
QuEChERS-based approach toward the analysis of two insecticides, methomyl and aldicarb, in blood and brain tissue
Patrick D.
DeArmond
*a,
Matthew K.
Brittain
a,
Gennady E.
Platoff
Jr.
b and
David T.
Yeung
c
aBattelle, 1425 Plain City-Georgesville Rd, West Jefferson, OH 43162, USA. E-mail: dearmondp@battelle.org; Fax: +1 614-458-4337; Tel: +1 614-424-4337
bNational Institute of Allergy and Infectious Diseases (NIH/NIAID), Bethesda, MD 20892, USA
cNational Institute of Neurological Disorders and Stroke (NIH/NINDS), Bethesda, MD 20892, USA
First published on 26th November 2014
Abstract
QuEChERS has been widely utilized for the analysis of pesticides in produce, but it has not been as widely used in clinical test specimens, especially for smaller, sub-gram sample sizes. This study describes the application of a miniaturized QuEChERS methodology toward the analysis of two insecticides, methomyl and aldicarb, in guinea pig blood and brain tissue. Matrix effects and absolute recoveries were investigated for both analytes in the two matrices. While the matrix effects of methomyl in both matrices were minimal at most levels (i.e., from −20% to 20%), aldicarb experienced signal suppression under the described conditions (mean of −47%). However, the matrix effects were not cause for concern due to the sensitivity of the method and the use of matrix-matched standards. The precision and accuracy of the method were excellent over a range of concentrations that spanned three orders of magnitude. The limits of detection (LOD) for both carbamates were determined to be 0.1 ng mL−1 in blood and 0.2 ng g−1 in brain. Other validation parameters, such as linearity, accuracy, precision, and recovery, were also satisfactory in the blood and brain tissue. This method was demonstrated to be sensitive and reproducible, and it should be applicable to the analysis of a wide range of compounds of interest in sub-gram- and sub-milliliter-sized clinical and toxicology specimens.
Introduction
Carbamate pesticides constitute a large class of effective agricultural insecticides, fungicides, and herbicides, among others. However, due to their anticholinesterase (anti-ChE) mechanism of action, they lack species specificity and thus pose a considerable threat to both humans and the environment.1 These pesticides act primarily by competitively inhibiting acetylcholinesterase (AChE) reversibly through the carbamylation of the active site serine residue. AChE inhibition subsequently leads to the overaccumulation of the neurotransmitter acetylcholine at synapses, which can result in the hyperstimulation of both muscarinic and nicotinic receptors, leading to miosis, salivation, lacrimation, urination, defecation, convulsions, and, if uncontrolled, death due to respiratory failure.1Due to their relatively high water solubility, the carbamate insecticides methomyl and aldicarb (see Fig. 1) are known potential groundwater contaminants, especially near their areas of application.2 The US Environmental Protection Agency (EPA) has set maximum contaminant levels for aldicarb in drinking water of 3 μg L−1,3 and proper management practices in the US has reduced the incidences of cases that exceed regulatory limits.4 However, as two of the more toxic carbamate pesticides available, the improper use of methomyl and aldicarb, whether intentional or accidental, can easily result in the poisoning and deaths of wildlife and humans.5
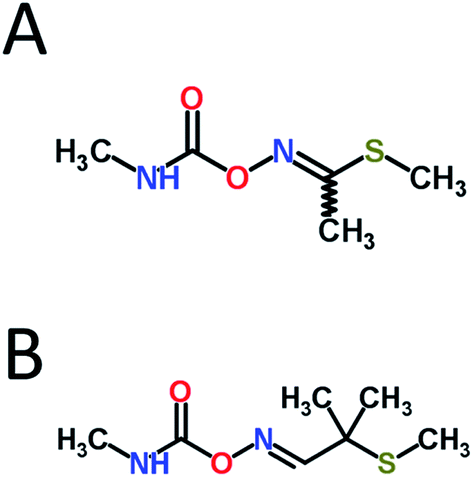 | ||
| Fig. 1 Molecular structures of (A) methomyl and (B) aldicarb. Structures obtained from ChemSpider (http://www.chemspider.com, accessed 6/24/14). | ||
In cases of possible human anti-ChE pesticide poisonings, in addition to measuring AChE activity, determination of the exact amount of pesticide residues present in the blood (as well as in other tissues) would likely be useful for diagnostic purposes. However, most of the currently available analytical methods that are used to determine pesticide levels in blood and brain utilize conventional solid-phase extraction (SPE) techniques,6–9 which are not only time-consuming, but also require large volumes of solvent. Originally developed for the multiresidue analysis of pesticides in produce,10 QuEChERS (quick, easy, cheap, effective, rugged, and safe) is a sample preparation technique that has now been used to analyze a broad spectrum of chemicals in various matrices. This method involves the extraction of the sample with acetonitrile, followed by liquid–liquid partitioning using salts such as MgSO4 and NaCl, and then a final cleanup step using dispersive solid-phase extraction (dSPE). QuEChERS is routinely used to quantify pesticide levels in products for human consumption.11–15 More specifically, the official methods AOAC 2007.01 (ref. 16) and European EN 15662 (ref. 17) both utilize QuEChERS for the analysis of pesticides in produce. However, fewer studies have utilized this technique to analyze pesticides in clinical test specimens, especially from small animals,18–22 and the constant demand for increases in sensitivity concurrent with decreases in sample size often necessitate the modification of existing techniques.23,24
As such, the purpose of this study was to evaluate a miniaturized QuEChERS-based methodology to quantify the anti-ChE carbamates methomyl and aldicarb in both guinea pig blood and brain tissue samples. This method involved extraction using acetonitrile, partitioning using MgSO4/sodium acetate (NaOAc), additional sample cleanup using C18 and amine sorbents, and analysis via liquid chromatography-tandem mass spectrometry (LC-MS/MS). Matrix effects, recoveries, precision, accuracy, linearity, and limits of detection and quantification were determined for this approach in both tissues.
