
Constructing a FRET-based molecular chemodosimeter for cysteine over homocysteine and glutathione by naphthalimide and phenazine derivatives†
Lin
Yang
,
Weisong
Qu
,
Xiao
Zhang
,
Yandi
Hang
and
Jianli
Hua
*
Key Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: jlhua@ecust.edu.cn
First published on 4th November 2014
Abstract
A molecular chemodosimeter PHSN, with a naphthalimide fluorophore connected to a newly designed phenazine energy acceptor, for the selective detection of cysteine was effectively synthesized. PHSN featured efficient intramolecular fluorescence resonance energy transfer (FRET) based on spectral overlap between the emission of naphthalimide and the absorption of phenazine. A cystamine chain with a S–S bond was applied to play the role of recognition moiety and the linker part. The specific reaction between the biological thiols and PHSN gave rise to an obvious fluorescence intensity enhancement at 540 nm, which is induced by cleavage of the disulfide bond followed by elimination of the FRET effect. High sensitivity and selectivity for cysteine over homocysteine and glutathione were also achieved. In addition, upon excitation at 400 nm, a relatively weak NIR emission provided an internal standard making PHSN a promising ratiometric sensor for cellular detection of cysteine.
Introduction
Biological thiols including cysteine (Cys), homocysteine (Hcy) and glutathione (GSH) play very important roles in biological processes.1,2 Cys contributes to amino acid mutations in genetic disease significantly more than other amino acids.3,4 Hcy concentration is related to cardiovascular and Alzheimer's diseases. GSH plays significant roles in intracellular redox activities. Therefore, the detection of biological thiols has attracted great attention during the past few decades.5–7Among various analytical methods for the detection of thiols, molecular probes especially fluorescent chemosensors and chemodosimeters have been developed as a significant area and attracted a tremendous amount of attention, due to their high sensitivity, selectivity and relatively rapid response.8–15 Although considerable fluorescent chemosensors and chemodosimeters have been carefully manipulated for the detection of biological thiols,16–29 limitations still exist. There are some requirements for a good chemosensor of biothiols. Firstly, a clear-cut observation of any changes in fluorescence is a favorable property of thiol sensors, which means that probes of switch-on30–32 or ratiometric types33–38 should be more valued. Secondly, the recognition mechanisms of most chemodosimeters are based on properties that are similar between different thiols, such as nucleophilic reactivity or redox susceptibility of the SH functional group. This means that sensors with the ability to distinguish between these three thiols are urgently required. However, only a few of the reported chemodosimeters meet both of these requirements.39–44
One of the most commonly used recognition methods in the design of a chemodosimeter for biological thiols is the cleavage of the disulfide group. This method relies on the specific disulfide–thiol exchange reaction, and chemodosimeters with a disulfide group as the receptor possess advantageous selectivity and always effectively suppress interference from other amino acids.45–48 Cystamine dihydrochloride contains two amino groups on both sides and can be easily used to construct a FRET system by connecting a fluorophore energy donor on one side and a suitable acceptor on the other side.49–57 As shown in Fig. 1, the cystamine chain provided the necessary Förster distance58 for efficient energy transfer from the fluorophore to the acceptor, therefore when excited, no fluorescence signal was observed. The sensor may therefore remain in an “off” state. With the introduction of thiols, cleavage of the disulfide was induced and followed by separation of the fluorophore from the acceptor, giving rise to the elimination of the energy transfer. Emission of the fluorophore will be uncovered and the sensor will turn to an “on” state.
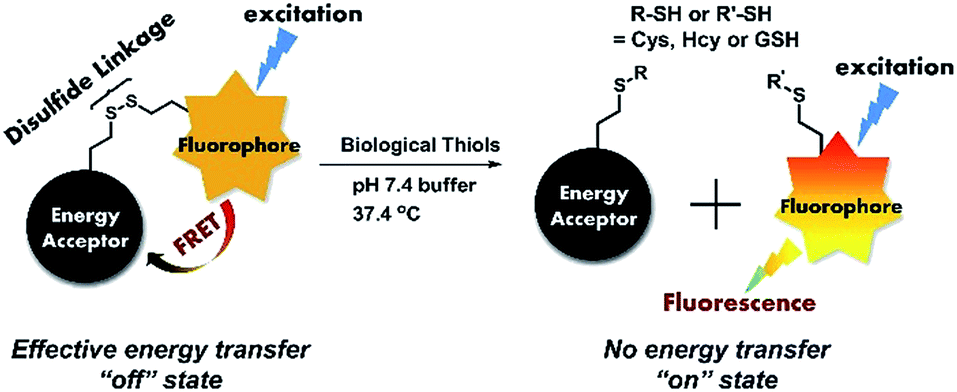 | ||
| Fig. 1 Demonstration of the recognition process of a FRET chemodosimeter based on a thiol participated disulfide cleavage reaction. | ||
Recently, we have reported a sensing platform based on phenazine derivatives (Fig. 2).59,60 Note that with a strong electron withdrawing group conjugated to the phenazine fluorophore, the sensor molecule remained in the turn-off state without any emission. We envision that this platform could be used as an energy acceptor for a FRET system. To improve the synthetic procedures, the methyl-indolium group was replaced by a cyanoacidic acid group for effective esterification, followed by amidation with a cystamine azyl terminal. The newly designed acceptor, phenazine-cyanoacidic acid (PHC), expressed a broad absorption from 400 nm to 600 nm with a maximum absorbance at 450 nm and weak emission at 650 nm, ascribed to the relatively reluctant election withdrawing effect of the cyanoacidic group compared with the indolium moiety (Fig. 2).
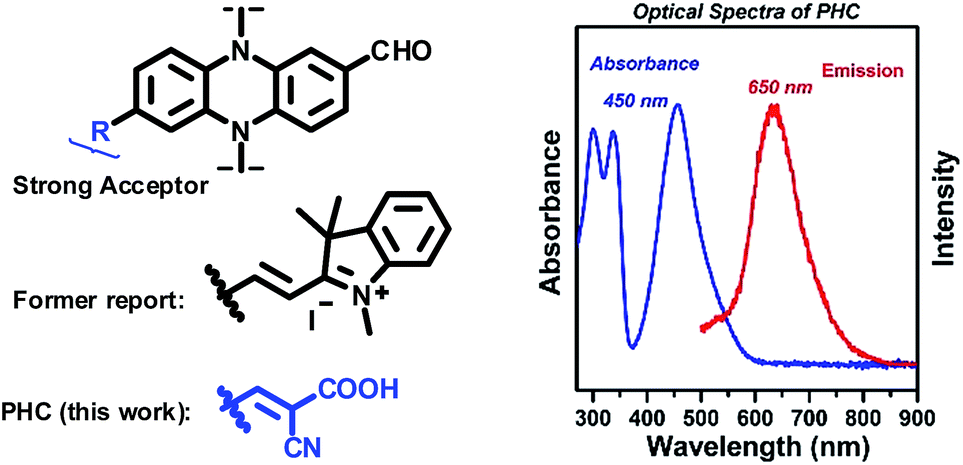 | ||
| Fig. 2 Left: the structures of our formerly reported phenazine derivatives and the newly designed phenazine-cyanoacidic compound PHC. Right: normalized absorbance and emission intensity spectra of PHC. | ||
To match this acceptor, there must be a fluorophore donor which has an effective spectral overlap with the absorption of the acceptor PHC. We chose naphthalimide as the fluorophore due to the wide application of naphthalimide derivatives as sensor fluorophores, and the effective overlap between the emission of naphthalimide (∼500 nm) and the absorption of PHC (400–600 nm).61–64 The cystamine chain of unilateral protection by (Boc)2O (di-tert-butyl dicarbonate) was directly and efficiently reacted with 4-morpholine-1,8-naphthalene anhydride and produced the precursor of the fluorophore (compound 3). Fig. 3 shows the normalized absorption of PHC and the emission of compound 3. The area surrounded by the green line shows the effective overlap between the absorption of the naphthalimide fluorophore 3 and acceptor PHC, which supports the design of the FRET-based chemodosimeter.
