
mRNA delivery using non-viral PCL nanoparticles†
Ilaria E.
Palamà
*a,
Barbara
Cortese
a,
Stefania
D'Amone
a and
Giuseppe
Gigli
abc
aNational Nanotechnology Laboratory, Institute Nanoscience CNR (NNL, CNR-NANO), via Arnesano, Lecce, Italy. E-mail: ilariaelena.palama@nano.cnr.it
bDept. Matematica e Fisica ‘Ennio De Giorgi’, University of Salento, via Monteroni, Lecce, Italy
cItalian Institute of Technology (IIT) – Center for Biomolecular Nanotechnologies, via Barsanti, Arnesano, Italy
First published on 17th September 2014
Abstract
Messenger RNA (mRNA) provides a promising alternative to plasmid DNA as a genetic material for delivery in non-viral gene therapy strategies. However, it is difficult to introduce mRNA in vivo mainly because of the instability of mRNA under physiological conditions. Here, mRNA-protamine complex encapsulated poly(ε-caprolactone) (PCL) nanoparticles (NPs) are proposed for the intracellular delivery of mRNA molecules. The nanoparticles with a size of about 247 nm in diameter have a core–shell structure with an mRNA-containing inner core surrounded by PCL layers, providing high stability and stealth properties to the nanoparticles. The partial neutralization of the negatively charged mRNA molecules with the cationic protamine allows one to modulate the release kinetics in a pH-dependent manner. At pH 7.4, mimicking the conditions found in the systemic circulation, only 25% of the mRNA is released after 48 hours post incubation, whereas at pH 5.0, recreating the cell endosomal environment, about 60% of the mRNA molecules are released within the same time window post incubation. These NPs show no cytotoxicity to NIH 3T3 fibroblasts, HeLa cells and MG63 osteoblasts up to 8 days of incubation. Given the stability, preferential release behavior, and well-known biocompatibility properties of PCL nanostructures, our non-viral PCL nanoparticles are a promising system that simultaneously resolved the two major problems of mRNA introduction and the instability, opening the door to various new therapeutic strategies using mRNA.
Introduction
Failure of clinical trials of non-viral vector-mediated gene therapy arises primarily from either an insufficient transgene expression level or immunostimulation concerns caused by the genetic information carrier. Neither of these issues could be addressed through engineering of sophisticated gene delivery vehicles. As a rapidly emerging class of nucleic acid therapeutics, there are key benefits in using mRNA that is frequently applied as a gene delivery molecule in the fields of inherited genetic disorders, cancer immunotherapy and stem cell-based biomedical research as an alternative to plasmid DNA (pDNA).1–3 As a direct source of gene products, mRNA has several advantages: first, mRNA contains no viral promoters (e.g. CMV) and bacterial sequences that can cause toxicity. Second, mRNA does not integrate into the host genome, which may lead to deleterious mutation.4 Third, gene expression via mRNA is relatively transient and therefore safer to use compared with DNA. Last but not least, as mRNA does not need to cross the nuclear envelop, it increases the chances of successfully transfecting quiescent cells. Indeed, mRNA can mediate a higher level of protein expression in vivo compared with DNA over shorter durations.5 Moreover, the cytoplasmic delivery of mRNA circumvented the nuclear envelope, which resulted in a higher gene expression level. A variety of nanoparticles as non-viral systems have been investigated for the effective in vivo delivery of genes. These include lipid-based and polymer-based nanoparticles where the polynucleotides are encapsulated within the hydrophilic core or adsorbed on the cationic surface. In polymeric complexes, the negatively charged acid nucleic molecules could be easily attached by surface-absorption on cationic polymers and surfactants6–9 or entrapped in the NP hydrophilic core prepared by double emulsion solvent evaporation methods.10,11 An attractive polymer for mRNA application is poly(ε-caprolactone) (PCL), approved by the Food and Drug Administration (FDA). Nanoparticles composed of PCL are promising for their high colloidal stability in a biological fluid, facile cellular uptake by endocytosis, low toxicity in vitro and in vivo, and controlled release of their cargo.12Therefore, we propose a new non-viral nanosystem for delivery of mRNA as an alternative to plasmid DNA in gene therapy. In this work, as a proof of concept, PCL nanoparticles have been developed as a non-viral vector for the delivery of GFP-mRNA. The PCL NPs, obtained by the emulsion–diffusion–evaporation method, comprise a hydrophilic core with the active agent, a PCL shell, and a PVA coating to increase the colloidal stability. Prior to particle assembly, GFP-mRNA RNA molecules are reacted with protamine (PRM), a polycation widely used in drug13,14 and gene delivery, to form an almost neutral mRNA-protamine complex. This leads to higher loading, better stability, and controlled release of the mRNA over time. In particular, protamine is an arginine-rich peptide with strong basic charge and it is a FDA approved nontoxic cationic peptide for use in humans as a heparin antagonist and as a long-acting delivery system for insulin. Protamine is also an excellent DNA condenser for in vitro cationic lipid-mediated gene transfer.15
The physicochemical properties of the NPs are characterized using various characterization techniques. The loading efficiency and release kinetics are analysed under different physiological conditions. The expression efficiency of mRNA is studied in vitro on transfected NIH 3T3 fibroblasts, HeLa cells and MG63 osteoblasts.
Experimental
Materials and methods
All tissue culture media and serums were purchased from Sigma-Aldrich, and cell lines were purchased from the American Tissue Type Collection (ATTC). The following chemicals were supplied by Sigma-Aldrich: thiazolyl blue tetrazolium bromide (MTT), phosphate buffered saline (PBS), Fluoroshield with DAPI, protamine sulphate, poly(ε-caprolactone) (PCL) with an average molecular weight (MW) of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 Da, and polyvinyl alcohol (PVA, MW 13–23 kDa, 87–80% hydrolyzed). Lipofectamine 2000, OptiMem®, MitoTracker, ERtracker, LysoTracker, pVAX1-lacZ plasmid, an mMessage mMachine® T7 Ultra kit and an Ambion MEGAclear™ kit were from Life Technology.
800 Da, and polyvinyl alcohol (PVA, MW 13–23 kDa, 87–80% hydrolyzed). Lipofectamine 2000, OptiMem®, MitoTracker, ERtracker, LysoTracker, pVAX1-lacZ plasmid, an mMessage mMachine® T7 Ultra kit and an Ambion MEGAclear™ kit were from Life Technology.
