
Microwave-assisted synthesis of resveratrol imprinted polymers with enhanced selectivity†
Lachlan J.
Schwarz
,
Mahesh K.
Potdar
,
Basil
Danylec
,
Reinhard I.
Boysen
and
Milton T. W.
Hearn
*
Department of Chemistry and Centre for Green Chemistry, Monash University, Melbourne, Victoria 3800, Australia. E-mail: milton.hearn@monash.edu
First published on 27th November 2014
Abstract
The microwave-mediated synthesis of resveratrol molecularly imprinted polymers is reported. The binding performance of these functional polymers has been evaluated and compared to the corresponding thermally-initiated polymers. The advantages of microwave-synthesis include a 20-fold decrease in polymerisation time, similar binding capacities and enhanced molecular selectivity for resveratrol. These binding data indicate microwave-mediated polymerisation provides superior binding site organization and access.
Introduction
Molecularly imprinted polymers (MIPs) are synthetic, highly cross-linked porous materials housing defined recognition sites that are complimentary to their molecular template. Their synthesis relies on the formation of a self-assembled or covalently assembled template–monomer complex. The three-dimensional arrangement, distances between interacting groups and spatial organization of the multiple binding site cavities, are subsequently fixed in space by polymerisation with an excess of cross-linking monomer in the presence of a suitable porogen. Removal of the molecular template reveals polymer-embedded binding sites which are complimentary both in shape and functionality, thereby affording selective rebinding of the molecular template or closely related structural analogues.MIPs are typically prepared via thermal- or photo-polymer-isation techniques. Thermal polymerisation methods inevitably result in binding site heterogeneity due to gains in conformational entropy. With photo-irradiation methods conducted at low temperatures, greater stabilization of the template–monomer complex can occur, leading to an improvement in binding site homogeneity. However, control of the non-uniform heat transfer and light penetration during photo-polymerisation can be difficult without access to expensive specialised hardware e.g. specially designed photo-reactors with semi-conductor (‘cold’) light sources such as light-emitting diodes.1 Microwave (MW) heating could – in principle – provide a superior synthetic platform for MIP preparations due to its ability to achieve uniform and rapid heating throughout a reaction mixture, leading to faster reaction rates and substantially reduced polymerisation times. Despite these obvious advantages, interest in microwave-assisted MIP syntheses has only recently begun to emerge,2–4 with the full potential of this alternative synthetic approach yet to be established.
We have previously reported the application of thermally-initiated (Th) imprinted polymers for the solid phase extraction of resveratrol, a valuable naturally-occurring bioactive polyphenol, and several structurally related compounds from complex wine and food waste matrices.5,6 In this investigation, we report the application of a microwave-mediated polymerisation for the improved synthesis of resveratrol imprinted polymers. Their binding properties have been compared to the corresponding thermally-initiated resveratrol imprinted polymers through detailed analyses of resveratrol static binding isotherms and competitive binding assays with several relevant closely-related structural analogues of resveratrol.
Materials and methods
Microwave MIP synthesis
Microwave MIPs were prepared using the following procedure: (E)-resveratrol (2 mmol, 456 mg) was dissolved in the porogen, CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH (5:1 v/v, 10 mL), with the functional monomer 4-vinylpyridine (4VP) (6 mmol, 642 μL), in a 20 mL microwave vial. The solution was sparged with Ar(g) for 1 min and sonicated for 20 min prior to standing on ice for 10 min. Ethylene glycol dimethacrylate (EGDMA) (30 mmol, 5.662 mL) and 2,2′-azobis(2-methylpropionitrile) (AIBN) (0.62 mmol, 105 mg) were added as cross-linker and initiator, respectively, after which the solution was sparged for 1 min with Ar(g) prior to sealing with a crimp cap. The pre-polymerisation solution was then irradiated using a Biotage Initiator MW at 60 °C for 63 min with microwave absorbance level set to high. The resulting polymer monolith was then rinsed with acetone and dried in vacuo at 60 °C. A non-imprinted reference polymer (NIP) was prepared in the same manner without the template (resveratrol) present. The dried polymers were then crushed and ground in a Retsch PM 200 Ball Mill (5 min, 300 rpm, 1 min intervals), sedimented in acetone to remove fines and collected using a 20–30 μm glass sintered funnel. The resveratrol template was extracted from the polymer matrix via a series of acidic washes with EtOH:AcOH (9:1, v/v) and continued until resveratrol could no longer be detected by RP-HPLC with UV-Vis detection at 321 nm. Polymers were then rinsed with EtOH and acetone until AcOH was no longer detectable and dried in vacuo at 40 °C.
