
An electrochemical and computational study for discrimination of D- and L-cystine by reduced graphene oxide/β-cyclodextrin
Erhan
Zor
*abc,
Haluk
Bingol
d,
Almira
Ramanaviciene
b,
Arunas
Ramanavicius
b and
Mustafa
Ersoz
e
aSelcuk University, Institute of Science, Department of Chemistry, Konya, Turkey. E-mail: zorerhan@gmail.com; Fax: +90 332 3238225; Tel: +90 332 3238220 - 5566
bVilnius University, Faculty of Chemistry, Centre of Nanotechnology and Materials Science, Vilnius, Lithuania. E-mail: almyra@imi.lt; arunas.ramanavicius@chf.vu.lt
cNecmettin Erbakan University, A.K. Education Faculty, Science and Technology Department, Konya, Turkey
dNecmettin Erbakan University, A.K. Education Faculty, Chemistry Department, Konya, Turkey. E-mail: halukbingol@gmail.com
eSelcuk University, Advanced Technology Research and Application Center, Konya, Turkey. E-mail: ersozm@gmail.com
First published on 21st October 2014
Abstract
Here, we report a novel enantioselective electrochemical biosensor for the discrimination of cystine enantiomers (D- and L-cystine) using a chiral interface for the specific recognition of D- and L-cystine. The biosensor is based on reduced graphene oxide modified by β-cyclodextrin (rGO/β-CD) at the GCE surface. During the preparation of rGO/β-CD/GCE, the modified electrode surfaces were characterized by cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and scanning electron microscopy (SEM). The electrochemical behaviours of the D- and L-cystine were investigated using the rGO/β-CD/GCE by CV and compared to bare GCE. A clear separation between the oxidation peak potentials of D- and L-cystine was observed at 1.32 and 1.42 V, respectively. The electrochemical discrimination performance of the fabricated chiral sensor was also examined by differential pulse voltammetry (DPV) in a mixed solution of D- and L-cystine. In addition, the DPV technique was used for the determination of D- and L-cystine at low concentration values in the range of 1.0–10.0 μM. To investigate the amperometric response of rGO/β-CD/GCE towards D- and L-cystine, the chronoamperometry technique was used in the concentration range of 10.0–100.0 μM. The interactions of the enantiomers with rGO/β-CD were modelled by molecular docking using AutoDock Vina, and the interaction energies were predicted to be −4.8 and −5.3 kcal mol−1 for D- and L-cystine, respectively. The corresponding values of binding constants were calculated to be 3.32 × 103 and 7.71 × 103 M−1, respectively. The experimental and molecular docking results indicate that the rGO/β-CD/GCE has a different affinity for each enantiomer.
1. Introduction
The investigation of chiral compounds has a fundamental significance for understanding the intrinsic properties of the biomolecular building blocks of life and broad classes of chemical processes occurring in living organisms.1–3 Because of this, the distinction of chiral interactions is crucial in drug discovery, pharmaceuticals and biochemical processes.4–6 Numerous compounds used in such areas are known to exist as a mixture of enantiomers. One enantiomer may be ineffective or frequently possess a different physiological role, which may cause serious side-effects, while the other may exhibit some desirable properties.7 In addition, the development of simple, rapid, sensitive, highly selective, and time-saving methods for the detection and quantification of one individual enantiomer in very complex samples containing a mixture of enantiomers still remains a challenge.8,9 Although various techniques, such as UV-vis spectroscopy,10 fluorescence spectroscopy,11 high-performance liquid chromatography,12 capillary electrophoresis13 and electrochemical investigations14,15 have been performed for the detection of the enantiomers of different compounds, a few studies have been successfully carried out for the electrochemical discrimination of enantiomers. For instance, Kataky and Lopes have achieved the discrimination of facilitated chiral transfer of the ephedrinium ion at liquid–liquid interface,4 Nie et al. have prepared cysteic acid modified glassy carbon electrodes to discriminate tyrosine enantiomers,16 and Zor et al. have developed an electrochemical biosensor based on the human serum albumin/graphene oxide/3-aminopropyltriethoxysilane modified indium tin oxide electrode for the discrimination of tryptophan enantiomers.7In recent years, the experimental studies combined with theoretical calculations have received increasing attention17 because theoretical approaches can provide an explanation of experimental observations.18,19 In this respect, molecular docking is one of the widely used molecular modeling methods to explain the molecular recognition of chiral molecules,17 and it can provide an insight into the preferred binding location.20
The key step for producing a chiral biosensor is to build a chiral surface having recognition sites with different affinities towards enantiomers.5,7 This surface serves to be a host for biologically active target compounds.21 Cyclodextrins (CDs) are a class of cyclic oligosaccharides, which are toroidal in shape with a hydrophobic inner cavity and a hydrophilic exterior.22–24
These interesting characteristics can enable them to selectively bind targeted molecules into their cavities through various types of intermolecular interactions, such as hydrophobic interaction, van der Waals force, electrostatic affinity, and hydrogen bonding.25 CDs can be attached on the surface of reduced graphene oxide (rGO) sheets not only to make graphene more hydrophilic but also to enable a high surface area and conductivity. Therefore, the integration of rGO and CD is expected to expand potential applications in various fields, such as sensors, electrocatalysis and biological probes.26 Recently, rGO-CDs modified electrodes have attracted significant interest because of their potential application as selective and sensitive electrochemical biosensors.22,23,25,27,28
Here, we present the discrimination of two enantiomeric forms of cystine (D- and L-cystine), which is a dimeric amino acid derivative and is a constituent of hair and nail keratin.29 The chemical oxidation of disulfides, such as cystine by bromine and other oxidizing agents, is a well-known commercial process.30 However, to the best of our knowledge, there is no prior report on the electrochemical enantiomeric discrimination of D- and L-cystine by CV and DPV techniques. To achieve this discrimination, we prepared and characterized a glassy carbon electrode surface modified with rGO/β-CD composite and electrochemically investigated the electrode performance in detail. The interactions between cystine enantiomers and rGO/β-CD were investigated by molecular docking studies because of the fact that it is a useful tool to confirm the binding mode, and the interaction energies were evaluated. The results show that the rGO/β-CD/GCE can be used as an effective electrochemical biosensor to discriminate D- and L-cystine.