Experimental
Animals
Hartley guinea pigs were used for this study and were purchased from Charles River Laboratories International, Inc. (Wilmington, MA). The experimental protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at Battelle. All procedures were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act of 1966 (P.L. 89-544), as amended.Materials
Methomyl (99.5%) was purchased from Chem Service, Inc. (West Chester, PA). Aldicarb (99.9%) was purchased from Sigma-Aldrich (St. Louis, MO). 13C2,d3-Aldicarb (99.1 atom% 13C, 99.4 atom% 2H, >99% compound purity) was purchased from Sigma-Aldrich, and 13C2,15N-methomyl (99 atom% 13C2, 98 atom% 15N, 98.9% compound purity) was purchased from Cambridge Isotope Laboratories, Inc. (Tewksbury, MA), both of which were used as internal standards (IS) during the extraction and analysis. Stock solutions of methomyl and aldicarb were prepared at a concentration of 1 mg mL−1 in acetonitrile. Subsequent working standards of methomyl and aldicarb were prepared in water from the stock solutions at concentrations of 5000 ng mL−1 and 50 ng mL−1. Stock solutions of the isotopically labeled carbamates were prepared at concentrations of 1000 ng mL−1 in water. All water, acetonitrile, and methanol used for standard preparation, extractions, and mobile phase preparation were Optima™ LC/MS-grade (Fisher Scientific, Hampton, NH).Naïve Hartley guinea pig whole blood was obtained in-house in EDTA tubes and was stored at ≤−70 °C. The whole blood was thawed unassisted at room temperature and spiked appropriately with the working standards to prepare the different standard levels described below. Naïve Hartley guinea pig brain cortexes were obtained in-house and were stored at ≤−70 °C prior to homogenization. The cortexes were allowed to thaw unassisted at room temperature, and an appropriate volume of acetonitrile was added to the tissue to result in 200 mg tissue per mL acetonitrile. Hardened 440C stainless steel balls (3/8 in. diameter, McMaster-Carr, Aurora, OH) were added, and the cortexes were then homogenized by vortexing. The homogenate was spiked appropriately with the working standards to prepare the different standard levels described below.
Sample preparation
The blood samples were vortexed thoroughly, and a 100 μL aliquot of each sample was added to a 1.5 mL microcentrifuge tube, followed by 10 μL of the 1000 ng mL−1 IS stock solution and 400 μL of acetonitrile. The tubes were vortexed and subsequently centrifuged at 1300×g for 10 min. The clear supernatant was added to approximately 0.2 g QuEChERS extraction salt (Restek, Bellefonte, PA), which consisted of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MgSO4/NaOAc, to partition the acetonitrile from the water layer. The final extracts were vortexed and subsequently centrifuged at 1300×g for 1 min. A total of 100 μL of the upper acetonitrile layer was then added to 100 μL of water, and the final extracts were directly analyzed using LC-MS/MS.
:1 MgSO4/NaOAc, to partition the acetonitrile from the water layer. The final extracts were vortexed and subsequently centrifuged at 1300×g for 1 min. A total of 100 μL of the upper acetonitrile layer was then added to 100 μL of water, and the final extracts were directly analyzed using LC-MS/MS.
A 500 μL aliquot of each brain homogenate extract was added to a 1.5 mL microcentrifuge tube, followed by 10 μL of the 1000 ng mL−1 IS stock solution. The tubes were vortexed and subsequently centrifuged at 1300×g for 10 min. The supernatant was added to approximately 0.2 g QuEChERS extraction salt. The tubes were vortexed and subsequently centrifuged at 1300×g for 1 min. The extract was further cleaned up by transferring the extract to approximately 0.05 g QuEChERS dSPE sorbent (Restek, Bellefonte, PA), which consisted of MgSO4/PSA exchange material/C18 (3:1:1). The tubes were again vortexed and subsequently centrifuged at 1300×g for 1 min, and a total of 100 μL of the extract was then added to 100 μL of water. The final extracts were then analyzed directly using LC-MS/MS.
