
Cooperative adsorption behavior of phosphopeptides on TiO2 leads to biased enrichment, detection and quantification†
A. I. K.
Eriksson
,
K.
Edwards
and
V.
Agmo Hernández
*
Department of Chemistry – BMC, Uppsala University, Husargatan 3, Box 579, 75123, Uppsala, Sweden. E-mail: victor.agmo@kemi.uu.se; Fax: +46 (0)18 471 3654; Tel: +46 (0)18 471 3635
First published on 15th October 2014
Abstract
The adsorption behavior of phosphopeptides onto TiO2 surfaces was studied using the quartz crystal microbalance with dissipation monitoring (QCM-D) as the main experimental technique. The main focus is the characterization of the emergence of positive cooperativity under conditions where the peptides have a positively charged C-term. It is shown that when carrying no net charge, small water-soluble peptides as a rule develop positive cooperativity. The impact of the adsorption mechanism on the outcome of TiO2 based enrichment methods was investigated with the help of matrix assisted laser desorption-ionization mass spectrometry (MALDI-MS). The data presented illustrate how the phosphopeptide profile in the enriched material may deviate from that in the native sample, as cooperative phosphopeptides are overrepresented in the former. Furthermore, commonly employed washing and elution solutions may facilitate preferential release of certain peptides, leading to further bias in the recovered sample. Taken together, the results of the present study demonstrate that thorough understanding of the mechanisms behind the adsorption of phosphopeptides on the enrichment material is necessary in order to develop reliable qualitative and quantitative methods for phosphoproteomics.
Introduction
The field of phosphoproteomics has grown rapidly in recent decades and mapping of the phosphoproteome is now recognized as essential for the complete characterization of proteins. Although the main focus to date has been the identification of new phosphorylation sites, recent research has also focused on characterizing the activity of already identified sites. The latter requires, in contrast to pure identification, accurate quantification of the amount of phosphorylated peptides both before and after triggering the activation. As a result, quantitative phosphoprotein analysis has become essential for the field.1–3The most crucial part of any phosphopeptide analysis, be it qualitative or quantitative, is the specific enrichment step, where the relatively low abundant phosphopeptides are isolated from the native protein digest in order to make their detection possible. There is a variety of different enrichment methods to choose from at the present time.4–11 However, poor overlap between the results obtained from different methods still remains a major problem.12–14 Some approaches seem to favor, for example, the enrichment of monophosphorylated peptides while others favor multiphosphorylated.12,15,16 In order to help devise an enrichment method that accurately reflects the phosphoproteome composition, it is first necessary to elucidate the mechanisms leading to enrichment in the methods most commonly used today.
In a recently published study we characterized the physical chemistry behind the phosphopeptide interaction with TiO2,17 which is one of the most commonly used enrichment materials (e.g. in metal oxide affinity chromatography (MOAC)15,18–20 and on-target methods10,11,13,21–32 for matrix assisted laser desorption-ionization mass spectrometry (MALDI-MS) analysis). In this study,17 the very interesting phenomenon of positive cooperativity was observed in the adsorption behavior of certain peptides. This means that, for the cited cases, there exists an attractive interaction between the peptides themselves, which implies that they can adsorb in multiple layers.33 The source of the cooperativity was determined to be electrostatic interactions between positive amino acids in at least one of the peptide end terms and negative phosphoryl groups. The electrostatic nature of the interaction suggests that the phenomena might be quite common in the digested samples normally used for analysis. The reason is that one of the most commonly used enzymes for protein digestion, trypsin, cleaves the amino acid chains C-terminally to lysine, K, or arginine, R, both of which are positively charged under the conditions that are normally used during the enrichment procedure. Phosphorylated peptides recovered after trypsination would therefore fulfill the condition previously observed for cooperativity to emerge.
In the present work, the adsorption behavior of synthetic monophosphorylated peptides in which cooperativity can be expected is studied with the help of the quartz crystal microbalance with dissipation monitoring (QCM-D) technique. The conditions under which cooperativity emerges are discussed. The impact that cooperativity may have on the reliability of the results obtained with common TiO2-based enrichment methods is also investigated and exemplified by studying on-target binding and results obtained from MALDI-MS. Even though it is virtually impossible to consider all possible variations in the structure and properties of phosphorylated peptides, this report illustrates phenomena which are likely to affect the output of most TiO2-based enrichment methods in use today. It also establishes a starting point from which future investigations on phosphopeptide enrichment behavior can benefit.
Materials and methods
Chemicals
Custom designed phosphopeptides were purchased from Thermo Scientific (Ulm, Germany). Trifluoroacetic acid (TFA >99%), sulfuric acid, hydrogen peroxide, acetonitrile (ACN), 2,5-dihydrobenzoic acid (DHB), salicylic acid (SA), ammonium dihydrogen phosphate (98%), ammonium hydroxide solution (25–28%), phosphoric acid (85%), acetic acid (HAc), terpineol, sodium phosphate (mono- and dibasic), and sodium chloride were obtained from Sigma-Aldrich (Steinheim, Germany). All aqueous solutions were prepared in deionized water (18.4 MΩ cm) obtained from a Milli-Q system (Millipore, Bedford, USA). Indium–tin oxide (ITO) coated glass slides were obtained from Hudson Surface Technology (Fort Lee, NJ, USA). TiO2 paste containing nanoparticles with a diameter of 18 nm and a pure anatase crystal structure was purchased from Dyesol (New South Wales, Australia). The paste was further diluted with four parts of terpineol before use. Titanium coated QCM-D sensors were purchased from Q-Sense (Gothenburg, Sweden).Peptides
Four custom designed phosphopeptides were used. These peptides are modifications of the EGFR peptide (DADE-pY-LIPQQG), whose interactions with TiO2 was characterized in our previous study.17 The peptides were designed so that they would fulfill the conditions that give rise to cooperativity according to our previous paper. First, the C-term amino acid (G) in all four custom peptides was changed to arginine (R), to simulate a tryptic digestion product. The N-term amino acid (D) was also changed to arginine (R) in two of the peptides. Finally, the phosphorylated amino acid was changed from tyrosine (Y) to serine (S) in two of the peptides. In this way all combinations of either tyrosine or serine phosphorylated peptides with either one or two R-substitutions were obtained. The sequences, mass and expected charge of all peptides at the pH used (2.6) are shown in Table 1. All peptide solutions were prepared by dilution with 0.01% TFA (aq.) from a stock solution of 1 mg mL−1 in TFA buffer (ACN/water/TFA 50/49.9/0.1 (v/v)).![[Y with combining low line]](https://www.rsc.org/images/entities/b_char_0059_0332.gif)
![[S with combining low line]](https://www.rsc.org/images/entities/b_char_0053_0332.gif)
![[R with combining low line]](https://www.rsc.org/images/entities/b_char_0052_0332.gif)
QCM-D
The quartz crystal microbalance with dissipation monitoring (QCM-D) is a technique that allows both the mass and viscoelastic properties of an adsorbed layer to be determined. The technique has been used to study the interactions, structure and kinetics of many different biological systems (lipids, DNA, proteins, cells, etc.).34–40 The core of the technique is a thin, circular quartz crystal with one of its faces (the sensing face) coated with the substrate material of interest. The other face is connected to an alternate potential which causes the crystal to oscillate due to the piezoelectric properties of quartz. During the QCM-D measurement, two parameters are monitored: the oscillation frequency (f) of the quartz crystal and the dissipation factor (D). Negative shifts in the oscillation frequency are related to the adsorption of the material on the surface of the sensor (the more mass adsorbed, the larger the shift). Changes in the dissipation factor, on the other hand, are related to the viscoelastic properties of the adsorbed film. For rigid films (ΔD ≈ 0), the frequency change obtained (Δf) can be directly related to the adsorbed mass (Δm) through the Sauerbrey relationship41| Δm = −xAΔfnn−1 | (1) |
All experiments were performed as described previously by Eriksson et al.17 Briefly, an E1 QCM-D model (Q-Sense, Gothenburg, Sweden) was used with a pump connected downstream in the flow system. The temperature was set to 21 °C and the pump flow was set to 100 μL min−1. Data were collected from the fundamental sensor oscillation frequency (5 MHz) as well as the 3rd, 5th, 7th, 9th, 11th and 13th overtones. The obtained data were used to build adsorption isotherms for the peptides studied.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 sulfuric acid–hydrogen peroxide) for 10–15 s in order to oxidize the surface. The clean sensors were then rinsed with water and left in 99% ethanol for at least 5 min to wash away any chemical residues. The sensors were dried with a gentle nitrogen flow before being mounted onto the instrument.