2. Experimental
2.1. Chemicals and apparatus
All the chemicals were of analytical grade and were used without further purification. Concentrated H3PO4 and H2SO4, H2O2 (30%), hydrazine hydrate solution (50 wt%), ammonia solution (28 wt%), graphite powder (99.99%), K2S2O8, KMnO4 (99%), P2O5, D-and L-cystine (cystine) and β-cyclodextrin were purchased from Sigma-Aldrich. All the aqueous solutions were freshly prepared using ultrapure water with a resistivity of 18.2 MΩ cm.Electrode morphologies were investigated by scanning electron microscopy (SEM), performed on a ZEISS EVO LS 10 SEM at an accelerating voltage of 20 kV and 5.00 k× magnification. UV-vis absorption spectra were obtained on a Shimadzu UV-1800 double beam spectrophotometer. Fourier transform infrared (FT-IR) spectra of the samples were recorded between 550 and 4000 cm−1 using an ATR FT-IR spectrometer (Perkin Elmer 100 FT-IR). Electrochemical impedance spectroscopy (EIS) measurements were conducted in A-PBS solution of pH 7.01 containing 1.0 mM [Fe(CN)6]−3/−4 redox couples. The impedance measurements were performed in the frequency range from 10 Hz to 100 kHz with 5 mV signal amplitude. Electrochemical measurements were performed in an electrochemical cell comprised of a three-electrode configuration system using an Autolab PGSTAT 30 Potentiostat/Galvanostat operated by the GPES software Eco Chemie (Utrecht, Netherlands). Glassy carbon electrode (GCE), Ag/AgCl in saturated KCl (Ag/AgCl/KClsat) and platinum wire were used as the working electrode, reference electrode and counter electrode, respectively. All the measurements were carried out in a mixed solution of 50 mM sodium acetate and 50 mM phosphate buffer (A-PBS) of pH 7.4 with 100 mM KCl at ambient temperature.
2.2. Preparation of β-cyclodextrin functionalized graphene
Graphene oxide (GO) was synthesized from graphite powder by the improved method,31 which has significant advantages over Hummers' method,32 with an additional preoxidation process. Firstly, 3.0 g graphite was pre-oxidized in a mixture containing 15 mL concentrated H2SO4, 1.5 g K2S2O8 and 1.5 g P2O5. The mixture was then diluted with ultrapure water, filtered and dried in a vacuum oven at 50 °C. Secondly, the preoxidized graphite was reoxidized by the improved method involving KMnO4 (18.0 g) in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of concentrated H2SO4/H3PO4 (360:40 mL) for the production of GO. β-Cyclodextrin functionalized graphene was synthesized as indicated in literature as follows:22 Briefly, a 10.0 mL graphene oxide solution (0.5 mg mL−1) was sonicated for 1 h to obtain a homogeneous dispersion. Then, it was mixed with 10.0 mL of 40 mg mL−1 aqueous solution of β-cyclodextrin and 150.0 μL of ammonia solution, followed by the addition of 10 μL of hydrazine solution. After shaking for few minutes, the solution was immersed in a water bath (60 °C) for 3.5 h to obtain a stable black dispersion. The dispersion was filtered to obtain reduced graphene oxide/β-cyclodextrin nanocomposite (rGO/β-CD), which can be readily redispersed in water by ultrasonication.
:1 mixture of concentrated H2SO4/H3PO4 (360:40 mL) for the production of GO. β-Cyclodextrin functionalized graphene was synthesized as indicated in literature as follows:22 Briefly, a 10.0 mL graphene oxide solution (0.5 mg mL−1) was sonicated for 1 h to obtain a homogeneous dispersion. Then, it was mixed with 10.0 mL of 40 mg mL−1 aqueous solution of β-cyclodextrin and 150.0 μL of ammonia solution, followed by the addition of 10 μL of hydrazine solution. After shaking for few minutes, the solution was immersed in a water bath (60 °C) for 3.5 h to obtain a stable black dispersion. The dispersion was filtered to obtain reduced graphene oxide/β-cyclodextrin nanocomposite (rGO/β-CD), which can be readily redispersed in water by ultrasonication.
2.3. Preparation of modified electrodes
Prior to modification, GCE surfaces were pre-cleaned with acetone, ethanol and ultra-pure water, respectively. Then, GCE surfaces were polished with 1.0, 0.3 and 0.05 μm alumina slurry (PACE Technologies, USA) on a felt pad, and washed with a copious amount of ultra-pure water. The GCE was then immersed in water and methanol for 15 minutes, respectively, in an ultrasonic bath (Sonorex Super RK 106, Germany) to remove residual alumina particles by sonication. The electrode was dried at room temperature before the modification step. After drying, the rGO/β-CD/GCE was prepared by casting 5.0 μL of rGO/β-CD suspension (0.2 mg mL−1). Finally, the obtained electrode was dried at room temperature overnight. The rGO/β-CD/GCE was then washed with ultra-pure water and dried before the use. A brief schematic diagram of producing an electrochemical biosensor for the discrimination of D- and L-cystine is presented in Scheme 1.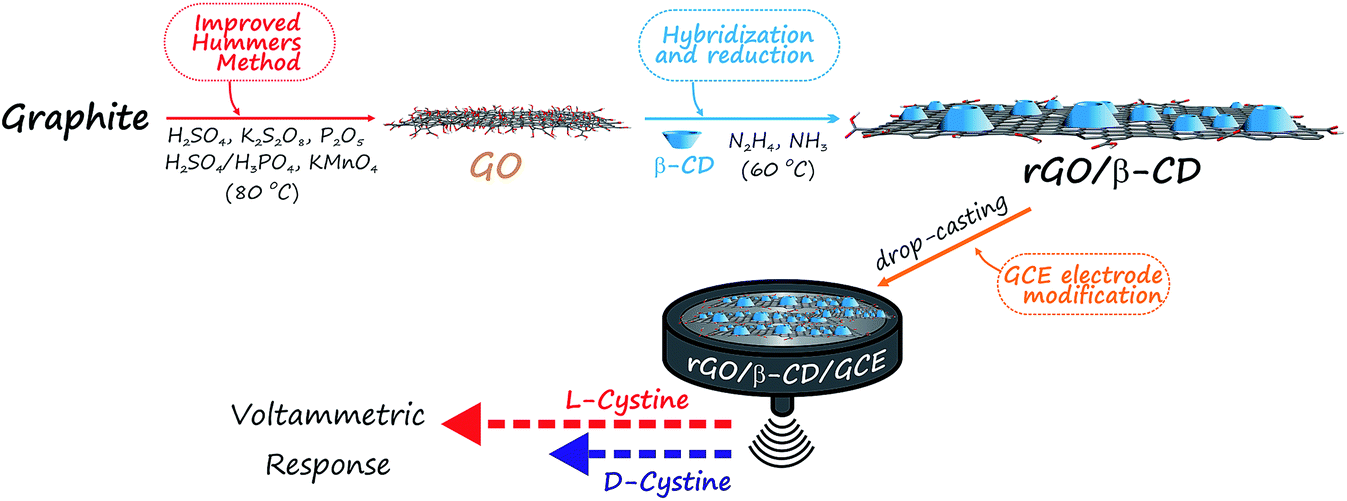 | ||
| Scheme 1 Schematic diagram of producing electrochemical biosensor for the discrimination of cystine enantiomers (D- and L-cystine). | ||
2.4. Procedure for molecular docking study
