-life of oxetenes at 70 °C. c At 70 °C. d Z/E stereoselectivity at 70 °C, and at 100 °C in parentheses. e Determined by 19F NMR analysis. f t-Bu was used instead of n-Bu in the 3-position of oxetene 5f. g At 100 °C. h Reaction did not proceed at 70 °C. | |||||
| 1 | m-MeOC6H4 | (b) | 140 | 5.0 × 10−3 | 98:2 (96:4) |
| 2 | Ph | (c) | 137 | 5.1 × 10−3 | 98:2 (96:4) |
| 3 | p-MeC6H4 | (d) | 106 | 6.6 × 10−3 | 98:2 (96:4) |
| 4 | p-MeOC6H4 | (e) | 51 | 1.4 × 10−2 | 98:2 (96:4) |
| 5 | Mes | (f) | 354 | 2.0 × 10−3 | 96:4 (96:4) |
| 6f | Mes | (g) | 275g | 2.5 × 10−3g | —h (98:2) |
| 7 |
|
(h) | 137 | 5.1 × 10−3 | 97:3 (96:4) |
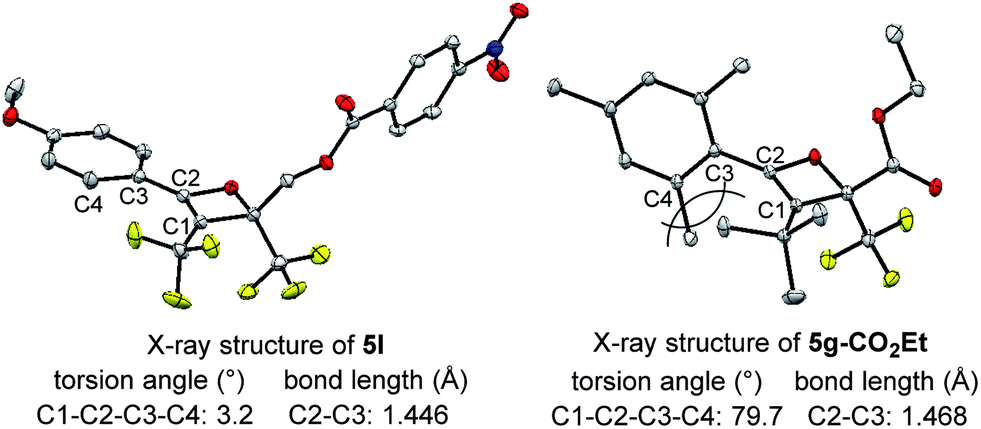 | ||
| Fig. 3 X-Ray structures of oxetenes. | ||
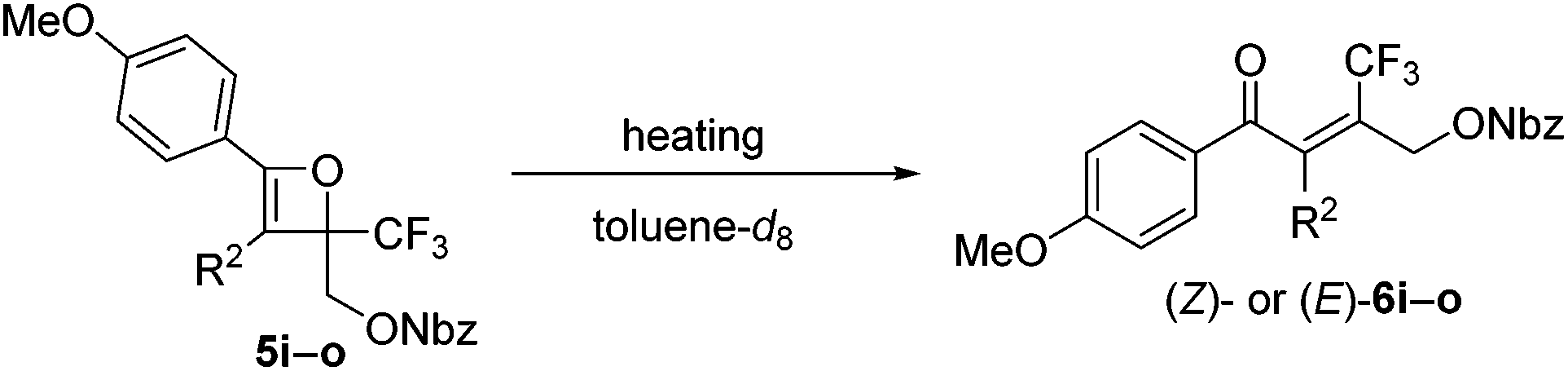
The effect of the R2-substituent on the torquoselectivity and thermal stability of oxetenes 5i–o with a para-methoxypheny group in the 4-position was also examined under the same reaction conditions (Table 4). Similarly to the effect of the R1-substituent (Table 3, entries 1–4), the reaction of oxetenes 5i–k with electron-withdrawing and -donating aromatic substituents afforded the corresponding Z-isomers with the same stereoselectivities (entries 1–3). While more electron-donating aromatic substituents decreased the thermal stability of the oxetenes, their influence was lower than that of the corresponding R1-substituents. Oxetenes 5l and 5m underwent the 4π ring-opening reaction with inward rotation to provide the E-isomers exclusively (entries 4 and 5). This complete torquoselectivity contributes to inhibition of the formation of the Z-isomer, due to the dipole repulsion in the TS between the strong electron-withdrawing R2-substituent (CF3 and ester) and the CF3 group (Fig. 4). The stereoselectivity of the reaction of oxetene 5n bearing a sterically demanding SiMe2Ph substituent, was similar to that of the reaction of unsubstituted 5o (entries 6 and 7). This result clearly shows that the stereoelectronic effect on the torquoselectivity of R1- and R2-substituents in the 3,4-positions of oxetene is extremely small, while the electronic properties strongly affect the thermal stability of oxetene. All of the reactions shown in Tables 3 and 4 provided the corresponding olefins 6 in almost quantitative yields.
|
|
|||||
|---|---|---|---|---|---|
| Entity | R2 | t 1/2 [h] | k [h−1] |
Z:Ed,e |
|
| a Toluene-d8 (1.0 M) was used as a solvent. b Half-life of oxetenes at 70 °C. c At 70 °C. d Z/E stereoselectivity at 70 °C, and at 100 °C in parentheses. e Determined by 19F NMR analysis. f Decomposition of 5n was observed at 100 °C. g Ph group was used instead of p-MeOC6H4 group in the 4-position of oxetene. | |||||
| 1 | p-NO2C6H4 | (i) | 3.2 | 2.2 × 10−1 | 99:1 (98:2) |
| 2 | Ph | (j) | 2.7 | 2.6 × 10−1 | 99:1 (98:2) |
| 3 | p-MeOC6H4 | (k) | 2.5 | 2.8 × 10−1 | 99:1 (98:2) |
| 4 | CF3 | (l) | 15 | 4.8 × 10−2 | 0:100 (0:100) |
| 5 | CO2Et | (m) | 10 | 6.9 × 10−2 | 0:100 (0:100) |
| 6 | SiMe2Ph | (n) | 50 | 1.4 × 10−2 | 1:99 (— f) |
| 7g | H | (o) | 3.9 | 1.8 × 10−1 | 99:1 (99:1) |
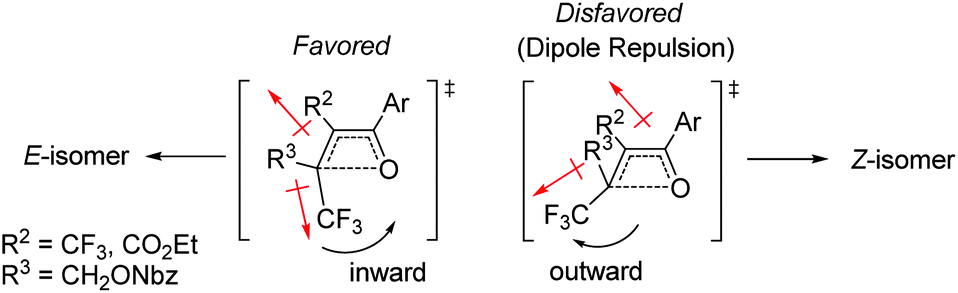 | ||
| Fig. 4 Effect of dipole repulsion on torquoselectivity. | ||
Conclusions
In conclusion, we have succeeded in the highly stereoselective synthesis of tetrasubstituted olefins bearing a CF3 group via the thermal 4π electrocyclic ring-opening reaction of various oxetenes. In this oxetene system, a remarkable inward rotation of the CF3 group was observed. It was suggested that the inward rotation originates from the orbital interactions between the breaking C–O σ orbital and the C–F σ* orbital in the transition state. It was demonstrated that the R1- and R2-substituents in the 3- and 4-positions of oxetenes scarcely affect the torquoselectivity, but rather affect the thermal stability of oxetenes in the reaction. Thus obtained, these tetrasubstituted olefins are essential as CF3-containing building blocks for pharmaceuticals and agrochemicals. The development of the catalytic asymmetric construction of trifluoromethylated tertiary and quaternary stereogenic carbon centers using these olefins is ongoing in our laboratory.Acknowledgements
This research was supported by Japan Science and Technology Agency (JST) (ACT-C: Creation of Advanced Catalytic Transformation for the Sustainable Manufacturing at Low Energy, Low Environmental Load), JSPS KAKENHI Grant Number 23750104, and Mizuho Foundation for the Promotion of Sciences. We thank Central Glass Co., Ltd. for the gift of ethyl trifluoropyruvate.Notes and references
- For recent reviews, see: (a) Z. Jin, G. B. Hammond and B. Xu, Aldrichimica Acta, 2012, 45, 67 CAS; (b) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (c) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (d) K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478 CrossRef CAS PubMed; (e) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455 CrossRef CAS PubMed; (f) K. Mikami, Y. Itoh and M. Yamanaka, Chem. Rev., 2004, 104, 1 CrossRef CAS PubMed.
- For selected recent examples, see: (a) M. Oishi, H. Kondo and H. Amii, Chem. Commun., 2009, 1909 RSC; (b) E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679 CrossRef CAS PubMed; (c) N. D. Ball, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 2878 CrossRef CAS PubMed; (d) X. Wang, L. Truesdale and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3648 CrossRef CAS PubMed; (e) L. Chu and F.-L. Qing, Org. Lett., 2010, 12, 5060 CrossRef CAS PubMed; (f) T. D. Senecal, A. T. Parsons and S. L. Buchwald, J. Org. Chem., 2011, 76, 1174 CrossRef CAS PubMed; (g) A. Zanardi, M. A. Novikov, E. Martin, J. Benet-Buchholz and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 20901 CrossRef CAS PubMed; (h) H. Morimoto, T. Tsubogo, N. D. Litvinas and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 3793 CrossRef CAS PubMed; (i) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224 CrossRef CAS PubMed; (j) Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95 CrossRef CAS PubMed.
- For selected recent examples, see: (a) A. T. Parsons, T. D. Senecal and S. L. Buchwald, Angew. Chem., Int. Ed., 2012, 51, 2947 CrossRef CAS PubMed; (b) M. Omote, M. Tanaka, A. Ikeda, S. Nomura, A. Tarui, K. Sato and A. Ando, Org. Lett., 2012, 14, 2286 CrossRef CAS PubMed; (c) G. K. S. Prakash, H. S. Krishnan, P. V. Jog, A. P. Iyer and G. A. Olah, Org. Lett., 2012, 14, 1146 CrossRef CAS PubMed.
- For examples of trisubstituted olefins bearing a CF3 group, see: (a) J. Duan, W. R. Dolbier, Jr and Q.-Y. Chen, J. Org. Chem., 1998, 63, 9486 CrossRef CAS; (b) M. Kimura, T. Yamazaki, T. Kitazume and T. Kubota, Org. Lett., 2004, 6, 4651 CrossRef CAS PubMed; (c) T. Konno, T. Takehana, M. Mishima and T. Ishihara, J. Org. Chem., 2006, 71, 3545 CrossRef CAS PubMed; (d) E. J. Cho and S. L. Buchwald, Org. Lett., 2011, 13, 6552 CrossRef CAS PubMed; (e) Z. He, T. Luo, M. Hu, Y. Cao and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 3944 CrossRef CAS PubMed; (f) K. Zine, J. Petrignet, J. Thibonnet and M. Abarbri, Synlett, 2012, 755 CAS; (g) L. Debien, B. Quiclet-Sire and S. S. Zard, Org. Lett., 2012, 14, 5118 CrossRef CAS PubMed; (h) P. G. Janson, I. Ghoneim, N. O. Ilchenko and K. J. Szabó, Org. Lett., 2012, 14, 2882 CrossRef CAS PubMed.
- For examples of tetrasubstituted olefins bearing a CF3 group, see: (a) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214 CrossRef CAS PubMed; (b) X. Liu, M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2004, 43, 879 CrossRef CAS PubMed; (c) Y. Takeda, M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2007, 46, 8659 CrossRef CAS PubMed; (d) M. Shimizu, Y. Takeda and T. Hiyama, Bull. Chem. Soc. Jpn., 2011, 84, 1339 CrossRef CAS.
- (a) H. Kawai, S. Okusu, E. Tokunaga, H. Sato, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2012, 51, 4959 CrossRef CAS PubMed; (b) Q.-H. Deng, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2012, 134, 10769 CrossRef CAS PubMed; (c) H. Kawai, S. Okusu, Z. Yuan, E. Tokunaga, A. Yamano, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2013, 52, 2221 CrossRef CAS PubMed; (d) J.-R. Gao, H. Wu, B. Xiang, W.-B. Yu, L. Han and Y.-X. Jia, J. Am. Chem. Soc., 2013, 135, 2983 CrossRef CAS PubMed.
- (a) W. R. Dolbier, Jr, H. Koroniak, K. N. Houk and C. Sheu, Acc. Chem. Res., 1996, 29, 471 CrossRef; (b) W. R. Dolbier, Jr, H. Koroniak, D. J. Burton, A. R. Bailey, G. S. Shaw and S. W. Hansen, J. Am. Chem. Soc., 1984, 106, 1871 CrossRef; (c) W. Kirmse, N. G. Rondan and K. N. Houk, J. Am. Chem. Soc., 1984, 106, 7989 CrossRef CAS; (d) N. G. Rondan and K. N. Houk, J. Am. Chem. Soc., 1985, 107, 2099 CrossRef CAS; (e) P. S. Lee, X. Zhang and K. N. Houk, J. Am. Chem. Soc., 2003, 125, 5072 CrossRef CAS PubMed; (f) Y.-h. Lam, S. J. Stanway and V. Gouverneur, Tetrahedron, 2009, 65, 9905 CrossRef CAS PubMed.
- (a) M. Murakami, Y. Miyamoto and Y. Ito, Angew. Chem., Int. Ed., 2001, 40, 189 CrossRef CAS; (b) M. Murakami, Y. Miyamoto and Y. Ito, J. Am. Chem. Soc., 2001, 123, 6441 CrossRef CAS; (c) M. Murakami and M. Hasegawa, Angew. Chem., Int. Ed., 2004, 43, 4874 CrossRef CAS PubMed; (d) M. Murakami, M. Hasegawa and H. Igawa, J. Org. Chem., 2004, 69, 587 CrossRef CAS PubMed; (e) M. Murakami, I. Usui, M. Hasegawa and T. Matsuda, J. Am. Chem. Soc., 2005, 127, 1366 CrossRef CAS PubMed.
- (a) M. Shindo and S. Mori, Synlett, 2008, 2231 CrossRef CAS PubMed; (b) M. Shindo and S. Mori, J. Synth. Org. Chem., Jpn., 2008, 66, 28 CrossRef CAS; (c) M. Shindo, K. Matsumoto, S. Mori and K. Shishido, J. Am. Chem. Soc., 2002, 124, 6840 CrossRef CAS PubMed; (d) S. Mori and M. Shindo, Org. Lett., 2004, 6, 3945 CrossRef CAS PubMed; (e) M. Shindo, T. Yoshikawa, Y. Itou, S. Mori, T. Nishii and K. Shishido, Chem.–Eur. J., 2006, 12, 524 CrossRef PubMed; (f) T. Yoshioka, S. Mori and M. Shindo, Tetrahedron, 2009, 65, 8832 CrossRef PubMed; (g) T. Yoshikawa, S. Mori and M. Shindo, J. Am. Chem. Soc., 2009, 131, 2092 CrossRef CAS PubMed.
- For examples of the ring opening reaction with amidines, see: (a) N. Shindoh, Y. Takemoto and K. Takasu, Chem.–Eur. J., 2009, 15, 7026 CrossRef CAS PubMed; (b) N. Shindoh, K. Kitaura, Y. Takemoto and K. Takasu, J. Am. Chem. Soc., 2011, 133, 8470 CrossRef CAS PubMed.
- (a) W. R. Dolbier, Jr, T. A. Gray, J. J. Keaffaber, L. Celewicz and H. Koroniak, J. Am. Chem. Soc., 1990, 112, 363 CrossRef; (b) W. R. Dolbier, Jr, H. Koroniak, D. J. Burton and P. Heinze, Tetrahedron Lett., 1986, 27, 4387 CrossRef; (c) W. R. Dolbier, Jr, H. Koroniak, D. J. Burton, P. Heinze, A. R. Bailey, G. S. Shaw and S. W. Hansen, J. Am. Chem. Soc., 1987, 109, 219 CrossRef.
- K. Aikawa, Y. Hioki, N. Shimizu and K. Mikami, J. Am. Chem. Soc., 2011, 133, 20092 CrossRef CAS PubMed.
- Perfluoroalkyl effect, see: (a) D. M. Lemal and L. H. Dunlap, Jr, J. Am. Chem. Soc., 1972, 94, 6562 CrossRef CAS; (b) D. M. Lemal, J. Org. Chem., 2004, 69, 1 CrossRef CAS PubMed.
- (a) W. J. Middleton, J. Org. Chem., 1965, 30, 1307 CrossRef CAS; (b) Y. Kobayashi, Y. Hanzawa, W. Miyashita, T. Kashiwagi, T. Nakano and I. Kumadaki, J. Am. Chem. Soc., 1979, 101, 6445 CrossRef CAS; (c) P. C. Martino and P. B. Shevlin, J. Am. Chem. Soc., 1980, 102, 5429 CrossRef CAS; (d) L. E. Friedrich and P. Y.-S. Lam, J. Org. Chem., 1981, 46, 306 CrossRef CAS; (e) M. Oblin, J.-L. Parrain, M. Rajzmann and J.-M. Pons, Chem. Commun., 1998, 1619 RSC.
- Theoretical studies on torquoselectivity of various oxetenes bearing fluoromethyl groups are ongoing in our laboratory in more detail.
- I. V. Alabugin and T. A. Zeidan, J. Am. Chem. Soc., 2002, 124, 3175 CrossRef CAS PubMed.
- Representative secondary orbital interactions are shown in Fig 2b. See ESI† for more details.
- In perfluoro systems, such as perfluoro-3-methyl- and -3,4-dimethylcyclobutenes, Dolbier and co-workers have already reported that the inward rotation of the CF3 group proceeded with the support of a dramatic outward preference of the fluorine substituents in the 3- and 4-positions; see ref. 7b and 11.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 948159–948162. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc52548a |
| This journal is © The Royal Society of Chemistry 2014 |