:1 sulfuric acid–hydrogen peroxide) for 10–15 s in order to oxidize the surface. The clean sensors were then rinsed with water and left in 99% ethanol for at least 5 min to wash away any chemical residues. The sensors were dried with a gentle nitrogen flow before being mounted onto the instrument.
The QCM-D flow system (excluding the sensor) was incubated in a PBS buffer (10 mM phosphate, 150 mM NaCl, pH 5.5) for 30 min before the experiment in order to saturate all surfaces with PO4− and avoid phosphopeptide loss in the loop. After mounting of the sensor, the system was loaded with the working solution (0.01% TFA) and allowed to stabilize.
Since the equilibrium is reached very slowly, the peptide solution was recirculated to avoid the use of large sample volumes. The equilibrium concentration was then calculated by subtracting the amount of adsorbed molecules from the total amount of molecules in the loading solution.
Phosphopeptide binding to TiO2-modified MALDI-MS targets
A 1:1:1:1 (0.25 μM each) mixture of the four peptides studied was loaded onto MALDI-MS targets modified with TiO2 spots according to the protocol for phosphopeptide enrichment reported by Eriksson et al.24 MALDI-MS experiments were performed after loading of the phosphopeptides on the substrate as well as after washing with different buffers/solutions that are often used in the washing or elution steps of common enrichment methods. The solutions used in this study were: (i) SA buffer (18 mg mL−1 SA in ACN/water/TFA 50/49.9/0.1 (v/v)), aqueous ammonium acetate (ii) pH 8, and (iii) pH 9, (iv) aqueous TFA (50 mM, pH 1.33), and (v) aqueous phosphate (10 mM NH4H2PO4, pH 2.6). Finally, the mixture was also analyzed with the combined enrichment and separation method (stripe target), developed by Eriksson et al.13
Results and discussion
Emergence of cooperativity
Adsorption isotherms were constructed from the QCM-D data by plotting the fraction of surface coverage (θ) versus the equilibrium bulk concentration (C). In agreement with our previous report,17 the dissipation changes were in all cases negligible and a proportional dependence of the adsorbed mass on the frequency shift can therefore be assumed (according to the Sauerbrey relationship given in eqn (1) above). The value of θ for each of the loaded concentrations can therefore be calculated directly as the ratio of the frequency shift Δf measured for each point divided by the frequency shift observed at saturation (Δfmax). With the obtained data, Scatchard plots (θ/C vs. θ) were built to better visualize any cooperativity. The different shapes obtained in the Scatchard plots are shown in Fig. 1. The initial positive slope of the Scatchard plots observed for the mod-Y-R (a) and mod-S-R (b) peptides (resulting in a characteristic “sad-mouth” shape of the curve), corresponds to the increase in surface affinity of the analyte upon increasing surface coverage expected for positive cooperativity.43 On the other hand, the Scatchard plots obtained for mod-S-2R (c) and mod-Y-2R (d) can be fitted to a straight line, indicating a Langmuir-like behavior.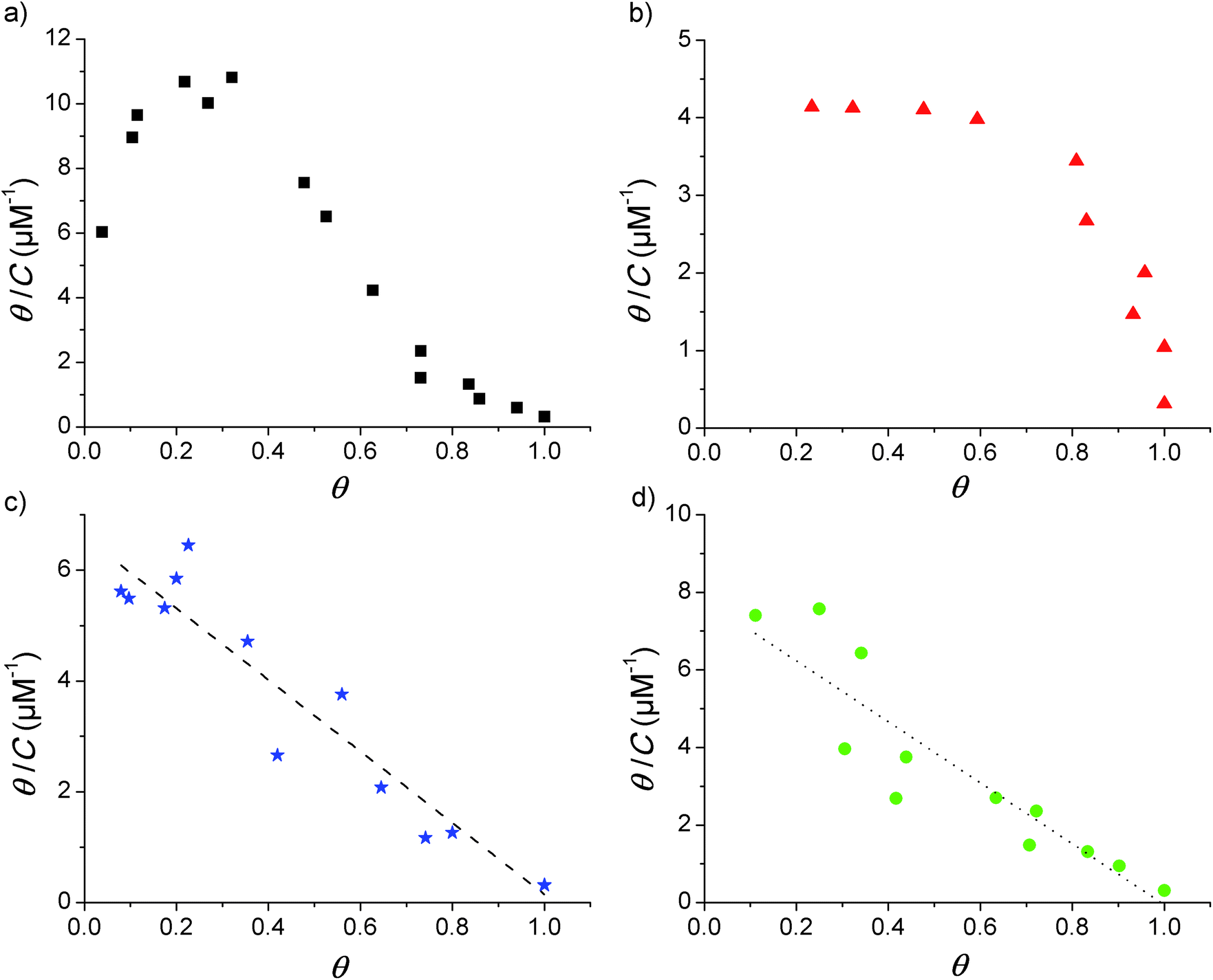 | ||
| Fig. 1 Scatchard plots obtained from the adsorption isotherms of the modified peptides. (a) mod-Y-R, (b) mod-S-R, (c) mod-Y-2R, and (d) mod-S-2R. The lines represent the linear fitting of the plots in (c) and (d): (c) y-intercept = −slope = K = 6.5 ± 0.33 μM−1, R2 = 0.920); (d) K = 7.8 ± 0.65 μM−1, R2 = 0.849. | ||