The molecular docking studies were performed by ADVina considering the algorithm that maintains a rigid macromolecule while allowing ligand flexibility.33 The crystal structure of β-CD was obtained from the protein data bank. All the hydrogen bonds for each rim of β-CD were oriented in the same direction.34 The two-dimensional (2D) structures of GO, rGO, and D- and L-cystine were drawn using ChemBioDraw v13.0. The starting geometries of GO and rGO were constructed considering the previous related literature.35–37 After the 2D sketches were converted to three dimensional (3D) images and the structures were energetically minimized, new coordinates were updated and recorded in PDB format with the help of Discovery Studio v3.5. The constructed graphene derivatives were assumed to be pristine and defect-, lesion-, and grain boundary-free.17 Before applying the molecular docking, the AutoDockTools version 1.5.6 (ADT) was used for the optimization of the guest compounds from the PDB files by adding Gasteiger charges, assigning polar hydrogen atoms and setting up rotatable bonds. The pdbqt format files (required as input ADVina) were generated using ADT. The docking procedure was performed in two parts: Part 1; β-CD was docked to rGO using the following cartesian coordinates: x = 0 Å, y = 0 Å, z = 0 Å. The molecular docking was carried out with a grid box size of 60 Å × 70 Å × 40 Å along the x, y, and z axes with a grid spacing of 1.000 Å. Finally, the lowest energy conformation was used for the docking analysis. Part 2; taking into account the docking conformation results of part 1, D- and L-cystine were docked to an rGO/β-CD structure using the following cartesian coordinates: x = −2.560 Å, y = −3.560 Å, z = −14.500 Å. A docking grid with a dimension of 14 Å × 14 Å × 20 Å and a grid spacing of 1.000 Å was applied for the second part of the molecular docking. All the other parameters were used as defined by ADVina for each docking step. The resulting nine binding models were further analyzed to find the most suitable binding model in each case. The preferred binding model having the minimum energy with the maximum number of poses clustered in that site was selected.37 The ADVina output results were used for the calculation of binding free energy change (ΔGbind), and the free energy change was further converted to binding constants (Kbind) using Kbind = exp(ΔGbind/RT) at 25 °C.3. Results and discussion
3.1. Characterization of the rGO/α-CD hybrid material
The obtained composite (rGO/β-CD) was confirmed by UV-vis and FT-IR spectral analysis. As shown in Fig. 1A, the GO dispersion displays the maximum absorption at 231 nm, which is because of the π–π* transition of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and a shoulder at 300 nm, which corresponds to the n–π* transition of the CO bond.38 The color of the dispersion changed from pale-brown to black (Fig. 1A inset), and the absorption peak of the GO dispersion at 231 nm shifted to 264 nm, suggesting that the conjugation within the rGO is formed because of the reduction by hydrazine.22,23,25
C bonds and a shoulder at 300 nm, which corresponds to the n–π* transition of the CO bond.38 The color of the dispersion changed from pale-brown to black (Fig. 1A inset), and the absorption peak of the GO dispersion at 231 nm shifted to 264 nm, suggesting that the conjugation within the rGO is formed because of the reduction by hydrazine.22,23,25
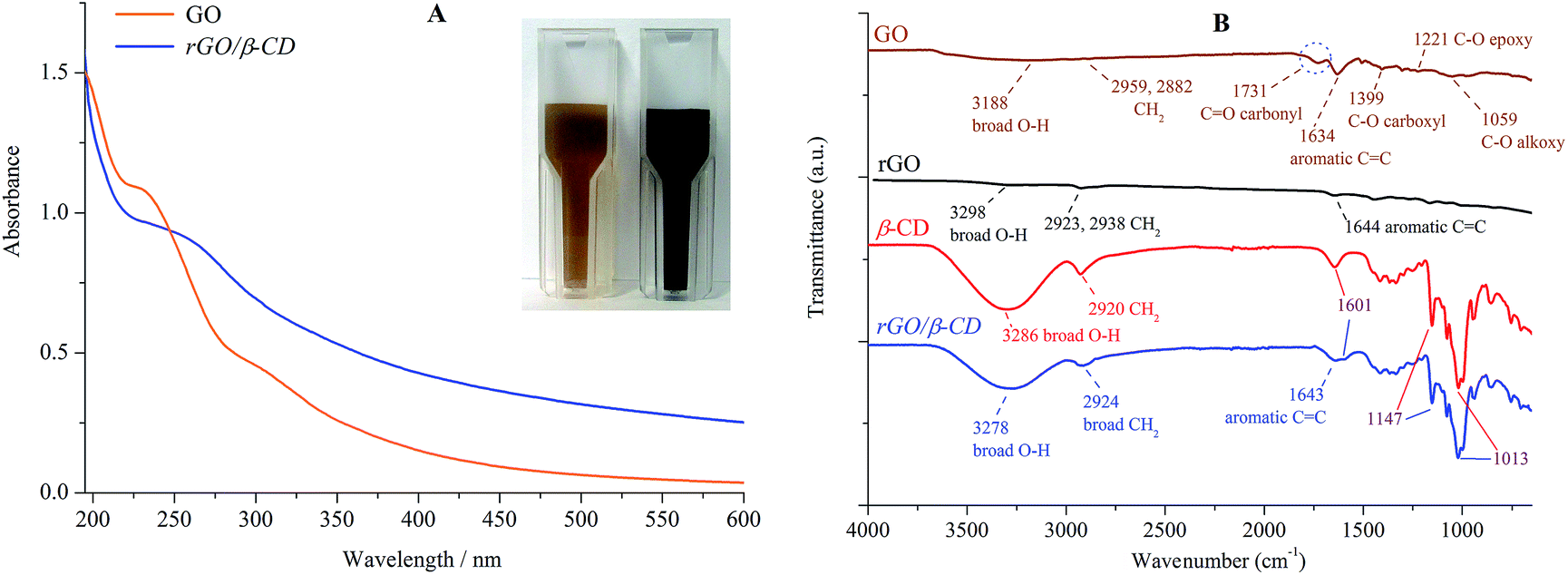 | ||
| Fig. 1 UV-vis absorption spectra of GO and rGO. The inset shows images of GO (pale-brown) and rGO/β-CD (black) (A). FT-IR spectra of GO, rGO, β-CD and rGO/β-CD (B). | ||
The comparable FT-IR spectral features of GO, β-CD, rGO and rGO/β-CD are presented in Fig. 1B. All the spectra of the graphene derivatives have characteristic broad bands in the range of 3188–3278 cm−1 for the O–H stretching vibration, and the characteristic stretching vibrational modes around 1595–1644 cm−1 for CC (aromatic) situated on the graphene sheet.22,39,40 After the reduction of GO to rGO sheet for rGO and rGO/β-CD, the peak intensity at 1731 cm−1 of CO (carbonyl) disappeared, the peaks at 1399, 1221 and 1059 cm−1 for GO corresponding to the C–O (carboxyl), C–O (epoxy) and C–O (alkoxy) functional groups, respectively, significantly decreased.40 These changes show that GO has been successfully reduced to rGO (or rGO/β-CD).41 Moreover, the spectrum of rGO/β-CD exhibits the characteristic β-CD absorption bands around 1601, 1147 and 1013 cm−1, indicating the ring vibrations, which clearly confirms that β-CD molecules are settled on the rGO sheet.22
3.2. Surface and electrochemical characterization of the modified electrodes
Scanning electron microscopy (SEM) was used to characterize the surface morphologies of bare GCE, rGO and rGO/β-CD modified GCE. The SEM image of rGO/GCE surface exhibits that rGO sheets surround the smooth surface of the bare GCE electrode (Fig. 2A), as can be seen in Fig. 2B. Similar to the rGO sheets, the rGO/β-CD modified GCE surface displays an efficient coverage of the hybrid material, as shown in Fig. 2C. | ||
| Fig. 2 SEM micrographs of GCE (A), rGO/GCE (B), and rGO/β-CD/GCE (C). | ||
CV and EIS were performed to investigate the electrochemical responses of the modified electrodes in A-PBS solution (pH 7.01) containing 1.0 mM [Fe(CN)6]−3/−4. Fig. 3A and B show the results of CV and EIS of the [Fe(CN)6]−3/−4 redox couple for bare GCE, rGO/GCE and rGO/β-CD/GCE modified surfaces, respectively. Although the bare GCE exhibits a well-defined redox peak for [Fe(CN)6]−3/−4 in accordance with the literature,42 the modifications of the GCE surface with rGO and rGO/β-CD result in significant change on the electrochemical behaviors with decreases in the values of peak currents ratio (Ip,a/Ip,c) and increases in the values of the separation of peak potentials (ΔEp).