Plasmid DNA preparation and GFP-mRNA production
The pVAX-GFP (3697 bp) plasmid was obtained by modification of the commercial plasmid pVAX1-lacZ (6050 bp, Life Technology) by replacement of the β-galactosidase reporter gene by the enhanced Green Fluorescent Protein (eGFP, referred to as GFP hereafter) gene. The details of the construction are described elsewhere.16 The plasmid contains the human cytomegalovirus (CMV) immediate-early promoter, a ColE1 type origin of replication and the kanamycin resistance gene for bacterial selection. Plasmid DNA (pDNA) was obtained by growing E. coli cultures (harbouring pVAX-GFP) overnight, in 2 L shake-flasks containing 250 mL LB medium and antibiotics (30 μg mL−1 of kanamycin). Then the plasmids were purified as described by means of EndoFree plasmid purification. The purified plasmid was diluted using Tris-EDTA (TE) buffer solution and stored at −20 °C. The integrity of the plasmid was confirmed by agarose gel electrophoresis. The purity and concentration of the plasmid were determined by UV absorbance at 260 nm.After linearizing the plasmid used as a template, mRNA was synthesized with an mMessage mMachine® T7 Ultra kit from Ambion (Life Technology) in accordance with the manufacturer's instructions. mRNA was purified using the Ambion MEGAclear™ kit from Ambion (Life Technology). The integrity of mRNA was confirmed by agarose gel electrophoresis. The purity and concentration of mRNA were determined by UV absorbance at 260/280 nm, respectively.
PCL NPs and GFP-mRNA/PRM PCL NPs preparation
PCL nanoparticles were prepared by an emulsion–diffusion–evaporation method. Briefly, GFP-mRNA was reconstituted in RNase-free water and was complexed with a protamine sulphate solution by incubating at room temperature for 30 minutes on a rotary shaker. GFP-mRNA (20 μg mL−1) was combined at various ratios, in particular 1:1, 1:3, 1:5, with the protamine sulphate (1 mg mL−1) previously dissolved in 0.1 M NaCl. A 100 mg PCL was dissolved in 10 mL organic phase consisting of 9 mL ethyl acetate and 1 mL acetone for 1 hour under mild heating at 30 °C. As for the aqueous phase, 100 mg PVA were stirred in 5 mL water for 2 hours at room temperature until a clear solution was obtained; then, GFP-mRNA/PRM complex solution was mixed. The organic phase was passed through a 0.22 μm syringe filter to remove any undissolved solids and subsequently added dropwise to the aqueous phase under constant stirring. The resulting microemulsion was kept under constant agitation on a magnetic stirrer at 1000 rpm for 1 hour and was subsequently sonicated for 30 minutes. This colloidal preparation was diluted to a volume of 50 mL by adding water dropwise under stirring conditions (1000 rpm, magnetic stirrer), which resulted in nanoprecipitation. In order to remove the organic solvent and to harden the PCL nanoparticles, the suspension was treated with a rotary evaporator at 50 mbar at 40 °C for 20 minutes. Next, the nanoparticle suspension was washed three times with RNase-free water by centrifugation at 12000 rpm for 10 minutes and then resuspended in water. The finished nanoparticle suspension was stored at 4 °C for further use. To determine the concentration of nanoparticles (weight per volume), this suspension was centrifuged at 12000 rpm for 10 minutes; the supernatant was removed and the pellet was allowed to dry under a nitrogen stream before weighing. For preparing fluorescent PRM-FITC PCL nanoparticles, 1 mg mL−1 PRM-FITC was added to the PCL solution and the formulation was carried out as described earlier. The labeled nanoparticles were stored in the dark at 4 °C until use.
Nanoparticles size, surface charge studies and stability
The average particle size and zeta potential of the prepared nanoparticles were determined by photon correlation spectroscopy using a Zetasizer Nano ZS90 (Malvern Instruments Ltd, USA) equipped with a 4.0 mW He–Ne laser operating at 633 nm and an avalanche photodiode detector. Measurements were made at 25 °C in aqueous solutions (pH 7). The PCL NPs solution (1 mg mL−1) was passed through a 0.45 μm pore size filter before measurements and appropriately diluted if necessary according to the instrument's requirements. The stability of mRNA/PRM PCL NPs was tested under physiological conditions. Briefly, nanoparticles were incubated in complete DMEM medium at 37 °C, and the size variation was measured over a period of 8 days by Dynamic Light Scattering (DLS) analysis. Representative measurements of three distinct sets of data have been reported (Student t-test, P < 0.05).Scanning electron microscopy (SEM) and atomic force microscopy (AFM)
For SEM analysis (RHAIT 150), samples were prepared by applying a drop of the particle suspension to a SiO2 wafer and then dried overnight. Prior to SEM observations, the samples were sputter-coated with a 10 nm gold layer to make them electronically conductive and to avoid electronic charging during SEM imaging. The morphological characterization has been performed by tapping mode AFM using a Solver PRO Scanning Probe Microscope (NT-MDT) in air at room temperature; we used TESPA (Veeco, USA) silicon cantilevers of 20–80 N m−1 spring constant and a resonance frequency of around 300 kHz. A drop of sample suspension was applied to a silicon support and then dried overnight.Encapsulation efficiency and in vitro mRNA release study
The encapsulation efficiency of the nanoparticles was determined by analysing the supernatant of the final emulsion once the nanoparticles were removed from it by centrifugation at 12 000 rpm for 10 minutes. For the estimation of mRNA present in the supernatant, absorbance was measured on a spectrophotometer at 260 nm. The amount of mRNA encapsulated and the percent encapsulation in the nanoparticles are given by the following equations:| mRNA encapsulated = total mRNA − filtrate mRNA |
| % Encapsulation = (Encapsulated mRNA)/(Total mRNA) × 100 |
The release of mRNA from PCL nanoparticles was measured in TE buffer. Briefly, a known amount of lyophilized GFP mRNA/PRM PCL nanoparticles (50 mg) was dispersed in 5 mL of phosphate buffer 1× (PBS) pH 7.4 and 500 μl of the sample was dialyzed in a large volume of PBS 1× pH 7.4 and pH 5.0, and maintained at 37 °C ± 0.5 °C under stirring at 50 rpm. At specified time intervals, the samples were centrifuged (12000 rpm for 10 minutes) and supernatants were collected. Samples were taken and analysed in triplicates. The concentration of GFP mRNA was determined from the corresponding absorbance measured on a spectrophotometer at 260 nm. Representative measurements of three distinct sets of data have been reported (Student t-test, P < 0.05).
Gel retardation assay
The electrophoretic mobility of the prepared complexes and nanoparticles with different weight ratios of PRM/GFP-mRNA (ratios 1:1, 1:3 and 1:5) was determined using 1% agarose gel in TBE 1× (Tris/borate/EDTA) buffer with 0.5 μg mL−1 of ethidium bromide at 110 V for 30 min. The gel was visualized under a UV transilluminator.