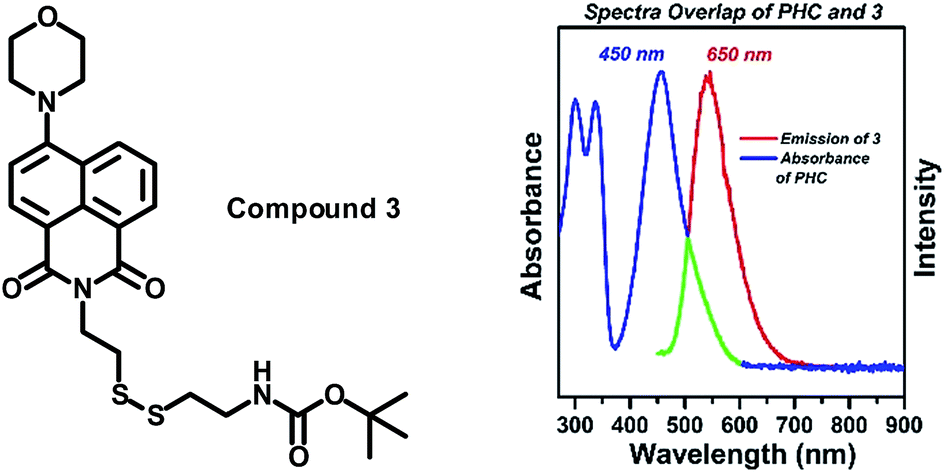 | ||
| Fig. 3 Left: the structure of our formerly reported phenazine derivatives and the newly designed phenazine-cyanoacidic compound PHC. Right: normalized absorbance spectra of PHC and the emission intensity of compound 3, the green line indicates the overlap area. | ||
After hydrolysis deprotection from (Boc)2O, the pre-fluorophore D was connected to the acceptor PHC through an NHS (N-hydroxy succinimide) activated acylation reaction under mild conditions, which resulted in the final product PHSN. PHSN consisted of two isolated moieties which were connected by a disulfide containing alkyl chain. A morpholine group was added to the 4-position of 1,8-naphthalimide to improve the water solubility of PHSN and to avoid other side reactions toward the highly active 4-position. The structure of PHSN was clearly confirmed by 1H NMR, 13C NMR and MALDI-TOF mass spectra (see ESI†).
As shown in Fig. 4, when excited at 400 nm, PHSN exhibits a very low intensity at 540 nm, because most of the energy of the excited naphthalimide fluorophore was transferred to the phenazine acceptor through a FRET transition. After cysteine joined in, the disulfide–thiol exchange reaction started. As the disulfide bond was disrupted, the distance between the donor and acceptor was elongated. Therefore, the overall transfer efficiency would infinitely decrease58 and the excited state fluorophore would decay through radiative transition leading to a turn-on fluorescence signal at 540 nm, which was exactly the original emission wavelength of the naphthalimide fluorophore.
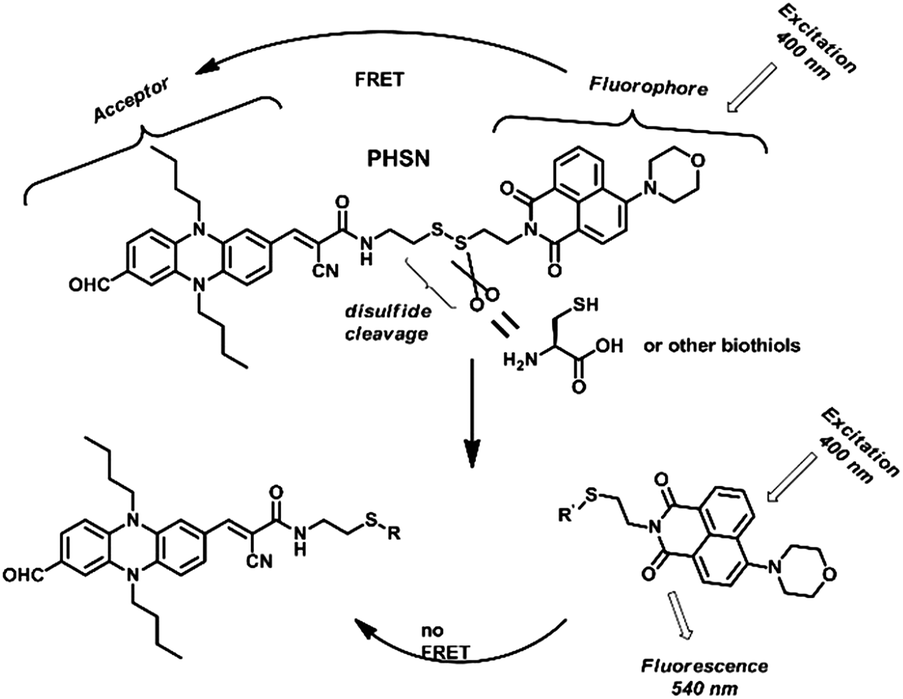 | ||
| Fig. 4 Proposed recognition process of PHSN as a fluorescent chemodosimeter towards cysteine and other biothiols. | ||
Results and discussion
Synthesis of PHSN
The synthetic route from phenazine to diformyl alkyl phenazine (5,10-dibutyl-5,10-dihydrophenazine-2,7-dicarbaldehyde) has been reported in our former literature.57 Phenazine was first reduced by Na2S2O4, then alkylated by butyl iodide followed by formylation through the Vilsmeier–Haack reaction. Cyanoacidic acid was then introduced by the Knoevenagel reaction. NHS esterification activated the carboxylic group, allowing the subsequent acylation to take place under mild condition. 4-morpholine-1,8-naphthalene65 reacted with one side protected cystamine dihydrochloride which was synthesized according to the literature,66 and turned into pre-donor compound 3. After hydrolysis by trifluoroacidic acid, compound 4 was achieved for further acylation. Finally PHSN was obtained by conjugation of 4 and 7 at room temperature with high yield. The details of synthesis are described in the Experimental section, and the structure characterization is formulated in the ESI.†Optical analysis
The absorption and emission spectra of PHSN were first explored before and after the addition of cysteine, homocysteine and glutathione. 