:EtOH (5:1 v/v, 10 mL), with the functional monomer 4-vinylpyridine (4VP) (6 mmol, 642 μL), in a 20 mL microwave vial. The solution was sparged with Ar(g) for 1 min and sonicated for 20 min prior to standing on ice for 10 min. Ethylene glycol dimethacrylate (EGDMA) (30 mmol, 5.662 mL) and 2,2′-azobis(2-methylpropionitrile) (AIBN) (0.62 mmol, 105 mg) were added as cross-linker and initiator, respectively, after which the solution was sparged for 1 min with Ar(g) prior to sealing with a crimp cap. The pre-polymerisation solution was then irradiated using a Biotage Initiator MW at 60 °C for 63 min with microwave absorbance level set to high. The resulting polymer monolith was then rinsed with acetone and dried in vacuo at 60 °C. A non-imprinted reference polymer (NIP) was prepared in the same manner without the template (resveratrol) present. The dried polymers were then crushed and ground in a Retsch PM 200 Ball Mill (5 min, 300 rpm, 1 min intervals), sedimented in acetone to remove fines and collected using a 20–30 μm glass sintered funnel. The resveratrol template was extracted from the polymer matrix via a series of acidic washes with EtOH:AcOH (9:1, v/v) and continued until resveratrol could no longer be detected by RP-HPLC with UV-Vis detection at 321 nm. Polymers were then rinsed with EtOH and acetone until AcOH was no longer detectable and dried in vacuo at 40 °C.
Surface area, porosity and SEM measurements
Nitrogen physisorption at 77 K was measured on a Micromeritics Tristar II 3020 instrument. Samples (0.1 g) were degassed at 80 °C to a vacuum of 0.05 bar using a Micromeritics VacPrep 061 degas station prior to analysis. BET surface areas were calculated at relative pressures between 0.05 and 0.35 bar. Scanning electron microscopic images were also obtained as described in the ESI.†Polymer swelling studies
Swelling studies were performed by packing the dry polymer (0.2 g) into a graduated glass syringe (1 mL). Excess solvent (acetonitrile) was then added and the syringe tapped until no further settling was observed. The volume measurements were made at 20 °C at 0, 3, 6 and 24 h. The swelling ratio was derived according to an established method and given as volume of swollen polymer/volume of dry polymer.7MIP binding evaluation: static binding isotherm
Single analyte static batch binding studies were conducted on 20 mg of MW-MIP, MW-NIP, Th-MIP and Th-NIP materials in acetonitrile solutions (1 mL) containing (E)-resveratrol at concentrations ranging from 0–4 mM. The binding solutions were incubated for a period of 2 h and then centrifuged at 13000 rpm (16200×g) for 15 min to pellet the polymer. Aliquots (200 μL) of the supernatants were removed for RP-HPLC analysis from which the concentration of unbound (E)-resveratrol was determined using a linear 5-point calibration curve. Subtraction of this value from the initial total analyte concentration gave the amount of analyte bound [B], expressed as μmol g−1 polymer.
Competitive static cross reactivity binding experiments were conducted as described above with the exception that the rebinding solution contained a mixture of the analytes resveratrol, catechin, quercetin, piceid, rosmarinic acid and apigenin each at 0.05 mM in acetonitrile (1 mL). Samples were treated as described above prior to RP-HPLC analysis.