LC-MS/MS analysis and quantification
All samples were analyzed for methomyl or aldicarb using LC-MS/MS. The analyses were performed on an AB Sciex Triple Quadrupole 5500 mass spectrometer (MS) that was interfaced with a Shimadzu Prominence Series HPLC system. The MS was equipped with the TurboIonSpray® (TIS) probe and utilized the TIS probe for positive-mode electrospray ionization (ESI+). The HPLC system consisted of a CBM-20A controller, two LC-20ADXR pumps, an SIL-20ACXR autosampler, and a CTO-20A column oven. The injection volume was set at 10 μL, which represented the injection of 1 μL of blood or 10 μg of brain for each sample. The carbamates were separated on a Dionex Acclaim Carbamate column (3 μm, 2.1 × 150 mm), using an Acclaim Carbamate guard column on the front end (5 μm, 2.1 × 10 mm). Mobile phase A was 0.25 mM ammonium formate in water, and mobile phase B was 0.25 mM ammonium formate in methanol. The initial mobile phase composition was 0% B (100% A), which was held for 2.5 min before ramping up to 90% B over 7 min. The composition was held at 90% B for 1.5 min before bringing back to 0% B over 30 s and holding for 2.5 min. The flow rate was 0.3 mL min−1, and the column temperature was maintained at 30 °C. To reduce the frequency of ion source cleaning, the mobile phase was redirected to waste during the first 7 min of the gradient, then to the mass spectrometer for the next 4 min, and then back to waste.The AB Sciex software Analyst version 1.6.2 was used for data acquisition and analysis. The MS parameters for each compound were optimized to ensure the most favorable ionization and ion transfer conditions and to attain optimum signal of both the precursor and fragment ions by infusing the analytes into 90% B at 0.3 mL min−1 and manually tuning the parameters. The source parameters were identical for all of the analytes: curtain gas, 25 psi; IonSpray voltage, 5500 V; source temperature, 500 °C; ion source gas 1 (nebulizer gas), 50 psi; ion source gas 2 (auxiliary gas), 25 psi. The ESI probe y-axis was set to 8.0 mm, and the x-axis was positioned at 3.0 mm. The declustering potential for methomyl and aldicarb was 50 V and 40 V, respectively, and the collision gas was set to a value of 8 for all of the analytes. The entrance potentials and the collision cell exit potentials were kept at 10 V for all the analytes, and the collision energy was optimized for each MRM transition. The MRM transitions and the collision energies for the carbamates investigated in this study are shown in Table 1. The precursor ion m/z of methomyl was due to protonation (i.e., [M + H]+), while the precursor ion m/z of aldicarb was due to the addition of an ammonium adduct (i.e., [M + NH4]+).
| Compound | MRM transition (m/z) | Collision energy (V) | |
|---|---|---|---|
| Methomyl | 163.1 > 88.0 | 11 | |
| 163.1 > 106.0 | (Qualitative) | 12 | |
| 13C2,15N-Methomyl (ISM) | 166.1 > 91.0 | 11 | |
| Aldicarb | 208.1 > 116.1 | 9 | |
| 208.1 > 89.0 | (Qualitative) | 18 | |
| 13C2,d3-Aldicarb (ISA) | 213.1 > 116.1 | 8 | |
The compounds were identified by their retention times and their specific MRM transitions. Quantification was performed using the isotopically labeled carbamates as internal standards. The analyte/IS peak area ratio was plotted as a function of the analyte concentration and was fitted to a linear regression (y = mx + b), with 1/x weighting.
Determination of matrix effects and recoveries
The matrix effects and absolute recoveries of the extraction and analysis methods for blood and brain were evaluated according to the following approach. Briefly, three sets of standards/samples were prepared, each consisting of 6 concentration levels ranging from 0.05 to 50 ng mL−1 carbamate (where each level contained 10 ng mL−1 IS). The first set consisted of reagent-only calibration standards that were prepared in water to evaluate the response of the carbamates in the absence of any type of extraction or biological matrix. While no extraction was performed on these standards, they were diluted by a factor of 2 with acetonitrile to mimic the solution compositions of the matrix-matched calibration standards and the spiked samples (vide infra). Next, matrix-matched calibration standards were prepared in extracts of naïve blood or brain homogenate (200 mg brain per mL acetonitrile). The extract (either from blood or brain), which consisted of an acetonitrile supernatant and which did not contain any analyte, was combined in equal parts with the reagent-only calibration standards that had been prepared in water. Finally, spiked blood and brain samples were prepared by spiking blood and brain homogenate (200 mg brain per mL acetonitrile) with the carbamates prior to extraction, performing the extraction, and then diluting the final extract by a factor of 2 with water. The final solution composition of the 3 sets consisted of 50:50 water/acetonitrile. Additionally, each set was prepared and analyzed in quintuplicate (n = 5).