Cell culture
Human osteosarcoma cells (MG63), human cervix carcinoma cells (HeLa) and mouse embryonic fibroblast cells (NIH 3T3) were maintained in DMEM supplemented with 10% FBS, 100 U mL−1 penicillin, 100 mg mL−1 streptomycin, 5% L-glutamine and 5% sodium pyruvate in a humidified incubator at 37 °C, 5% CO2, and 95% relative humidity. Upon reaching confluence, cells were passaged using 0.25% trypsin-EDTA. Cells at passages 3–10 were used in the following experimental assays.Cytotoxicity assay
Cytotoxicity viability was evaluated by the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay against mouse embryonic fibroblast cells (NIH 3T3), human cervix carcinoma cells (HeLa) and human osteosarcoma cells (MG63) incubated with PCL nanoparticles (1 mg mL−1). Cells were seeded on 24-well plates at a density of 105 cells per well in complete culture media. After 24 h, the media without FBS were used to replace the usual ones, and PCL nanoparticles were resuspended in complete medium and were added to each well. Untreated samples were used as the control groups. After an appropriate incubation period, the cultures were removed from the incubator and the MTT solution was added in an amount equal to 10% of the culture volume. Then the cultures were returned to the incubator and incubated for 3 hours. After the incubation period, the cultures were removed from the incubator and the resulting MTT formazan crystals were dissolved with acidified isopropanol solution to an equal culture volume. The plates were read within 1 hour after adding acidified isopropanol solution. The absorbance was spectrophotometrically measured at a wavelength of 570 nm and the background absorbance measured at 690 nm was subtracted. The percentage viability is expressed as the relative growth rate (RGR) by the equation:| RGR = (Dsample/Dcontrol) × 100% |
Intracellular localization study
To determine the cellular uptake of the PRM-FITC PCL NPs, we seeded 105 cells mL−1 in a sterile glass-culture slide. The cells were incubated with the PRM-FITC PCL NP dispersions at a concentration of 0.05 mg mL−1. After 3 hours of incubation at 37 °C, the culture medium was removed, and the cells were washed three times with phosphate buffered saline. For fluorescent microscopic observation, cells were fixed in situ for 5 minutes in 3.7% formaldehyde and mounted with Fluoroshield with DAPI. To study the intracellular localization of PRM-FITC PCL NPs, immunostaining with LysoTracker Red (Life Technology), MitoTracker Red (Life Technology) and ER-Tracker Red (Life Technology) were performed in accordance with the manufacturer's instructions, to label lysosomes, mitochondria and endoplasmic reticulum, respectively. Confocal micrographs were taken with a Leica confocal scanning system mounted into a Leica TCS SP5 (Leica Microsystem GmbH, Mannheim, Germany), equipped with a 63× oil immersion objective and a spatial resolution of approximately 200 nm in x–y and 100 nm in z. The 3D confocal scanning was performed by reconstructing the photoluminescence coming from different focalized slices with a sequential image acquisition. The optical sections were collected in the transverse x–z and y–z planes.GFP mRNA/PRM PCL nanoparticles transfection and GFP detection
For in vitro transfection study, the mouse embryonic fibroblast cells (NIH 3T3), human cervix carcinoma cells (HeLa) and human osteosarcoma cells (MG63) were split one day prior to transfection and plated at a density of 105 cells. Before transfection, the culture medium was replaced with OptiMem. The cells were transfected with mRNA/PRM PCL NPs containing 1 μg of GFP mRNA at 37 °C for 4 hours. Then the GFP mRNA/PRM PCL NPs were removed and the cells were incubated in fresh DMEM with 10% FBS at 37 °C, 5% CO2, and 95% relative humidity for another 24 hours. GFP mRNA and Lipofectamine 2000 (Life technology) complexes were used as the positive control of mRNA efficacy according to the manufacturer's instructions. For fluorescent microscopic observations, cells were fixed in situ for 5 minutes in 3.7% formaldehyde and mounted with Fluoroshield with DAPI. Confocal micrographs were taken with a Leica confocal scanning system mounted into a Leica TCS SP5 (Leica Microsystem GmbH, Mannheim, Germany), equipped with a 63× oil immersion objective and a spatial resolution of approximately 200 nm in x–y and 100 nm in z.For quantitative measurement of transfection with GFP mRNA/PRM PCL NPs or Lipofectamine 2000, the cells were lysed using 50 mL 0.1% Triton X-100 in 0.2 M NaOH. Cell-associated GFP fluorescence was quantified by analysing the cell lysates in a fluorescence plate reader (λ excitation 488 nm, λ emission 508 nm). The quantity of GFP was expressed as the percentage of the fluorescence associated with cells vs. that present in the non-transfected cells. Representative measurements of three distinct sets of data have been reported (Student t-test, P < 0.05).
Results and discussion
Messenger RNA has a high potential to produce proteins or peptides for therapeutic purposes in a safe manner without any risk of random integration into the genome. Despite pioneering studies to transfect mRNA into cells using a non-viral method, the interest in the clinical use of mRNA has been limited for a long time. There are two major problems associated with mRNA introduction: mRNA is considered to be unstable to obtain sufficient protein expression in clinical settings17 and mRNA induces strong immune reactions through recognition by Toll-like receptors (TLRs),18,19 hampering repeated mRNA administration. Thus, the requirement of an effective mRNA non-viral delivery system to overcome the instability of mRNA should be further explored to realise in vivo mRNA administration. Although some strategies have been reported for non-viral in vivo mRNA administration, including injection of naked mRNA20 in combination with physical pressure such as electroporation or the gene gun21 and the usage of synthetic carriers based on cationic lipids and polymers,22,23 low efficiency and short duration of protein expression remain significant problems to be solved. These issues motivated us to apply a new methodology using our non-viral polymeric carrier for in vitro mRNA administration.Synthesis and characterization of PCL nanoparticles loaded with GFP mRNA
The GFP mRNA loaded PCL nanoparticles were prepared by an adapted emulsion–diffusion–solvent evaporation method as shown in Scheme 1. GFP mRNA-protamine complexes were formed in aqueous solution with the objective of enhancing the loading of the mRNA molecules; for this aim, we have performed a partial neutralization of the mRNA molecules with the positively charged protamine. Then GFP mRNA-PRM complexes were mixed with an aqueous solution of PVA and this aqueous core (water phase) was coated with the oil phase containing the PCL molecules.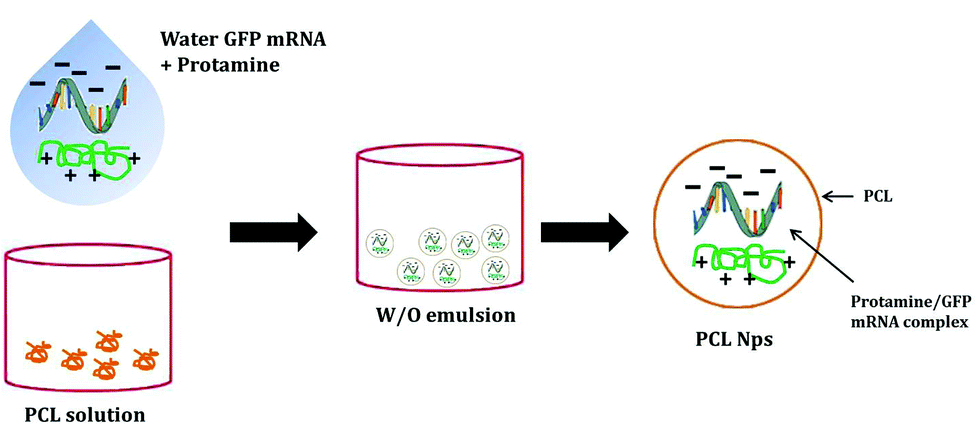 | ||
| Scheme 1 Schematic illustration of the fabrication process of PCL nanoparticles incorporating GFP-mRNA/protamine complexes. An emulsion-diffusion-evaporation method is used for the synthesis of the nanostructures. W and O represent the water phase and the oil phase, respectively. | ||
The synthesis protocol was optimized to obtain non-aggregating, spherical in shape with smooth surface NPs as shown by the AFM and SEM images (Fig. 1A and C, respectively). The nanoparticles observed by AFM and SEM have a spherical geometry with a good size distribution. The surface of these nanoparticles as observed under SEM was free from any pores or cracks and reasonably monodisperse nanoparticles form by polymers, all exhibiting a smooth surface morphology (Fig. 1C). The resulting NPs were characterized for their physicochemical properties, such as size and surface zeta potential. As shown in Fig. 1D, PCL nanoparticles had a zeta potential of −4.72 mV ± 0.402 mV and an average diameter of 247.43 nm ± 0.577 nm (Fig. 1E and B) with a polydispersion index (PdI) of 0.334 ± 0.040. The negative charge of mRNA loaded PCL NPs confirms that the mRNA-PRM complexes were engulfed in the PCL NPs hydrophilic core and were fully covered by the PCL and PVA composite shell.
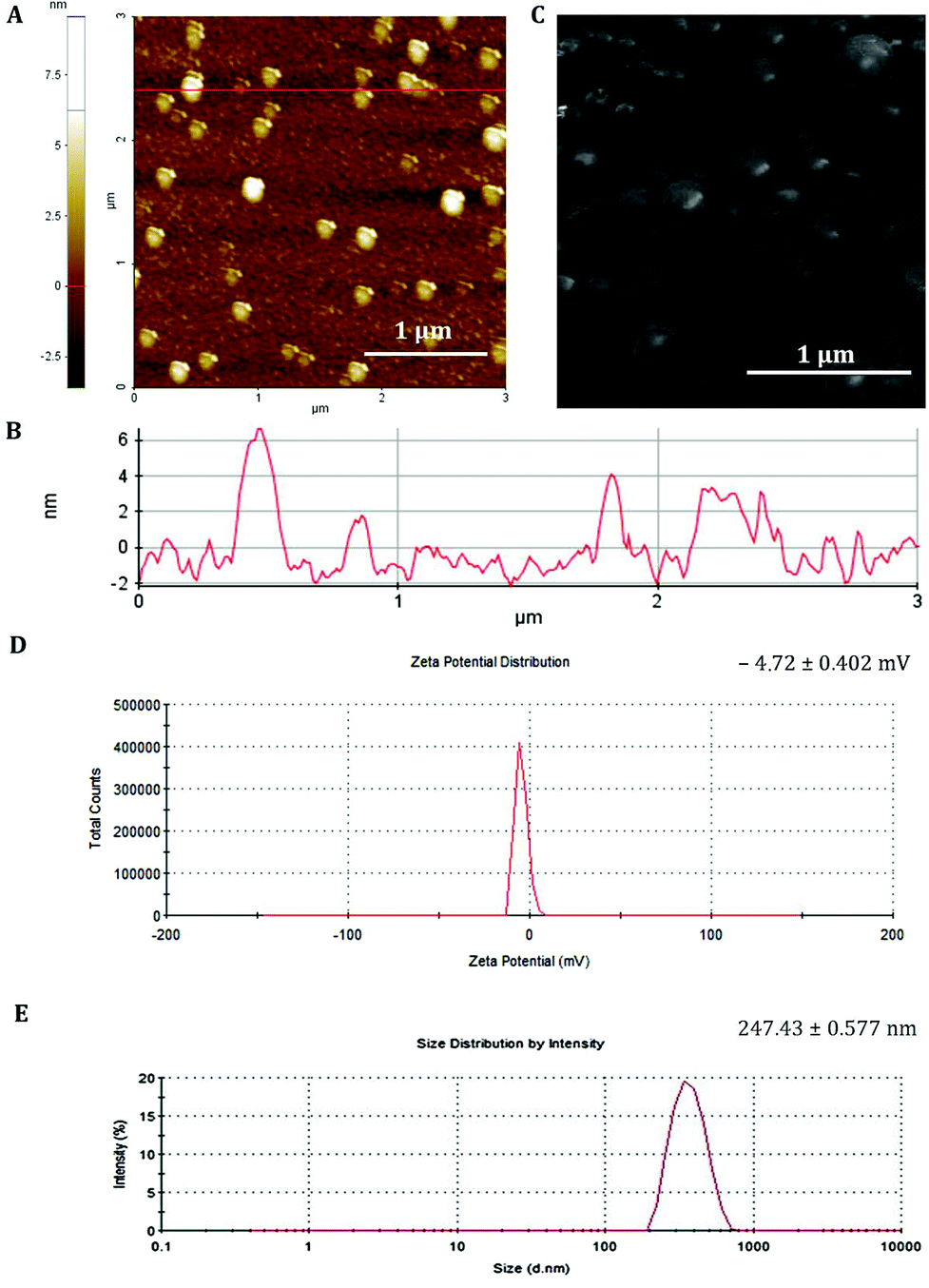 | ||
| Fig. 1 Physicochemical characterization of GFP-mRNA-loaded PCL NPs. (A) AFM image of dried PCL NPs. (B) Red line profile of the AFM image. (C) SEM image showing the size and morphology of the dried sample. Scale bars: 1 μm. (D) Surface zeta potential distribution demonstrating the uniformity of the sample population. Mean ± standard deviation is presented in the upper corner of the panel. (E) Size distribution from DLS analysis showing the mean ± standard deviation of PCL NPs. Values represent the mean ± standard deviation of four independent experiments. | ||
The stability of the mRNA loaded NPs was tested under physiological conditions. NPs were incubated in complete DMEM medium at 37 °C, and the size variation was measured over a period of 8 days by DLS analysis. As shown in Fig. 2A, over a period of 8 days, PCL NPs maintain their hydrodynamic size of about 247 nm, demonstrating no significant aggregation. In addition, no significant change in PdI was observed, supporting evidence of particle stability under physiological conditions.
