
pH and glucose responsive nanofibers for the reversible capture and release of lectins†
Yinan
Wang
ab,
Yohei
Kotsuchibashi
c,
Koichiro
Uto
d,
Mitsuhiro
Ebara
d,
Takao
Aoyagi
d,
Yang
Liu
b and
Ravin
Narain
*a
aDepartment of Chemical and Materials Engineering, University of Alberta, 116 St and 85 Ave, Edmonton, AB T6G 2G6, Canada. E-mail: narain@ualberta.ca; Fax: +1 (780)492 2881; Tel: +1 (780) 492 1736
bDepartment of Civil and Environmental Engineering, University of Alberta, 116 St and 85 Ave, Edmonton, AB T6G 2G6, Canada
cInternational Center for Young Scientists (ICYS), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
dBiomaterials Unit, International Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), Tsukuba, Ibaraki 305-0044, Japan
First published on 17th September 2014
Abstract
A dual pH and glucose responsive boronic acid containing nanofiber was constructed for the reversible capture and release of lectins. The effects of surface groups and pH values on selective lectin capture were investigated by fluorescence microscopy. Compared to the pristine nanofibrous membrane, glucose and galactose functionalized nanofiber surfaces showed significantly higher capture of ConA and Jacalin, under alkaline conditions. On the other hand, treatment of the modified nanofibers with an acidic solution resulted in the detachment of both the lectins and glycopolymers from the nanofiber surface. As expected, once the glycopolymers are displaced, no lectins were adhered to the nanofiber surface under alkaline conditions. These functional nanofibers can therefore be easily modified and hence can be used for quick removal of selective proteins or toxins from the solution.
1. Introduction
Over the past few decades, nanofibers of various polymers or inorganic materials with diameters ranging from a few tens of nanometers to a few micrometers have been produced by relatively simple and low-cost electrospinning techniques.1 The obtained nanofibrous mats have shown great success in various fields, including tissue engineering,2 drug delivery,3 membrane filtration,4,5 and sensors,6,7 due to their extremely large surface area and high porosity.1,5 Among the numerous electrospun materials, glycopolymers are believed to be one of the most attractive, not only because of the important roles that carbohydrate–protein interactions play in many biological processes,8,9 but also due to the multiple copies of sugar residues attached to the polymer backbones that can enhance the binding affinity with proteins by the “glycoside cluster effect”.10However, the electrospun nanofibers based on the bulk glycopolymers remain a challenge, since most of the synthetic glycopolymers are water soluble, which leads to the instability of their nanofibers under aqueous conditions. Moreover, nanofibers made from glycopolymers (homopolymers) often suffer from lower efficacy towards molecular recognition processes due to the low availability of the sugar residues on the nanofiber surfaces.11 To solve these problems, researchers have grafted sugar residues onto water-insoluble electrospun nanofibrous surfaces12,13 or performed the copolymerization of sugar monomers with water insoluble monomers to obtain water insoluble copolymers which formed water insoluble electrospun nanofibrous mats after electrospinning.11,14
Recently, it was also noticed that the increased surface area on electrospun nanofibers can enhance the sensitivity of the stimuli-responsive materials to the external stimuli, and therefore result in dynamically and reversibly tunable “smart” nanofibrous structures that can be potentially used for delivery of drugs or cells.3,15 Boronic acids and their ester derivatives are a class of important stimuli-responsive materials, which can reversibly interact with diols at a pH value higher than their pKa.16,17 This unique property has made these materials attractive in a wide range of biomedical fields, such as controlled release of insulin,18–20 capture and release of circulating tumor cells (CTCs),21 glucose sensing,22–24 and tissue engineering.25 However, electrospun nanofibers containing boronic acids are not very common26 due to the cost associated in making those nanofibers. We propose here a low cost version of boronic acid based photo-crosslinked nanofibers using a copolymer derived from 3-acrylamidophenylboronic acid (AAPBA) and 2-hydroxyethyl methacrylate (HEMA) and the subsequent modification of the nanofibers with glycopolymers allowed the selective binding of specific lectins.
2. Materials and methods
2.1. Materials
All chemicals were purchased from Sigma-Aldrich Chemicals (Oakville, ON, Canada) and the organic solvents were from Wako Pure Chemical Industries, Ltd (Japan). The glycomonomers were synthesized as previously described.27–30 The structures of the monomers and initiator (4,4′-azobis(4-cyanovaleric acid) (ACVA)) are shown in Scheme 1. | ||
| Scheme 1 Chemical structures of the monomers and ACVA. | ||
2.2. Methods
The 1H NMR spectra of the monomers and polymers were recorded on a Varian 500 MHz spectrometer using D2O or DMSO-d6 as the solvent. The number average molecular weight (Mn) and polydispersity (Mw/Mn) were determined using polystyrene standards (Mw = 5900–788![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) at room temperature and a Viscotek model 250 dual detector (refractometer/viscometer) in DMF eluents (containing 10 mM LiBr) at a flow rate of 1.0 mL min−1. The capture and release of FITC-labeled lectins (Vector Laboratories, USA) on glycopolymer modified boronic acid based nanofibers were studied by fluorescence microscopy (Microscope Axio Imager.M2, Carl Zeiss, Germany) with a wide-field fluorescence microscope excitation light source (X-cite® 120Q, Lumen Dynamic, ON, Canada).
000 g mol−1) at room temperature and a Viscotek model 250 dual detector (refractometer/viscometer) in DMF eluents (containing 10 mM LiBr) at a flow rate of 1.0 mL min−1. The capture and release of FITC-labeled lectins (Vector Laboratories, USA) on glycopolymer modified boronic acid based nanofibers were studied by fluorescence microscopy (Microscope Axio Imager.M2, Carl Zeiss, Germany) with a wide-field fluorescence microscope excitation light source (X-cite® 120Q, Lumen Dynamic, ON, Canada).
 | ||
| Scheme 2 Synthesis of GMA modified photo-crosslinkable P(HEMA-st-AAPBA). | ||
To introduce the free double bonds onto P(HEMA-st-AAPBA) chains, 0.5 g of the polymer was dissolved in 20 mL DMF with an exceeding amount (1000×) of glycidyl methacrylate (GMA). After the addition of a small amount of triethylamine, the solution was left to stir for 24 h in the dark, precipitated and purified by repeated washing with a large amount of diethyl ether. The conversion of the GMA modified P(HEMA-st-AAPBA) was again determined using a JNM-GSX300 1H NMR spectrometer (JEOL, Tokyo, Japan) with DMSO-d6 as the solvent.
For lectin capture, the deprotected nanofibers were incubated in 10 mg mL−1 glycopolymer (PLAEMA (with galactose pendent groups) or PGAPMA (with glucose pendent groups)) Tris-0.1 M buffer solution (pH 9.0) for 15 min to make the glycopolymers deposit on nanofibers. The free glycopolymers were washed away from the nanofiber surfaces by rinsing with Tris-0.1 M HCl buffer (pH 9.0), followed by immersing the nanofibers in 100 μL FITC-lectin (Jacalin or ConA, 20 μL mL−1) Tris-0.1 M HCl buffer solution (pH 9.0) for 15 min at room temperature. The nanofibers were washed again with pH 9.0 Tris-0.1 M HCl buffer several times before observing the florescence of the captured FITC-lectins in a fluorescence microscope.
For lectin release, the FITC-lectin/NF loaded 24-well plates were filled with 0.1 M HCl and incubated at room temperature in the dark for 15 min. The NFs were then rinsed with pH 9.0 Tris-0.1 M HCl buffer three times and imaged by fluorescence microscopy. At least three lectin capture and release cycles were evaluated in the current study (Scheme 3, FITC-lectin capture and release on a photo-crosslinked polymer NF).
 | ||
| Scheme 3 FITC-lectin capture and release on a photo-crosslinked polymer NF. | ||
The glycopolymer modification as well as lectin capture and release efficiencies on the NF surfaces were evaluated by the % fluorescence area value, which is determined as  . The fluorescence area was measured using ImageJ, whereas the total microscopy image area was always fixed as 1443520 pixels. The % fluorescence area values were obtained from three independent repeats.
. The fluorescence area was measured using ImageJ, whereas the total microscopy image area was always fixed as 1443520 pixels. The % fluorescence area values were obtained from three independent repeats.
3. Results and discussion
3.1. Polymer synthesis
3-Acrylamidophenylboronic acid (AAPBA) was first protected with 1,4-butanediol in the presence of triethylamine. The protected monomer was then copolymerized with 2-hydroxyethyl methacrylate (HEMA) via conventional free radical polymerization, and the molecular weight details are shown in Table 1. Since only 1,2-, 1,3-, and 1,4-diols can form a complex with boronic acids,16 the adjacent hydroxyl groups in PHEMA should not have any interaction with boronic acids during the copolymerization.31 The resulting copolymer was subsequently reacted with glycidyl methacrylate (GMA) so that free vinyl groups could be introduced onto the copolymers32,33 (Scheme 2). The 1,4-butanediol protection on boronic acid is expected to prevent unnecessary reactions of the epoxy rings from GMA with the hydroxyl groups on boronic acid,33 and additionally this could interfere with the complex formation between boronic acid and carbohydrates.| N GMA | M n (Da) | M w/Mn | |
|---|---|---|---|
| a The number of GMA in each polymer (NGMA) was calculated as NHEMA × GMA mol%. | |||
| PHEMA321 | 64 | 50100 |
1.98 |
| Diol protected P(HEMA760-st-AAPBA38) | 69 | 108200 |
1.64 |
The 1H NMR spectra of PHEMA and 1,4-butanediol protected P(HEMA-st-AAPBA) before and after the introduction of GMA are shown in Fig. S1.† It was found that ∼20 mol% of hydroxyl groups on the PHEMA homopolymer successfully reacted with GMA as evidenced from the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH chemical shifts at δ = 5.5–6.5 (Fig. S1a†). On the other hand, in Fig. S1b,† the proportion of phenyl groups in P(HEMA-st-AAPBA) was found to be around 5 mol%, which was identical to the amount of 1,4-butanediol present in polymers, indicating that all boronic acids had been successfully protected. However, from the 1H NMR spectra of the GMA modified P(HEMA-st-AAPBA) (Fig. S1b†), only 10 mol% of the hydroxyl groups on copolymer chains had been successfully modified by the GMA molecules (based on the signal to noise in the spectra (Fig. S1b†), the actual proportion of the GMA in the polymer might be lower than 10 mol%). Although the amount of double bonds introduced onto polymers was relatively low in this study, according to the work of Aoyagi et al.,3 10 mol% of the photo-crosslinkable moiety present in electrospun nanofibers were high enough for photochemical crosslinking.
CH chemical shifts at δ = 5.5–6.5 (Fig. S1a†). On the other hand, in Fig. S1b,† the proportion of phenyl groups in P(HEMA-st-AAPBA) was found to be around 5 mol%, which was identical to the amount of 1,4-butanediol present in polymers, indicating that all boronic acids had been successfully protected. However, from the 1H NMR spectra of the GMA modified P(HEMA-st-AAPBA) (Fig. S1b†), only 10 mol% of the hydroxyl groups on copolymer chains had been successfully modified by the GMA molecules (based on the signal to noise in the spectra (Fig. S1b†), the actual proportion of the GMA in the polymer might be lower than 10 mol%). Although the amount of double bonds introduced onto polymers was relatively low in this study, according to the work of Aoyagi et al.,3 10 mol% of the photo-crosslinkable moiety present in electrospun nanofibers were high enough for photochemical crosslinking.
Similarly, for the glycopolymers synthesized by the conventional free radical polymerization, polymers’ molecular weights and structures were characterized by GPC (Table 2) and NMR (Fig. S1c†), respectively. It was found that both glycopolymers (PLAEMA and PGAPMA) had been successfully synthesized by free radical polymerization. The polymers’ molecular weights were found to be over 80 kDa with very wide molecular weight distribution (PDIs are over 4.0). Based on this information, the high molecular weight glycopolymers are capable of interacting on the boronic acid containing nanofiber surface by boronate–diol interactions,19 while part of the carbohydrate residues on the polymer chains are still available to capture the lectins in aqueous medium.
| M n (Da) | M w/Mn | |
|---|---|---|
| PLAEMA231 | 108400 |
4.24 |
| PGAPMA270 | 86400 |
4.38 |
3.2. Electrospinning and photo-crosslinking of the nanofibers
The photo-crosslinkable P(HEMA760-st-AAPBA38) nanofibers were fabricated by electrospinning under optimized conditions (Fig. 1a). To ensure that UV light penetrates completely through the electrospun nanofibrous mats during the photo-crosslinking process, we have controlled the thickness of the electrospun nanofibrous mats by electrospinning the polymer solution for a short period (1 h)34,35 at a low polymer concentration (10 wt%),36 slow pumping rate (0.5 mL h−1) and large needle-to-collector distance (19 mm).1 As shown in Fig. 1, nanofibers, with an average diameter of ∼400 nm, were randomly distributed and formed a continuous fibrous structure on either aluminum foil (Fig. 1b) or the glass surface (Fig. 1c). Therefore, the morphology of the electrospun nanofibers is independent of the collector materials. On the other hand, the as-spun P(HEMA760-st-AAPBA38) nanofibrous mats showed high porosity and therefore light can easily penetrate through, resulting in a bright background (Fig. 1c). Therefore, we believe that nanofibrous mats should be completely crosslinked during the photo-crosslinking process. | ||
| Fig. 1 (a) Fabrication of photo-crosslinkable polymer nanofibers by electrospinning. SEM images of P(HEMA760-st-AAPBA38) nanofibers deposited on different substrates, (b) aluminum foil, (c) glass slides. (d) Optical microscopy images of the P(HEMA760-st-AAPBA38) nanofibers before photo-crosslinking, and photo-crosslinked P(HEMA760-st-AAPBA38) nanofibers after incubation in PBS (e) and pH 9.0 Tris-0.1 M HCl buffer (f) for 24 h. Optical microscopy images of the photo-crosslinked P(HEMA321) nanofibers after incubation in pH 9.0 Tris-0.1 M HCl buffer for 24 h (g). Scale bar = 20 μm. | ||
When irradiated with the UV light, the photoinitiator distributed on the surface or bulk of the nanofibers generated free radicals and chemically crosslinked the alkene groups on the P(HEMA760-st-AAPBA38) nanofibers (Scheme 2).3 The photo-crosslinked nanofibers were water insoluble even after incubation in PBS buffer for 24 h (Fig. 1e). Interestingly, in pH 9.0 Tris-0.1 M HCl buffer, the nanofibers were found to be swollen (Fig. 1f), which could be explained by the increasing water adsorption on nanofibers when boronic acids changed to the anionic (B(OH)3−) in the basic aqueous environment.16,37
The P(HEMA321) nanofibers were electrospun and crosslinked in a similar way to the P(HEMA760-st-AAPBA38) nanofibers. Interestingly, after 24 h incubation in pH 9.0 Tris-0.1 M HCl buffer, the photo-crosslinked P(HEMA321) nanofibers were found to be extremely swollen (Fig. 1g) possibly due to the adsorption and retention of a large amount of water.
3.3. pH and glucose dual responsiveness of the boronic acid containing nanofiber surface
Since the boronate–diol interaction only occurs under basic conditions,16,19,37 the photo-crosslinked P(HEMA760-st-AAPBA38) can be deprotected by incubation in a 24-well plate that is loaded with 0.4 mL of 0.1 M HCl. On the other hand, the binding affinity between the boronic acid and 1,4-diol is much weaker than 1,2 and 1,3-diols.16,37 Therefore, in the present study, if there are residual 1,4-butanediol left on the nanofiber surface after the acid treatment, it should be completely replaced by the FITC-glycopolymers; images of the fluorescence signals uniformly distributed along the nanofibers are shown in Fig. 2. During the HCl deprotection and FITC-glycopolymer modification, we believe chemical reactions occurred not only on the surface but also in the bulk of the nanofibrous mats as evidenced by the swelling (Fig. 1f) and FITC staining (Fig. 2 and 3) in the bulk of the mats. | ||
| Fig. 2 FITC-PLAEMA (a) and FITC-PGAPMA (b) modified photo-crosslinked P(HEMA760-st-AAPBA38) nanofiber surfaces. Fluorescence could be removed after incubating glycopolymer modified NF in 500 mg mL−1 glucose solution (pH 9.0) for 48 h. (c) Reversible % fluorescence areas change when FITC-PLAEMA (solid line) and FITC-PGAPMA (dash line) modified NFs were incubated in 500 mg mL−1 glucose and 10 mg mL−1 FITC glycopolymer solutions (pH 9.0) for 48 h and 15 min, respectively. Scale bar = 20 μm. | ||
 | ||
| Fig. 3 FITC-PLAEMA (a) and FITC-PGAPMA (b) modified photo-crosslinked P(HEMA760-st-AAPBA38) nanofiber surfaces. Fluorescence could be removed after rinsing the glycopolymer modified NFs with 0.1 M HCl. (c) Reversible % fluorescence areas change when FITC-PLAEMA (solid line) and FITC-PGAPMA (dash line) modified NFs were incubated in 10 mg mL−1 FITC glycopolymer solutions (pH 9.0) for 15 min, followed by rinsing with 0.1 M HCl. Scale bar = 20 μm. | ||
The responsive nature of the resulting glycopolymer modified nanofiber to glucose was then studied. Immersion in glucose solution resulted in the displacement of the glycopolymer from the nanofiber surfaces, however long incubation time (48 h) was required for the complete displacement of the glycopolymers as shown in Fig. 2a and b. Interestingly, compared to the FITC-PGAPMA modified NFs, the one modified with FITC-PLAEMA showed larger areas of fluorescence and some residual fluorescence could be spotted on the NF surface even after 48 h incubation at high glucose concentrations (Fig. 2c). These observations could be explained by the higher association constant (pKa) between galactose and boronic acids.38
The pH responsiveness of the photo-crosslinked P(HEMA760-st-AAPBA38) nanofibers was then studied as shown in Fig. 3. Before fluorescence microscopy observations, the acid treated nanofibers were washed with Tris-0.1 M HCl buffer (pH 9.0) to prevent the fluorescence quenching under the acidic conditions.39 The results clearly indicated that FITC-glycopolymers were able to adsorb on the boronic acid containing nanofiber surfaces under basic conditions (pH 9.0), whereas rapid dissociation of glycopolymers occurred when the nanofibers were rinsed with 0.1 M HCl.
3.4. Lectin binding on different surfaces
The adsorption of FITC labeled lectins on photo-crosslinked nanofibrous membrane deposited glass slides was first studied by fluorescence microscopy. The glass slides were treated with an acidic solution to deprotect the boronic acid groups in the electrospun nanofibers. After rinsing with DI water and incubation with glycopolymers in Tris-0.1 M HCl buffer solution (pH 9.0) for 15 min, the resulting glycopolymer modified nanofibers were incubated with FITC labeled lectins in Tris-0.1 M HCl buffer solution (pH 9.0) for another 15 min and washed with Tris-0.1 M HCl buffer (pH 9.0) again to remove the free FITC-lectins. The results are shown in Fig. 4.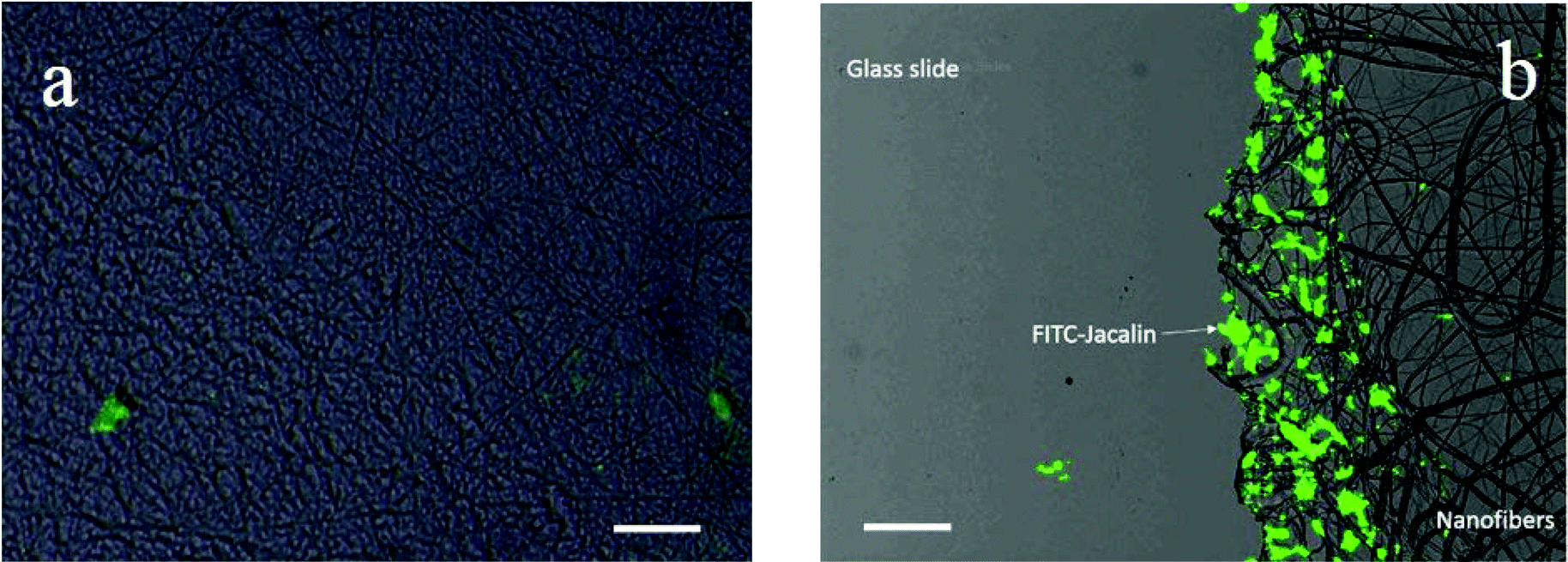 | ||
| Fig. 4 FITC-Jacalin adhered on photo-crosslinked (a) P(HEMA321) and (b) P(HEMA760-st-AAPBA38) nanofiber surfaces. At the edge of the nanofibrous mat on a glass slide, most of the FITC-Jacalin adsorbed on PLAEMA modified photo-crosslinked P(HEMA760-st-AAPBA38) nanofibers, whereas negligible FITC-Jacalin could be spotted on glass slides. Scale bar = 20 μm. | ||
It was found that the FITC-Jacalin (galactose specific lectin) interaction on photo-crosslinked P(HEMA321) nanofiber (Fig. 4a) surfaces was negligible as compared to that on the PLAEMA modified photo-crosslinked P(HEMA780-st-AAPBA38) nanofiber surface (Fig. 4b). As compared to the results obtained by Wang et al. that no lectin adsorption could be observed on glycopolymer free nanofibers,11 our results might suggest that neither glycopolymers nor lectin can strongly adsorb on the photo-crosslinked P(HEMA321) homopolymer nanofibrous surfaces, whereas a significantly larger amount of FITC-Jacalin could be captured on the glycopolymer (PLAEMA) modified photo-crosslinked P(HEMA780-st-AAPBA38) nanofiber surface through the lectin–carbohydrate interactions (Fig. 4).
Incubation of the pristine and glycopolymer modified photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers in FITC-lectin Tris-0.1 M HCl buffer solutions (pH 9.0) for 15 min (Fig. 3) showed that the lectins can only interact with the glycopolymer functionalized nanofiber surface, and not on the boronic acid modified nanofiber surface (Fig. 5a and e). Nanofibers with PLAEMA (galactose containing glycopolymer) and PGAPMA (glucose containing polymer) modification can capture Jacalin (Fig. 5b) and ConA (Fig. 5d), respectively. No fluorescence signals could be observed when FITC-ConA was incubated with the PLAEMA modified photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers (Fig. 5c and e). From these observations, the nanofibers could be used to selectively capture different lectins when the surface was functionalized with different glycopolymers.
 | ||
| Fig. 5 Fluorescence microscopy images for FITC-Jacalin adsorption on pristine (a) and PLAEMA modified photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers (b), and FITC-ConA on PLAEMA (c) and PGAPMA (d) modified photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers. Scale bar = 20 μm. (e) % Fluorescence areas of FITC-ConA or Jacalin on different nanofiber surfaces. | ||
On the other hand, compared to the FITC-glycopolymer modified photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers (Fig. 2c and 3c), less fluorescence could be observed when FITC-lectins were captured on the glycopolymer modified P(HEMA780-st-AAPBA38) nanofiber surface (Fig. 5e). These observations might be explained by the lower concentration of FITC-lectins (20 μL mL−1) used for the lectin capture assay or the weaker affinity between carbohydrates and lectins in the aqueous environment.40 Interestingly, unlike the FITC-glycopolymers that were uniformly distributed along the photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers (Fig. 2 and 3), FITC-lectins were found to be aggregated when adsorbed on the glycopolymers modified nanofibers surfaces (Fig. 5). We believe that the latter case could be explained by the electrical double layer depression and the absolute zeta potential decrease of the proteins in the Tris-buffered saline.41
3.5. Reversible capture and release of lectins on the glycopolymer modified nanofiber surface
The reversible capture and release of lectins on glycopolymers modified photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers was studied (Scheme 3) and the results are shown in Fig. 6. It was found that fluorescently labeled Jacalin and ConA could interact with PLAEMA (Fig. 6a) and PGAPMA (Fig. 6b) modified photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers under basic conditions. After incubation with 0.1 M HCl for 15 min and rinsing with pH 9 buffer solution, no fluorescence was observed even after re-incubating the nanofibers with lectins (Fig. 6a and b). Considering that a negligible amount of lectins could interact with pristine photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers (Fig. 5a), these results suggest that the glycopolymers from the nanofiber surface were removed during the acid solution treatment.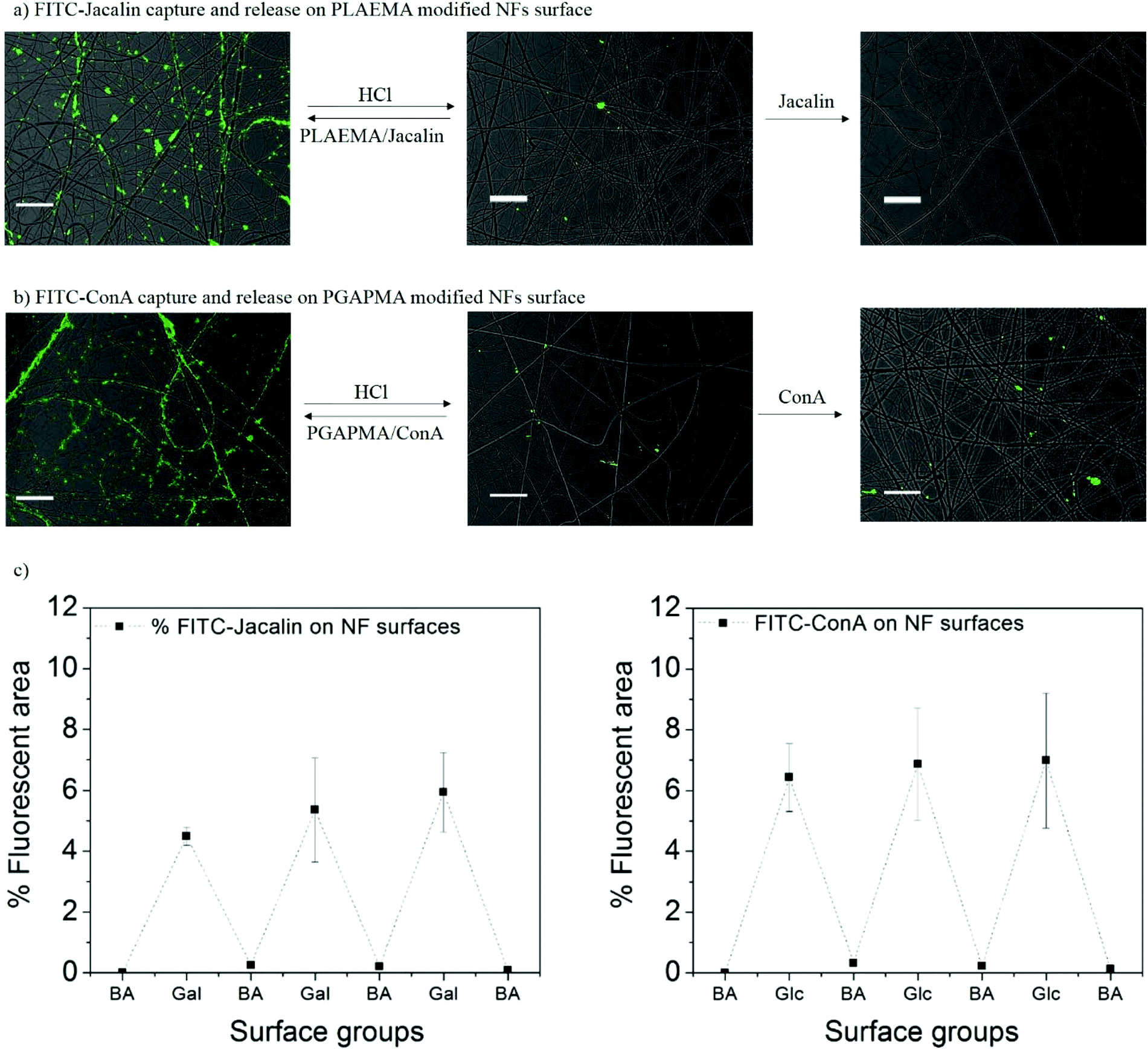 | ||
| Fig. 6 Fluorescence microscopy images for FITC-Jacalin (a) and FITC-ConA (b) reversibly captured and released from the photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers. Scale bar = 20 μm. (c) % Fluorescence areas of FITC-ConA and FITC-Jacalin on the nanofiber surface in response to alternations of surface groups between carbohydrates and boronic acid. | ||
Fig. 6c shows the % fluorescence area changes on the photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers in response to alterations of surface groups between carbohydrates and boronic acid. When glycopolymer (PLAEMA) modified nanofibers were incubated with FITC-Jacalin for 15 min, the lectin was captured by the galactose groups on nanofibers and showed ∼6% coverage with fluorescence signals in microscopy images (Fig. 6a and c). Once the nanofibers were immersed in 0.1 M HCl for 15 min and rinsed with pH 9 buffer solution, the fluorescence areas were reduced to ∼0%, indicating that PLAEMA and glycopolymer–lectin conjugates were removed from the nanofiber surface, and the surface groups were restored to boronic acids (Fig. 6a and c). These observations could be repeated by alternatively incubating the photo-crosslinked PLAEMA modified P(HEMA780-st-AAPBA38) nanofibers in FITC-Jacalin and acidic solutions (Fig. 6c), suggesting that our materials could be used for reversible capture and release of Jacalin. We also evaluated the FITC-ConA capture and release on glucose containing glycopolymer (PGAPMA) modified nanofibers. The results were similar to those for the FITC-Jacalin capture and release on PLAEMA modified nanofiber surface (Fig. 6c), suggesting that using different glycopolymers to modify the nanofiber surface, our photo-crosslinked P(HEMA780-st-AAPBA38) nanofibers could be used for reversible capture and release of various lectins. Moreover, the nanofiber fabricated in the present study could also be used for the reversible capture of virus or even bacteria from contaminated water. Compared to other platforms such as carbon nanotubes,42 surfaces43 and nanoparticles,44,45 for pathogen capture/detection, we believe our nanofibers are easier to handle, reusable and less expensive.
4. Conclusions
This study presents the first example of using a boronic acid containing photochemically crosslinked polymer nanofiber membrane for the reversible capture and release of lectins. The pH and glucose dual responsive behavior, as well as the adsorption of lectins (FITC-Jacalin and FITC-ConA) on different surfaces (pristine, galactose and glucose containing polymer modified nanofibers) were studied by fluorescence microscopy. The FITC labeled glycopolymers could adsorb on the nanofiber surface under basic conditions (pH 9.0) and are released at either high glucose concentrations or under acidic conditions. FITC-Jacalin and FITC-ConA were successfully captured on the galactose and glucose containing polymer modified nanofiber surface, respectively, whereas no lectin adsorption can be observed on the pristine nanofibers. Immersion of the FITC-lectins conjugated nanofibers in acidic solution for 15 min resulted in the rapid release of both the lectins and the glycopolymers from the nanofiber surfaces. Therefore, such a type of nanofiber can find application in the quick removal of specific proteins or toxins in solution.Acknowledgements
This research was supported by research grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada, the Canada Foundation for Innovation (CFI), and the National Institute for Materials Science (NIMS) internship program.Notes and references
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS PubMed.
- H. Yoshimoto, Y. M. Shin, H. Terai and J. P. Vacanti, Biomaterials, 2003, 24, 2077–2082 CrossRef CAS.
- Y.-J. Kim, M. Ebara and T. Aoyagi, Angew. Chem., Int. Ed., 2012, 51, 10537–10541 CrossRef CAS PubMed.
- K. Namekawa, M. T. Schreiber, T. Aoyagi and M. Ebara, Biomater. Sci., 2014, 2, 674–679 RSC.
- X. Wang, X. Chen, K. Yoon, D. Fang, B. S. Hsiao and B. Chu, Environ. Sci. Technol., 2005, 39, 7684–7691 CrossRef CAS.
- J. Shin, S.-J. Choi, I. Lee, D.-Y. Youn, C. O. Park, J.-H. Lee, H. L. Tuller and I.-D. Kim, Adv. Funct. Mater., 2013, 23, 2357–2367 CrossRef CAS PubMed.
- D.-J. Yang, I. Kamienchick, D. Y. Youn, A. Rothschild and I.-D. Kim, Adv. Funct. Mater., 2010, 20, 4258–4264 CrossRef CAS PubMed.
- D. R. Bundle and N. M. Young, Curr. Opin. Struct. Biol., 1992, 2, 666–673 CrossRef CAS.
- N. Sharon, FEBS Lett., 1987, 217, 145–157 CrossRef CAS.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321–327 CrossRef CAS.
- L. Wang, G. R. Williams, H.-l. Nie, J. Quan and L.-m. Zhu, Polym. Chem., 2014, 5, 3009–3017 RSC.
- S. Patel, K. Kurpinski, R. Quigley, H. Gao, B. S. Hsiao, M.-M. Poo and S. Li, Nano Lett., 2007, 7, 2122–2128 CrossRef CAS PubMed.
- T. Uyar, R. Havelund, J. Hacaloglu, F. Besenbacher and P. Kingshott, ACS Nano, 2010, 4, 5121–5130 CrossRef CAS PubMed.
- Q. Yang, J. Wu, J.-J. Li, M.-X. Hu and Z.-K. Xu, Macromol. Rapid Commun., 2006, 27, 1942–1948 CrossRef CAS PubMed.
- Y.-J. Kim, M. Ebara and T. Aoyagi, Adv. Funct. Mater., 2013, 23, 5753–5761 CrossRef CAS PubMed.
- S. D. Bull, M. G. Davidson, J. M. H. van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2012, 46, 312–326 CrossRef PubMed.
- S. L. Wiskur, J. J. Lavigne, H. Ait-Haddou, V. Lynch, Y. H. Chiu, J. W. Canary and E. V. Anslyn, Org. Lett., 2001, 3, 1311–1314 CrossRef CAS PubMed.
- Q. Guo, Z. Wu, X. Zhang, L. Sun and C. Li, Soft Matter, 2014, 10, 911–920 RSC.
- Y. Kotsuchibashi, R. V. C. Agustin, J.-Y. Lu, D. G. Hall and R. Narain, ACS Macro Lett., 2013, 2, 260–264 CrossRef CAS.
- A. Matsumoto, K. Yamamoto, R. Yoshida, K. Kataoka, T. Aoyagi and Y. Miyahara, Chem. Commun., 2010, 46, 2203–2205 RSC.
- H. Liu, Y. Li, K. Sun, J. Fan, P. Zhang, J. Meng, S. Wang and L. Jiang, J. Am. Chem. Soc., 2013, 135, 7603–7609 CrossRef CAS PubMed.
- T. Hoare and R. Pelton, Macromolecules, 2007, 40, 670–678 CrossRef CAS.
- M. B. Lerner, N. Kybert, R. Mendoza, R. Villechenon, M. A. Bonilla Lopez and A. T. Charlie Johnson, Appl. Phys. Lett., 2013, 102 CrossRef CAS PubMed , 183113.
- C. Zhang, M. D. Losego and P. V. Braun, Chem. Mater., 2013, 25, 3239–3250 CrossRef CAS.
- T. Aoki, Y. Nagao, K. Sanui, N. Ogata, A. Kikuchi, Y. Sakurai, K. Kataoka and T. Okano, J. Biomater. Sci., Polym. Ed., 1997, 9, 1–14 CrossRef CAS PubMed.
- E. Baştürk and M. V. Kahraman, Polym. Compos., 2012, 33, 829–837 CrossRef PubMed.
- M. Benaglia, E. Rizzardo, A. Alberti and M. Guerra, Macromolecules, 2005, 38, 3129–3140 CrossRef CAS.
- Z. Deng, H. Bouchékif, K. Babooram, A. Housni, N. Choytun and R. Narain, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4984–4996 CrossRef CAS PubMed.
- Z. Deng, M. Ahmed and R. Narain, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 614–627 CrossRef CAS PubMed.
- Z. Deng, S. Li, X. Jiang and R. Narain, Macromolecules, 2009, 42, 6393–6405 CrossRef CAS.
- Y.-J. Lee, S. A. Pruzinsky and P. V. Braun, Langmuir, 2004, 20, 3096–3106 CrossRef CAS.
- W. N. E. van Dijk-Wolthuis, O. Franssen, H. Talsma, M. J. van Steenbergen, J. J. Kettenes-van den Bosch and W. E. Hennink, Macromolecules, 1995, 28, 6317–6322 CrossRef CAS.
- A. V. Reis, A. R. Fajardo, I. T. A. Schuquel, M. R. Guilherme, G. J. Vidotti, A. F. Rubira and E. C. Muniz, J. Org. Chem., 2009, 74, 3750–3757 CrossRef CAS PubMed.
- G. Kim, H. Yoon and Y. Park, Appl. Phys. A, 2010, 100, 1197–1204 CrossRef CAS.
- J. Lee, Y. H. Jeong and D.-W. Cho, Macromol. Mater. Eng., 2014, 229, 1052–1061 Search PubMed.
- S. V. Fridrikh, J. H. Yu, M. P. Brenner and G. C. Rutledge, Phys. Rev. Lett., 2003, 90, 144502 CrossRef.
- D. G. Hall, in Boronic Acids, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 1–99 Search PubMed.
- J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769–774 CrossRef CAS.
- D. Fangkai, M. Yunhao, Z. Fang, Y. Changmin and W. Shuizhu, Nanotechnology, 2013, 24, 365101 CrossRef PubMed.
- C. Ke, H. Destecroix, M. P. Crump and A. P. Davis, Nat. Chem., 2012, 4, 718–723 CrossRef CAS PubMed.
- P. Attard, in Adv. Chem. Phys., John Wiley & Sons, Inc., 2007, pp. 1–159 Search PubMed.
- L. Gu, T. Elkin, X. Jiang, H. Li, Y. Lin, L. Qu, T.-R. J. Tzeng, R. Joseph and Y.-P. Sun, Chem. Commun., 2005, 874–876 RSC.
- Q. Yang, M. Strathmann, A. Rumpf, G. Schaule and M. Ulbricht, ACS Appl. Mater. Interfaces, 2010, 2, 3555–3562 CAS.
- G. Pasparakis and C. Alexander, Angew. Chem., Int. Ed., 2008, 47, 4847–4850 CrossRef CAS PubMed.
- R. Ribeiro-Viana, M. Sánchez-Navarro, J. Luczkowiak, J. R. Koeppe, R. Delgado, J. Rojo and B. G. Davis, Nat. Commun., 2012, 3, 1303 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of P(HEMA321) and 1,4-butanediol protected P(HEMA780-st-AAPBA38) before and after the introduction of GMA. See DOI: 10.1039/c4bm00269e |
| This journal is © The Royal Society of Chemistry 2015 |