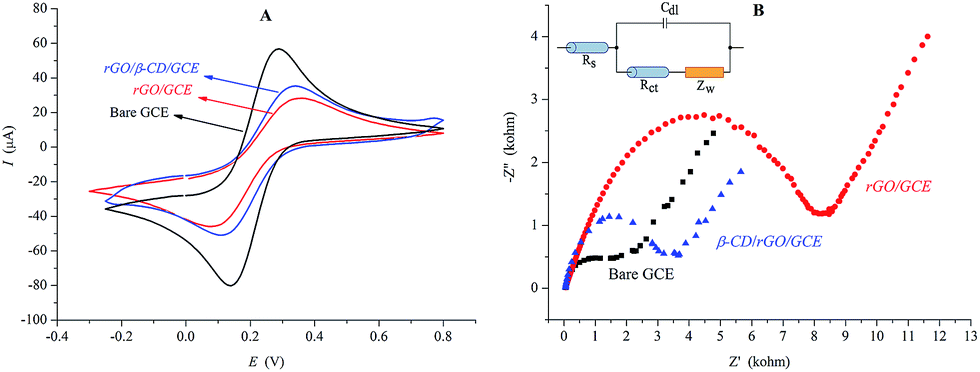 | ||
| Fig. 3 Cyclic voltammograms (A) and Nyquist diagrams obtained at bare GCE, rGO/GCE and rGO/β-CD/GCE electrode in the presence of 1 mM [Fe(CN)6]−3/−4 in A-PBS solution (pH 7.01) (B). The inset is Randles equivalent circuit used to model impedance data in the presence of the redox couples. | ||
According to the related studies, these changes can be attributed to the presence of a film of graphene derivatives on the electrode surface, which acts as an insulating layer, resulting in difficult interfacial electron transfer because of their disrupted sp2 bonding networks.43–45 After the modifications of the GCE surface, the semicircle parts on the impedance spectrums increased, as can be seen in Fig. 3B. The obvious changes in the diameter in each modification indicate that rGO decelerates the electron transfer of [Fe(CN)6]−3/−4 because graphene derivatives act as a hindering layer for electron transfer, as described above. The impedance values in the results of EIS were also fitted to the standard Randles equivalent circuit (the inset of Fig. 3B) comprising the charge transfer resistance (Rct) on the electrode surfaces. The analyzed results also are tabulated in Table 1.
| Electrode | E 1/2 (mV) | ΔEp (mV) | I p,a/Ip,c | R ct (kohm) |
|---|---|---|---|---|
| Bare GCE | 214 | 134 | 1.03 | 1.558 |
| rGO/GCE | 218 | 282 | 0.87 | 6.911 |
| rGO/β-CD/GCE | 223 | 232 | 0.93 | 2.114 |
3.3. Enantioselective discrimination of D- and L-cystine at rGO/β-CD/GCE by voltammetry
Fig. 4 shows the CV measurements performed with rGO/β-CD/GCE. No well-defined electrochemical response for the D- and L-cystine were obtained at bare GCE (the inset), which is in accordance with the previous report.16 After the modification of the surface with rGO/β-CD, strong irreversible oxidation peaks of D- and L-cystine were observed at different potential values of 1.32 and 1.42 V, respectively. The possible oxidation mechanism is provided in Scheme 2 as previously reported.30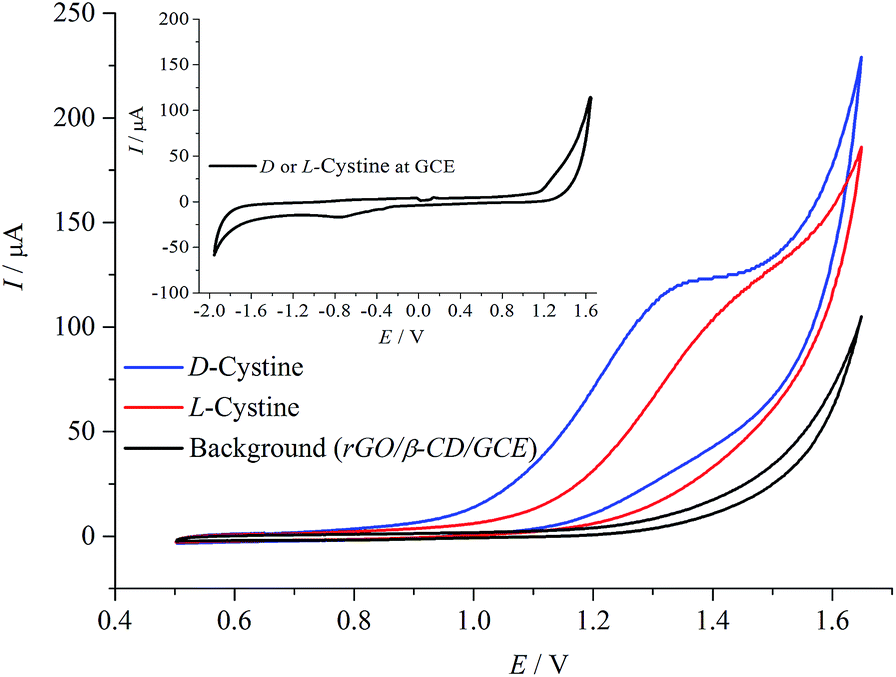 | ||
| Fig. 4 Cyclic voltammograms in the absence and presence of D- and L-cystine (50 μM) in A-PBS solution at rGO/β-CD/GCE. Potential sweep rate was 0.1 V s−1. | ||
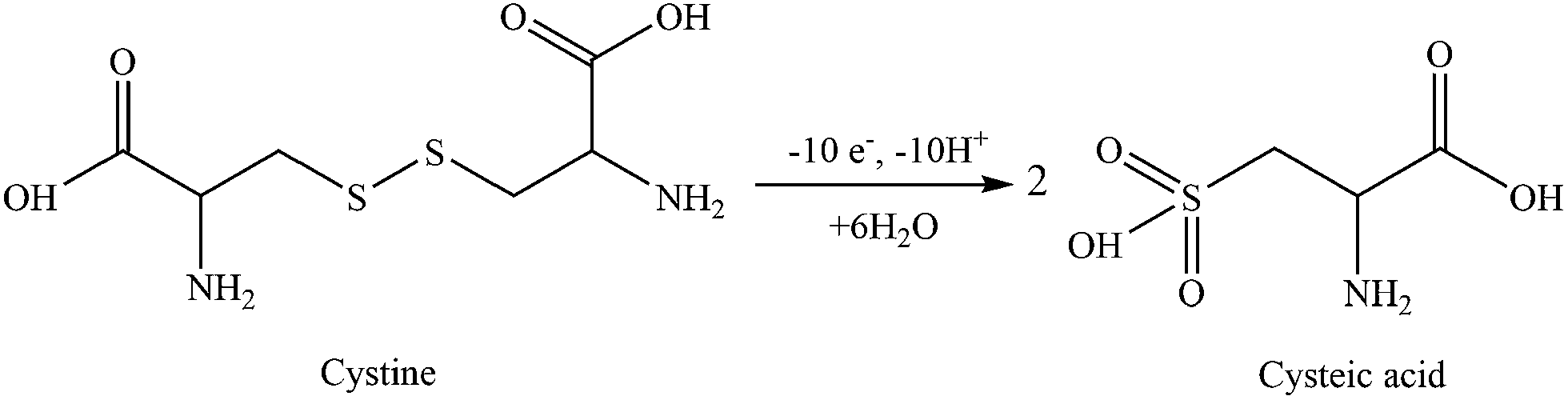 | ||
| Scheme 2 Possible oxidation mechanism of cystine to cysteic acid at rGO/β-CD/GCE. | ||