40.0 equivalents of each thiol were added respectively to the 5 μM PHSN buffer solution (HEPES buffer with 50% EtOH v/v, pH 7.4) at 37.4 °C. PHSN showed weakly emissive fluorescence at 540 nm and 670 nm before the addition of the thiols. After the reaction of PHSN with cysteine for 1 h, the intensity at 540 nm quickly increased to more than 5-fold. After 2 h, a nearly 10-fold increase was obtained. The obvious appearance and increase of yellow fluorescence can be easily detected by the naked eye (Fig. 5a and b). The change in the emission spectra is in accordance with the change in excitation spectra, in which the intensity at 400 nm drastically increased at the same time when the emission was set at 540 nm (Fig. S1†). On the other hand, the absorption spectra of PHSN remained unchanged during the recognition reaction (Fig. S2†), therefore, no change in color of the PHSN solution was observed before and after the treatment with cysteine (Fig. 5b). Compared with the emission spectrum of the naphthalimide donor (Fig. 3), this fluorescence enhancement could be definitely contributed to the recovery of the original emission of compound 3. Along with the unchanged absorption spectra, these phenomena clearly confirmed the FRET effect from naphthalimide to phenazine, and exclude the possibility of static quenching induced by π–π stacking. On the other hand, upon treatment with Hcy (40 eq.) for 1 h, only a 2-fold increase of emission intensity was detected and a similar result was obtained for GSH treatment. As shown in Fig. 5c, PHSN exhibited an impressive ability to distinguish Cys over Hcy and GSH in buffer solution. We ascribed this ability to the specific redox susceptibility7 of Cys, over Hcy and GSH, toward the PHSN disulfide group.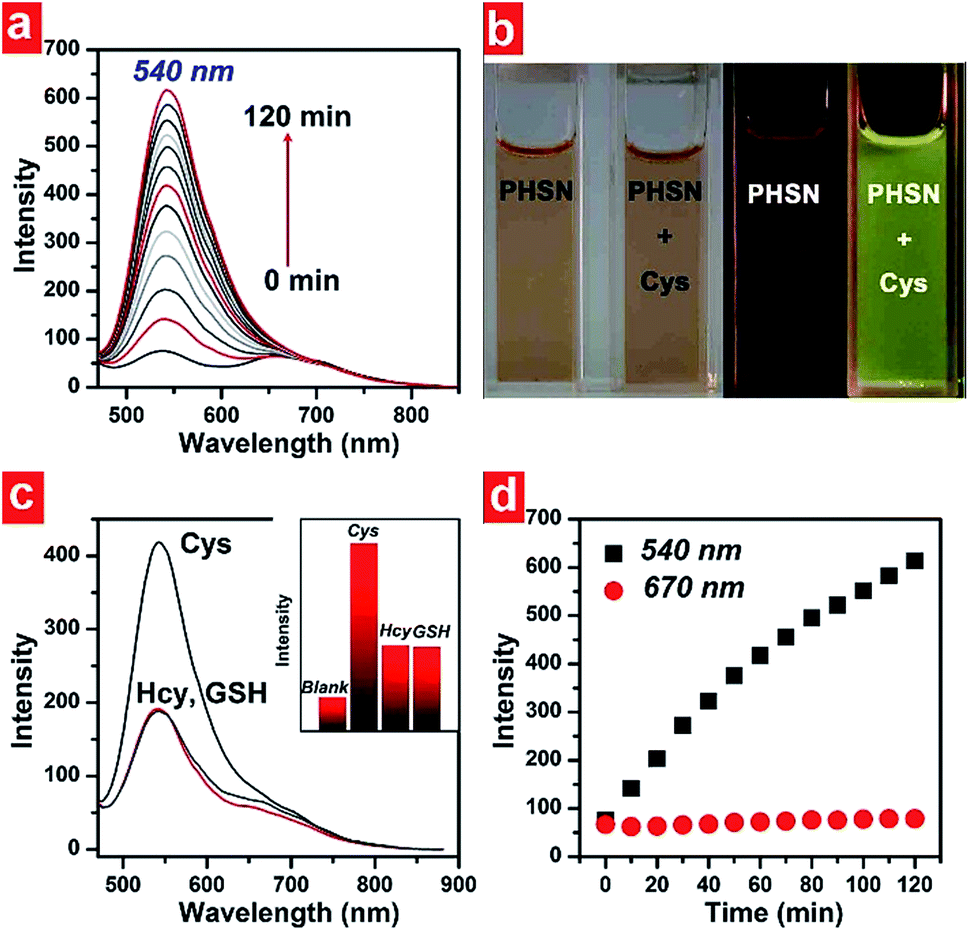 | ||
| Fig. 5 (a) Time-dependent fluorescence emission spectra of PHSN (5 μM) toward cysteine, from 0–120 min in HEPES buffer (50% EtOH v/v, pH 7.4) at 37.4 °C; (b) photographs of PHSN before and after the reaction with cysteine, under natural light and UV radiation respectively; (c) comparison of the emission intensity of PHSN (5 μM) toward Cys, Hcy and GSH after 1 h reaction, respectively. Inset: corresponding histogram of the intensity at 540 nm; (d) corresponding plot of the intensity at 540 nm and 670 nm against a reaction time from 0–60 min (λex = 400 nm, λem = 540 nm, 670 nm respectively). | ||
The time-dependent fluorescence intensity change at 540 nm and 670 nm are plotted in Fig. 5d. Before adding cysteine, the intensity ratio of 540 nm and 670 nm was about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. When cysteine was added, the emission at 670 nm remained relatively constant, as the intensity at 540 nm gradually increased. However, due to the overlap of these two emission bands, the emission of 670 nm did not influence the recognition results.
:1. When cysteine was added, the emission at 670 nm remained relatively constant, as the intensity at 540 nm gradually increased. However, due to the overlap of these two emission bands, the emission of 670 nm did not influence the recognition results.
1H NMR analysis
1H NMR titration experiments were conducted for further investigation into the reaction process. Constrained by the poor solubility of cysteine in deuterated DMSO solvent, we resorted to mercaptopropionic acid (MPA) instead of cysteine, as it has a similar structure but much better solubility in DMSO. In the 1H NMR spectrum of PHSN, two groups of peaks can be identified for the relatively independent donor and acceptor moieties (Fig. S4†). Upon the addition of 1.0 equivalent of MPA for 30 min, a small signal peak at 12.3 ppm emerged, which could be ascribed to the proton of the carboxyl group (Fig. S5†). As the reaction continued, little change happened to the coupling splitting of the peaks around the low field. However, no obvious change in chemical shift could be observed. As shown in Fig. 6, all of the peaks nearly retained their original position, and the integrated number of protons also remained unchanged. These results indicate that the disulfide–thiol reaction did not disturb the conjugation structure of the naphthalimide fluorophore and the phenazine acceptor. All the evidence combined with the above mentioned optical spectral measurements, strongly attest to the proposed mechanism of recognition toward biological thiols, which is dominated by the disulfide–thiol exchange reaction.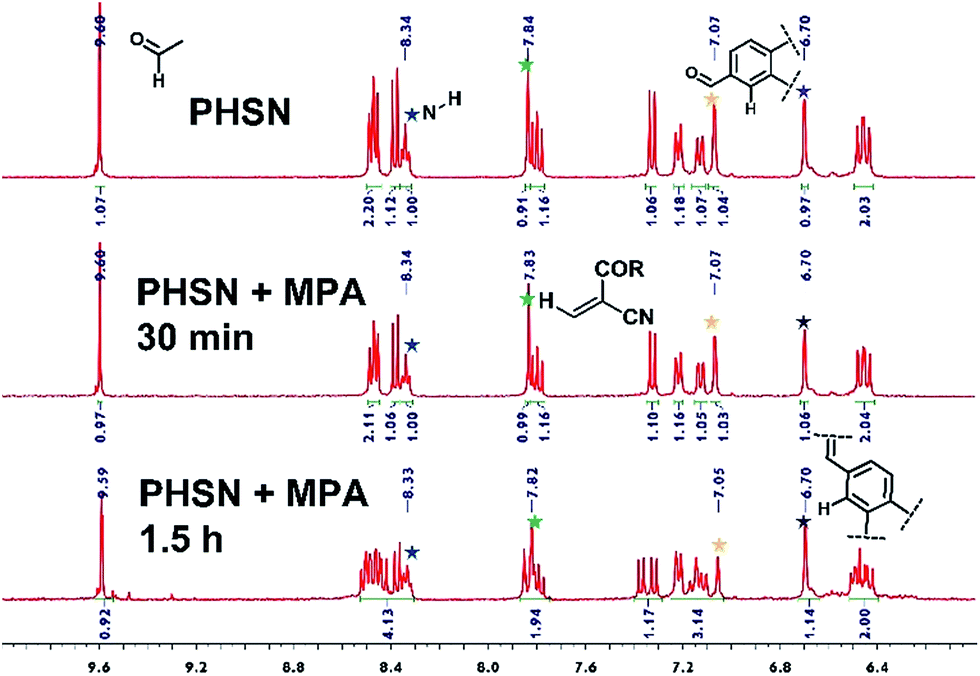 | ||
| Fig. 6 Segmental 1H NMR spectra of PHSN (5 mM) reacted with 1.0 eq. MPA for 0 s (no MPA), 30 min, and 1.5 h in DMSO-d6. The colorful star marked peaks are representative NMR signals of the feature protons. | ||
Linear response toward cysteine
In order to demonstrate the linearity of the response of PHSN toward cysteine, PHSN (5 μM) was treated with different concentrations of Cys in HEPES buffer solution (50% EtOH v/v, pH 7.4) at 37.4 °C (Fig. 7). The corresponding intensity at 540 nm was measured after 1 h from 0–16 eq. cysteine. The intensity change at 540 nm was plotted against the equivalents of Cys. According to the calibration curve, good linear correlation was obtained (R2 =0.99) which indicates a good linear relationship between the fluorescence intensity of the probe and the concentration of Cys. This result is in line with our expectation that PHSN could be used for the quantitative measurement of Cys.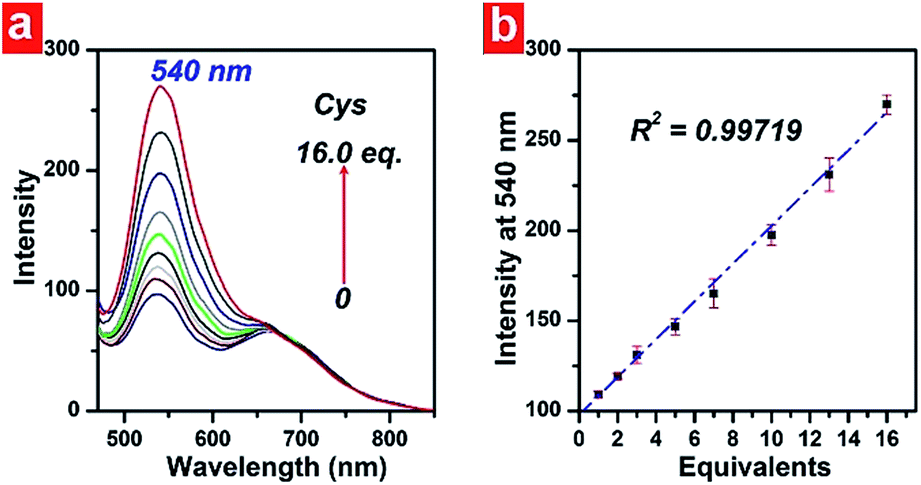 | ||
| Fig. 7 (a) Emission spectra of PHSN (5 μM) reacted with 0–16.0 eq. Cys in HEPES buffer (50% EtOH v/v, pH 7.4) at 37.4 °C. Each spectrum was recorded after 1 h titration. (b) Corresponding linear fitted plot of intensity at 540 nm against Cys equivalents. | ||
To further test the sensitivity of PHSN (5 μM) toward Cys, fluorescence titration was conducted at a low concentration of Cys from 0 to 0.6 equivalents (3 μM) in HEPES buffer solution. As shown in Fig. 8 and S6a,† the intensity-equivalent plot indicates that PHSN was sensitively responsive to only 0.1 equivalent (500 nM) Cys (PHSN/Cys = 10/1). This concentration is much lower than the normal levels of Cys (0.25–0.38 mM) in biological systems.
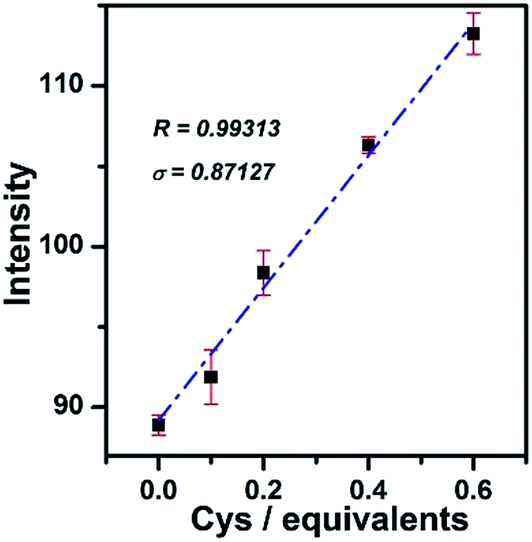 | ||
| Fig. 8 Linear fitted plot of intensity at 540 nm of PHSN (5 μM) against Cys, from 0.0–0.6 equivalents in HEPES buffer (50% EtOH v/v, pH 7.4) at 37.4 °C. The red bar represents the standard deviation of three parallel measurements. σ represented the standard deviation of the fluorescence spectrometer, which was derived from 7 parallel measurements. | ||
Selectivity
Selectivity is another key factor which determines the possible applicability of a chemodosimeter in complicated real samples. To examine the selectivity of our FRET-based chemodosimeter, PHSN was treated with three biothiols and various other common amino acids, including Ala, Arg, Asp, Glu, Gly, Leu, Met, Pro, Ser, Thr and Try, which did not induced any apparent intensity enhancement at 540 nm even at 40.0 eq. for 1 h (Fig. 9). The same result was also revealed by the excitation spectra (Fig. S7†), demonstrating that PHSN is efficiently selective for biothiols, especially for cysteine over homocysteine, glutathione and other amino acids.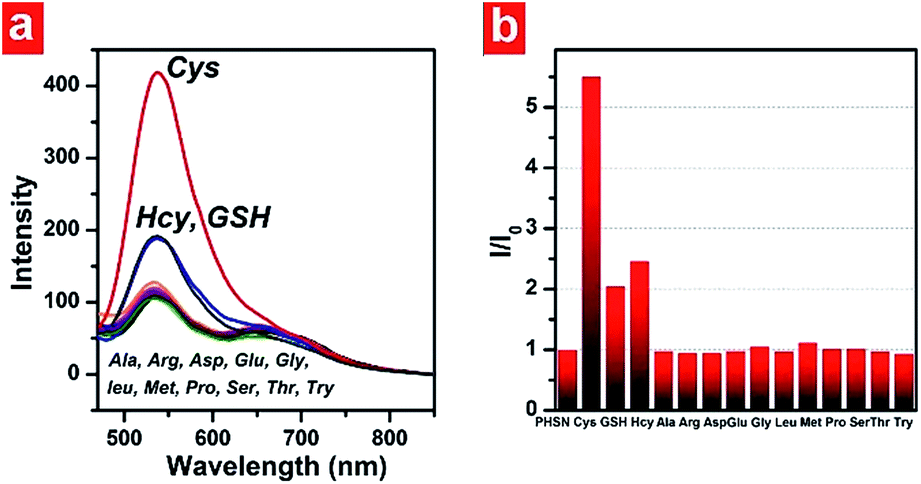 | ||
| Fig. 9 (a) Fluorescence emission spectra of PHSN (5 μM) against 40 equivalents of Cys, Hcy, GSH and various other amino acids in HEPES buffer (50% EtOH v/v, pH 7.4) at 37.4 °C. (b) Corresponding histogram indicating the selectivity of PHSN for Cys over other amino acids. | ||
Intracellular detection
In this regard, we applied PHSN to the fluorescent sensing of biothiols in Hela cells using confocal laser scanning microscopy. The Hela cells were first incubated with PHSN (10 μM) for 40 min, as shown in Fig. 10a, and barely any fluorescence could be observed in the green channel (500–600 nm). Subsequently, Cys (10 μM) was added into the system and was incubated for another 60 min; an apparent intensity enhancement appeared in the green channel (Fig. 10d). The results indicate that PHSN could be used as an effective tool for the fluorescent sensing of intracellular cysteine (Fig. 10).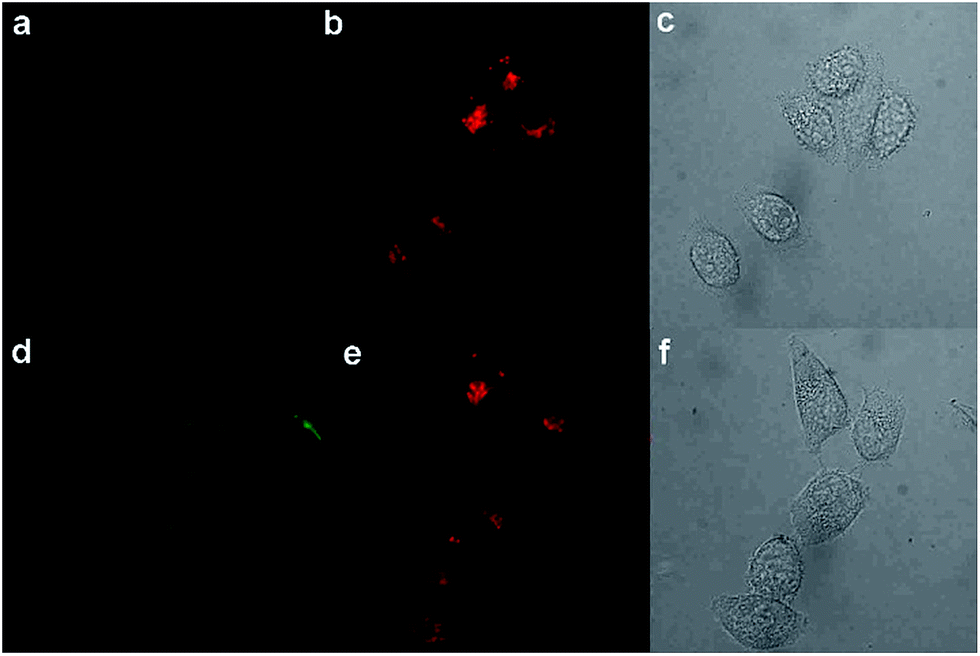 | ||
| Fig. 10 Confocal laser scan images of Hela cells incubated with PHSN (10 μM) in PBS buffer pH 7.4 at 37.4 °C. (a)–(c) Show the green channel (λex = 488 nm, λem = 525–575 nm), red channel (λex = 561 nm, λem = 600–700 nm) and bright field images of the Hela cells incubated with PHSN (10 μM) for 40 min. (d)–(f) Show the green channel, red channel and bright field images when incubated with PHSN (10 μM) for 40 min, then added and incubated with cysteine (10 μM) for another 60 min. | ||
Meanwhile, the fluorescence signal in the red channel remained unchanged during the process. It was introduced as an internal standard to realize a ratiometric imaging of the intracellular cysteine.67,68 As shown in Fig. 11, a distinct change in the intensity ratio of the green/red channel could be obtained. The Z-scan of the confocal imaging shows that PHSN could be well distributed in the cytoplasm (Fig. S8†).
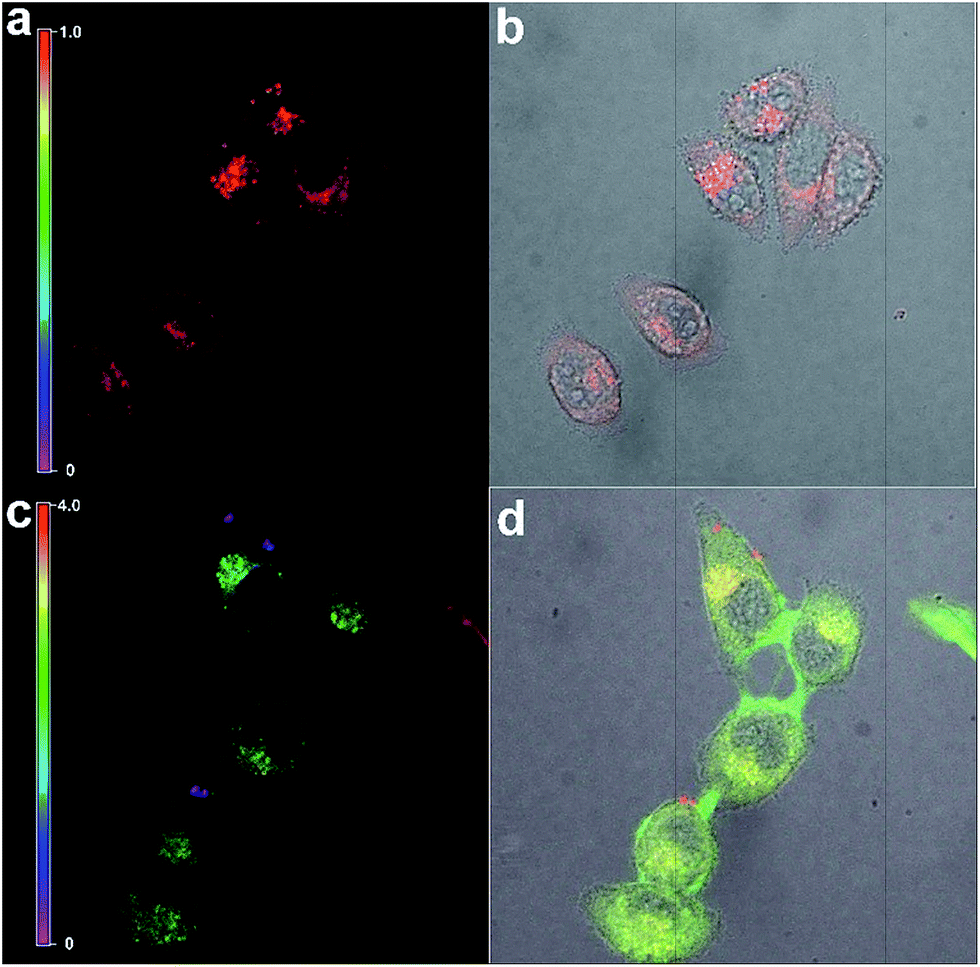 | ||
| Fig. 11 Confocal laser scan fluorescence ratio images of Hela cells incubated with PHSN (10 μM) in PBS buffer pH 7.4 at 37.4 °C for 40 min (above); then added to and incubated with cysteine (10 μM) for another 60 min (below). (a) and (c) are the ratio channel (green/red) images; (b) and (d) are the bright field images merged with both the green and red channel images. Green channel (λex = 488 nm, λem = 525–575 nm), and red channel (λex = 561 nm, λem = 600–700 nm). The scale bar is from 0 to 1.0 for (a) and 0 to 4.0 for (c). | ||
The same procedure was conducted for Hcy and GSH. According to the literature, ratiometric probes always take advantage of a better resolved signal. Benefiting from the ratiometric fluorescence change of PHSN towards the biothiols, the differentiation of Cys from Hcy and GSH in a ratiometric manner could be achieved. As shown in Fig. S9,† when PHSN was treated with 10 μM Cys for 60 min, an obvious ratio change signal appeared, whilst a relatively smaller change in ratio signal was detected when PHSN was treated with Hcy and GSH. This result indicates that PHSN could successfully detect cysteine over homocysteine and glutathione in Hela cells.
Conclusions
In summary, we constructed a FRET-type chemodosimeter PHSN for biological thiols, based on the fluorescence energy transfer from a 4-morpholine naphthalimide fluorophore to our newly designed phenazine acceptor, through a cystamine alkyl chain with a disulfide bond as the linkage. Depending on the specific redox susceptibility of the cystamine disulfide group toward the biothiols, PHSN exhibited impressive sensitivity and selectivity towards the biothiols, especially for cysteine over homocysteine, glutathione and various other amino acids. The detection limit was lower than 500 nM and the recognition time was less than 1 h. PHSN was successfully applied to the turn-on detection of intracellular cysteine. Specifically, depending on the constant emission at 670 nm of the phenazine acceptor, a ratiometric fluorescence signal was achieved and greatly contributed to distinguishing Cys from Hcy and GSH in Hela cells. With a convenient synthetic route, FRET chemodosimeters based on a phenazine acceptor are a promising platform for the detection of biological thiols.Experimental section
Synthesis of compound 3
A mixture of 1 (0.85 g, 3.0 mmol) and 2 (0.75 g, 3.0 mmol) was dissolved in 30 mL ethanol. The reaction process was traced using a TLC plate. After stirring overnight under reflux, the mixture was cooled to room temperature. The solvent was evaporated and the crude product was purified using silicon column chromatography, using a mixed solvent of CH2Cl2/ethyl acetate (20:1, v/v) as the eluent to isolate the pure compound 3 (0.55 g, 35% yield) as a yellow crystal. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.61 (d, J = 7.1 Hz, 1H), 8.55 (d, J = 8.0 Hz, 1H), 8.44 (d, J = 8.3 Hz, 1H), 7.72 (m, 1H), 7.24 (d, J = 8.1 Hz, 1H), 5.34 (s, 1H), 4.53 (m, 2H), 4.03 (m, 4H), 3.47 (m, 2H), 3.28 (m, 4H), 3.03 (m, 2H), 2.90 (m, 2H), 1.43 (s, 9H). 13C NMR (101 MHz, DMSO) δ (ppm): 163.31, 162.77, 155.39, 132.13, 130.61, 130.49, 128.98, 125.94, 125.08, 122.28, 115.53, 114.91, 77.72, 66.15, 52.97, 37.53, 35.10, 28.15. MS (ESI, m/z): calcd for [M + Na]+ C25H31N3O5S2Na+: 540.1603; found: 540.1603.
Synthesis of compound 4
Compound 3 was dissolved in TFA (trifluoroacidic acid) (0.5 mL) and stirred under an argon atmosphere at room temperature. TLC plate analysis was used to trace the reaction process. After about 10 min, the hydrolysis reached equilibrium, and TFA was evaporated. The crude product was dissolved in CH2Cl2 and purified using a silicon column with CH2Cl2/EtOH (3:1 v/v) as the eluent. Pure compound 4 was originally a bright yellow crystal then quickly become sticky under room temperature. 1H NMR (400 MHz, DMSO) δ (ppm): 8.51 (t, J = 8.0 Hz, 1H), 8.43 (d, J = 8.1 Hz, 1H), 7.99 (s, NH), 7.83 (t, J = 7.9 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 4.37 (t, J = 7.1 Hz, 2H), 3.92 (m, 4H), 3.23 (m, 4H), 3.13 (m, 2H), 3.01 (m, 4H). 13C NMR (101 MHz, DMSO) δ (ppm): 163.57, 163.02, 155.63, 132.37, 130.82, 130.77, 129.15, 126.15, 125.21, 122.39, 115.56, 115.08, 66.12, 56.01, 52.98, 37.85, 34.06, 18.44. MS (ESI, m/z): calcd for [M + H]+ C20H24N3O3S2+: 418.1259; found: 418.1258.
Synthesis of compound 6 (PHC)
Diformyl butyl phenazine (5, 300 mg, 0.85 mmol) and 73 mg cyanoacidic acid (0.85 mmol) were mixed and dissolved in CH3CN. Two drops of piperidine was added under an argon atmosphere. The mixture was then heated to 80 °C and stirred for 12 h. After cooling to room temperature, the solvent was evaporated. The crude product was purified using silicon column chromatography, with CH2Cl2/EtOH (5:1, v/v) as the eluent, to obtain the dark red solid product 6 (110 mg, 30%). 1H NMR (400 MHz, DMSO) δ (ppm): 9.57 (s, 1H), 7.75 (s, 1H), 7.19 (d, J = 8.7 Hz, 1H), 7.15 (s, 1H), 7.11 (d, J = 8.4 Hz, 1H), 6.66 (s, 1H), 6.46 (m, 2H), 3.45 (m, 4H), 1.58 (m, 4H), 1.42 (m, 4H), 0.96 (q, J = 7.3 Hz, 6H). 13C NMR (101 MHz, DMSO) δ (ppm): 188.85, 169.21, 163.79, 148.20, 140.77, 138.38, 133.45, 132.97, 128.70, 128.14, 124.22, 117.98, 109.49, 109.16, 108.67, 107.03, 77.83, 58.62, 53.75, 18.18, 17.40, 12.54. MS (ESI, m/z): calcd for [M − H]− C25H26N3O3−: 416.1974; found: 416.1980.
Synthesis of compound 7
A mixture of 6 (100 mg, 0.24 mmol), EDCHCl (46 mg, 0.24 mmol) and a catalytic amount of DMAP were dissolved in DMF under an ice bath below 0 °C and stirred for 15 min. The mixture was then moved to an oil bath and warmed to room temperature. After stirring for 6 h, the solvent was evaporated. Using silicon column chromatography with CH2Cl2/EtOH (50:1, v/v) as the eluent, the product was achieved in 75% yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.61 (s, 1H), 7.90 (s, 1H), 7.31 (d, J = 1.4 Hz, 1H), 7.17 (dd, J = 8.2, 1.4 Hz, 1H), 7.05 (dd, J = 8.4, 1.6 Hz, 1H), 6.80 (d, J = 1.3 Hz, 1H), 6.34 (d, J = 8.3 Hz, 1H), 6.25 (d, J = 8.5 Hz, 1H), 3.46 (m, 4H), 2.89 (s, 4H), 1.93 (dd, J = 12.5, 3.2 Hz, 2H), 1.50 (m, 4H), 1.04 (m, 6H). 13C NMR (101 MHz, DMSO) δ (ppm): 190.52, 170.61, 162.77, 160.53, 156.77, 144.14, 141.86, 135.74, 134.65, 133.46, 130.33, 130.22, 124.81, 116.58, 111.45, 111.32, 110.54, 109.87, 87.12, 45.70, 44.99, 36.25, 31.23, 26.53, 26.11, 25.97, 19.70, 14.15, 14.11. MS (ESI, m/z): calcd for [M + H]+ C29H31N4O5+: 515.2294; found: 515.2291.
Synthesis of PHSN
Compound 7 (50 mg, 0.1 mmol) was dissolved in DMF (3 mL) and stirred while in an ice bath. A 10 mL DMF solution of compound 4 was added in a dropwise fashion. 2 droplets of Et3N was added to provide a basic environment. After stirring for 30 min below 0 °C, the ice bath was replaced by an oil bath and the stirring continued for 4 h. Then DMF was evaporated. The crude product was purified using a silicon column with CH2Cl2/EA (4:1, v/v). A red solid product was finally obtained with 25% yield. 1H NMR (400 MHz, DMSO) δ (ppm): 9.58 (s, 1H), 8.44 (m, 2H), 8.34 (m, 2H), 7.79 (m, 2H), 7.29 (d, J = 8.1 Hz, 1H), 7.19 (d, J = 7.2 Hz, 1H), 7.09 (d, J = 6.4 Hz, 1H), 7.04 (s, 1H), 6.66 (s, 1H), 6.41 (m, 2H), 4.33 (m, 2H), 3.87 (m, 4H), 3.51 (m, 2H), 3.18 (m, 4H), 2.98 (m, 4H), 1.44 (m, 8H), 0.98 (t, J = 6.8 Hz, 3H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ (ppm): 189.76, 164.42, 163.92, 161.91, 155.87, 151.35, 142.52, 141.30, 134.93, 134.83, 132.89, 131.49, 131.07, 130.33, 130.25, 130.16, 129.96, 126.13, 125.89, 124.97, 123.07, 118.28, 116.82, 115.00, 110.16, 110.00, 109.84, 108.26, 97.68, 66.95, 53.43, 46.07, 45.31, 39.60, 39.50, 37.92, 35.72, 29.69, 26.32, 20.03, 19.96, 13.84, 13.80. MS (MALDI-TOF, m/z): calcd for [M] C45H48N6O5S2: 816.3128; found: 816.3133.
Acknowledgements
This work was supported by the National Basic Research 973 Program (2013CB733700), NSFC/China (21372082, 2116110444, 91233207 and 21172073), the Science Fund for Creative Research Groups (21421004). J.-L. Hua very much appreciates Prof. H. Tian’s helpful discussions and valuable comments.Notes and references
- Y. Tay, X. Dou, Z. T. Luo, D. T. Leong and J. P. Xie, Anal. Chem., 2013, 85, 1913 CrossRef PubMed.
- H. S. Jung, X. Q. Chen, J. S. Kim and J. Y. Yoon, Chem. Soc. Rev., 2013, 42, 6019 RSC.
- X. Q. Chen, Y. Zhou, X. J. Peng and J. Y. Yoon, Chem. Soc. Rev., 2010, 39, 2120 RSC.
- G. L. Wang, H. J. Jiao, X. Y. Zhu, Y. M. Dong and Z. J. Li, Analyst, 2013, 138, 2000 RSC.
- X. D. Jiang, J. Zhang, X. M. Shao and W. L. Zhao, Org. Biomol. Chem., 2012, 10, 1966 CAS.
- V. Hong, A. A. Kislukhin and M. G. Finn, J. Am. Chem. Soc., 2009, 131, 9986 CrossRef CAS PubMed.
- N. J. Pace and E. Weerapana, ACS Chem. Biol., 2013, 8, 283 CrossRef CAS PubMed.
- M. Isik, T. Ozdemir, I. S. Turan, S. Kolemen and E. U. Akkaya, Org. Lett., 2013, 15, 216 CrossRef CAS PubMed.
- S. Madhu, R. Gonnade and M. Ravikanth, J. Org. Chem., 2013, 78, 5056 CrossRef CAS PubMed.
- M. Li, X. M. Wu, Y. Wang, Y. S. Li, W. H. Zhu and T. D. James, Chem. Commun., 2014, 50, 1751 RSC.
- X. H. Cheng, H. Z. Jia, J. Feng, J. G. Qin and Z. Li, Sens. Actuators, B, 2014, 199, 54 CrossRef CAS PubMed.
- M. L. Wei, P. Yin, Y. M. Shen, L. L. Zhang, J. H. Deng, S. Y. Xue, H. T. Li, B. Guo, Y. Y. Zhang and S. Z. Yao, Chem. Commun., 2013, 49, 4640 RSC.
- J. Liu, Y. Q. Sun, X. Lv and W. Guo, Anal. Methods, 2013, 5, 3642 RSC.
- D. P. Murale, H. Kim, W. S. Choi and D. G. Churchill, Org. Lett., 2013, 15, 3630 CrossRef CAS PubMed.
- S. Lim, J. O. Escobedo, M. Lowry, X. Y. Xu and R. Strongin, Chem. Commun., 2010, 46, 5707 RSC.
- H. Y. Shiu, H. C. Chong, Y. C. Leung, M. K. Wong and C. M. Che, Chem.–Eur. J., 2010, 16, 3308 CrossRef CAS PubMed.
- H. S. Jung, K. C. Ko, G. H. Kim, A. R. Lee, Y. C. Na, C. Kang, J. Y. Lee and J. S. Kim, Org. Lett., 2011, 13, 1498 CrossRef CAS PubMed.
- L. G. Wang, Q. Zhou, B. C. Zhu, L. G. Yan, Z. M. Ma, B. Du and X. L. Zhang, Dyes Pigm., 2012, 95, 275 CrossRef CAS PubMed.
- Z. Xu, L. Xu, J. Zhou, Y. F. Xu, W. P. Zhu and X. H. Qian, Chem. Commun., 2012, 48, 10871 RSC.
- W. Y. Lin, L. Yuan, Z. M. Cao, Y. M. Feng and L. L. Long, Chem.–Eur. J., 2009, 15, 5096 CrossRef CAS PubMed.
- S. Madhu, R. Gonnade and M. Ravikanth, J. Org. Chem., 2013, 78, 5056 CrossRef CAS PubMed.
- A. Shibata, K. Furukawa, H. Abe, S. Tsuneda and Y. Ito, Bioorg. Med. Chem. Lett., 2008, 18, 2246 CrossRef CAS PubMed.
- J. Bouffard, Y. Kim, T. M. Swager, R. Weissleder and S. A. Hilderbrand, Org. Lett., 2008, 10, 37 CrossRef CAS PubMed.
- Y. Q. Sun, M. L. Chen, J. Liu, X. Lv, J. F Li and W. Guo, Chem. Commun., 2011, 47, 11029 RSC.
- T. K. Kim, D. N. Lee and H. J. Kim, Tetrahedron Lett., 2008, 49, 4879 CrossRef CAS PubMed.
- Z. G. Yang, N. Zhao, Y. Sun, F. Miao, Y. Liu, X. Liu, Y. H. Zhang, W. T. Ai, G. F. Song, X. Y. Shen, X. Q. Yu, J. Z. Sun and W. Y. Wong, Chem. Commun., 2012, 48, 3442 RSC.
- J. H. Lee, C. S. Lim, Y. S. Tian, J. H. Han and B. R. Cho, J. Am. Chem. Soc., 2010, 132, 1216 CrossRef CAS PubMed.
- J. Yin, Y. H. Kwon, D. B. Kim, D. Y. Lee, G. Kim, Y. Hu, J. H. Ryu and J. Yoon, J. Am. Chem. Soc., 2014, 136, 5351 CrossRef CAS PubMed.
- H. Y. Lee, Y. P. Choi, S. Kim, T. Yoon, Z. Guo, S. Lee, K. M. K. Swamy, G. Kim, J. Y. Lee, I. Shin and J. Yoon, Chem. Commun., 2014, 50, 6967 RSC.
- X. Li, S. J. Qian, Q. J. He, B. Yang, J. Li and Y. Z. Hu, Org. Biomol. Chem., 2010, 8, 3627 CAS.
- J. Youziel, A. R. Akhbar, Q. Aziz, M. E. B. Smith, S. Caddick, A. Tinker and J. R. Baker, Org. Biomol. Chem., 2014, 12, 557 CAS.
- M. Zhang, M. X. Yu, F. F. Li, M. W. Zhu, M. Y. Li, Y. H. Gao, L. Li, Z. Q. Liu, J. P. Zhang, D. Q. Zhang, T. Yi and C. H. Huang, J. Am. Chem. Soc., 2007, 129, 10322 CrossRef CAS PubMed.
- L. Y. Niu, Y. S. Guan, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, J. Am. Chem. Soc., 2012, 134, 18928 CrossRef CAS PubMed.
- G. J. Kim, K. Lee, H. Kwon and H. J. Kim, Org. Lett., 2011, 13, 2799 CrossRef CAS PubMed.
- L. Deng, W. T. Wu, H. M. Guo, J. Z. Zhao, S. M. Ji, X. Zhang, X. L. Yuan and C. L. Zhang, J. Org. Chem., 2011, 76, 9294 CrossRef CAS PubMed.
- X. W. Cao, W. Y. Lin and Q. X. Yu, J. Org. Chem., 2011, 76, 7423 CrossRef CAS PubMed.
- B. C. Zhu, X. L. Zhang, Y. M. Li, P. F. Wang, H. Y. Zhang and X. Q. Zhuang, Chem. Commun., 2010, 46, 5710 RSC.
- X. L. Zhang, Y. Xiao and X. H. Qian, Angew. Chem., Int. Ed., 2008, 47, 8025 CrossRef CAS PubMed.
- L. Y. Niu, Y. S. Guan, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, Chem. Commun., 2013, 49, 1294 RSC.
- X. D. Jiang, J. Zhang, X. M. Shao and W. L. Zhao, Org. Biomol. Chem., 2012, 10, 1966 CAS.
- L. Y. Niu, Y. S. Guan, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, J. Am. Chem. Soc., 2012, 134, 18928 CrossRef CAS PubMed.
- X. F. Yang, Y. X. Guo and R. M. Strongin, Angew. Chem., Int. Ed., 2011, 50, 10690 CrossRef CAS PubMed.
- H. S. Jung, J. H. Han, T. Pradhan, S. Kim, S. W. Lee, J. L. Sessler, T. W. Kim, C. Kang and J. S. Kim, Biomaterials, 2012, 33, 945 CrossRef CAS PubMed.
- H. L. Wang, G. D. Zhou, H. W. Gai and X. Q. Chen, Chem. Commun., 2012, 48, 8341 RSC.
- X. M. Wu, X. R. Sun, Z. Q. Guo, J. B. Tang, Y. Q. Shen, T. D. James, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2014, 136, 3579 CrossRef CAS PubMed.
- P. K. Pullela, T. Chiku, M. J. Carvan and D. S. Sem, Anal. Biochem., 2006, 352, 265 CrossRef CAS PubMed.
- A. M. Piggott and P. Karuso, Anal. Chem., 2007, 79, 8769 CrossRef CAS PubMed.
- K. G. Reddie, W. H. Humphries, C. P. Bain, C. K. Payne, M. L. Kemp and N. Murthy, Org. Lett., 2012, 14, 680 CrossRef CAS PubMed.
- G. Zlokarnik, P. A. Negulescu, T. E. Knapp, L. Mere, N. Burres, L. X. Feng, M. Whitney, K. Roemer and R. Y. Tsien, Science, 1998, 279, 84 CrossRef CAS.
- J. Y. Shao, H. Y. Sun, H. M. Guo, S. M. Ji, J. Z. Zhao, W. T. Wu, X. L. Yuan, C. L. Zhang and T. D. James, Chem. Sci., 2012, 3, 1049 RSC.
- H. B. Yu, Y. Xiao, H. Y. Guo and X. H. Qian, Chem.–Eur. J., 2011, 17, 3179 CrossRef CAS PubMed.
- H. Y. Shiu, M. K. Wong and C. M. Che, Chem. Commun., 2011, 47, 4367 RSC.
- F. E. Jernigan and D. S. Lawrence, Chem. Commun., 2013, 49, 6728 RSC.
- Z. G. Zhou, M. X. Yu, H. Yang, K. Huang, F. Y. Li, T. Yi and C. H. Huang, Chem. Commun., 2008, 3387 RSC.
- J. L. Fan, M. M. Hu, P. Zhan and X. J. Peng, Chem. Soc. Rev., 2013, 42, 29 RSC.
- K. Kikuchi, Chem. Soc. Rev., 2010, 39, 2048 RSC.
- A. Chevalier, C. Massif, P. Y. Renard and A. Romieu, Chem.–Eur. J., 2013, 19, 1686 CrossRef CAS PubMed.
- L. Yuan, W. Y. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462 CrossRef CAS PubMed.
- L. Yang, X. Li, J. B. Yang, Y. Qu and J. L. Hua, ACS Appl. Mater. Interfaces, 2013, 5, 1317 CAS.
- L. Yang, X. Li, Y. Qu, W. S. Qu, X. Zhang, Y. D. Hang, H. Agren and J. L. Hua, Sens. Actuators, B, 2014, 203, 833 CrossRef CAS PubMed.
- G. Y. Jiang, S. Wang, W. F. Yuan, L. Jiang, Y. L. Song, H. Tian and D. B. Zhu, Chem. Mater., 2006, 18, 235 CrossRef CAS.
- H. B. Yu, M. Y. Fu and Y. Xiao, Phys. Chem. Chem. Phys., 2010, 12, 7386 RSC.
- J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 1316 CrossRef PubMed.
- C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 12668 CrossRef PubMed.
- J. C. Wu, T. Yi, T. M. Shu, M. X. Yu, Z. G. Zhou, M. Xu, Y. F. Zhou, H. J. Zhang, J. T. Han, F. Y. Li and C. H. Huang, Angew. Chem., Int. Ed., 2008, 47, 1063 CrossRef CAS PubMed.
- K. A. Jacobson, B. Fischer and X. D. Ji, Bioconjugate Chem., 1995, 6, 255 CrossRef CAS.
- Z. Q. Guo, S. W. Nam, S. Park and J. Y. Yoon, Chem. Sci., 2012, 3, 2760 RSC.
- T. Myochin, K. Hanaoka, T. Komatsu, T. Terai and T. Nagano, J. Am. Chem. Soc., 2012, 134, 13730 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectral data, supplementary figures and more details about cell imaging. See DOI: 10.1039/c4an01732c |
| This journal is © The Royal Society of Chemistry 2015 |