The two latter peptides follow an almost ideal Langmuir behavior, as suggested by the linear Scatchard plots obtained. Unfortunately, the QCM-D technique is not sensitive enough to resolve the adsorption behavior at surface coverage values lower than those shown in the figure. However, the observed wide linear range recorded for mod-S-2R and mod-Y-2R serves as a reliable indication of a Langmuir-like behavior. It is noteworthy that, for these samples, only the peptides substituted with a single arginine (R) show unequivocally positive cooperativity. Notably, in our previous work17 we demonstrated that IR (a tri-phosphorylated peptide derived from the insulin receptor and with three positive residues at or close to the ends) also shows cooperativity in spite of the multiple positive residues. The common characteristic of all peptides showing cooperative behavior, besides the presence of positive residues at the end of the sequence, is a net zero-charge of the molecule. Therefore, it can be speculated that electrostatic attraction forces between negative phosphoryl groups and positive amino acid residues alone are not enough for cooperativity to emerge. It also appears to be important that electrostatic repulsive forces between the peptides are absent or minimal.
The plots in Fig. 1 further suggest that the cooperativity appears irrespective of whether the phosphorylated amino acid residue is tyrosine or serine. However, the overall shapes of the Scatchard plots obtained for the two cooperative peptides are quite different, which indicates that even though both are cooperative, the parameters describing the interaction are different. It is beyond the scope of this report to study the mechanisms behind this variance, but it is an interesting observation for future studies.
It is also necessary to point out that, although all the peptides in the present study contain an arginine end; other positively charged C-term amino acids may give rise to cooperative adsorption behavior. As an example, we previously showed that the IR peptide,17 in which lysine is found at the C-term, presents a distinct cooperative behavior. Given the evidence gathered up to this point, it would appear that a positive charge at the C-term and an overall neutral charge of the peptide are the only requirements for cooperativity to emerge. Whether the same would be observed if the positive charge is located in the N-term, and whether cooperativity would arise from a positive charge just right next to the end-term amino acids, are matters for future research.
In summary, the observations described above suggest that positive cooperativity might be a common phenomenon during phosphopeptide enrichment from biologically relevant samples, since most monophosphorylated tryptic peptides (considering no missed cleavages) will present a positively charged C-term and display a zero-net charge under the conditions commonly used in these studies. This may bias the analysis, as cooperative phosphopeptides can be overrepresented in the enriched sample, as will be shown in the following sections.
Impact of cooperativity on TiO2-based enrichment methods
:1:1:1 mixture of the four custom modified peptides on a normal steel target (Fig. 2a) and after equilibration on a TiO2 spot (Fig. 2b). The figure is the result of adding several spectra obtained at different random positions on different spots.
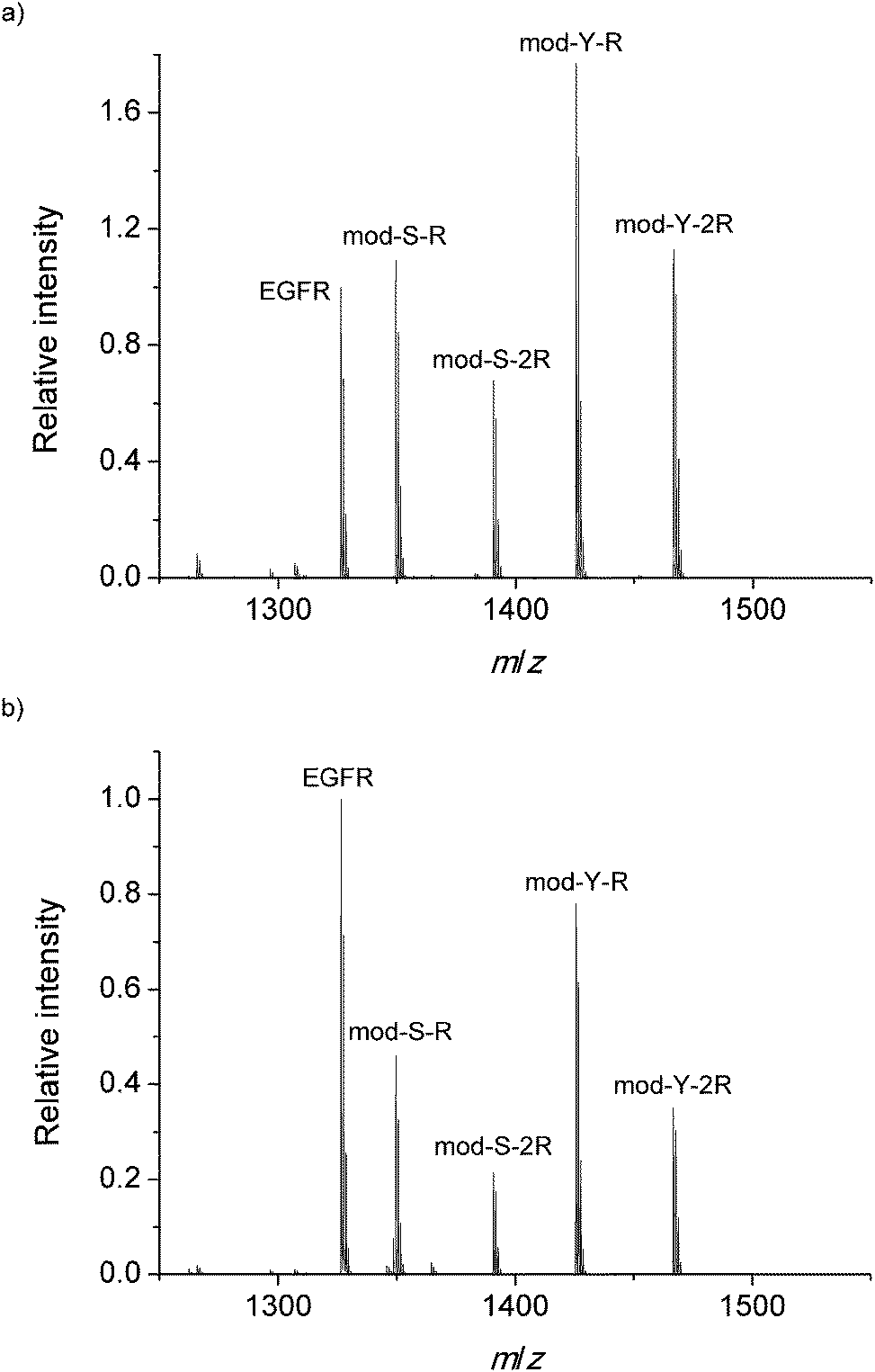 | ||
| Fig. 2 Internal standard normalized MALDI-MS spectrum obtained (a) on the steel target and (b) after binding on TiO2. | ||
In order to be able to estimate the efficiency of the phosphopeptide i.e. the proportion of phosphopeptides in the sample that bind to the TiO2 surface, a known concentration (1 μM) of native phosphorylated EGFR peptide was added to the DHB matrix in both experiments. This serves as a standard to monitor the extent of binding. Interestingly, the cooperative peptides present a higher signal intensity in both measurements, despite the results of earlier research which have shown that the ionization efficiency is related to the amount of R residues.44 The MS spectrum obtained on the steel target is used as a reference to monitor any bias during the adsorption on TiO2. Fig. 2b presents the mean mass spectrum obtained after equilibration on the TiO2 spots, which shows a clear decrease in all phosphorylated signals with respect to the EGFR standard. This decrease in the relative signal may arise from complete saturation of the surface with phosphopeptides and subsequent removal of the excess peptides during the 10 second immersion in the 0.01% TFA bath described in the methods.
However, even though EGFR fulfills the requirements for a suitable standard (similar MW, structure, etc.),45,46 the results should be treated with caution as MALDI-MS is not completely reliable as a quantitative technique45 given the irregular distribution of the analyte and the matrix on the spots. The trend observed is merely an indication of a likely adsorption behavior. More interestingly, the spectra obtained after equilibration on the TiO2-modified target show a pronounced decrease in the signal of the non-cooperative peptides with respect to the cooperative peptides. This is clearly appreciated in Fig. 3, which shows the signal from phosphopeptides adsorbed on TiO2 normalized against the signals on the steel target.
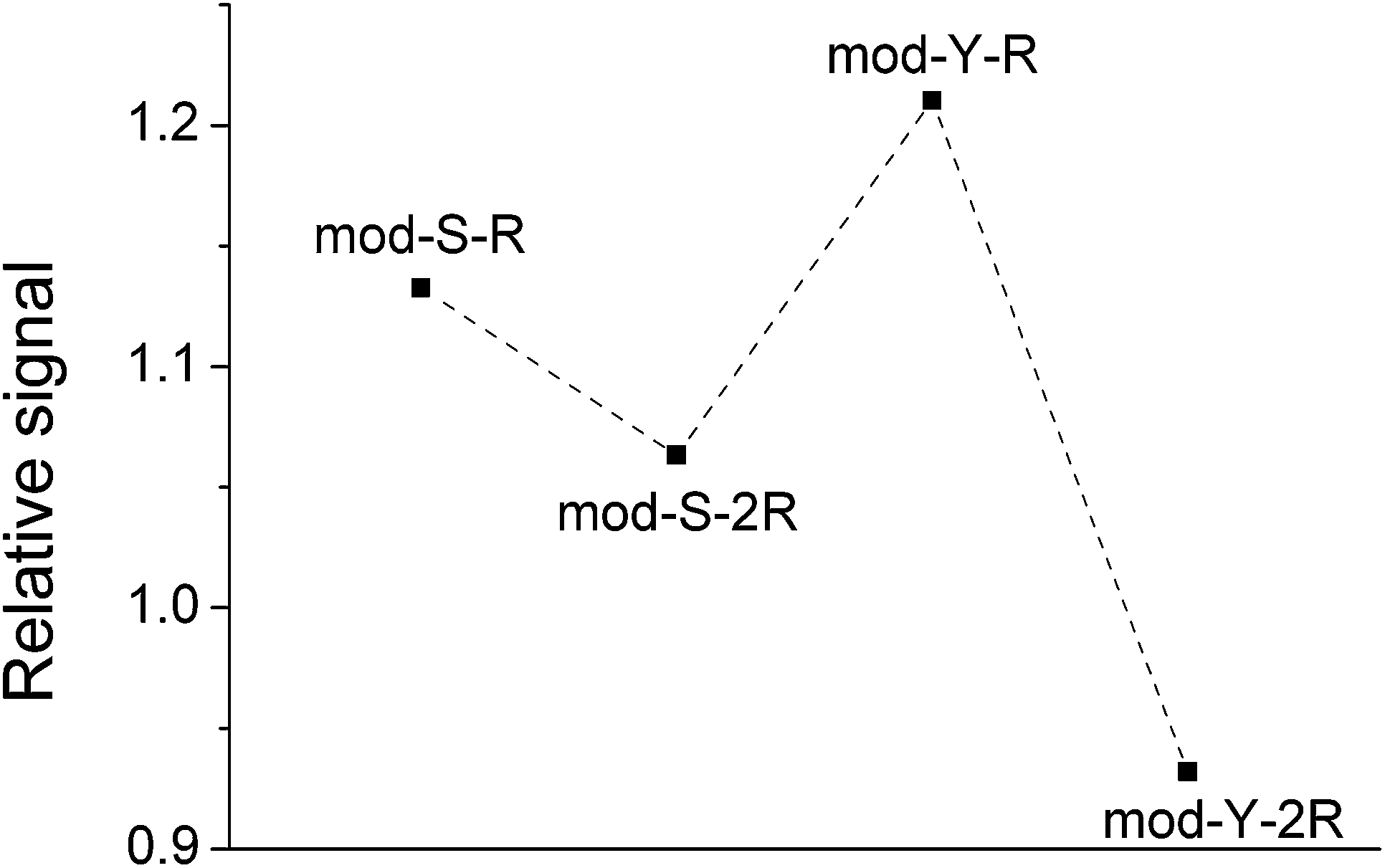 | ||
| Fig. 3 Diagram showing the average of the ratio of the signal areas obtained on TiO2 spots and on steel. The data presented is the average of the ratios calculated for every single pair of experiments (y-axis = 〈(Signal TiO2)/(Signal steel)〉) and comprises 18 spectra obtained on 10 spots on steel and 23 spectra obtained on 10 TiO2 spots (total = 18 × 23 = 414 ratios were calculated for each spot). | ||
The same trend was observed in all repetitions of the experiment. In the case of a non-biased adsorption, all points in the figure should appear at similar y-values. However, here it is apparent that the signals from the non-cooperative peptides decrease significantly more than those from the cooperative peptides, meaning that the binding under these conditions would be biased towards the latter. Paired t-tests corroborated that the relative signals of mod-Y-R and mod-S-R are indeed significantly larger than those of mod-Y-2R (p ≪ 0.001) and mod-S-2R (p = 0.007), respectively. This result agrees with the notion of multiple adsorbed layers being formed in the case of cooperative peptides, which in principle can adsorb even when there are no sites left on the TiO2 surface (a condition that is likely fulfilled given that most of the loaded phosphopeptides remain in solution). Both the raw experimental data and the supporting statistical analysis are included as Appendix A in the ESI.†
These findings clearly show that phosphopeptide enrichment methods may be intrinsically biased, as cooperative phosphopeptides will be overrepresented in the enriched sample. In qualitative studies, in which the aim is to determine the amino acid residues that can be found in a phosphorylated protein, the non-cooperative peptides may be missed as their concentration will be lower. In quantitative phosphoproteomics, the consequences may be even more severe, as, even if the peptides are successfully detected, the quantification will only reflect the relative composition of what was enriched, and not necessarily that of what was originally present in the sample. The co-enrichment of known concentrations of isotopically labeled peptides could in principle serve as an internal standard to reflect and correct for this bias. However, this would only be reliable if the concentrations of labeled and unlabeled peptides are in the same range or if the adsorption of the peptide follows pseudo-first-order kinetics. The first condition requires previous knowledge concerning the concentration in the sample, which makes the quantification experiment pointless. The second condition may be achieved if samples with sufficient peptide concentrations are used, or when the number of binding sites on the surface is excessively large. The former option requires a highly concentrated sample while the latter may result in a highly diluted sample for analysis. Even though the right conditions could be achieved to account for the preferential enrichment of cooperative peptides, it is in any case necessary to, first and foremost, characterize the adsorption mechanism of the peptide.
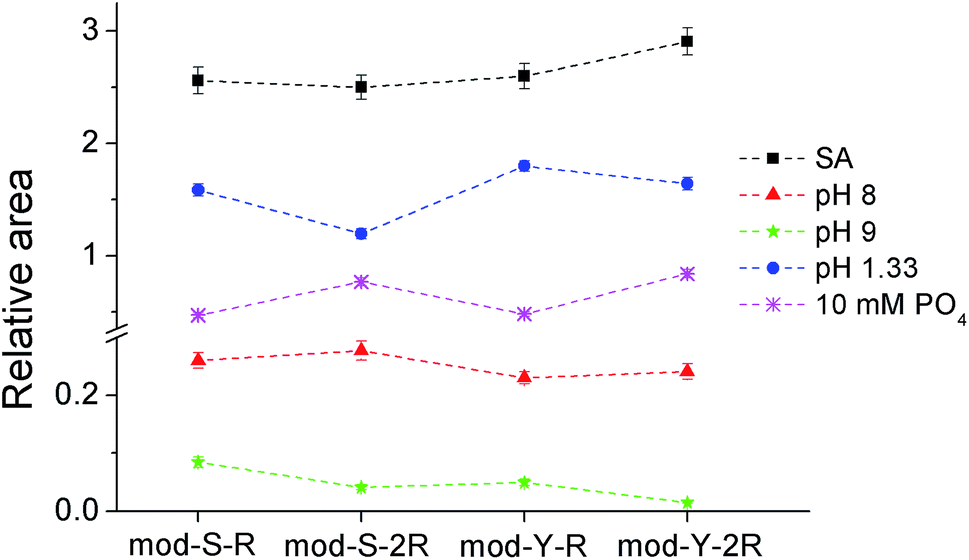 | ||
| Fig. 4 Diagram showing the average of the ratio of the signal areas obtained after washing on a TiO2 spot and on the spot without washing treatment. The washing solutions used are indicated in the legend. The data presented are the average of the ratios calculated for every single pair of experiments (y-axis = 〈(Signal after washing)/(Signal on TiO2)〉) and comprises 23 spectra obtained on 10 TiO2 spots and at least 9 spectra obtained on at least 3 washed spots. The error bars represent the standard error. | ||
The results from the treatment with pH 8 (Fig. 4, red triangles) show that the serine substituted peptides are slightly overrepresented with respect to the tyrosine substituted peptides (p < 0.05). On the other hand, cooperativity does not seem to play a role (raw data and the results from the paired t-test can be found in Appendix C in the ESI†). At pH 9 (green stars), the excess of serine substituted peptides is still present, and a significant excess of cooperative peptides is observed (raw data and the paired t-tests are provided in Appendix D in the ESI†). In fact, non-cooperative peptides were not observed at all in 80% of the experiments performed. It can be speculated that the preferential release of non-cooperative peptides arises due to the lower pKa of TiO2 (∼5.8)50 as compared to that of the side chain of the arginine residues (∼12.8), meaning that the interaction assumed to give rise to cooperativity is much less affected than the one keeping the peptides bound to the surface. It may seem counterintuitive that a non-affected peptide–peptide interaction can prevent the release of peptides from the TiO2 surface when the peptide–TiO2 interaction is decreased. However, it is necessary to take into account the fact that the peptide–TiO2 and the peptide–peptide binding reactions are simultaneous and reversible and therefore codependent. It can be demonstrated from the extended 2-layer BET-isotherm model, which describes the cooperative behavior,17 that the amount of cooperative peptides that would remain at the surface is in fact larger than that of non-cooperative if only, or primarily, the TiO2–peptide interaction is reduced.
Another interesting feature of the pattern obtained at basic pH values is the relative increase in the serine containing peptide signals compared to the equivalent tyrosine containing versions. This indicates that tyrosine phosphorylated peptides are more easily washed away by basic pH than the corresponding serine phosphorylated peptides. It should be considered, however, that, as shown in Fig. 4 and in Appendix C,† the measured signals are very low, suggesting that a great majority of the peptides are washed away at this moderately high pH.
The results obtained at pH 1.33 (blue circles) show a similar trend concerning cooperativity to that observed at pH 9, i.e. cooperative peptides are significantly overrepresented after the washing treatment. In this case, the results are far more trustworthy given the high signal intensities (raw data and the paired t-test included in Appendix E in the ESI†). Under the used acidic conditions, it is expected that a significant proportion (∼40%) of the phosphoryl groups will be protonated, thus losing their negative charge, which should affect both the binding to TiO2 and the interaction between peptides. The decrease in the signals of non-cooperative relative to the cooperative peptides can be expected from this assumption and is in line with the 2-layer BET isotherm model used to describe the cooperative behavior. Noteworthy, in contrast to the behavior found at basic pH values, is the fact that the relative signal from serine substituted peptides decreases with respect to the signal from the equivalent tyrosine substituted versions. This observation indicates that the binding of phosphorylated tyrosine is less affected at acidic pH values, suggesting the possibility that the phosphoryl group in phosphotyrosine has a lower pKa than in phosphorylated serine.
In principle, the bias obtained from phosphate washing can help compensate for the bias of the enrichment method itself as they are opposite. However, it is not clear which of the effects will be predominant.
| Washing/elution solution | Preferent binding | Preferent release (biasa for column MOAC methods) |
|---|---|---|
| a Not considering the bias from the adsorption. | ||
| SA | None | None |
| Ammonium acetate, pH 8 | None | None |
| Ammonium acetate, pH 9 | Serine-phosphorylated peptides | Non-cooperative peptides |
| TFA, pH 1.33 | Cooperative peptides | Non-cooperative peptides |
| Phosphate 10 mM, pH 2.6 | Non-cooperative peptides | Cooperative peptides |
First, peptides that show cooperative adsorption will be overrepresented after TiO2 enrichment, regardless of the experimental approach used (on-target or in-column enrichment). Secondly, the washing/elution solution may cause further bias. With regard to on-target methods, in which usually SA or similar buffers are used for washing, this second problem can be diminished or avoided. On the other hand, in the column-based MOAC approach, where pH changes are usually used to release the bound peptides, the results will be dependent on the eluent used. This is a problem already addressed by Aryal et al.20 who discovered that the phosphopeptides recovered from TiO2-MOAC columns varied significantly with the eluent used, as the results in this report also show.
The results of the experiment are shown in Fig. 5, where the MALDI mass spectra obtained from different sections of the stripe (see the Methods section) are shown.
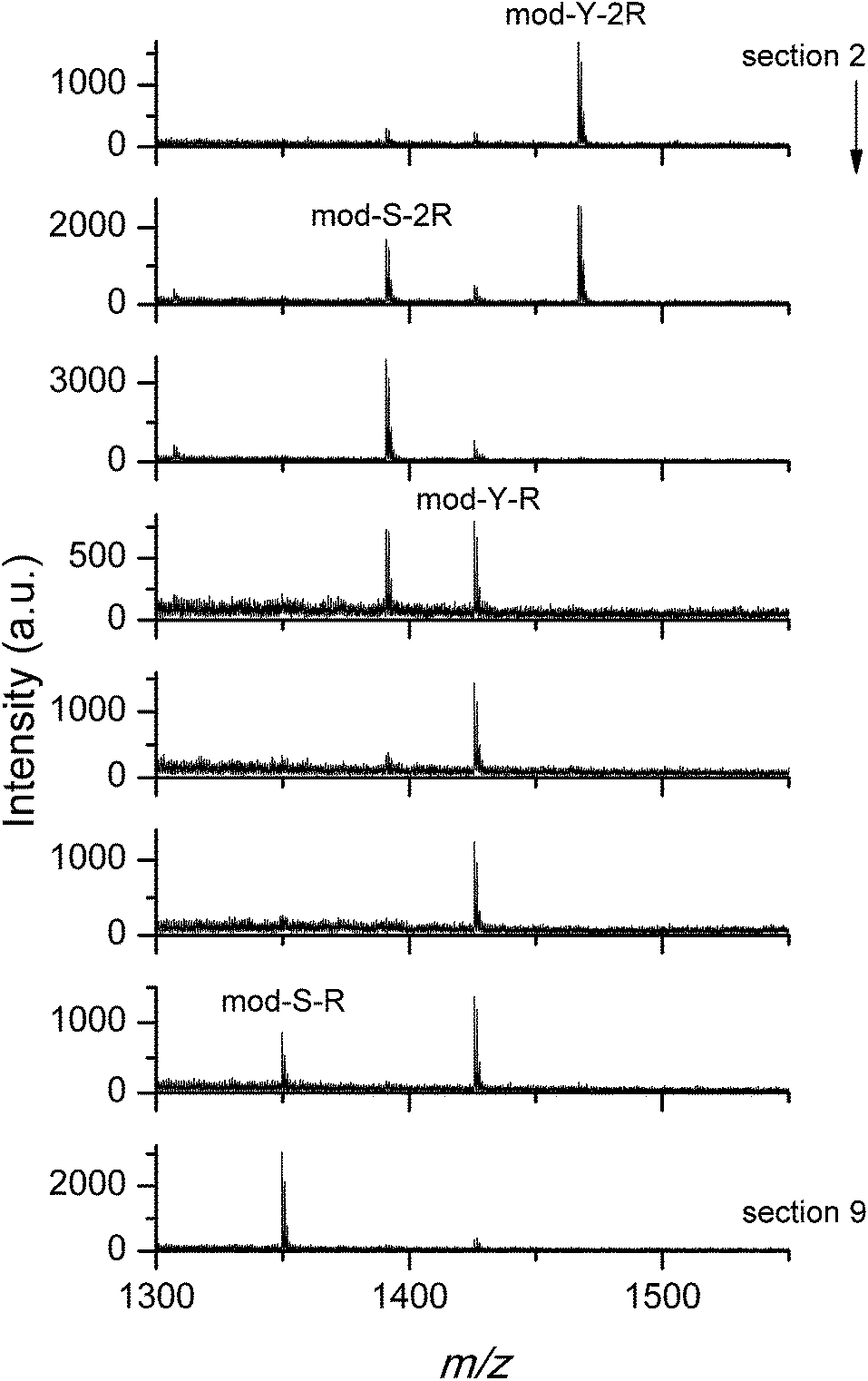 | ||
| Fig. 5 MALDI mass spectra for sections 2 (start)–9 (end) of the stripe target. The first occurrence of each peptide peak is indicated. The arrow indicates the direction of the elution. | ||
As can be seen from the figure, the loaded peptides were successfully separated. In agreement with the results discussed in the previous section, the elution with phosphate anions favored the release of cooperative peptides, which are found further up the stripe. The small difference between serine and tyrosine substituted peptides observed in Fig. 4 also affects the elution behavior, as the serine peptides are more easily eluted than the corresponding tyrosine substituted versions. Similar experiments were performed using basic buffers (pH value between 6.5 and 9) as eluents instead of the phosphate solution used above, with the purpose of confirming a behavior opposite to that shown in Fig. 5. Unfortunately, the almost instant release of the peptides at high pH is a problem, given the slow elution rate. The peptides diffuse in the available volume and are lost in the pool of eluent. The small traces detected in the stripe do not allow for further analysis.
In any case, the good agreement between the QCM-D, the on-target spot binding/washing, and, in the case of phosphate washing, the stripe separation experiments, is remarkable. Taken together, our results provide solid evidence concerning the preferential binding/washing/elution of different sets of peptides depending on their adsorption mechanism and the eluent/washing solution used.
Conclusions
The presence of competing adsorption mechanisms can result in significant overrepresentation of certain phosphopeptides in the enriched sample. The results from this study suggest that positive cooperativity, i.e. the positive interaction between adsorbed peptides and peptides in the bulk solution, might be a common phenomenon during the enrichment of tryptic peptides. It is, however, important to note that the peptides studied here are rather small and water-soluble. Further investigations are therefore needed to elucidate if the predictions from the present study also hold for larger and more hydrophobic peptides, where more complex interactions may come into play. Furthermore, phosphopeptide enrichment from complex samples (such as tryptic digests from large proteins) is likely to involve several other adsorption mechanisms, and whether cooperativity is significantly relevant in such a scenario needs to be explored further.However, it is clear that unless the adsorption mechanisms of each peptide are known and taken into account, the analysis of the eluted fraction in chromatography, or of the remaining peptides bound in on-target methods, may give a skewed picture of the distribution of phosphorylation. Even if the purpose is to perform a comparative analysis, complex adsorption behavior may lead to erroneous conclusions concerning the extent of the increase or decrease in the phosphopeptide concentration. Data collected in the present study also illustrate that the washing and elution steps needed to recover the phosphopeptides, or to wash away the remaining non-phosphorylated peptides, may lead to further bias. These observations may, at least partially, explain the lack of overlap between the phosphoproteomes determined from different methods. This problem clearly needs to be resolved in order to obtain reliable quantitative phosphopeptide analysis methods. Given the potential complexity of the adsorption behavior in a relevant peptide mixture, it is likely that the only option to perform a reliable quantitative analysis is to enrich or isolate 100% of all the phosphopeptides present.
In summary, it can be concluded that in-depth knowledge of the various mechanisms governing the affinity of phosphopeptides to the enrichment material is necessary for the further development of reliable enrichment protocols. In the present study we have clarified some important factors relevant to the interaction between small, water-soluble phosphopeptides and TiO2-based materials.
Acknowledgements
Financial support from the Swedish Research Council is acknowledged. A. Hagfeldt (Department of Chemistry – Ångström, Uppsala University) is acknowledged for providing the TiO2 and J. Bergquist (Department of Chemistry-BMC, Uppsala University) for assistance with MALDI TOF/TOF MS.References
- A. Nita-Lazar, H. Saito-Benz and F. M. White, Proteomics, 2008, 8, 4433–4443 CrossRef CAS PubMed.
- T. B. Schreiber, N. Mausbacher, S. B. Breitkopf, K. Grundner-Culemann and H. Daub, Proteomics, 2008, 8, 4416–4432 CrossRef CAS PubMed.
- C. L. Nilsson, Anal. Chem., 2012, 84, 735–746 CrossRef CAS PubMed.
- A. Tichy, B. Salovska, P. Rehulka, J. Klimentova, J. Vavrova, J. Stulik and L. Hernychova, J. Proteomics, 2011, 74, 2786–2797 CrossRef CAS PubMed.
- F. Delom and E. Chevet, Proteome Sci., 2006, 4, 15 CrossRef PubMed.
- T. E. Thingholm, O. N. Jensen and M. R. Larsen, Proteomics, 2009, 9, 1451–1468 CrossRef CAS PubMed.
- K. Engholm-Keller and M. R. Larsen, Proteomics, 2013, 13, 910–931 CrossRef CAS PubMed.
- J. Fila and D. Honys, Amino Acids, 2012, 43, 1025–1047 CrossRef CAS PubMed.
- I. L. Batalha, C. R. Lowe and A. C. A. Roque, Trends Biotechnol., 2012, 30, 100–110 CrossRef CAS PubMed.
- M. L. Stolowitz, Proteomics, 2012, 12, 3438–3450 CrossRef CAS PubMed.
- Y. Zhang, L. L. Li, P. Y. Yang and H. J. Lu, Anal. Methods, 2012, 4, 2622–2631 RSC.
- B. Bodenmiller, L. N. Mueller, M. Mueller, B. Domon and R. Aebersold, Nat. Methods, 2007, 4, 231–237 CrossRef CAS PubMed.
- A. Eriksson, J. Bergquist, K. Edwards, A. Hagfeldt, D. Malmstrom and V. A. Hernandez, Anal. Chem., 2011, 83, 761–766 CrossRef CAS PubMed.
- A. I. K. Eriksson, M. Bartsch, J. Bergquist, K. Edwards, S. B. Lind and V. Agmo Hernández, J. Chromatogr. A, 2013, 1317, 105–109 CrossRef CAS PubMed.
- S. S. Jensen and M. R. Larsen, Rapid Commun. Mass Spectrom., 2007, 21, 3635–3645 CrossRef CAS PubMed.
- T. E. Thingholm, O. N. Jensen, P. J. Robinson and M. R. Larsen, Mol. Cell. Proteomics, 2008, 7, 661–671 CAS.
- A. I. K. Eriksson, K. Edwards, A. Hagfeldt and V. A. Hernandez, J. Phys. Chem. B, 2013, 117, 2019–2025 CrossRef CAS PubMed.
- M. W. H. Pinkse, P. M. Uitto, M. J. Hilhorst, B. Ooms and A. J. R. Heck, Anal. Chem., 2004, 76, 3935–3943 CrossRef CAS PubMed.
- M. R. Larsen, T. E. Thingholm, O. N. Jensen, P. Roepstorff and T. J. D. Jorgensen, Mol. Cell. Proteomics, 2005, 4, 873–886 CAS.
- U. K. Aryal and A. R. S. Ross, Rapid Commun. Mass Spectrom., 2010, 24, 219–231 CrossRef CAS PubMed.
- L. Qiao, C. Roussel, J. J. Wan, P. Y. Yang, H. H. Girault and B. H. Liu, J. Proteome Res., 2007, 6, 4763–4769 CrossRef CAS PubMed.
- F. Torta, M. Fusi, C. S. Casari, C. E. Bottani and A. Bachi, J. Proteome Res., 2009, 8, 1932–1942 CrossRef CAS PubMed.
- M. L. Niklew, U. Hochkirch, A. Melikyan, T. Moritz, S. Kurzawski, H. Schluter, I. Ebner and M. W. Linscheid, Anal. Chem., 2010, 82, 1047–1053 CrossRef CAS PubMed.
- A. Eriksson, J. Bergquist, K. Edwards, A. Hagfeldt, D. Malmstrom and V. A. Hernandez, Anal. Chem., 2010, 82, 4577–4583 CrossRef CAS PubMed.
- J. D. Dunn, E. A. Igrisan, A. M. Palumbo, G. E. Reid and M. L. Bruening, Anal. Chem., 2008, 80, 5727–5735 CrossRef CAS PubMed.
- F. Tan, Y. J. Zhang, J. L. Wang, J. Y. Wei, P. B. Qin, Y. Cai and X. H. Qian, Rapid Commun. Mass Spectrom., 2007, 21, 2407–2414 CrossRef CAS PubMed.
- L. A. Qiao, H. Y. Bi, J. M. Busnel, M. Hojeij, M. Mendez, B. H. Liu and H. H. Girault, Chem. Sci., 2010, 1, 374–382 RSC.
- P. Kouvonen, E. M. Rainio, V. Suni, P. Koskinen and G. L. Corthals, Mol. BioSyst., 2011, 7, 1828–1837 RSC.
- S. Ekstrom, L. Wallman, G. Helldin, J. Nilsson, G. Marko-Varga and T. Laurell, J. Mass Spectrom., 2007, 42, 1445–1452 CrossRef PubMed.
- H. Wang, J. C. Duan and Q. Cheng, Anal. Chem., 2011, 83, 1624–1631 CrossRef CAS PubMed.
- Z. F. Zeng, Y. D. Wang, S. L. Shi, L. F. Wang, X. H. Guo and N. Lu, Anal. Chem., 2012, 84, 2118–2123 CrossRef CAS PubMed.
- L. Krasny, P. Pompach, M. Strohalm, V. Obsilova, M. Strnadova, P. Novak and M. Volny, J. Mass Spectrom., 2012, 47, 1294–1302 CrossRef CAS PubMed.
- F. Gritti and G. Guiochon, J. Chromatogr. A, 2004, 1028, 197–210 CrossRef CAS PubMed.
- M. Rodahl, F. Hook, C. Fredriksson, C. A. Keller, A. Krozer, P. Brzezinski, M. Voinova and B. Kasemo, Faraday Discuss., 1997, 107, 229–246 RSC.
- C. Fredriksson, S. Kihlman, M. Rodahl and B. Kasemo, Langmuir, 1998, 14, 248–251 CrossRef CAS.
- N.-J. Cho, C. W. Frank, B. Kasemo and F. Hook, Nat. Protoc., 2010, 5, 1096–1106 CrossRef CAS PubMed.
- B. Reiss, A. Janshoff, C. Steinem, J. Seebach and J. Wegener, Langmuir, 2003, 19, 1816–1823 CrossRef CAS.
- G. A. McCubbin, S. Praporski, S. Piantavigna, D. Knappe, R. Hoffmann, J. H. Bowie, F. Separovic and L. L. Martin, Eur. Biophys. J. Biophys. Lett., 2011, 40, 437–446 CrossRef CAS PubMed.
- V. A. Hernandez, J. Samuelsson, P. Forssen and T. Fornstedt, J. Chromatogr. A, 2013, 1317, 22–31 CrossRef PubMed.
- V. A. Hernandez, K. Reijmar and K. Edwards, Anal. Chem., 2013, 85, 7377–7384 CrossRef PubMed.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- S. Kjellstrom and O. N. Jensen, Anal. Chem., 2004, 76, 5109–5117 CrossRef PubMed.
- G. Guiochon, A. Felinger, D. G. Shirazi and A. M. Katti, Fundamentals of Preparative and Nonlinear Chromatography, Elsevier, The Netherlands, 2006 Search PubMed.
- T. Nishikaze and M. Takayama, Rapid Commun. Mass Spectrom., 2006, 20, 376–382 CrossRef CAS PubMed.
- E. Szajli, T. Feher and K. F. Medzihradszky, Mol. Cell. Proteomics, 2008, 7, 2410–2418 CAS.
- W. Wilkinson, A. Gusev, A. Proctor, M. Houalla and D. Hercules, Fresenius' J. Anal. Chem., 1997, 357, 241–248 CrossRef CAS.
- T. E. Thingholm, T. J. D. Jorgensen, O. N. Jensen and M. R. Larsen, Nat. Protoc., 2006, 1, 1929–1935 CrossRef CAS PubMed.
- B. D. Halligan, V. Ruotti, W. Jin, S. Laffoon, S. N. Twigger and E. A. Dratz, Nucleic Acids Res., 2004, 32, W638–W644 CrossRef CAS PubMed.
- S. Gauci, B. van Breukelen, S. M. Lemeer, J. Krijgsveld and A. J. R. Heck, Proteomics, 2008, 8, 4898–4906 CrossRef CAS PubMed.
- C. E. McNamee, Y. Tsujii and M. Matsumoto, Langmuir, 2005, 21, 11283–11288 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Both the raw experimental data and the statistical analyses upon which the discussion in the section “Impact of cooperativity on TiO2-based enrichment methods” is based are included in Appendixes A–F. See DOI: 10.1039/c4an01580k |
| This journal is © The Royal Society of Chemistry 2015 |