The voltammetric response of cystine oxidation to cysteic acid is changed according to enantiomeric structure. The high sensitivity and selective recognition capability of rGO/β-CD/GCE can be attributed to three factors: (i) the rGO/β-CD/GCE has a larger surface area; (ii) the attached β-CD prevents the rGO from agglomeration; thus, rGO/β-CD/GCE has more accessible active sites than bare GCE and considerably higher catalytic activity;46 (iii) these active sites of β-CD have a different binding affinity towards target cystine enantiomers that might lead to the differences in the free Gibbs energy; this effect is observed as a potential shift in electrochemical measurements.16 The CV measurements illustrate that by the combination of the unique electronic properties of rGO with the high selective β-CD compound, the rGO/β-CD nanocomposite shows significantly improved electrochemical sensing performance compared to that of bare GCE.
Fig. 5A and B show the additional CV measurements carried out to investigate the relationship between the peak current and the concentration in the range of 1–100 μM of D- and L-cystine. The oxidation current values increase with the increasing concentration of D- and L-cystine. High oxidation potential values for corresponding concentrations were observed for L-cystine. This can be attributed to the fact that β-CD attached on rGO exhibits stronger interaction7 with L-cystine than D-cystine because of the more suitable matching size, which has significant importance for enantioselective recognition.
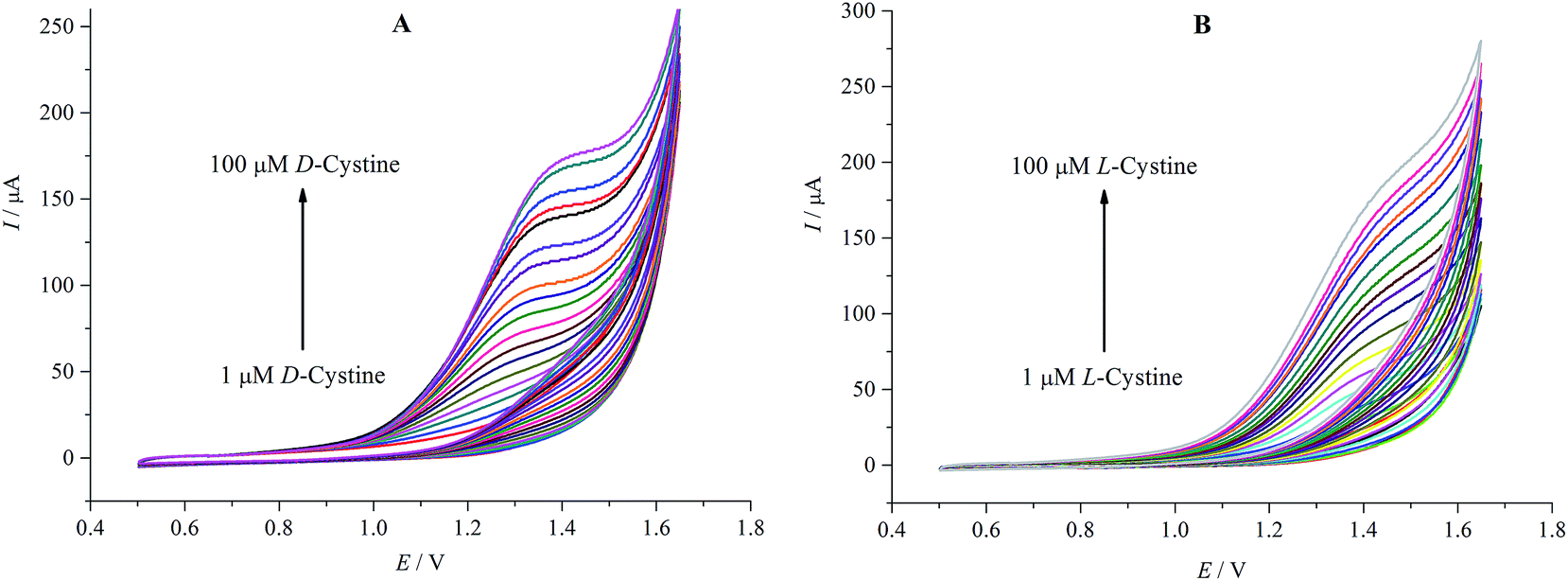 | ||
| Fig. 5 Cyclic voltammograms at increasing concentration of D-cystine (A) and L-cystine (B) (1–100 μM) in A-PBS solution at rGO/β-CD/GCE; potential sweep rate was 0.1 V s−1. | ||
Fig. 6A shows the DPV measurements for D- and L-cystine at rGO/β-CD/GCE. In accordance with the CV measurements, the well-defined peaks of D- and L-cystine were observed at different potential values of 1.22 and 1.36 V, respectively. Fig. 6B and C show the current change with the increasing concentration of D- and L-cystine, respectively. It can also be easily seen that the oxidation peak current of D- and L-cystine also linearly increases with the increasing concentration of D- and L-cystine in the range of 1.0-10.0 μM.