RP-HPLC analysis conditions
RP-HPLC analysis was performed using an Agilent Technologies HPLC 1100 series (Agilent Technologies, Waldbronn, Germany) equipped with ChemStation software, a degasser, a binary gradient pump, an auto-sampler with a 900 μL sample loop and an UV-DAD. Separations were performed with a Zorbax Eclipse XDB C18 (4.6 mm × 150 mm, 5 μm particle size) (Agilent Technologies, Melbourne, Australia) double end-capped column operated at ambient temperature. The UV-DAD was set to 200–600 nm at a spectral acquisition rate of 2 nm scans per step with simultaneous monitoring at 280, 321, 340 and 370 nm for all compounds. The mobile phase composition was a mixture of H2O (0.1% formic acid) (mobile phase A) and 70:30 CH3CN:H2O (0.1% formic acid) (mobile phase B) and was delivered with the following gradient elution profile: 28.5% B, 0–1 min; 28.5–100% B, 1–10 min, 100% B 10–15 min; 28.5% B 15.01 min; 2 min post time at a flow rate of 0.5 mL min−1 and injection volume of 5 μL to the RP-HPLC column. The mobile phases were filtered through a polypropylene membrane filter (0.2 μm pore size, 47 mm diameter) purchased from Pall Corp. (Melbourne, Australia) and was degassed for 20 min in an ultrasonic bath (Elmasonic, Singen, Germany) prior to use.
Results and discussion
From initial observations, it was immediately apparent that MW irradiation significantly increased the rate of polymerisation, with an approximate 20-fold decrease in the time required for the formation of the bulk MIP polymer monoliths. The corresponding thermal polymerisation took 24 h compared to just 63 minutes by the MW approach.8Nitrogen sorption isotherms (Fig. 1) were obtained for each MIP and NIP material, from which Brunauer–Emmett–Teller (BET) surface area and porosity data were derived (Table 1) in order to compare physical characteristics. The isotherms display similar trajectories for both MW- and Th-MIP preparations, with the latter demonstrating superior N2 sorption capacity. A similar trend was observed for the non-imprinted counterparts (see ESI†Fig. 1(A)). Barrett–Joyner–Halenda (BJH) pore size distribution plots demonstrated little difference between polymer materials, regardless of preparation type, and in each case revealed a narrow distribution of pores with diameters in the range of 24–28 Å (see ESI†Fig. 1(B)). A clear difference in the BET surface area was observed based on polymer preparation method, with those prepared via thermal means resulting in approximately 25% greater surface areas than their MW counterparts (Table 1). When considering the total pore volume MW-MIP, MW-NIP and Th-NIP all exhibited similar volumes while Th-MIP demonstrated up to 23% greater pore volume (Table 1).
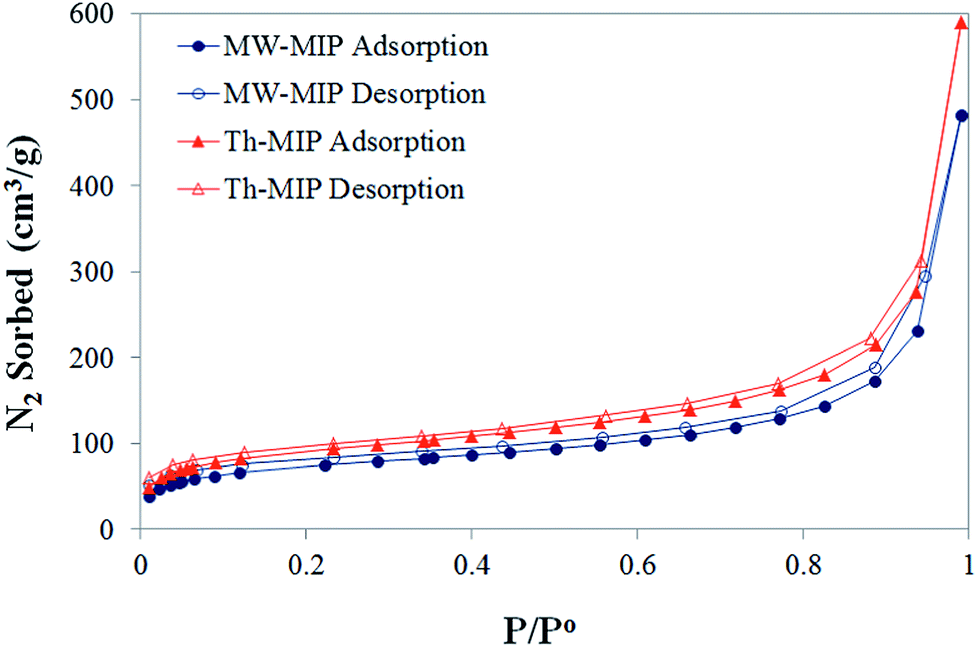 | ||
| Fig. 1 Nitrogen physisorption isotherms for the MW- and Th-MIP materials conducted at 77 K. | ||
| Polymer | BET surface area (m2 g−1) | BJH total pore volume (cm3 g−1) |
|---|---|---|
| MW-MIP | 237.9073 | 0.744687 |
| Th-MIP | 297.4240 | 0.912279 |
| MW-NIP | 241.3138 | 0.791013 |
| Th-NIP | 306.8645 | 0.740610 |
Initial studies into the binding characteristics of microwave-mediated (MW-MIP) and thermally-initiated (Th-MIP) MIPs involved single analyte static batch binding assays from which the adsorption capacity, total number of binding sites (N) and binding site dissociation constants (KD) were determined. Briefly, triplicate samples of the MW-MIP and non-imprinted reference polymer (MW-NIP) were incubated in CH3CN containing resveratrol at concentrations ranging from 0.01 mM to 4 mM for 2 h, centrifuged and the supernatant analysed by RP-HPLC. The amount of bound resveratrol ([B]) was determined by subtracting the supernatant concentration from the initial resveratrol concentration and plotted versus the resveratrol concentration to give the static binding isotherms. The static binding isotherm for the Th-MIPs was found to be similar to results obtained with five different batches of these polymers as reported elsewhere.8 Comparison of the relative static binding isotherms confirmed that the MW-MIPs and Th-MIPs had similar resveratrol binding capacities (Fig. 2(a)).
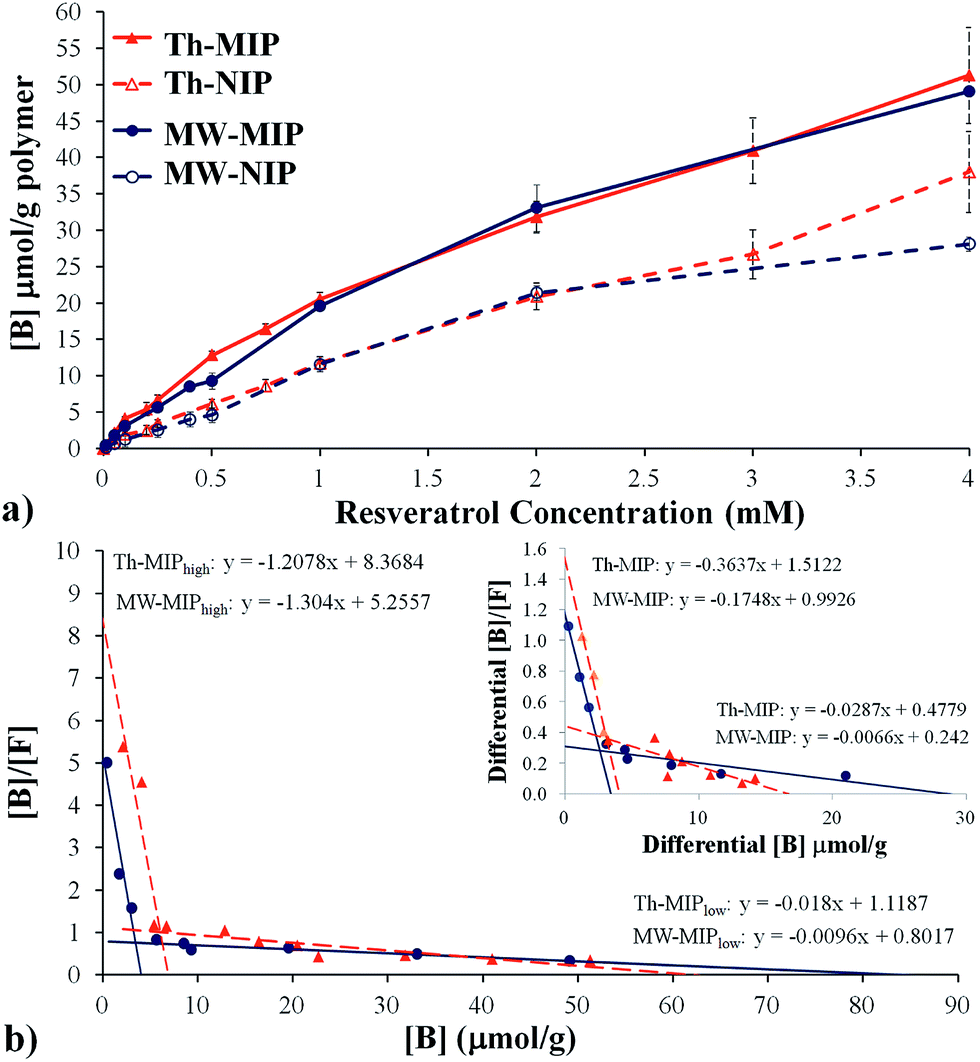 | ||
| Fig. 2 Resveratrol binding data from (a) static batch binding isotherm assays with bound [B] vs. resveratrol concentration for the imprinted (MIP, solid lines) and the non-imprinted (NIP, dashed lines) polymers; and (b) associated bimodal Scatchard analyses of bound [B]/free [F] vs. [B] resveratrol for both MW (●) and thermal (▲) MIP preparations. The inset shows the Scatchard plot as a function of differential binding (MIP-NIP). All analyses were performed in triplicate with polymer samples (20 mg) with different concentrations of resveratrol in acetonitrile (1 mL). | ||
A more detailed analysis of the isothermal binding data was undertaken to investigate whether the respective polymerisation processes had resulted in differing cavity distributions and affinities despite the apparent similarity in resveratrol binding capacities. This was achieved through detailed examination of the derived Scatchard plots, e.g. from plots of [B]/[F] vs. [B], where [B] and [F] are the concentrations of the bound and free analyte respectively, (Fig. 2(b)). These data were subsequently fitted by regression analysis to two distinct linear regions as described by the equation [B]/[F] = (−1/KD)N + N/KD, where KD is the dissociation constant and N is the total number of binding sites, and indicated the presence of two distinct binding site classes, i.e. the MIPs are composed of both high and low affinity binding sites. The determined KD and N values of these two classes of binding sites are summarised in Table 2.
| Binding site class | K D M−1 | K D(low)/KD(high) | N μmol g−1 | N (low)/N(high) |
|---|---|---|---|---|
| a Refers to differential binding ([B]MIP − [B]NIP) data. | ||||
| MWhigh | 0.77 | 135.29 | 4.03 | 20.63 |
| MWlow | 104.17 | 83.15 | ||
| Thhigh | 0.83 | 67.10 | 6.93 | 8.97 |
| Thlow | 55.56 | 62.15 | ||
| MWhigha | 2.91 | 31.82 | 3.40 | 8.40 |
| MWlowa | 92.59 | 28.57 | ||
| Thhigha | 2.66 | 13.10 | 4.08 | 4.08 |
| Thlowa | 34.84 | 16.65 | ||
Scatchard analyses revealed large differences in KD and N values for the high and low affinity binding site classes between the MW-MIP and Th-MIP materials. Examination of the ratios of these descriptors for the low and high affinity sites moreover indicate that the observed binding was primarily due to interactions via low affinity binding sites, which were proportionally 20.63 and 8.97 times more prevalent for the MW-MIPs than the Th-MIPs, respectively. Despite the lower incidence of high affinity binding sites for the MW-MIP, their lower KD and ratio of affinity constants, e.g. KD(low)/KD(high), potentially indicate higher selectivity, which would allow greater discrimination of resveratrol from analogues compared to their thermally produced counterparts.4 This outcome can be attributed to MW-mediated effects that result in better dipole alignment within the non-covalent template-monomer assemblies and diminished exposure time for thermally induced destabilisation prior to their covalent ‘positional fixing’ via polymerisation.9 As such, these MW irradiation effects are manifested as significantly reduced polymerisation time, absence of ‘wall effects’,10 and rapid, more homogenous heating throughout the entire polar MW reaction medium.
To examine the impact of non-selective interactions of the analyte and polymer on the KD and N descriptor values, the binding differential ([B]MIP − [B]NIP) was examined. Shea et al. have shown that some Th-MIPs bimodal Scatchard plots can be reduced to a single linear representation if the Scatchard analysis was performed on the binding differential between MIP and NIP.11 These workers concluded that the low affinity binding sites observed in the original bimodal plot were solely due to non-selective interactions. With the MIP systems reported herein, the bimodal character persisted when Scatchard analysis was based on differential binding (Fig. 2(b) inset), indicating that analyte binding to the low affinity sites cannot be solely attributed to non-selective binding.
Interestingly, the N value for the MW-MIP changed only marginally (from 4.03 to 3.40 μmoles g−1), suggesting minimal influence from non-selective or non-template derived binding site interactions, while the Th-MIP showed an approximate 40% reduction (from 6.93 to 4.08 μmoles g−1). This finding suggests that a proportion of the high affinity binding sites in the Th-MIP are randomly generated without influence from the template. This result is consistent with the proposal put forward by Turner et al. that the increased capacity of thermally derived MIPs may in part be due to their larger surface area arising from the equilibrium driven formation of more micro-/meso-porous structures, which in turn leads to a greater number of binding sites with high and low affinity.4 If so, then it is conceivable that this increase in porosity may be accompanied by the formation of numerous non-template derived cavities which should also arise in the non-imprinted polymer. This outcome would account for the large reduction in N that was observed for Th-MIP (40%) based upon consideration of the differences in the binding differential.
The molecular selectivity (α) of each type of MIP was then established from investigations conducted under competitive batch binding conditions with several resveratrol structural analogues. The same amount of MIP and NIP materials were incubated in triplicate for 2 h at constant temperature (22 °C) in a 0.05 mM mixed solution of resveratrol, catechin, quercetin, piceid, rosmarinic acid and apigenin in acetonitrile, the structures of which are shown in Fig. 3. The amount of analyte bound [B] to the respective polymers was determined from the difference between the initial and final concentrations of the supernatant as determined by RP-HPLC. From these data the selectivity (α) was derived using the equation α = ([B]MIP − [B]NIP)/[B]NIP, for each of the competing analytes (Fig. 4). A substantial increase in selectivity towards resveratrol was observed for the MW-MIP under these competitive binding conditions compared to that of the Th-MIP.
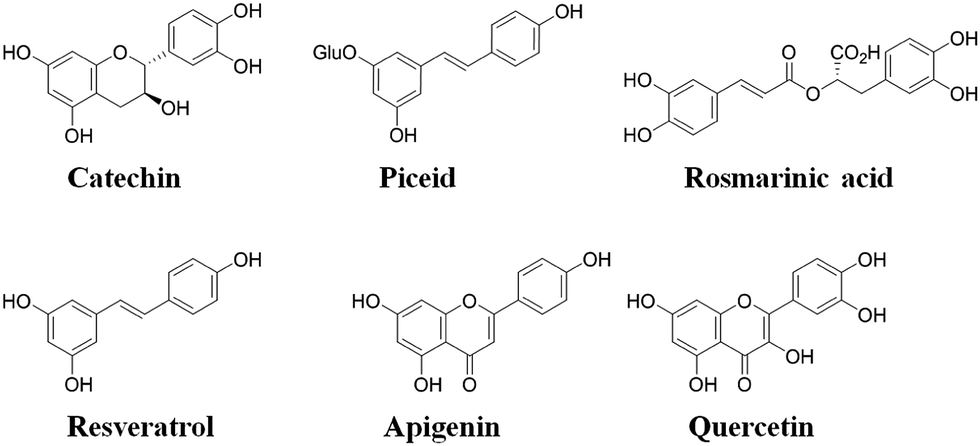 | ||
| Fig. 3 Analytes employed in competitive binding assays. | ||
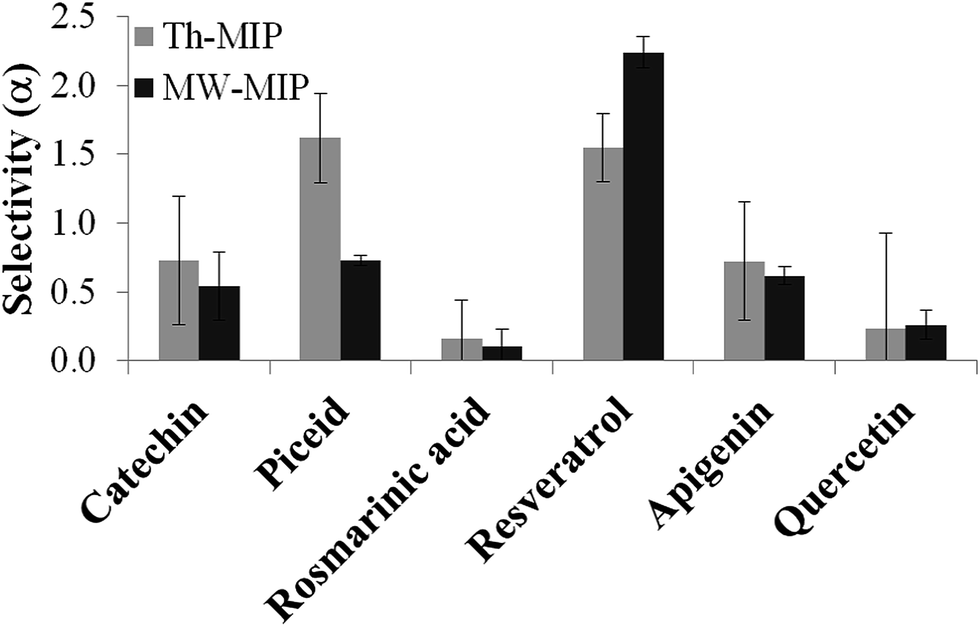 | ||
| Fig. 4 The selectivity α, of both MW-MIP and Th-MIP preparations is displayed after incubation with a mixture of six similarly structured polyphenols, each at a concentration of 0.05 mM in acetonitrile. | ||
The enhanced selectivity displayed by the MW-MIP may be attributed to the greater organisational clustering of the high affinity binding sites and lower non-selective binding, than that observed for the Th-MIP counterpart. This property is highlighted by the relatively small change in N value for MW-MIP as evident from the analysis of differential binding descriptors (Table 2). It was demonstrated that this efficient MW methodology led to a higher degree of cross-linking, indicated by the swelling ratio (volume of wet polymer/volume of dry polymer) of 1.20 for the MW-MIP compared to 1.44 for the Th-MIP, resulting from a more complete polymerisation compared to that of the thermal method. This outcome explains the increased selectivity and reduced binding capacity of the MW-MIP compared to its Th-MIP counterpart.12
An interesting observation regarding the Th-MIP was the large variation in the uptake of the competing analytes when the reproducibility of several batches of the Th-MIP polymer was examined, demonstrating batch-to-batch variability in binding site fidelity and selectivity. Notably, this variability had only a relatively minor impact upon the uptake of resveratrol when examined under non-competitive conditions. In comparison, microwave-assisted synthesis, which allows greater control over the heat transfer, is likely to minimise this batch-to-batch variability. Since the binding site formation relies on the self-assembly of the template and functional monomer species, the rapid MW-mediated polymerisation can be anticipated to mitigate or minimize this variability, resulting in more reproducible preparations from batch to batch. Moreover, MW effects arising from dielectric heating mechanisms, wherein dipoles are constantly aligning with the oscillating electric field, may also directly influence the imprinting procedure. For example, Mata et al. have demonstrated that the application of an electric field with a vector opposite in direction to that of the dipole moment of a hydrogen bonded dimer can strengthen the covalent character of the interaction due to mutually increased polarisation and thereby stabilisation of hydrogen-bonded complexes.13 In the context of molecular imprinting, this behaviour would be manifested as stabilisation of the pre-polymerisation complexes associated with the specific high and low affinity binding sites, i.e. those containing multiple hydrogen-bonding interactions, and being sufficiently stable, they behave as a single molecular construct during the dipole realignment, thereby retaining the morphological integrity required for the construction of the specific binding sites.
Conclusions
Resveratrol imprinted polymers have been prepared using microwave-mediated irradiation and compared with their thermally prepared counterparts. Despite both preparation types exhibiting similar binding characteristics with regard to resveratrol affinity and capacity, subtle differences were observed when a more detailed study of the binding descriptors was undertaken, especially those associated with the binding differential. These analyses revealed the propensity for microwave-mediated irradiation to confer a different organisation of clusters of high affinity binding sites which result in significantly enhanced selectivity towards the resveratrol template in comparison to several competitive binding and closely related analogues.These results indicate that microwave-mediated irradiation with rapid and more homogenous heating can be successfully employed to more preferentially ‘lock’ the pre-polymerisation complexes preventing their destabilisation due to thermal equilibrium processes. Moreover, these results provide a rationale for the earlier divergent reports about the adsorption capacities associated with MW-MIPs and their thermally prepared counterparts.2,4 It is anticipated that these procedures will enable greater control over batch-to-batch reproducibility compared to that currently observed with thermally produced MIP systems thus enabling many different classes of MIPs with improved selectivity to be prepared. Accordingly, additional studies to examine these effects, optimise the microwave polymerisation procedures further and fully characterise and compare the MW-MIP and Th-MIP physical properties, are underway and will be reported in due course.
Acknowledgements
Authors would like to acknowledge the assistance of Dr Christian Vogt with the nitrogen physisorption measurements and the funding support of the Australian Research Council.Notes and references
- S. Landgraf, Spectrochim. Acta, Part A, 2001, 57, 2029–2048 CrossRef CAS.
- H. Zhu, Y. Wang, Y. Yuan and H. Zeng, Anal. Methods, 2011, 3, 348–355 RSC.
- Z. Zhang, W. Tan, Y. Hu, G. Li and S. Zan, Analyst, 2012, 137, 968–977 RSC.
- N. W. Turner, C. I. Holdsworth, S. W. Donne, A. McCluskey and M. C. Bowyer, New J. Chem., 2010, 34, 686–692 RSC.
- S. N. N. S. Hashim, L. J. Schwarz, R. I. Boysen, Y. Yang, B. Danylec and M. T. W. Hearn, J. Chromatogr. A, 2013, 1313, 284–290 CrossRef CAS PubMed.
- L. J. Schwarz, B. Danylec, Y. Yang, S. J. Harris, R. I. Boysen and M. T. W. Hearn, J. Agric. Food Chem., 2011, 59, 3539–3543 CrossRef CAS PubMed.
- S. A. Piletsky, A. Guerreiro, E. V. Piletska, I. Chianella, K. Karim and A. P. F. Turner, Macromolecules, 2004, 37, 5018–5022 CrossRef CAS.
- L. J. Schwarz, B. Danylec, S. J. Harris, R. I. Boysen and M. T. W. Hearn, J. Chromatogr. A, 2011, 1218, 2189–2195 CrossRef CAS PubMed.
- C. O. Kappe and A. Stadler, in Microwaves in Organic and Medicinal Chemistry, Wiley-VCH Verlag, 2005, vol. 25, ch. 2, pp. 9–28 Search PubMed.
- Wall effects refer to the uneven heating of a bulk liquid due to a hot reactor surface. In microwave heating this is not observed as the reactor surface does not absorb microwave energy while the bulk liquid does, thereby ensuring a more even heating process.
- K. J. Shea, D. A. Spivak and B. Sellergren, J. Am. Chem. Soc., 1993, 115, 3368–3369 CrossRef CAS.
- A. Rosengren, B. Karlsson and I. Nicholls, Int. J. Mol. Sci., 2013, 14, 1207–1217 CrossRef CAS PubMed.
- I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Chem. Phys., 2009, 130, 044104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Nitrogen sorption isotherms for NIP materials and BJH pore size distribution plots; RP-HPLC chromatographs of polyphenol mixtures after incubation with either MIP or NIP materials. See DOI: 10.1039/c4ay02518k |
| This journal is © The Royal Society of Chemistry 2015 |