Matrix effects (%ME) and absolute recovery (%RE) were then calculated as:
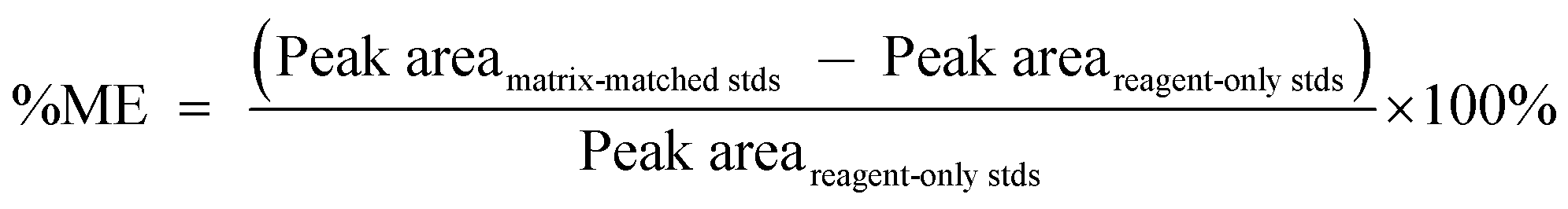 | (1) |
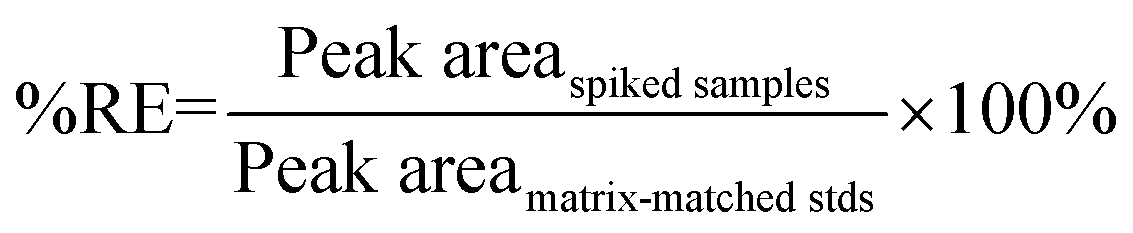 | (2) |
“Peak area” in eqn (1) and (2) refer to the mean peak areas obtained at each concentration level (n = 5). It should be noted that although the final actual concentrations levels in each set were 0.05, 0.25, 0.5, 2.5, 12.5, and 50 ng mL−1, these levels corresponded to initial concentrations in blood and brain that ranged from 0.5 to 500 ng mL−1 and from 0.5 to 500 ng g−1, respectively. Therefore, the 6 different concentration levels are referred to by their respective concentrations in blood and brain.
Determination of limit of detection and limit of quantification
The limit of detection (LOD) was determined by measuring the signal from 20 blank injections and calculating the concentration at:| LOD = meanblank + 3(SDblank) | (3) |
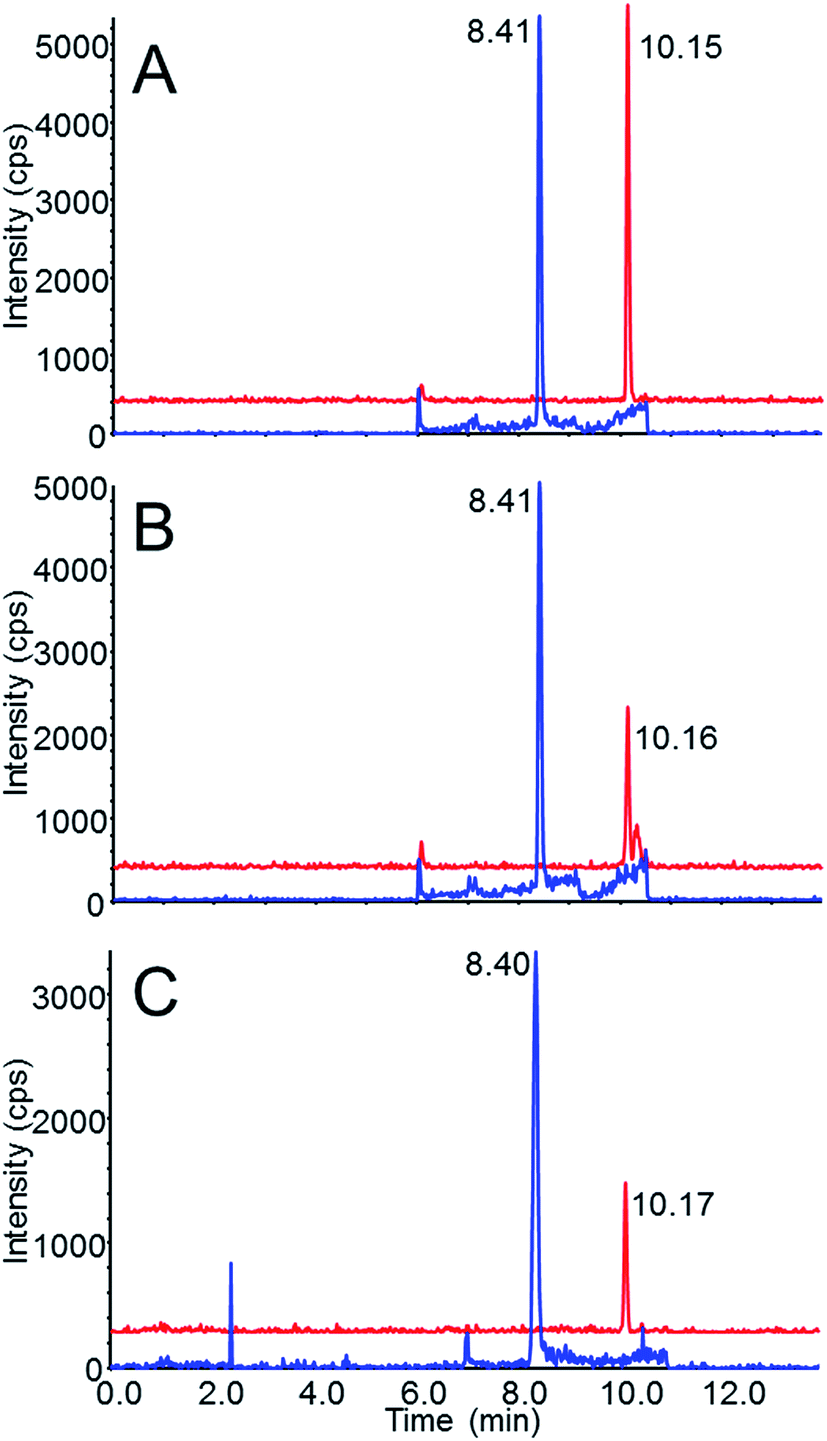 | ||
| Fig. 2 (A) Chromatogram of 0.05 ng mL−1 carbamates in 50/50 water/acetonitrile (equivalent to 0.5 ng mL−1 in blood or 0.5 ng g−1 in brain following extraction) (B) chromatogram of 0.5 ng nL−1 carbamates in blood following extraction (C) chromatogram of 0.5 ng g−1 carbamates in brain homogenate following extraction. Blue trace: methomyl; red trace: aldicarb. The red trace (aldicarb) is artificially raised above the blue trace to show more detail. The numbers next to the peaks refer to the retention times (min). | ||
The limit of quantification (LOQ) was not empirically derived but was rather determined at a “functionally relevant” concentration. In other words, the LOQ was the matrix-matched calibration standard that consistently provided a signal-to-noise ratio that was greater than 10 and that resulted in both the accuracy and precision between replicate injections, which was measured in terms of %RSD, being less than 20%.
Results and discussion
Miniaturized QuEChERS technique in blood and brain
The QuEChERS approach toward sample preparation and analysis is one that is simple and rapid, and it has been demonstrated to be effective towards a broad range of chemicals in various foodstuffs. Therefore, this technique was selected for the analysis of methomyl and aldicarb in guinea pig blood and brain tissue. The basic QuEChERS approach utilizes an extraction with acetonitrile, liquid–liquid partitioning with a polar salt, and then a final cleanup step using C18 and/or primary/secondary amine (PSA) sorbents. The sample then is typically evaporated to dryness and reconstituted prior to gas chromatography (GC)-MS or LC-MS/MS analysis. Because it has been demonstrated to be a robust method, an evaluation of different partitioning salt compositions (e.g., MgSO4 alone, MgSO4 + NaCl, MgSO4 + NaOAc, etc.) was not conducted to determine which, if any, composition performed best in terms of extraction efficiency. For this study, a pre-weighed mixture of 4:1 MgSO4/NaOAc was used as the extraction salt. Similarly, different dSPE sorbent compositions were not evaluated. A pre-weighed mixture of 3:1:1 MgSO4/PSA/C18 was used to dry/clean up the final extract. Because of the small sample sizes and low solvent volumes, only 0.2 g and 0.05 g of extraction salt and dSPE sorbent, respectively, were used instead of the typical ∼5 g and ∼0.2 g,10 respectively. No dSPE step was performed for blood, as initial tests determined that the cleanup step in blood samples was no more effective in decreasing matrix effects or increasing absolute recovery through the use of the aforementioned dSPE sorbent composition (data not shown) than in the absence of this final cleanup step. The use of an acidic extraction (i.e., 1% acetic acid in acetonitrile as specified by AOAC Method 2007.01 (ref. 16)) was also briefly evaluated, but there was no difference in the extraction efficiency between extractions with and without acid (data not shown). Therefore, the extractions were performed only in acetonitrile.
No deterioration of reagent-only standards were observed after 2 weeks storage at 4 °C, and recoveries of 87% and 93% for methomyl and aldicarb, respectively, were obtained after 8 weeks storage at 4 °C. No stability was determined for the carbamates in blood or brain samples, i.e., the spiked samples. The LOD values for methomyl and aldicarb in blood were determined to be 0.1 ng mL−1, and the LOD values for methomyl and aldicarb in brain were 0.2 ng g−1.
Evaluation of precision, accuracy, and linearity
Three different sets of samples were prepared for comparison purposes (reagent-only calibration standards, matrix-matched calibration standards, and spiked blood/brain homogenate samples). The precision of the method was evaluated by comparing the repeatability (%RSD, n = 5) of the extraction at 6 different levels ranging from 0.5 to 500 ng mL−1 in blood and 0.5 to 500 ng g−1 in brain (Tables 2 and 3, respectively). The precision of the extraction with regard to the peak areas of the carbamates in the spiked samples was generally less than 15% (%RSD). However, at the lowest concentration in blood, the analyte response produced a %RSD value greater than 20% for both carbamates. These precision data demonstrate the excellent repeatability of the extraction method for methomyl and aldicarb in both matrices over a range of concentrations spanning 3 orders of magnitude.| Concentration of methomyl in blood (ng mL−1) | %MEa | Absolute recoveryb (%) | Method recoveryc (%) | Accuracyd (%) | Precisione (%) (n = 5) |
|---|---|---|---|---|---|
| a Calculated using eqn (1). b Calculated using eqn (2). c Absolute recovery of methomyl normalized by the recovery of the internal standard. d Calculated as: (mean observed concentration)/(nominal concentration) × 100%. e %RSD of the methomyl peak area at each concentration level (SD/mean × 100%). | |||||
| 0.5 | 14 | 107 | 91 | 88 | 22 |
| 2.5 | 5.9 | 123 | 100 | 106 | 12 |
| 5.0 | −0.9 | 110 | 94 | 103 | 6.7 |
| 25 | −2.0 | 103 | 90 | 96 | 18 |
| 125 | −0.3 | 112 | 101 | 109 | 4.0 |
| 500 | 4.3 | 108 | 101 | 98 | 4.2 |
| Concentration of aldicarb in blood (ng mL−1) | %MEa | Absolute recoveryb (%) | Method recoveryc (%) | Accuracyd (%) | Precisione (%) (n = 5) |
|---|---|---|---|---|---|
| a Calculated using eqn (1). b Calculated using eqn (2). c Absolute recovery of aldicarb normalized by the recovery of the internal standard. d Calculated as: (mean observed concentration)/(nominal concentration) × 100%. e %RSD of the aldicarb peak area at each concentration level (SD/mean × 100%). | |||||
| 0.5 | −42 | 99 | 88 | 116 | 22 |
| 2.5 | −48 | 116 | 97 | 100 | 9.1 |
| 5.0 | −50 | 120 | 98 | 97 | 4.7 |
| 25 | −49 | 124 | 87 | 86 | 9.4 |
| 125 | −51 | 138 | 101 | 100 | 1.9 |
| 500 | −47 | 121 | 98 | 101 | 0.8 |
The analyte/IS response ratio was linear (R2 > 0.99) from 0.5 to 500 ng mL−1 in blood and from 0.5 to 500 ng g−1 in brain for both analytes. The accuracy of the measured values at each level, expressed as (mean observed concentration)/(nominal concentration) × 100%, was within 85–115% at all levels, except for the low methomyl level in brain (0.5 ng g−1). Therefore, the linear calibration range of methomyl in brain was narrowed to the range of 0.5 to 125 ng g−1, which brought the accuracy of the low end to within the 85–115% range, to ensure accuracy at all levels. Additionally, the slopes of the curves were consistent between matrices (e.g., 10.05 × 10−3vs. 9.92 × 10−3 for methomyl in brain and blood, respectively, and 8.20 × 10−3vs. 8.97 × 10−3 for aldicarb in brain and blood, respectively). For reference, the slopes of methomyl and aldicarb in reagent-only standards were 10.18 × 10−3 and 8.51 × 10−3, respectively.
Matrix effects and recoveries
The matrix effects and absolute recoveries of the extraction method were evaluated by comparing the mean instrument responses (n = 5) of the reagent-only calibration standards, the matrix-matched calibration standards, and the spiked samples using eqn (1) and (2). Tables 2 and 3 present the results for the carbamates in both matrices. The matrix effects of methomyl in blood were generally minimal, as %ME ranged from −2 to 14% for methomyl (where %ME < −20% indicates signal suppression and %ME > 20% indicates signal enhancement). The absolute recoveries ranged from 103 to 123% for methomyl across the range of concentrations. Similarly, the matrix effects for methomyl in brain were also minimal, as %ME ranged from −11 to 21% for methomyl. The absolute recoveries of methomyl ranged from 93–103% across the range of concentrations. Taken together with the results from the blood, the extraction of methomyl from blood and brain using this approach provided excellent absolute recoveries and virtually no matrix effects due to the biological matrices.As shown in Table 3, significant signal suppression was observed for aldicarb. The %ME ranged from −51 to −42% in blood (mean = −48%) and −50 to −44% in brain (mean = −47%). The absolute recoveries of aldicarb in the blood were slightly high, as they ranged from 99–138%. The lots of blood used to prepare the matrix-matched calibration standards and the spiked blood samples were different. Therefore, as recoveries by their definition cannot be greater than 100%, the high recoveries obtained here indicated the presence of matrix effects that were a result of the different lots of blood. However, the high absolute recoveries were normalized using the recoveries of the internal standards (i.e., the method recovery, see Tables 2 and 3). The absolute recoveries of aldicarb in brain ranged from 69–88%.
Chromatograms of the carbamates in reagent-only standards and of the spiked samples following extraction are shown in Fig. 2. Fig. 2A shows both methomyl and aldicarb at 0.05 ng mL−1 in 50/50 water/acetonitrile (equivalent to the 0.5 ng mL−1 or the 0.5 ng g−1 extract level in blood and brain, respectively). Both methomyl and aldicarb presented similar intensities at this concentration. In Fig. 2B and C, which are chromatograms of the carbamates in the spiked samples following extraction, the aldicarb response decreases by a factor of 2 while the response of methomyl is relatively unchanged, which resulted mainly from the aforementioned matrix effects.