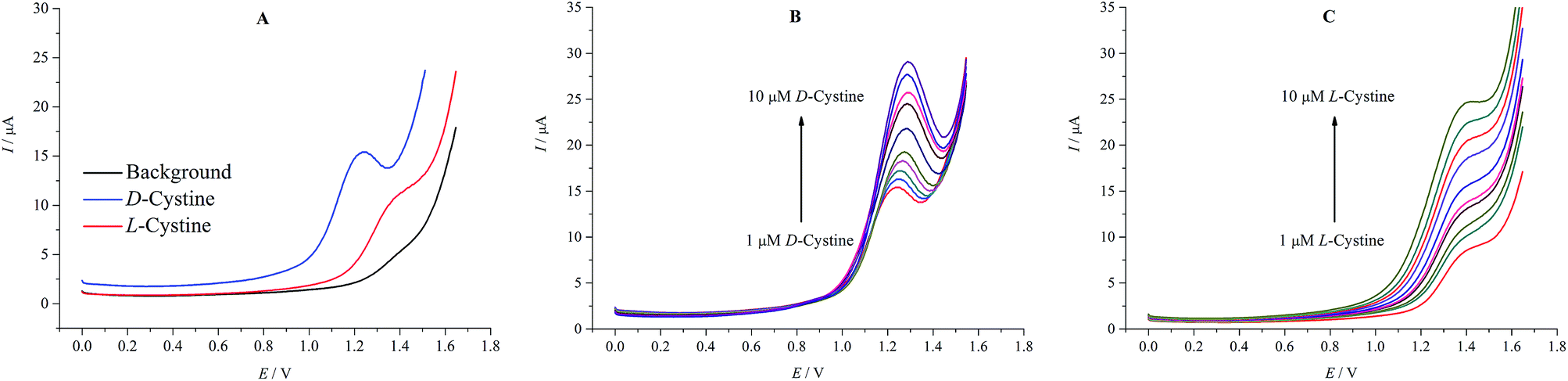 | ||
| Fig. 6 Differential pulse voltammograms in the absence and presence of D- and L-cystine in A-PBS solution (A). Differential pulse voltammograms at increasing concentration of D-cystine (B) and L-cystine (C) in low concentration range (1–10 μM) at rGO/β-CD/GCE; potential sweep rate was 0.05 V s−1. | ||
Fig. 7 shows differential pulse voltammograms for mixtures of D- and L-cystine in different ratios of 1:0, 1:1, and 0:1 c/c. As can be easily seen, the oxidation of 1:1 (c/c) enantiomeric mixture occurs at 1.27 V. This potential value is the approximate mean of the oxidations of D- and L-cystine enantiomers, indicating clearly that there is a racemic mixture in the medium.
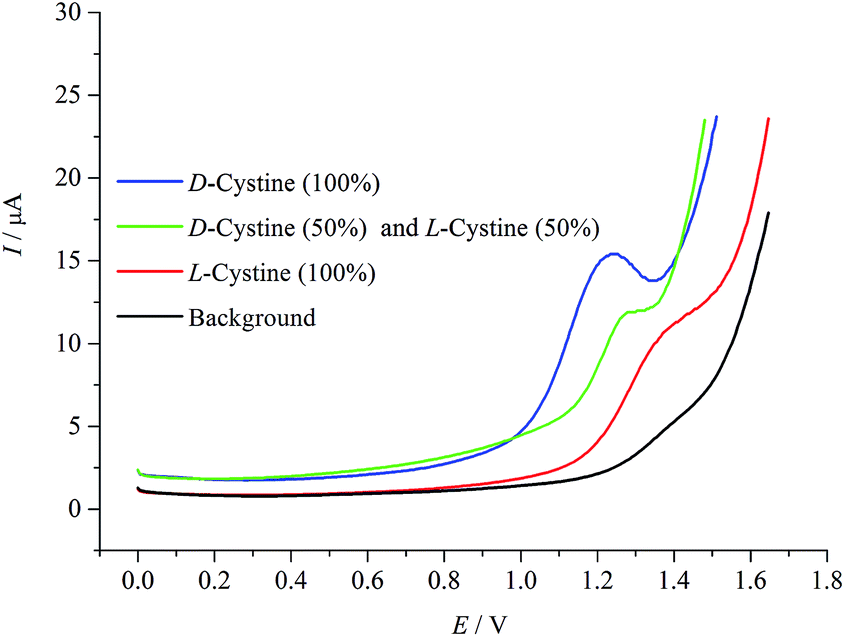 | ||
| Fig. 7 Differential pulse voltammograms of the enantiomeric mixtures of cystine at different ratios of D- and L-cystine (1:0, 1:1, and 0:1 c/c). Potential sweep rate was 0.05 V s−1. | ||
3.4. Amperometric responses of rGO/β-CD/GCE for D-and L-cystine
The amperometric responses of the rGO/β-CD/GCE versusD- and L-cystine concentration (10.0–100.0 μM) were investigated by chronoamperometry technique and the results are shown in Fig. 8A and B. As can be easily seen, rGO/β-CD/GCE exhibited a good amperometric response to the increasing concentration of D-and L-cystine with the ΔImax of 303.01 μA and 288.04 μA, respectively. In accordance with the CV and DPV measurements, the amperometric results show that the oxidation peak current of D- and L-cystine also linearly increases with the increasing concentration of D- and L-cystine in the range of 10.0–100.0 μM, as presented in Fig. 8C and D.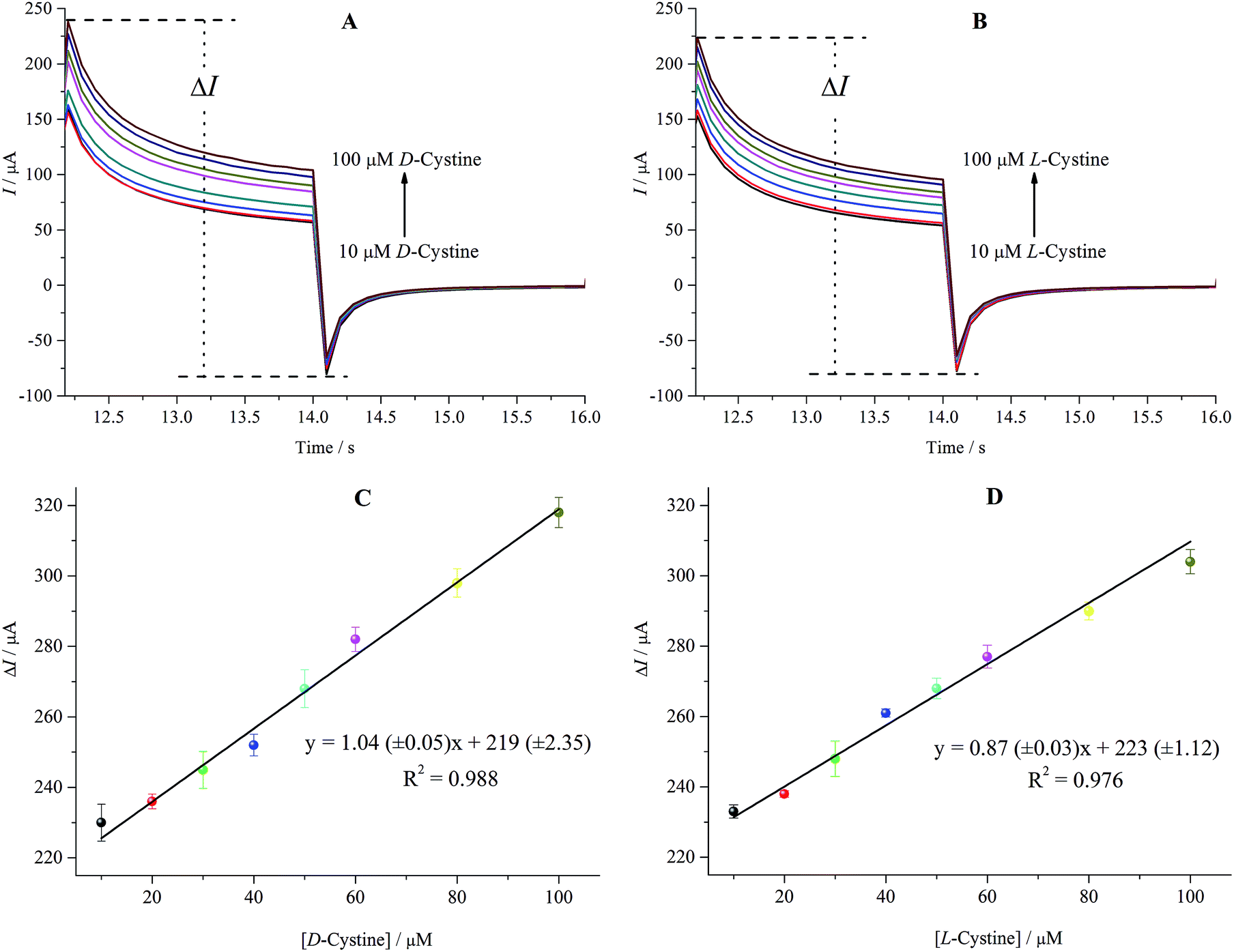 | ||
| Fig. 8 Chronoamperograms at increasing concentration of D-cystine (A) and L-cystine (B) (10–100 μM) at rGO/β-CD/GCE. The applied potentials are 0.00 and 1.32 V for D-cystine, 0.00 and 1.42 V vs. Ag/AgCl (3 M KCl) for L-cystine. (C) and (D) show the linear relationship between the enantiomer concentrations of cystine and the oxidation peak currents. | ||
3.5. The stability, reproducibility and repeatability of the rGO/β-CD/GCE
The reproducibility of the rGO/β-CD/GCE was investigated by the successive detection of 50.0 μM D- and L-cystine with three independently prepared electrodes, and relative standard deviation (RSD) was calculated to be 2.45% and 4.03%, respectively. The repeatability of modified electrodes was evaluated by the analysis of voltammetric responses three times using the same electrode, and relative standard deviations (RSD) were calculated to be 3.28% and 5.33%, respectively.The stability of the rGO/β-CD/GCE was investigated by CV in a constant D- and L-cystine concentration (50.0 μM) over a period of 2 weeks. It was observed that during the first week the current responses remained almost stable and showed a decrease in the current density of about 12.13% and 13.29% of the initial values at the end of 2 weeks. The decrease of current response may be attributed to the degradation of rGO/β-CD sensing layer.
3.6. Molecular docking results
In addition to the experimental studies of D- and L-cystine with rGO/β-CD, a molecular modeling simulation was also performed. Recently, there has been an increasing interest in molecular modeling studies for the recognition of a variety of molecules with cyclodextrins for inclusion complexes and other aspects of supramolecular chemistry.19 In this respect, Zhang et al. depicted that the molecular modeling studies of host–guest interactions between CDs and enantiomers should provide better insights into these interactions and elucidate chiral recognition processes.19 Alvira has studied the separation of alanine enantiomers using β-CD by a molecular dynamics simulation and stated that β-CD is a good enantioselective chiral selector.47 Herein, we aimed to theoretically study the inclusion complex formation and to predict the preferentially recognition of D- and L-cystine to rGO/β-CD. The molecular docking studies were performed in two parts by employing ADV. In the first part of the docking process, β-CD was docked to GO, as indicated in our previous paper for α-CD.17 Similar to α-CD, β-CD also preferentially settled on the basal plane of GO from the wide rim, which has a distinctly hydrophilic character17,18 (Fig. 9A). The overall binding energy of β-CD on GO for this docking process was calculated to be −9.5 kcal mol−1. Fig. 9B shows the rGO/β-CD structure after the reduction of the residual oxygen containing groups of GO by a reducing agent, as indicated in the experimental part. | ||
| Fig. 9 Overall structures of β-CD in complexes with GO (A) and rGO (B) represented as stick models. Side view stick model of schematic drawing for the rGO/β-CD complex with D-cystine (C) and L-cystine (D), respectively. | ||
In the second part, the docking models were evaluated for the preferential recognition of D- and L-cystine by rGO/β-CD. Fig. 9C and D show the docked models in which D- and L-cystine penetrate through the cavity of β-CD. The overall binding energies for the recognition of D- and L-cystine to rGO/β-CD were predicted to be −4.8 and −5.3 kcal mol−1, respectively. Considering the overall binding energies, the binding constants were calculated to be 3.32 × 103 and 7.71 × 103 M−1, respectively, resulting in a high chiral discrimination with a ratio of KL/KD = 2.32. According to the “three-point interaction” theory suggested by Dalgliesh in 1952,48 at least three configuration-dependent points are needed for the recognition between chiral selectors and enantiomeric analytes, and one of them must be an enantioselective interaction.49 Herein, the different binding constants values can be attributed to the enantioselective binding affinity of β-CD. Therefore, β-CD provides a chiral environment exhibiting various noncovalent interactions, such as van der Waals interactions, hydrophobic interactions and hydrogen bonding, which play a pivotal role for chiral discrimination. As a result of considerably more interactions that occurred for L-cystine, rGO/β-CD has a higher binding energy than D-cystine. The higher binding energy and binding constant for the interactions occurring between L-cystine and rGO/β-CD are consistent with the electrochemical results, indicating the higher binding affinity of rGO/β-CD for L-cystine, which also provide a better insight into the interactions between cystine enantiomers and rGO/β-CD on a molecular level.
4. Conclusions
In the present work, we report a simple and fast method for the discrimination of D- and L-cystine by rGO/β-CD/GCE electrode. In the experimental part of the study, CV, DPV and chronoamperometry techniques were employed to investigate the electrochemical response of the modified electrode for the discrimination of D- and L-cystine. The enantioselective properties of β-CD provide an opportunity for the discrimination of D- and L-cystine. The rGO layer provides a large surface area for the deposition of β-CD. The described method is very simple, cheap and time saving. To the best of our knowledge, it is the first report on an electrochemical chiral sensor for the discrimination of D- and L-cystine. In the second part of the study, molecular interactions were investigated by molecular docking to compare the experimental results with the computational results. The molecular docking results supported the electrochemical results, indicating the different enantioselective affinity of rGO/β-CD towards D- and L-cystine. As a consequence, this study is expected to be a useful and promising platform in the sensor area for the electrochemical discrimination of some chiral analytes.Acknowledgements
This work was supported by TÜBİTAK (113Z664) and was produced from a part of E. Zor's PhD thesis. We also express our deep thanks to the Turkish Academy of Sciences (TUBA).Notes and references
- M. Trojanowicz and M. Kaniewska, Electroanalysis, 2009, 21, 229–238 CrossRef CAS.
- L. Wu and F. G. Vogt, J. Pharm. Biomed. Anal., 2012, 69, 133–147 CrossRef CAS PubMed.
- L. Song, S. Wang, N. A. Kotov and Y. Xia, Anal. Chem., 2012, 84, 7330–7335 CrossRef CAS PubMed.
- R. Kataky and P. Lopes, Chem. Commun., 2009, 12, 1490–1492 RSC.
- Q. Zhang, L. Guo, Y. Huang, Y. Wang, Q. Han and Y. Fu, Anal. Methods, 2013, 5, 4397–4401 RSC.
- Y. Tao, N. R. Quebbemann and R. R. Julian, Anal. Chem., 2012, 84, 6814–6820 CrossRef CAS PubMed.
- E. Zor, I. Hatay Patir, H. Bingol and M. Ersoz, Biosens. Bioelectron., 2013, 42, 321–325 CrossRef CAS PubMed.
- D. Patterson, M. Schnell and J. M. Doyle, Nature, 2013, 497, 475–477 CrossRef CAS PubMed.
- L. Challier, F. Mavré, J. Moreau, C. Fave, B. Schollhorn, D. Marchal, E. Peyrin, V. Noel and B. Limoges, Anal. Chem., 2012, 84, 5415–5420 CrossRef CAS PubMed.
- J. Athilakshmi, M. Mohan and D. K. Chand, Tetrahedron Lett., 2013, 54, 427–430 CrossRef CAS PubMed.
- Z. Huang, S. Yu, K. Wen, X. Yu and L. Pu, Chem. Sci., 2014, 5, 3457–3462 RSC.
- L. Qin, X.-W. Hea, W.-Y. Li and Y.-K. Zhang, J. Chromatogr. A, 2008, 1187, 94–102 CrossRef CAS PubMed.
- L. Sánchez-Hernández, M. L. Marina and A. L. Crego, J. Chromatogr. A, 2011, 1218, 4944–4951 CrossRef PubMed.
- Y. Wang, L. Luo, Y. Ding, X. Zhang, Y. Xu and X. Liu, J. Electroanal. Chem., 2012, 667, 54–58 CrossRef CAS PubMed.
- E. Khaled, M. S. Kamel, H. N. A. Hassan and H. Y. Abou-Enein, J. Electroanal. Chem., 2011, 661, 239–244 CrossRef CAS PubMed.
- R. Nie, X. Bo, H. Wang, L. Zeng and L. Guo, Electrochem. Commun., 2013, 27, 112–115 CrossRef CAS PubMed.
- E. Zor, M. E. Saglam, S. Alpaydin and H. Bingol, Anal. Methods, 2014, 6, 6522–6530 RSC.
- K. B. Lipkowitz, Chem. Rev., 1998, 98, 1829–1873 CrossRef CAS PubMed.
- X.-H. Zhang, H.-L. Wu, X.-L. Yin, L.-H. Li, J.-Y. Wang, Y. Chen, C. Kang and Ru-Q. Yu, Anal. Methods, 2013, 5, 710–717 RSC.
- X. Li, G. Wang, D. Chen and Y. Lu, RSC Adv., 2014, 4, 7301–7312 RSC.
- L. Szente and J. Szemán, Anal. Chem., 2013, 85, 8024–8030 CrossRef CAS PubMed.
- Y. Guo, S. Guo, J. Ren, Y. Zhai, S. Dong and E. Wang, ACS Nano, 2010, 4, 4001–4010 CrossRef CAS PubMed.
- Y. Guo, S. Guo, J. Li, E. Wang and S. Dong, Talanta, 2011, 84, 60–64 CrossRef CAS PubMed.
- Z. Wang, S. Xiao and Y. Chen, J. Electroanal. Chem., 2006, 589, 237–242 CrossRef CAS PubMed.
- D. Lu, S. Lin, L. Wang, X. Shi, C. Wang and Y. Zhang, Electrochim. Acta, 2012, 85, 131–138 CrossRef CAS PubMed.
- M. Chen, Y. Meng, W. Zhang, J. Zhou, J. Xie and G. Diao, Electrochim. Acta, 2013, 108, 1–9 CrossRef CAS PubMed.
- C. Xu, X. Wang, J. Wang, H. Hu and L. Wan, Chem. Phys. Lett., 2010, 498, 162–167 CrossRef CAS PubMed.
- Q. Han, Y. Wang, Y. Huang, L. Guo and Y. Fu, Analyst, 2013, 138, 2051–2056 RSC.
- A. Nosal-Wiercinska, Electrochim. Acta, 2013, 92, 397–403 CrossRef CAS PubMed.
- K. Firoz Babu, R. Sivasubramanian, M. Noel and M. Anbu Kulandainathan, Electrochim. Acta, 2011, 56, 9797–9801 CrossRef PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- J. Köhler and N. Grczelschak-Mick, Beilstein J. Org. Chem., 2013, 9, 118–134 CrossRef PubMed.
- S. Mao, H. Pu and J. Chen, RSC Adv., 2012, 2, 2643–2662 RSC.
- Y. Zhang, C. Wu, S. Guo and J. Zhang, Nanotechnology Reviews, 2013, 2, 27–45 CAS.
- G. P. Kotchey, B. L. Allen, H. Vedala, N. Yanamala, A. A. Kapralov, Y. Y. Tyurina, J. Klein-Seetharaman, V. E. Kagan and A. Star, ACS Nano, 2011, 5, 2098–2108 CrossRef CAS PubMed.
- J.-L. Chen, X.-P. Yan, K. Meng and S.-F. Wang, Anal. Chem., 2011, 83, 8787–8793 CrossRef CAS PubMed.
- E. Zor, M. E. Saglam, I. Akin, A. O. Saf, H. Bingol and M. Ersoz, RSC Adv., 2014, 4, 12457–12466 RSC.
- V. Chandra and K. S. Kim, Chem. Commun., 2011, 47, 3942–3944 RSC.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CAS.
- F. C. Moraesa, I. Cesarinoa, V. Cesarinoa, L. H. Mascarob and S. A. S. Machado, Electrochim. Acta, 2012, 85, 560–565 CrossRef PubMed.
- Y. Hu, K. Wang, Q. Zhang, F. Li, T. Wu and L. Niu, Biomaterials, 2012, 33, 1097–1106 CrossRef CAS PubMed.
- Y. Hu, F. Li, X. Bai, D. Li, S. Hua, K. Wang and L. Niu, Chem. Commun., 2011, 47, 1743–1745 RSC.
- E. Zor, A. O. Saf, H. Bingol and M. Ersoz, Anal. Biochem., 2014, 449, 83–89 CrossRef CAS PubMed.
- L. Tan, K.-G. Zhou, Y.-H. Zhang, H.-X. Wang, X.-D. Wang, Y.-F. Guo and H.-L. Zhang, Electrochem. Commun., 2010, 12, 557–560 CrossRef CAS PubMed.
- E. Alvira, Tetrahedron: Asymmetry, 2013, 24, 1198–1206 CrossRef CAS PubMed.
- C. E. Dalgliesh, J. Chem. Soc., 1952, 3940–3942 RSC.
- C. Chang, X. Wang, Y. Bai and H. Liu, TrAC, Trends Anal. Chem., 2012, 39, 195–206 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |