
Synthesis and evaluation of a silica-bonded concanavalin A material for lectin affinity enrichment of N-linked glycoproteins and glycopeptides†
Yujie
Liu
a,
Dongmei
Fu
ab,
Yuansheng
Xiao
a,
Zhimou
Guo
a,
Long
Yu
*ab and
Xinmiao
Liang
*a
aKey Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: yulong@dicp.ac.cn; liangxm@dicp.ac.cn
bNational Glycoengineering Research Center, Shandong University, China
First published on 25th November 2014
Abstract
A lectin affinity chromatography material was synthesized by covalent bonding of concanavalin A onto silica gel (named SG Con A) via a rapid, simple and robust method. The material exhibited high selectivity and efficiency as well as low sample loss for the selective enrichment of N-linked glycoproteins and glycopeptides.
Lectin affinity chromatography (LAC) is an important enrichment technique that has been widely employed in glycoproteomic analysis.1 Taking advantage of the specific interactions between carbohydrates and the immobilized lectins,2 LAC is capable of recognizing and enriching the peptides or proteins attached with certain types of glycans and thus depleting those non-glycosylated counterparts. Due to their narrow specificities,3 combined use of different types of lectins either sequentially or in parallel has been adopted in LAC enrichment to avoid the loss of the glycosylated peptides or proteins.4 Such enrichment procedures are usually performed in solid-phase extraction or batch mode, since most of the LAC materials are prepared by using soft porous gel supports like agarose or more recently, by using magnetic nanoparticles, for the immobilization of lectins.5,6 Despite their advantages such as chemical inertness and hydrophilicity, gel supports usually exhibit relatively low mechanical stability and could not withstand the high backpressure in high performance liquid chromatography (HPLC). In comparison to those low-pressure matrices, functionalized silica gel has been utilized as LAC materials in the separation studies of glycoproteins on account of its mechanical and chemical stability properties.7 Covalent bonding of lectins on silica gel was traditionally performed via the reaction between the ε-amino groups of lysine residues and the silica resins activated by tresyl chloride, carbodiimidazole, or (3-mercaptopropyl)trimethoxy-silane, as well as by forming a Schiff-base between the aldehyde activated silica and the primary amine groups of the ligand followed by reduction with sodium cyanoborohydride to yield a stable alkyl amide.8 Given the loss of lectin's bioactivity caused by the use of highly toxic reagents and longer reaction times, in this research, we introduce a rapid and simple method to synthesize a silica-bonded concanavalin A material named SG Con A for the efficient enrichment of certain types of glycoproteins and glycopeptides.
As shown in Scheme 1, silica gel was first modified with a hydrophilic spacer arm 3 containing an amide bond and carboxyl residues obtained from the reaction of silicon reagent 1 and glutaric anhydride 2. The structure of silicon reagent 3 was confirmed by LC-MS analysis, in which its m/z ratio was in agreement with that of the MS peak 337 (M + H)+ in Fig. S1.† After elemental analysis (EA) (Table S1†) and Fourier-transform infrared spectroscopy (FT-IR) characterization (Fig. S2a†), the modified silica particles 4 were activated by N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxylsuccinimidyl (NHS) to obtain silica particles 5. As shown in Fig. S2b,† the bands at 1716 cm−1 (silica particles 4) and 1699 cm−1, and 1653 cm−1 (silica particles 5) were assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of amides, which demonstrates that the carboxylic groups have been activated by the EDC/NHS reagent.9 Subsequently, lectin Con A was immobilized onto EDC/NHS activated silica gel in hydroxyethyl piperazine ethanesulfonic acid (HEPES) buffer. Followed by blocking the remaining active NHS groups with ethanolamine, and washing off the unbound lectin Con A with HEPES buffer that contained a high salt concentration to reduce non-specific binding, the synthesis of SG Con A was completed, with 99.8 mg Con A bound to per gram of silica gel measured by the BCA method. Afterwards, SG Con A was packed into a column to enrich glycoproteins or glycopeptides.
O stretching vibration of amides, which demonstrates that the carboxylic groups have been activated by the EDC/NHS reagent.9 Subsequently, lectin Con A was immobilized onto EDC/NHS activated silica gel in hydroxyethyl piperazine ethanesulfonic acid (HEPES) buffer. Followed by blocking the remaining active NHS groups with ethanolamine, and washing off the unbound lectin Con A with HEPES buffer that contained a high salt concentration to reduce non-specific binding, the synthesis of SG Con A was completed, with 99.8 mg Con A bound to per gram of silica gel measured by the BCA method. Afterwards, SG Con A was packed into a column to enrich glycoproteins or glycopeptides.
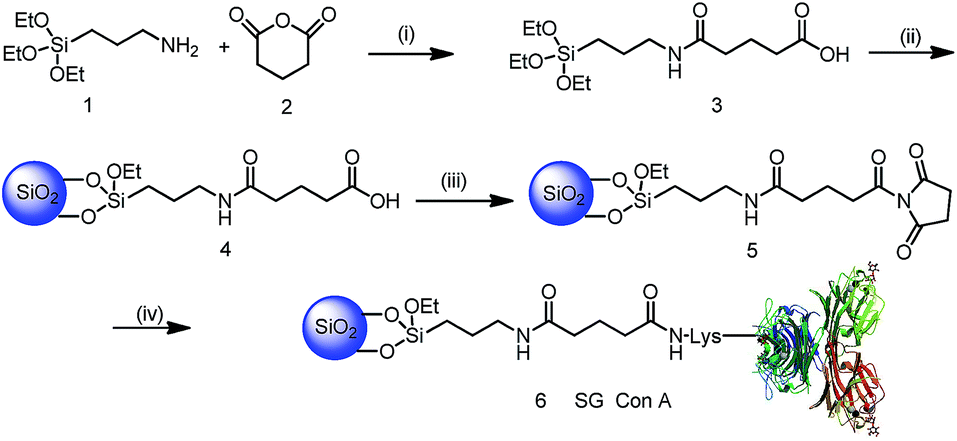 | ||
| Scheme 1 Synthesis of SG Con A, conditions: (i) tetrahedron furan, rt; (ii) silica particles, toluene, 90 °C reflux; (iii) EDC/NHS, 20 mM HEPES buffer (pH 7.2) containing 0.15 M sodium chloride, 1 mM calcium chloride, 1 mM manganese chloride, 1 mM magnesium chloride, rt; (iv) Con A, 0.1M methyl α-D-mannopyranoside in HEPES buffer (pH 7.2), rt. rt means room temperature. | ||
For the investigation of the selectivity and efficiency of SG Con A, the enrichment of glycoproteins and glycopeptides was performed in high performance liquid affinity chromatography (HPLAC) mode and SPE mode, respectively. It is known that concanavalin A (molecular mass of 104–112 kDa) is a lectin with relatively broad specificity that mainly targets the mannose core of N-glycans,10 and also binds hybrid-type and bi-antennary complex-type to a lesser extent but does not bind more highly branched complex-type N-glycans.11a Herein, non-glycosylated protein human serum albumin (HSA, molecular mass of 66.5 kDa), high-mannose-type glycoprotein bovine pancreatic ribonuclease B (RNase B, molecular mass of 14.9 kDa) and chicken ovalbumin (molecular mass of 45 kDa) with high-mannose and hybrid-type glycans were selected as model proteins to verify the specificity of SG Con A. As shown in Fig. 1a, HSA was washed out from the matrix during the void time indicating that non-glycosylated proteins cannot be retained on SG Con A, whereas RNase B attached with high-mannose glycans showed strong affinity with the material and could only be eluted from the column with the competitive solvent containing 0.2 M methyl α-D-mannopyranoside (Fig. 1b). For ovalbumin, as shown in Fig. 1c, it displayed two chromatographic peaks after SG Con A separation. The first peak emerged in the void time and the other emerged after the competitive solvent has been applied, which indicates the different binding affinity of the two peaks separated by SG Con A. In order to profile the glycan compositions of the two peaks, we further fractionated the isolated proteins by injecting 50 μL of a 10 mg mL−1 solution of ovalbumin (Fig. 1d), which fell in the maximum sample loading amount of 63.4 mg mL−1 measured by frontal affinity chromatography.11b Then, the obtained fractions were dialysed with the recovery ratio of 88%, and deglycosylated with PNGase F prior to MS analysis. Under positive ion mode conditions, sixteen high-mannose and hybrid type of the released glycans, together with some of their sodium adducts, were identified from the second fraction (Fig. 2), which was possibly responsible for the higher affinity of the glycoproteins in this fraction. In contrast, the majority of the ovalbumin in the first fraction were non-glycosylated (data not shown).
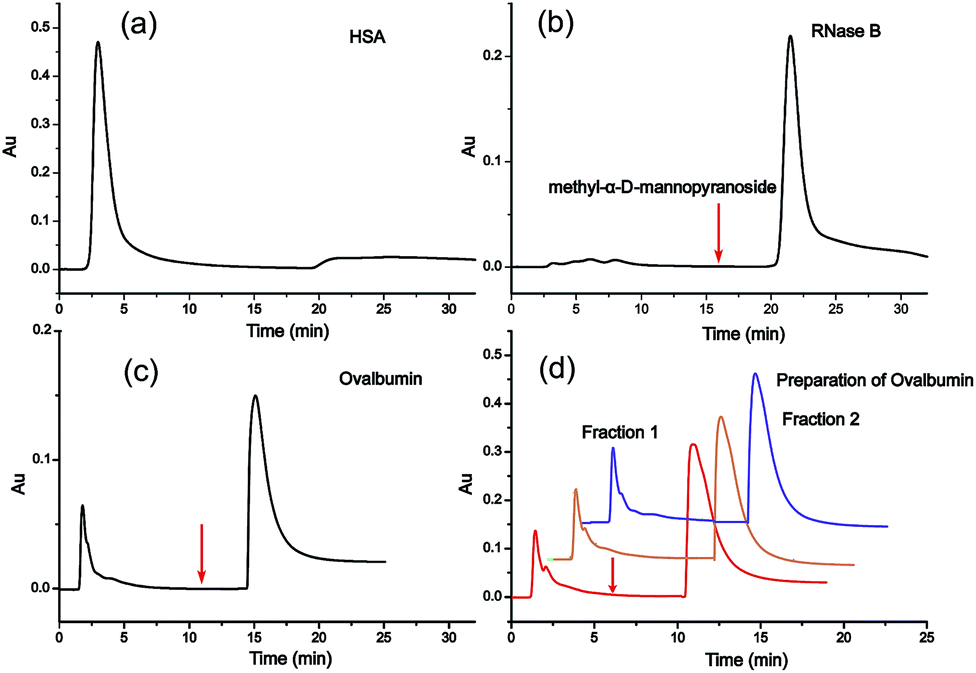 | ||
| Fig. 1 (a) The separation of HSA; (b) the separation of RNase B; (c) the separation of ovalbumin; (d) the preparation of ovalbumin. (Note that the preparation was performed one year after the column was packed, which indicates the reproducibility and stability of the column.) | ||
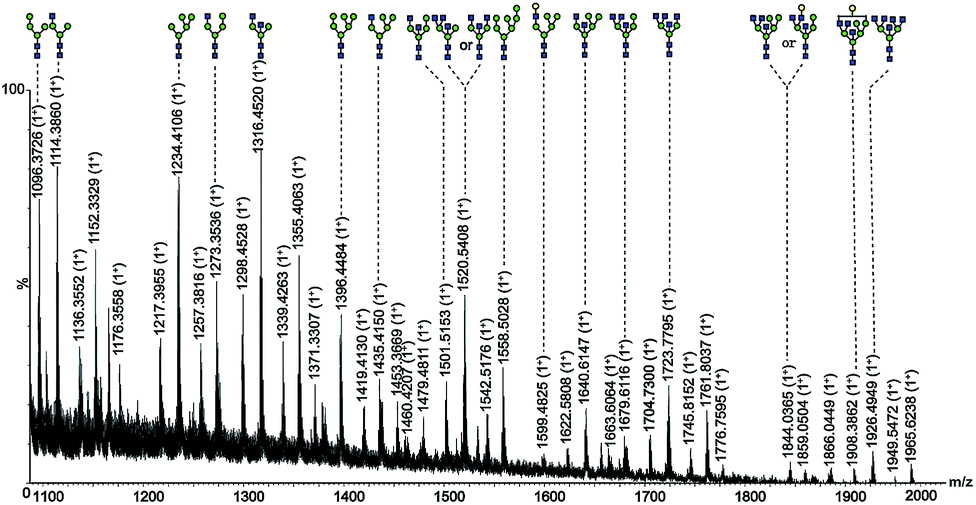 | ||
Fig. 2 Nano-ESI-MS spectrum of ovalbumin glycans in fraction 2 deglycosylated with PNGase F.  mannose; mannose;  N-acetylglucosamine; N-acetylglucosamine;  galactose. galactose. | ||
For further identification of the glycan structures, the ions at m/z 1257.3833 (M + Na)+ and 1745.8517 (M + Na)+ were selected for MS2 analysis as shown in Fig. 3. The observation of ions at m/z 1257.3, m/z 1036.3, m/z 874.3, m/z 712.2, and m/z 671.2 confirmed the presence of mannose residues at the non-reducing end.12 And the retention characteristics of the model proteins on SG Con A proved the specific enrichment ability of the material as well as the effectiveness of the synthetic method.
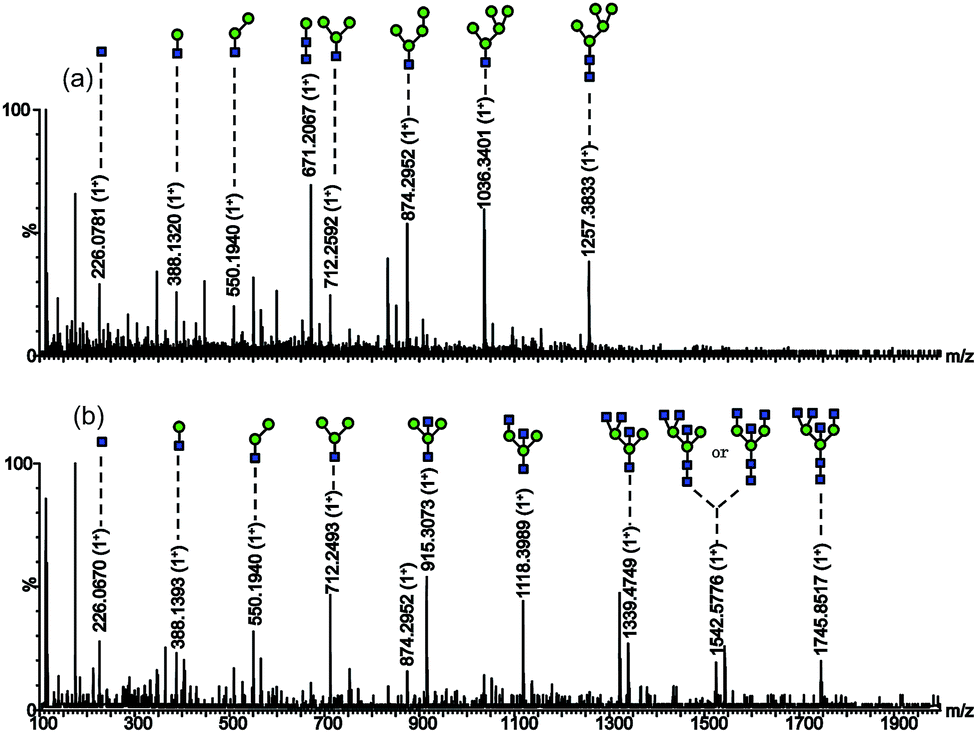 | ||
Fig. 3 Nano-ESI-MS/MS spectrum of the glycans with (a) m/z 1257.3833 (M + Na)+; (b) m/z 1745.8517 (M + Na)+.  mannose; mannose;  N-acetylglucosamine. N-acetylglucosamine. | ||
In addition, the selectivity of SG Con A for the enrichment of N-linked glycopeptides from RNase B was also evaluated in comparison to that of agarose Con A commercially available. As shown in Fig. 4a, only 2 glycopeptides were detected with peptides when no enrichment procedure was performed.13 After the digests were treated with SG Con A and agarose Con A, respectively, all high mannose-type glycans at the only glycosylated site of RNase B could be detected as shown in Fig. 4b and c. The great difference between the two affinity matrices was that a few of the glycopeptides could be found during the washing step for agarose Con A (Fig. S3†), while no sample loss was found during SG Con A enrichment (Fig. S4†). The enrichment results demonstrated SG Con A as a selective and efficient material for trapping of N-linked glycopeptides.
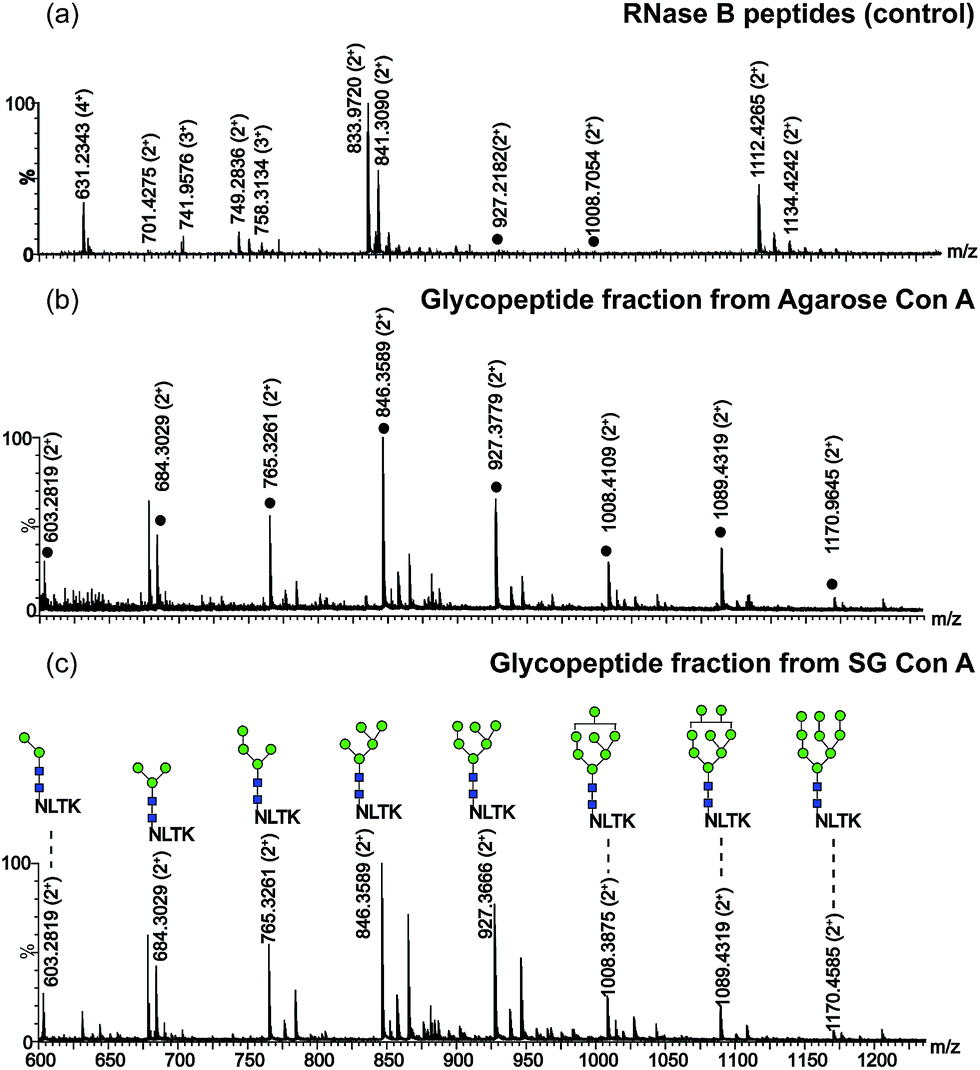 | ||
Fig. 4 Nano-ESI-MS spectrum of RNase B tryptic digests treated with different materials in SPE mode: (a) peptide fraction desalted with C18 material (control). The peptides were eluted from the column with the solutions of ACN–H2O–FA (60/40/0.1 (v/v)) after loading and rinsing steps; (b) glycopeptide fraction enriched with agarose Con A. Glycopeptide fraction was eluted from the column with 0.2 M methyl α-D-mannopyranoside in 10 mM HEPES buffer after the non-glycosylated peptides were removed by using the HEPES buffer; (c) glycopeptide fraction enriched with SG Con A. The enrichment condition was identical to that of agarose Con A enrichment.  mannose; mannose;  N-acetylglucosamine. N-acetylglucosamine. | ||
To design lectin affinity material targeting for specific glycoconjugate enrichment was rather difficult due to the intrinsic low affinity of carbohydrates, as well as the reduction of lectin activity during immobilization.14 To avoid the drastic loss of lectin's bioactivity during immobilization, we introduced a mild bonding method based on EDC/NHS activation. The attractive aspect of the method lies in the introduction of a hydrophilic spacer arm, which can not only bridge the lectin and the activated silica particle to form SG Con A, but also considerably reduce the non-specific adsorption when enriching glycoproteins or glycopeptides. In comparison to other traditional methods for the covalent bonding of lectins or proteins,8a less synthetic steps were required in our route. Meanwhile, no more chemical treatment of the resulting material was needed after concanavalin A was linked, which reduces the influence on the configuration and bioactivity of the immobilized lectins. Another advantage of the proposed synthesis approach consists in that it is applicable not limited to Con A but other lectins or affinity ligands, and the use of a SG Con A column a year later for chromatographic preparation of ovalbumin showed superior reproducibility and stability of the material. When compared to that of agarose Con A, the advantageous feature of the prepared SG Con A exists in that there is no sample loss during the enrichment of glycopeptides, which indicates that it is a potentially applicable matrix for quantitative analysis in glycoproteomic research.
Conclusions
In summary, we introduced a simple and robust method for rapid preparation of a silica-bonded Con A material named SG Con A, which has been utilized successfully for selective and efficient enrichment of N-linked glycoproteins and glycopeptides. High binding capacity, excellent affinity selectivity and low sample loss make SG Con A a promising material for application in affinity chromatography enrichment.Acknowledgements
We gratefully acknowledge the financial support from the National High Technology Research and Development Program of China (863 Program, 2012AA020203) and the National Natural Science Foundation of China (Grant no. 21135005 and no. 21205116).Notes and references
- (a) R. Qiu and F. E. Regnier, Anal. Chem., 2005, 77, 2802–2809 CrossRef CAS PubMed; (b) J. R. Yates, C. I. Ruse and A. Nakorchevsky, Annu. Rev. Biomed. Eng., 2009, 11, 49–79 CrossRef CAS PubMed.
- (a) W. I. Weis and K. Drickamer, Annu. Rev. Biochem., 1996, 65, 441–473 CrossRef CAS PubMed; (b) C. Zuber and J. Roth, The Sugar Code. Fundamentals of Glycosciences, 2009, pp. 87–110 Search PubMed.
- (a) X. Fang and W.-W. Zhang, J. Proteomics, 2008, 71, 284–303 CrossRef CAS PubMed; (b) M. Kullolli, W. S. Hancock and M. Hincapie, Anal. Chem., 2009, 82, 115–120 CrossRef PubMed; (c) Y. Mechref, M. Madera and M. V. Novotny, Lectins: Analytical Technologies, 2007, pp. 267–298 Search PubMed.
- (a) T. Plavina, E. Wakshull, W. S. Hancock and M. Hincapie, J. Proteome Res., 2007, 6, 662–671 CrossRef CAS PubMed; (b) W.-C. Lee and K. H. Lee, Anal. Biochem., 2004, 324, 1–10 CrossRef CAS PubMed.
- (a) P. Gemeiner, M. Polakovič, D. Mislovičová and V. Štefuca, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 715, 245–271 CrossRef CAS; (b) G. Palmisano, S. E. Lendal, K. Engholm-Keller, R. Leth-Larsen, B. L. Parker and M. R. Larsen, Nat. Protoc., 2010, 5, 1974–1982 CrossRef CAS PubMed.
- (a) D. S. Hage and J. Cazes, Handbook of affinity chromatography, CRC Press, 2005 Search PubMed; (b) M. Urh, D. Simpson and K. Zhao, Methods Enzymol., 2009, 463, 417–438 CAS; (c) J. Tang, Y. Liu, P. Yin, G. P. Yao, G. Q. Yan, C. H. Deng and X. M. Zhang, Proteomics, 2010, 10, 2000–2014 CrossRef CAS PubMed.
- (a) M. Madera, Y. Mechref and M. V. Novotny, Anal. Chem., 2005, 77, 4081–4090 CrossRef CAS PubMed; (b) A. F. Bergold and P. W. Carr, Anal. Chem., 1989, 61, 1117–1128 CrossRef CAS.
- (a) A. Monzo, G. K. Bonn and A. Guttman, TrAC, Trends Anal. Chem., 2007, 26, 423–432 CrossRef CAS PubMed; (b) P.-O. Larsson, Methods Enzymol., 1984, 104, 212–223 CAS.
- (a) A.-F. Che, Z.-M. Liu, X.-J. Huang, Z.-G. Wang and Z.-K. Xu, Biomacromolecules, 2008, 9, 3397–3403 CrossRef CAS PubMed; (b) S. Velasco-Lozano, F. López Gallego, R. Vázquez-Duhalt, J. C. Mateos-Díaz, J. M. Guisán and E. Favela-Torres, Biomacromolecules, 2014, 15, 1896–1903 CrossRef CAS PubMed.
- H.-J. Gabius, The sugar code: fundamentals of glycosciences, John Wiley & Sons, 2011 Search PubMed.
- (a) D. N. Moothoo and J. H. Naismith, Glycobiology, 1998, 8, 173–181 CrossRef CAS PubMed; (b) Y. Ohyama, K.-i. Kasai, H. Nomoto and Y. Inoue, J. Biol. Chem., 1985, 260, 6882–6887 CAS.
- D. Harvey, D. Wing, B. Küster and I. Wilson, J. Am. Soc. Mass Spectrom., 2000, 11, 564–571 CrossRef CAS.
- L. Yu, X. Li, Z. Guo, X. Zhang and X. Liang, Chem.–Eur. J., 2009, 15, 12618–12626 CrossRef CAS PubMed.
- (a) M. N. Gupta, Methods for affinity-based separations of enzymes and proteins, Springer, 2002 Search PubMed; (b) A. Verdoliva, F. Pannone, M. Rossi, S. Catello and V. Manfredi, J. Immunol. Methods, 2002, 271, 77–88 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ay02286f |
| This journal is © The Royal Society of Chemistry 2015 |