Evaluation of method
Ideally, analytical methods will be free from matrix effects and will result in absolute recoveries near 100% of the analytes of interest. As an example, the described miniaturized QuEChERS method for methomyl in blood and brain exhibited these characteristics at most levels. In contrast, signal suppression was more prominent following the extraction of aldicarb. However, the excellent agreement between analyte and internal standard with regard to the matrix effect and absolute recovery values ensured that the measured values were accurate regardless of signal suppression or low recoveries.This described method was applied to the analysis of methomyl and aldicarb that was present in both the blood and brain cortex of Hartley guinea pigs after the subcutaneous administration of the two carbamates.25 The %RSD values between triplicate extractions of blood and cortex samples from animals that had been challenged with aldicarb ranged from 2.2–27% (median 7.1%) and 0.5–13% (median 3.0%), respectively. The %RSD values between triplicate extractions of blood and cortex samples from animals that had been challenged with methomyl ranged from 2.7–46% (median 6.7%) and 1.6–9.0% (median 3.7%),25 respectively, indicating good precision for the majority of measurements using this method.
Based on the data presented here, this method was demonstrated to be highly sensitive and reproducible. This paper supports the increasing numbers of works that suggest the use of QuEChERS could easily be applied to the analysis of many toxicants in any number of body fluids or clinical test specimens.18–22 Additionally, it is likely that this approach may also be applicable for screening any number of small molecule classes, such as drugs of abuse, pharmaceuticals, and other compounds of interest as well.
Analytical method miniaturization typically results in more environmentally friendly approaches to sample analysis due to reductions in solvent and chemical use. It also enables the analysis of smaller sample sizes with increased sensitivity. A technique closely related to QuEChERS, salting-out assisted liquid–liquid extraction (SALLE),26 has previously been reported for use with small body fluid samples. For example, Wabuyele et al. used SALLE to determine securinine in mouse plasma,27 and Mohamed et al. analyzed anti-glaucoma drugs in aqueous humor samples.28 Recent examples of miniaturized QuEChERS have also been reported. For example, Gonzalo-Lumbreras et al. miniaturized the QuEChERS technique to determine triclosan and its metabolite in fish roe and surimi,23 while Walorczyk and Drozdzynski used miniaturized QuEChERS to determine 140 pesticides in fruits and vegetables.24 Our report confirms the application of miniaturized QuEChERS for the determination of pesticides in small clinical test specimens. Because clinical test specimens from animals or humans are often limited with regard to the volume or mass of sample that can be acquired, the commercial availability of smaller QuEChERS packets or wellplates would facilitate the rapid analysis of sub-gram- or sub-milliliter-sized clinical and toxicology specimens.
Acknowledgements
This work was supported by the National Institutes of Health (NIH) Office of the Director through an interagency agreement (OD#: Y1-OD-0387-01) between the National Institute of Allergy and Infectious Diseases (NIAID) and Department of Defense (DoD) and prepared under the auspices of the NIH and the DoD Defense Technical Information Center (DTIC) under the Chemical, Biological, Radiological & Nuclear Defense Information Analysis Center (CBRNIAC) program, Contract no. SP0700-00-D-3180, Delivery Order Number 0687, CBRNIAC Task 832/CB-IO-OOI2. The views expressed in this article are those of the authors and do not reflect the official policy of the NIH, Department of Health and Human Services, or the U.S. Government. No official support or endorsement of this article by the NIH or DoD is intended or should be inferred. The authors thank Brent McCracken and Jessica Schimmoeller (Battelle) for their critical review of the manuscript.References
- R. C. Gupta, Reproductive and Developmental Toxicology, ed. R. C. Gupta, Academic Press, London, 2011, pp. 471–486 Search PubMed.
- R. L. Jones and T. L. Estes, Summary of aldicarb monitoring and research programs in the U.S.A., J. Contam. Hydrol., 1995, 18, 107–140 CrossRef CAS.
- U. S. EPA, 2012 Edition of the Drinking Water Standards and Health Advisories, EPA 822-S-12-001, Office of Water, Washington, D.C, 2012.
- R. L. Jones and R. Allen, Summary of potable well monitoring conducted for aldicarb and its metabolites in the United States in 2005, Environ. Toxicol. Chem., 2007, 26, 1355–1360 CrossRef CAS.
- L. R. Goldman, M. Beller and R. J. Jackson, Aldicarb food poisonings in California, 1985–1988: toxicity estimates for humans, Arch. Environ. Health, 1990, 45, 141–147 CrossRef CAS.
- U. S. Gill, H. M. Schwartz and B. Wheatley, Development of a method for the analysis of PCB congeners and organochlorine pesticides in blood/serum, Chemosphere, 1996, 32, 1055–1076 CrossRef CAS.
- B. L. Tan and M. Ali Mohd, Analysis of selected pesticides and alkylphenols in human cord blood by gas chromatograph-mass spectrometer, Talanta, 2003, 61, 385–391 CrossRef CAS.
- R. M. Whyatt, D. B. Barr, D. E. Camann, P. L. Kinney, J. R. Barr, H. F. Andrews, L. A. Hoepner, R. Garfinkel, Y. Hazi, A. Reyes, J. Ramirez, Y. Cosme and F. P. Perera, Contemporary-use pesticides in personal air samples during pregnancy and blood samples at delivery among urban minority mothers and newborns, Environ. Health Perspect., 2003, 111, 749–756 CrossRef CAS.
- P. Li and M. G. Bartlett, A review of sample preparation methods for quantitation of small-molecule analytes in brain tissue by liquid chromatography tandem mass spectrometry (LC-MS/MS), Anal. Methods, 2014, 6, 6183–6207 RSC.