 | ||
| Fig. 2 (A) Size distribution from DLS analysis showing the effect of the incubation of PCL NPs in complete DMEM medium at 37 °C on the stability of the NPs. Size distribution of the NPs was determined by DLS analysis performed each time point. (B) Zeta potential of the PRM alone, mRNA alone, mRNA-PRM complexes at different ratios (PRM:mRNA, in particular 1:1, 1:3 and 1:5), PCL nanoparticles and mRNA/PRM complexes loaded in PCL NPs. The PRM:mRNA ratio of 1:1 was chosen for NPs synthesis. Representative measurements of three distinct sets of data have been reported (Student t-test, P < 0.05). | ||
GFP mRNA/PRM loading in PCL NPs and mRNA release kinetics under different physiological conditions
The materials used in the preparation of the mRNA loaded PCL NPs were mostly negatively charged, except the positively charged protamine. The PCL and mRNA show a negative zeta potential (see Fig. 2B). Therefore to form stable NPs, the mRNA molecules were partially neutralized by forming complexes with protamine. This is a cationic polyelectrolyte, which can efficiently form stable complexes with the negatively charged mRNA. As shown in Fig. 2B, protamine and mRNA were mixed at different ratios. By increasing the PRM:mRNA ratio from 1:1 to 1:5, the zeta potential of the complex was modulated. An PRM:mRNA ratio of 1:1 was chosen for the synthesis of the mRNA loaded PCL NPs. In addition, the binding capacity of cationic protamine and subsequently of PCL NPs for mRNA was investigated by assessing the complexes’ electrophoretic mobility in an agarose gel. As shown in Fig. 3, the agarose gel electrophoresis retardation assay was performed with different weight ratios of PRM/GFP-mRNA, ratios 1:1, 1:3 and 1:5. If a mRNA-PRM complex or mRNA/PRM-PCL NPs were formed efficiently, that is, all mRNA is bound to the nanoparticles, no band for free mRNA can be observed, and the immobile complex remains in the starting zone of the gel.
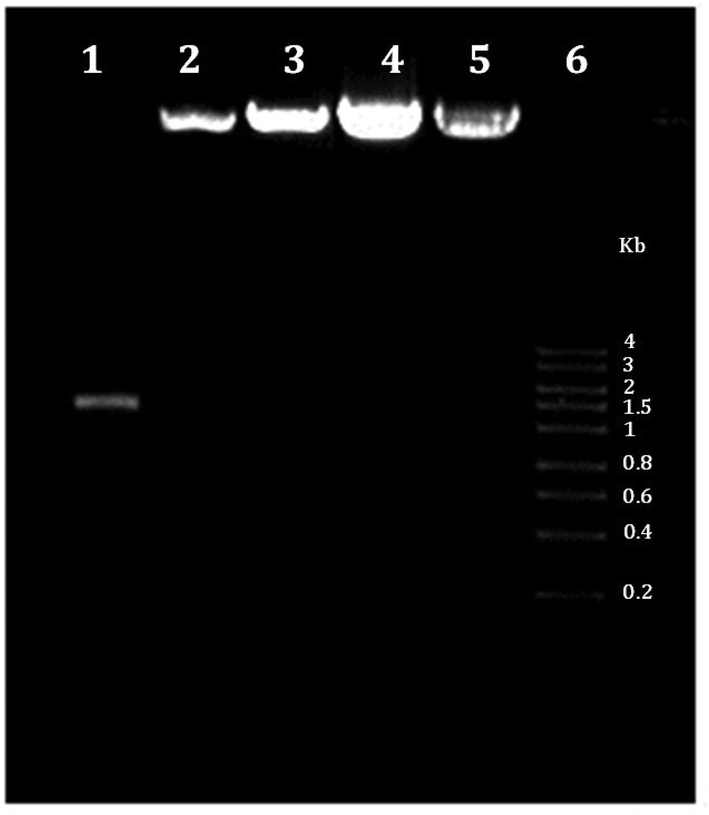 | ||
| Fig. 3 Agarose gel electrophoresis retardation assay. Lane 1: mRNA GFP free; lane 2: PRM/GFP-mRNA ratio 1:1; lane 3: PRM/GFP-mRNA ratio 1:3; lane 4: PRM/GFP-mRNA ratio 1:5; lane 5: PRM/GFP-mRNA loaded in PCL NPs; lane 6: RNA marker. | ||
The encapsulation efficiency of the PCL NPs was found to be 58.47 ± 0.65% and the release of mRNA molecules from the PCL NPs was studied over time at two different pHs (7.4 and 5.0) that were selected to mimic the physiological conditions that NPs would find in the circulation (pH 7.4) and in the acidic environment of the cell endosomes (pH 5.0), respectively. The percentage of mRNA released over time is shown in Fig. 4. For circulating PCL NPs (pH 7.4), the release rate is extremely low within the first few hours, and after 48 hours, only about 25% of the loaded mRNA is released. In contrast, within a cell endosome (pH 5.0), about 60% of the mRNA molecules are released within the same time window post incubation. These different release kinetics are indeed associated with different release mechanisms. At pH 7.4, the mRNA release is mostly governed by the diffusion of the molecules out of the NP hydrophilic core, through the pores of the PCL/PVA shell, and eventually in the surrounding environment, thus confirming that in neutral pH the mRNA release is mostly driven by passive diffusion through the pores of the polymeric shell. In contrast, the rapid release observed at pH 5.0 is most likely associated with the hydrolysis of the PCL/PVA shell under acid conditions that would progressively increase the nanoparticle porosity with consequent rapid release of its payload.
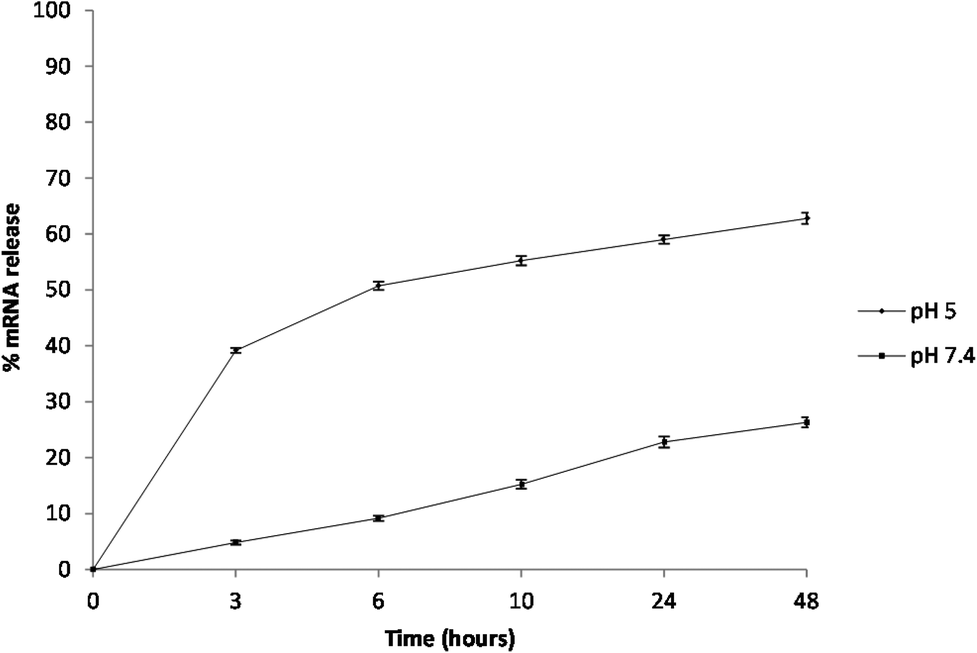 | ||
| Fig. 4 In vitro cumulative percent of release of GFP mRNA from PCL nanoparticles over time. Measurements were performed at 37 °C and pH 5.0 or pH 7.4. Representative measurements of three distinct sets of data have been reported (Student t-test, P < 0.05). | ||
Cytotoxicity of PCL nanoparticles and intracellular localization
In vitro cytotoxicity of the PCL nanoparticles was estimated by performing a MTT test on mouse embryonic fibroblast cells (NIH 3T3), human cervix carcinoma cells (HeLa) and human osteosarcoma cells (MG63). In particular, it was important to assess the cytotoxicity of the nanoparticles upon 24 hours of incubation since the cells would be in an exponential growth phase during this period and any toxicity that reflects the inhibition of proliferation and/or cell death would be clearly visible.24As shown in ESI Fig. S1,† the PCL NPs did not show any significant effect on cell viability for the time frames that we have tested.
To identify the intracellular distribution of PRM-FITC PCL nanoparticles (green channel in CLSM images of ESI Fig. S2, S3 and S4†), we performed co-localization assays on NIH 3T3 fibroblasts, HeLa cells and MG63 osteoblasts with MitoTracker, ERtracker and Lysotracker markers (red channel in CLSM images of ESI Fig. S2–S4†) for mitochondria, endoplasmic reticulum and lysosomes, respectively. As shown in ESI Fig. S2–S4† high co-localization (yellow present in the merge channel of CLSM images of ESI Fig. S2–S4†) of green fluorescence of PRM-FITC PCL nanoparticles and red fluorescence of MitoTracker marker was observed and its released mRNA were primarily transported to mitochondria. Conversely, low co-localization signals with red fluorescence of ERtracker and Lysotracker were observed, and this indicates that the mRNA is not typically localized within the endoplasmic reticulum and lysosomes. Thanks to the presence of protamine, it is possible to overcome the mRNA degradation inside the lysosomes/endosomes and more mRNA following escape from endosomes resulting in high expression efficiency. The positively charged arginine groups of protamine could bind to the acidic phospholipid groups in the membrane of the lysosomes and induced lysosomal leakage in vitro.25 The polycationic protamine-coated nanoparticles might exert sufficient buffering capacity in lysosomes to facilitate lysosomal escape,26,27 which resulted in cytoplasm release of exogenous molecules.
In vitro transfection efficiency
PCL NPs loaded with GFP mRNA/PRM were incubated with NIH 3T3 fibroblasts, HeLa cells and MG63 osteoblasts for 4 hours. After 24 hours post transfection, cells were fixed, and their nuclei were stained with DAPI. As shown in Fig. 5, lateral, front, and top views show the fluorescent GFP molecules expressed by the transfected mRNA accumulating within the cell and localizing in either the cytoplasm or perinuclear regions.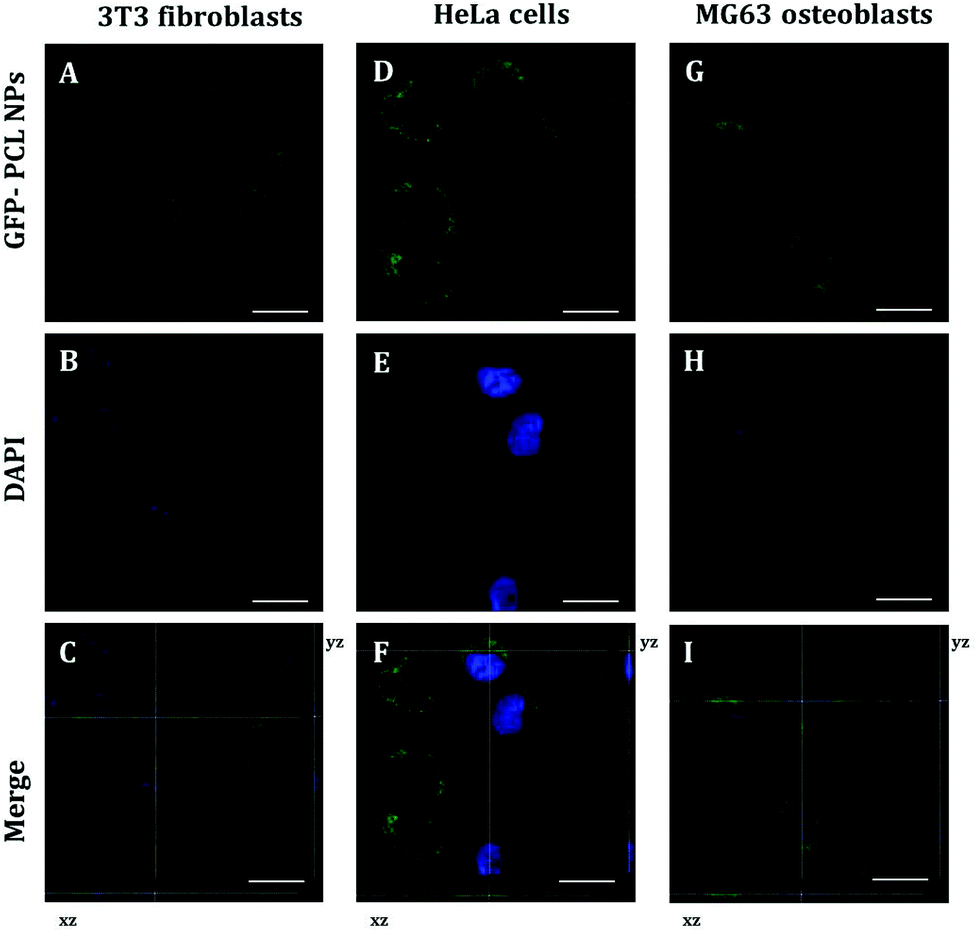 | ||
| Fig. 5 CLSM images of GFP-mRNA expression delivered by PCL NPs of the transfected NIH 3T3 fibroblasts (A, B, C), HeLa cells (D, E, F) and MG63 osteoblasts (G, H, I) after 24 hours of incubation. CLSM images A, D, G show the expression of GFP (green). Cell nuclei were counterstained with DAPI (blue) as shown in the CLSM images B, E, H. Each merge of confocal images (C, F, I) reports the corresponding Z-stack optical sections (yz and xz) obtained through photoluminescence reconstruction in the z-direction, with a z-resolution of 200 nm to confirm the spatial GFP expression inside cells. Scale bars: 25 μm. | ||
To better characterize the PCL NPs contribution to the intracellular delivery of mRNA molecules, the internalization efficiencies of different systems were compared, namely, GFP mRNA-PCL NPs and GFP mRNA-lipofectamine. In particular, Lipofectamine 2000 is a cationic liposome formulation that functions by complexing with nucleic acid molecules, allowing them to overcome the electrostatic repulsion of the cell membrane and to be taken up by the cell.28 The resulting CLSM images are shown in Fig. 6.
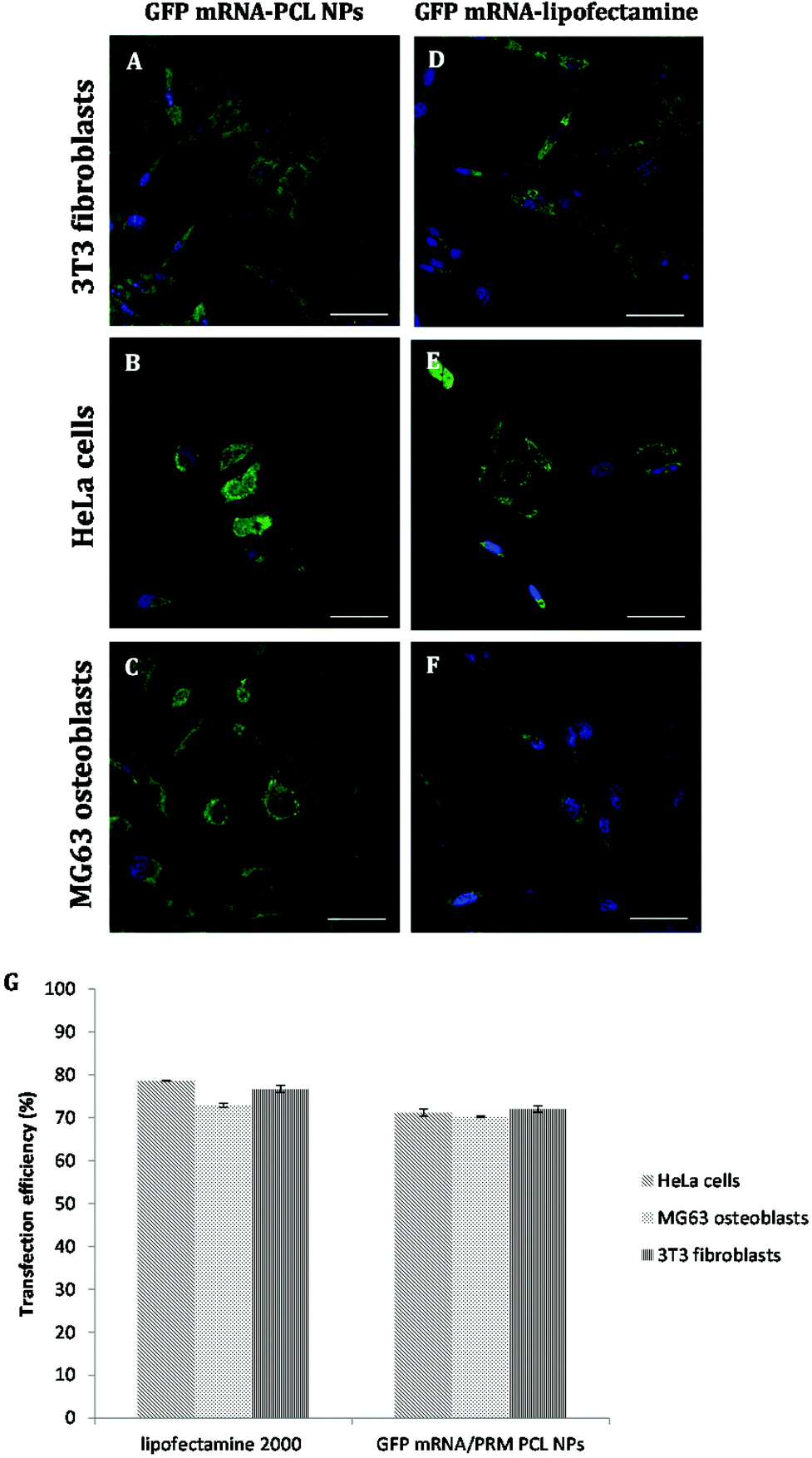 | ||
| Fig. 6 CLSM images of differential GFP-mRNA expression (green) delivered by PCL NPs (A, B, C) and Lipofectamine 2000 (C, D, E) of the transfected NIH 3T3 fibroblasts (A, D), HeLa cells (B, E) and MG63 osteoblasts (C, F) after 24 hours of incubation. Cell nuclei were counterstained with DAPI (blue). Scale bars: 50 μm. (G) Transfection efficiency (%) of mRNA-GFP delivery by PCL NPs or Lipofectamine 2000. Representative measurements of three distinct sets of data have been reported (Student t-test, P < 0.05). | ||
As expected, the commercially available system designed for in vitro cell transfection, GFP mRNA-lipofectamine, delivered the mRNA molecules with a high efficiency, as demonstrated by the amount of green fluorescence due to the expression of GFP accumulating within the cells. Notably, the GFP mRNA-PCL NPs and GFP mRNA-lipofectamine complexes presented comparable delivering efficiency, demonstrating the high transfection efficiency of our NPs, as is evident in Fig. 6G. It is evident from the CLSM images that our PCL NPs mediated transfection efficiency is substantially higher as seen from the number of cells expressing GFP as a result of translation of mRNA as compared to the transfection efficiency obtained with lipofectamine, used as the positive control. This finding confirms the functional integrity of the mRNA during its encapsulation and subsequent release from the nanoparticles.
Conclusions
Transcript replacement therapy using non-viral vectors could be a promising approach for the treatment of inherited genetic disorders for which other treatment options are limited or unavailable. Moreover, administration of mRNA may have potential in the field of regenerative medicine and for the treatment of degenerative diseases. A new method to improve the transfection efficiency of GFP mRNA, used as an example, loaded in PCL NPs as non-viral vectors is discussed in this work. Our non-viral system presents a characteristic core–shell architecture based on the assembly of mRNA molecules and the protamine complex and PCL. The PCL NPs have a strong potential to function as an effective mRNA-containing carrier with high stability and stealth properties, thereby addressing the issue of mRNA instability. In the future, the retention and release kinetics of mRNA inside the nanoparticles should be highly controlled by the molecular design of the nanoparticles. In this way, we believe that an even more prolonged expression from mRNA will be achieved using the PCL nanoparticle system to fulfil the various needs of treatments and administration routes, opening the door to various new therapeutic strategies using mRNA.Acknowledgements
This study was supported by MAAT – Nanotecnologie molecolari per la salute dell'uomo e l'ambiente (PON R&C 2007–2013, project code PON02_00563_3316357). No writing assistance was utilized in the production of this manuscript. The authors have no competing interests to disclose.References
- V. F. Van Tendeloo, P. Ponsaerts and Z. N. Berneman, Curr. Opin. Mol. Ther., 2007, 9, 423 CAS.
- R. M. Steinman and H. Hemmi, Curr. Top. Microbiol. Immunol., 2006, 311, 17 CAS.
- L. Warren, P. D. Manos, T. Ahfeldt, Y. H. Loh, H. Li and F. Lau, Cell Stem Cell, 2010, 7, 618 CrossRef CAS PubMed.
- H. Wurtele, K. C. Little and P. Chartrand, Gene Ther., 2003, 10, 1791 CrossRef CAS PubMed.
- B. Probst, B. Weide, B. J. Scheel, I. Pichler, H. G. Hoerr, S. Rammensee and S. Pascolo, Gene Ther., 2007, 14, 1175 CrossRef PubMed.
- Z. Amoozgar, J. Park, Q. Lin and Y. Yeo, Mol. Pharm., 2012, 9, 1262 CAS.
- I. S. Kim, S. K. Lee, Y. M. Park, Y. B. Lee, S. C. Shin, K. C. Lee and I. J. Oh, Int. J. Pharm., 2005, 298, 255 CrossRef CAS PubMed.
- I. E. Palamà, M. Musarò, A. M. L. Coluccia, S. D'Amone and G. Gigli, J. Drug Delivery, 2011, 1 CrossRef PubMed.
- I. E. Palamà, A. M. L. Coluccia and G. Gigli, Nanomedicine, 2013 DOI:10.2217/NNM.13.147.
- A. Alshamsan, A. Haddadi, S. Hamdy, J. Samuel, A. O. El-Kadi, H. Uludag and A. Lavasanifar, Mol. Pharm., 2010, 5, 1643 CrossRef PubMed.
- K. A. Woodrow, Y. Cu, C. J. Booth, J. K. Saucier-Sawyer, M. J. Wood and W. M. Saltzman, Nat. Mater., 2009, 8, 526 CrossRef CAS PubMed.
- M. D. Bhavsar and M. M. Amiji, AAPS PharmSciTech, 2008, 9(1), 288–294 CrossRef CAS PubMed.
- I. E. Palamà, A. M. L. Coluccia, A. Della Torre, V. Vergaro, E. Perrone, R. Cingolani, R. Rinaldi and S. Leporatti, Sci. Adv. Mater., 2010, 2, 1 CrossRef PubMed.
- I. E. Palamà, S. Leporatti, E. De Luca, C. Gambacorti-Passerini, N. Di Renzo, M. Maffia, R. Rinaldi, G. Gigli, R. Cingolani and A. M. L. Coluccia, Nanomedicine, 2010, 5, 419 CrossRef PubMed.
- F. L. Sorgi, S. Bhattacharya and L. Huang, Gene Ther., 1997, 4, 961 CAS.
- A. R. Azzoni, S. C. Ribeiro, G. A. Monteiro and D. M. F. Prazeres, J. Gene Med., 2007, 9, 392 CrossRef CAS PubMed.
- G. Tavernier, O. Andries, J. Demeester, N. N. Sanders and S. C. De Smedt, et al. , J. Controlled Release, 2011, 150, 238 CrossRef CAS PubMed.
- F. Heil, H. Hemmi, H. Hochrein, F. Ampenberger and C. Kirschning, et al. , Science, 2004, 303, 1526 CrossRef CAS PubMed.
- K. Kariko, H. Ni, J. Capodici, M. Lamphier and D. Weissman, J. Biol. Chem., 2004, 279, 12542 CrossRef CAS PubMed.
- J. Probst, B. Weide, B. Scheel, B. J. Pichler and I. Hoerr, et al. , Gene Ther., 2007, 14, 1175 CrossRef CAS PubMed.
- P. Qiu, P. Ziegelhoffer, J. Sun and N. S. Yang, Gene Ther., 1996, 3, 262 CAS.
- S. Zou, K. Scarfo, M. H. Nantz and J. G. Hecker, Int. J. Pharm., 2010, 389, 232 CrossRef CAS PubMed.
- X. Su, J. Fricke, D. G. Kavanagh and D. J. Irvine, Mol. Pharm., 2011, 8, 774 CrossRef CAS PubMed.
- N. Nafee, M. Schneider, U. F. Schaefer and C. M. Lehr, Int. J. Pharm., 2009, 381, 130 CrossRef CAS PubMed.
- E. V. Rukavishnikova, T. A. Korolenko, T. Sassa, T. Oka, S. Horiuchi and Y. Natori, FEBS Lett., 1995, 369, 217 CrossRef CAS.
- T. Storni, T. M. Kündig, G. Senti and P. Johansen, Adv. Drug Delivery Rev., 2005, 57, 333 CrossRef CAS PubMed.
- S. Fischer, E. Uetz-von Allmen, Y. Waeckerle-Men, M. Groettrup, H. P. Merkle and B. Gander, Biomaterials, 2007, 28, 994 CrossRef CAS PubMed.
- B. Dalby, S. Cates, A. Harris, E. C. Ohki, M. L. Tilkins, P. J. Price and V. C. Ciccarone, Methods, 2004, 33, 95 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00242c |
| This journal is © The Royal Society of Chemistry 2015 |