- M. Anastassiades, S. J. Lehotay, D. Stajnbaher and F. J. Schenck, Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce, J. AOAC Int., 2003, 86, 412–431 CAS.
- A. Albert, A. Kramer, S. Scheeren and C. Engelhard, Rapid and quantitative analysis of pesticides in fruits by QuEChERS pretreatment and low-temperature plasma desorption/ionization orbitrap mass spectrometry, Anal. Methods, 2014, 6, 5463–5471 RSC.
- C. Diez, W. A. Traag, P. Zommer, P. Marinero and J. Atienza, Comparison of an acetonitrile extraction/partitioning and “dispersive solid-phase extraction” method with classical multi-residue methods for the extraction of herbicide residues in barley samples, J. Chromatogr. A, 2006, 1131, 11–23 CrossRef CAS.
- U. Koesukwiwat, K. Sanguankaew and N. Leepipatpiboon, Rapid determination of phenoxy acid residues in rice by modified QuEChERS extraction and liquid chromatography-tandem mass spectrometry, Anal. Chim. Acta, 2008, 626, 10–20 CrossRef CAS.
- J. M. Lee, J. W. Park, G. C. Jang and K. J. Hwang, Comparative study of pesticide multi-residue extraction in tobacco for gas chromatography-triple quadrupole mass spectrometry, J. Chromatogr. A, 2008, 1187, 25–33 CrossRef CAS.
- A. Kruve, A. Kunnapas, K. Herodes and I. Leito, Matrix effects in pesticide multi-residue analysis by liquid chromatography-mass spectrometry, J. Chromatogr. A, 2008, 1187, 58–66 CrossRef CAS.
- AOAC, Pesticide Residues in Foods by Acetonitrile Extraction and Partitioning with Magnesium Sulfate, AOAC Official Method 2007.01, 2007.
- E. N. 15662, Foods of plant origin – Determination of pesticide residues using GC-MS and/or LC-MS/MS following acetonitrile extraction/partitioning and cleanup by dispersive SPE – QuEChERS-method, 2008.
- D. Vudathala, M. Cummings and L. Murphy, Analysis of multiple anticoagulant rodenticides in animal blood and liver tissue using principles of QuEChERS method, J. Anal. Toxicol., 2010, 34, 273–279 CrossRef CAS.
- G. Reiter, J. Mikler, I. Hill, K. Weatherby, H. Thiermann and F. Worek, Simultaneous quantification of VX and its toxic metabolite in blood and plasma samples and its application for in vivo and in vitro toxicological studies, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 2704–2713 CrossRef CAS.
- K. Usui, Y. Hayashizaki, M. Hashiyada and M. Funayama, Rapid drug extraction from human whole blood using a modified QuEChERS extraction method, Leg. Med., 2012, 14, 286–296 CrossRef CAS.
- F. Plossl, M. Giera and F. Bracher, Multiresidue analytical method using dispersive solid-phase extraction and gas chromatography/ion trap mass spectrometry to determine pharmaceuticals in whole blood, J. Chromatogr. A, 2006, 1135, 19–26 CrossRef.
- B. Kinsella, S. J. Lehotay, K. Mastovska, A. R. Lightfield, A. Furey and M. Danaher, New method for the analysis of flukicide and other anthelmintic residues in bovine milk and liver using liquid chromatography-tandem mass spectrometry, Anal. Chim. Acta, 2009, 637, 196–207 CrossRef CAS.
- R. Gonzalo-Lumbreras, J. Sanz-Landaluze and C. Cámara, Analytical performance of two miniaturized extraction methods for triclosan and methyltriclosan, in fish roe and surimi samples, Food Chem., 2014, 146, 141–148 CrossRef CAS.
- S. Walorczyk and D. Drozdzynski, Development and validation of a routine multiresidue method for determining 140 pesticides in fruits and vegetables by gas chromatography/tandem quadrupole mass spectrometry, J. AOAC Int., 2011, 94, 1625–1642 CrossRef CAS.
- M. K. Brittain, K. G. McGarry, R. A. Moyer, M. C. Babin, D. A. Jett, G. E. Platoff and D. T. Yeung, Efficacy Assessments of a Standard Countermeasure Regimen against the Toxic Effects of Carbamate Poisoning in the Hartley Guinea Pig, unpublished work, 2014.
- J. Zhang, H. Wu, E. Kim and T. A. El-Shourbagy, Salting-out assisted liquid/liquid extraction with acetonitrile: a new high throughput sample preparation technique for good laboratory practice bioanalysis using liquid chromatography-mass spectrometry, Biomed. Chromatogr., 2009, 23, 419–425 CrossRef CAS.
- S. L. Wabuyele, D. Wald and Y. Xu, Development and validation of LC-MS/MS method for quantitative determination of (−)-securinine in mouse plasma, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 960, 19–26 CrossRef CAS.
- A. M. I. Mohamed, H. M. Abdel-Wadood and H. S. Mousa, Simultaneous determination of dorzolomide and timolol in aqueous humor: A novel salting out liquid–liquid microextraction combined with HPLC, Talanta, 2014, 130, 495–505 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |