
Synthesis and characterization of vinyl-functionalized magnetic nanofibers for protein imprinting†
Yanxia
Li
ab,
Qiu
Bin
b,
Zhenyu
Lin
b,
Yiting
Chen
a,
Huanghao
Yang
b,
Zongwei
Cai
*c and
Guonan
Chen
*b
aDepartment of Chemistry and Chemical Engineering, Minjiang University, Fuzhou, Fujian 350108, China
bMinistry of Education Key Laboratory of Analysis and Detection Technology for Food Safety (Fuzhou University), Fujian Province Key Laboratory of Analysis and Detection for Food Safety, Department of Chemistry, Fuzhou University, Fuzhou, 350002, China. E-mail: gnchen@fzu.edu.cn
cDepartment of Chemistry, Hong Kong Baptist University, Hong Kong, China. E-mail: zwcai@hkbu.edu.hk
First published on 22nd September 2014
Abstract
One kind of surface protein imprinting method was developed by a more convenient, simpler and cheaper approach based on vinyl-functionalized magnetic nanofibers (NFs).
Molecular imprinting has become an attractive method in terms of predetermination, specific recognition, and practicality.1–3 The quick formation and high stability of the imprinted materials enable molecularly imprinted polymers (MIPs) to be used in extensive applications,4 as for example affinity chromatographic supports, catalysts, and sensor materials.5–7 However, there are still many disadvantages of traditional imprinted materials, such as low adsorption capacity, slow mass transfer, poor specificity, low sensitivity and instability, limiting application scopes of separation and purification. To address these problems, nanotechnology has been explored for the novel molecularly imprinted materials.8,9 Nano-structured imprinted materials have a high surface-to-volume ratio and most of the template molecules are situated at or close to the material surface,10 to reduce the mass transfer resistance of template protein.
The molecular imprinting substrates of bulk polymers,11,12 Fe3O4 (ref. 13, 14) and silica15,16 have been widely used due to the low cost and easy preparation. Among them, magnetic Fe3O4 nanomaterials were considered as new, efficient and reusable catalysts in molecular imprinting techniques.17,18 Au coating on a magnetic core Fe3O4@Au could attain both the advantages of chemical stability and biocompatibility of Au and the magnetic properties of Fe3O4. The Au layer could be expected to protect the magnetite from etching in a harsh environment. Furthermore, the Au surface can be readily functionalized through Au–S binding.19 These features of Fe3O4@Au NPs extended the applications in bio-functional materials.20,21 Polyaniline (PA) can be used as the imprinted polymer layer and functional substrate.22,23 The imino group of polyaniline surface, which can generate electrostatic adsorption with the template molecule, could be functionalized as the substrate. Liang et al.24 reported a monomer strategy for imprinting of 1,3-dinitrobenzene molecules at the surface of the vinyl-functional PA NF layer which can be copolymerized with a functional monomer.25,26
Protein imprinting is a highly efficient method for protein separation and purification. However, protein imprinting is a very challenging task because of the diverse structures of proteins.27,28 Herein, we synthesized a new composite of protein-imprinted NFs with binding sites on the surface. First, PA NFs composited with Fe3O4@Au nanoparticles (NPs) were functionalized by introducing vinyl bonds, followed by copolymerization of functional monomers to prepare surface imprinted polymer layers by acrylamide (AAm) precipitation polymerization. Compared to the traditional MIPs, the protein-imprinted NFs showed rigid fibrous morphology, high selectivity, binding capacity, chemical stability as well as faster mass transfer rates.
Surface imprinting is one of the most attractive approaches to prepare MIPs with cavities at the surface or close to the surface, facilitating mass transfer. The surface modifications of the substrate, however, would be the challenge of surface imprinting. To address this problem, various methods have been developed, such as formation of a monolayer29 and functionalization.30 Of particular interest, one method namely vinyl functionalization has been successfully applied in surface imprinting.27,31 In our previous work,32 we reported a kind of surface glycoprotein imprinting over magnetic Fe3O4@Au multifunctional NFs which proved to be effective by introducing aminophenylboronic acid and the vinyl group on the substrates, but the harsh acidic or basic conditions of extraction and incubation reduced the stability and selectivity to some special proteins. Here, we present a simple method for preparing uniform surface-imprinted polymer NFs capable of capturing the template noncovalently, rather than covalently, onto the support surface which would overcome the disadvantage, while maintaining the advantages to some degree. The general scheme for the proposed formation mechanisms of Fe3O4@Au functional MIPs is illustrated in Fig. 1. In the first step, Fe3O4 NPs were doped in the MIPs because they have unique magnetic properties that enable them to be handled by magnetic field, and facilitate separation and purification of nanomaterials in the synthesis process. Secondly, p-amino-thiophenol (PATP) capped Fe3O4@Au NPs were formed in order to introduce aniline groups for copolymerization with aniline. Thirdly, a vinyl group was introduced onto the Fe3O4@Au@PA NF surface with amide reaction of acryloyl chloride which produced a fast response and high-yield. The template Lys has a relatively small molecular size, and through multiple hydrogen bonding interactions with the monomer of acrylamide (AAm) and methacrylic acid (MAA) imprinted sites in the polymer are achievable.
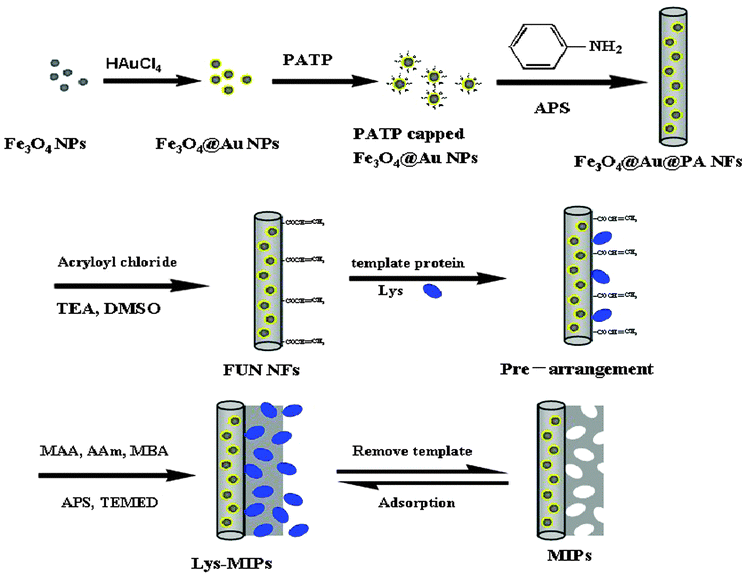 | ||
| Fig. 1 Synthesis route towards MIPs. APS = ammonium persulfate; DMSO = dimethyl sulfoxide; TEA = triethylamine; MBA = N,N-methylenebisacrylamide; TEMED = N,N,N,N-tetramethyl-ethylenediamine. | ||
The morphological structures of Fe3O4@Au@PA NFs and MIPs were detected by TEM (see Fig. S1 in ESI†). The average diameter of Fe3O4@Au@PA NFs was about 20 nm (Fig. S1a, ESI†) with a length of about 600 nm. It may be observed that MIPs also exhibited nanofiber structure (Fig. S1b, ESI†) with an average diameter of 50 nm which is larger than that of Fe3O4@Au@PA NFs, indicating that a MIP layer of about 15 nm thickness was attached on the surface of the substrate successfully. As can be seen, it showed a rigid linear structure compared to traditional NFs or nanowires which usually exhibited toughness and curves in MIP technology.10,33 Furthermore, the NFs were interrupted in the imprinting polymerization process which may be due to their rigidity and brittleness. At the same time, Fe3O4@Au NPs were not found in TEM images. All of the above-mentioned phenomena indicate the participation of Fe3O4@Au NPs in polymerization.
To further determine the characteristics of the MIPs, FT-IR spectra of the naked Fe3O4 NPs, Fe3O4@Au MNPs, Fe3O4@Au@PA NFs, FUN-NFs, NIPs, and MIPs were compared in Fig. S2 (see ESI†). The peaks at 584 cm−1 and 677 cm−1 observed in curves a–f were related to the Fe–O group, and the peak around 3465 cm−1 was assigned to the –OH vibrations on the surface of magnetite.34 In curve c, peaks at 1236 and 1148 cm−1 were assigned to C–N and the in-plane bending of C–H which indicates that doped polyaniline was successfully synthesized.10 In curve d, 1600 cm−1 stretch vibrations were assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, which clearly indicates that the vinyl groups of CH2CHCOCl were successfully introduced onto the polyaniline surface. The characteristic peaks at 1502 cm−1 (curves c–f) corresponded to the CC stretching of benzenoid rings. The absorption peaks of curves e and f are not obvious, which may be due to interference with infrared absorption by the acrylamide polymer layer and the template Lys.
C, which clearly indicates that the vinyl groups of CH2CHCOCl were successfully introduced onto the polyaniline surface. The characteristic peaks at 1502 cm−1 (curves c–f) corresponded to the CC stretching of benzenoid rings. The absorption peaks of curves e and f are not obvious, which may be due to interference with infrared absorption by the acrylamide polymer layer and the template Lys.
The TGA curves of Fe3O4@Au NPs (a), FUN-NFs (b), and MIPs (c) are given in Fig. S3 (ESI†). As shown in Fig. S3 (ESI†), compared to Fe3O4@Au NPs (curve a), FUN-NFs (curve b) displayed about 25% weight-loss when the temperature was increased to 800 °C and a sharp decline under 400 °C which was responsible for the weight loss of the polyaniline layer. At the same time, MIPs (curve c) displayed about 40% weight-loss which fully demonstrated the existence of the Lys imprinted gel polymer layer. When the temperature exceeded 600 °C, a platform of all the curves proved that the organic polymer layer is completely decomposed, leaving relatively stable inorganic constituents.
To further investigate the characteristics of the synthetic NFs, electrochemical technology is another effective analytical method. Electrically conductive polymer materials were adopted in the synthesis of MIPs, which can provide effective comparison of the nature of the NIP and MIP materials by electrochemical technology. Cyclic voltammetry (CV) can effectively evaluate the impact of these materials on the electron transfer speed on the surface of electrodes. Three kinds of nanomaterials (NIPs, MIPs, MIPs rebinding 0.5 mg mL−1 Lys) with a similar composition (Fe3O4@Au, PA, polyacrylamide) were immobilized on the electrode surface respectively using chitosan solution with higher conductivity. The difference among the three kinds of materials is the amount of adsorbed protein which greatly influences electron transfer at the electrode surface. As can be seen from Fig. S4 (ESI†), the NIP electrode (Fig. S4a, ESI†) having the highest peak current response shows good conductive properties of NIPs, which can effectively promote the electron transfer in the surface of the electrode. But the peak current of the MIP electrode (Fig. S4b, ESI†) slightly decreased, which may be due to the small amount of protein residues in MIP which often hinder the electron transfer of the electrode surface. A clearly decreased peak current of MIPs after rebinding of the Lys electrode shows that a large number of Lys were imprinted to the MIPs successfully (Fig. S4c, ESI†).
Fig. 2 shows the adsorption isotherm of MIPs and NIPs, which indicates that the MIPs had a higher binding capacity for Lys than NIPs. When the initial concentration of Lys is 0.5 mg mL−1, a saturation adsorption was achieved for MIPs. The adsorption isotherm of NIPs has the same trend as that of MIPs and reaches an adsorption plateau at 0.5 mg mL−1. The experimental maximum adsorption capacities are calculated to be 76.8 mg g−1 for MIPs and 32.48 mg g−1 for NIPs respectively, which are significantly higher than those reported in the literature for Lys imprinting.27,28 This obvious disparity between MIPs and NIPs suggests that the molecular recognition sites were generated on the surface of the MIPs by Lys involved in the polymerization process.
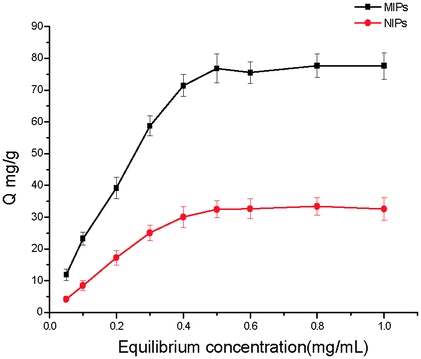 | ||
| Fig. 2 Adsorption isotherms of Lys on the NIPs and MIPs. Adsorption conditions: V = 1.0 mL, m = 3.0 mg, Ci = 0.01–1.0 mg mL−1, time 2 h, temperature RT, PBS (10 mM, pH 7.4). | ||
The adsorption equilibrium of Lys on MIPs and NIPs was found to obey the Freundlich isotherm:35
| Qe = QfCe1/n | (1) |
A linear equation can be obtained by taking the logarithm of Freundlich equation:
lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qe = 1/nlgCe + lgQf Qe = 1/nlgCe + lgQf | (2) |
| K0 = Qnf | (3) |
MIPs were tested and utilized under subsaturation conditions. This region is the linear portion of the binding isotherm when plotted in a log–log format and is well fit by the Freundlich isotherm.36,37 By fitting the experimental data with the equation (see Fig. S5 in ESI†), the values of the four parameters (Qf, 1/n, K0, and R2) are listed in Table S1 (see ESI†) for the adsorption of Lys on MIPs and NIPs. As we can see, the value of K0 on MIPs is 0.69, being considerably greater than 0.37 on NIPs. Moreover, Qf for MIPs is 0.73, being greater than 0.40 for NIPs. Therefore, MIPs have a higher adsorption capacity compared to NIPs. According to the values of correlation coefficients (R2), the Freundlich model is suitable for describing the adsorption process, indicating that the adsorption of Lys on MIPs and NIPs is heterogeneous.
To gain further insight into the protein-binding mechanism, a kinetic study was carried out with a time course of binding after bringing an optimized concentration of protein in contact with MIPs. Fig. 3a shows that 0.5 mg mL−1 Lys binding in all cases reached equilibrium within 240 min. For MIPs, there always exist two kinds of imprinted forms: one is surface imprinting and the other is being embedded into the polymer. The MIPs took up 79% of the equilibrium amount during 40 min. The latter showed a slow dynamic binding process and the total equilibrium time was 90 min. The rebinding rate of the proposed MIPs is reasonable because part of the protein binding process is carried out inside the polymer.
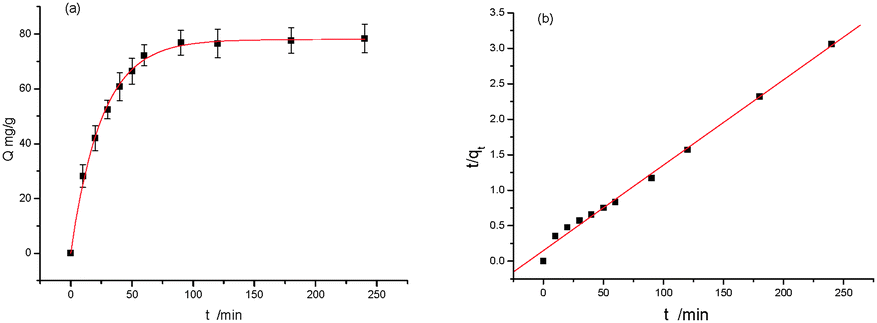 | ||
| Fig. 3 Dynamic curves of Lys on the MIPs with pseudo-first order (a) and pseudo-second order (b). V = 1.0 mL, m = 3.0 mg, C0 = 0.5 mg mL−1, temperature RT. | ||
Adsorption kinetic studies in liquid–solid systems are often conducted under batch conditions where the transient adsorbate concentration in the solution is fitted by a suitable kinetic model. Among these models the pseudo-first order and pseudo-second order models have been used most frequently.38,39 These two rate equations are shown below as eqn (4) and (5), respectively.
| qt = qe[1 − e(−k1t)] | (4) |
The linear form of the pseudo-second-order model is expressed as follows:
| t/qt = t/qe + 1/k2qe2 | (5) |
The two quasi-models consider all of the adsorption processes, such as membrane diffusion, adsorption, and molecular diffusion. The fitting curves were obtained from eqn (4) and (5) (Fig. 3a and b). Table S2 (see ESI†) presents the kinetic parameters of MIPs calculated from eqn (4) and (5). It shows that the value of R2 (R2 > 0.99) was high, suggesting that both models are in line with the experimental results. But it is clear that for the pseudo-first-order model the calculated qe values were in better agreement with the experimental values.
The special selectivity test of MIPs was carried out using BHb (Mw 64.5 kDa, pI 6.8), HRP (Mw 40 kDa, pI 7.2), BSA (Mw 68 kDa, pI 4.6), and Cyt c (Mw 12.4 kDa, pI 10.2) as comparative substrates. The selected comparative proteins possess large differences in molecular mass and isoelectric points (pI). The amounts of adsorption of these proteins to MIPs and NIPs were determined with the equilibrium adsorption method, as shown in Fig. S6 (ESI†). The results showed that the amounts of template protein adsorbed on MIPs were more than those of comparative proteins.
The selectivity can be evaluated by calculating the imprinting factor (IF).28
| IF = QMIP/QNIP | (6) |
Using Cyt c as the competitor, the binary protein competitive adsorption experiments further confirmed the rebinding selectivity for Lys against Cyt c. The use of such similar proteins as references which have Mw and pI rather close to those of Lys presents a genuine challenge to the imprinting process, since both the size of cavities and the nature of physical forces responsible for the recognition are supposed to be approximately the same. The IF for Lys and Cyt c are 2.55 and 1.10, respectively, with no obvious difference with the selectivity test. But the absorption capacity was decreased from 76.80 to 58.04, due to interference from Cyt c coexistence. In our previous work,35 covalent molecular imprinting technology has been shown to have greatly improved selectivity to some proteins, but low selectivity to Cyt c due to some harsh experimental conditions. In the present non-covalent molecular imprinting, mild experimental conditions are suitable for more protein coexisting environment. Through investigation of stability and regeneration, the MIPs and NIPs were stable for up to three adsorption cycles. The results (Fig. S7, ESI†) indicate that the MIPs show an outstanding regeneration ability, being more stable under acidic conditions for elution.35
In summary, the synthesized novel vinyl-functionalized magnetic NFs with a homogeneous MIP outer layer for selective recognition of Lys have been characterized. The synthesis of MIPs is simple, convenient, with high-yield and low-cost. The acquired MIPs exhibit high loading capacity, rational adsorption dynamics, and excellent rebinding selectivity due to the specific interactions between the imprinted sites and target species. Moreover, the imprinted materials show high stability and are reusable for good repeatability. The strategy reported herein can be further expected to be used to fabricate various molecular-recognition systems for sensing or separation science.
This project was financially supported by NSFC (21275031, 41076059, 20928005, 20905034, 21175025, 21405075), the Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1116), and Young teachers' Teaching and Scientific Research Project of Fujian Province (JA14257).
Notes and references
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739 CrossRef CAS PubMed.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783 CrossRef CAS.
- P. Alonso-González, P. Albella, M. Schnell, J. Chen, F. Huth, A. García-Etxarri, F. Casanova, F. Golmar, L. Arzubiaga and L. Hueso, Nat. Commun., 2012, 3, 684 CrossRef PubMed.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828 CrossRef CAS PubMed.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102 CrossRef CAS PubMed.
- R. Zhang, Y. Zhang, Z. Dong, S. Jiang, C. Zhang, L. Chen, L. Zhang, Y. Liao, J. Aizpurua and Y. Luo, Nature, 2013, 498, 82 CrossRef CAS PubMed.
- J. R. Lombardi and R. L. Birke, Acc. Chem. Res., 2009, 42, 734 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou and D. Y. Wu, Nature, 2010, 464, 392 CrossRef CAS PubMed.
- A. R. Guerrero and R. F. Aroca, Angew. Chem., Int. Ed., 2011, 50, 665 CrossRef CAS PubMed.
- M. D. Furtaw, D. L. Steffens, T. M. Urlacher and J. P. Anderson, Anal. Chem., 2013, 85, 7102 CrossRef CAS PubMed.
- C. B. Milojevich, D. W. Silverstein, L. Jensen and J. P. Camden, J. Am. Chem. Soc., 2011, 133, 14590 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp and K. Kneipp, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17149 CrossRef CAS PubMed.
- G. F. Walsh and L. Dal Negro, Nano Lett., 2013, 13, 3111 CrossRef CAS PubMed.
- F. M. Geiger, Annu. Rev. Phys. Chem., 2009, 60, 61 CrossRef CAS PubMed.
- J. Kozuch, C. Steinem, P. Hildebrandt and D. Millo, Angew. Chem., Int. Ed., 2012, 51, 8114 CrossRef CAS PubMed.
- L. V. Brown, K. Zhao, N. King, H. Sobhani, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2013, 135, 3688 CrossRef CAS PubMed.
- R. Bukasov and J. S. Shumaker-Parry, Anal. Chem., 2009, 81, 4531 CrossRef CAS PubMed.
- J. Zhang, Z. Gryczynski and J. R. Lakowicz, Chem. Phys. Lett., 2004, 393, 483 CrossRef CAS PubMed.
- M. H. Chowdhury, S. N. Malyn, K. Aslan, J. R. Lakowicz and C. D. Geddes, Chem. Phys. Lett., 2007, 435, 114 CrossRef CAS PubMed.
- J. Wang, Y. Shan, W.-W. Zhao, J.-J. Xu and H.-Y. Chen, Anal. Chem., 2011, 83, 4004 CrossRef CAS PubMed.
- Y. Shan, J.-J. Xu and H.-Y. Chen, Chem. Commun., 2009, 905 RSC.
- M. M. Richter, Chem. Rev., 2004, 104, 3003 CrossRef CAS PubMed.
- W. Miao, Chem. Rev., 2008, 108, 2506 CrossRef CAS PubMed.
- Y. Liang, L. Gu, X. Liu, Q. Yang, H. Kajiura, Y. Li, T. Zhou and G. Shi, Chem. – Eur. J., 2011, 17, 5989–5997 CrossRef CAS PubMed.
- Z. Chen, Y. Liu, Y. Wang, X. Zhao and J. Li, Anal. Chem., 2013, 85, 4431 CrossRef CAS PubMed.
- H. Wei, J. Liu, L. Zhou, J. Li, X. Jiang, J. Kang, X. Yang, S. Dong and E. Wang, Chem. – Eur. J., 2008, 14, 3687 CrossRef CAS PubMed.
- X. Wang, W. Yun, P. Dong, J. Zhou, P. He and Y. Fang, Langmuir, 2008, 24, 2200 CrossRef CAS PubMed.
- L. Hu, Z. Bian, H. Li, S. Han, Y. Yuan, L. Gao and G. Xu, Anal. Chem., 2009, 81, 9807 CrossRef CAS PubMed.
- L. Zhao, S. Krishnan, Y. Zhang, J. B. Schenkman and J. F. Rusling, Chem. Res. Toxicol., 2009, 22, 341 CrossRef CAS PubMed.
- N. P. Sardasai, J. C. Barron and J. F. Rusling, Anal. Chem., 2011, 83, 6698 CrossRef PubMed.
- M. Jebb, P. K. Sudeep, P. Pramod, K. G. Thomas and P. V. Kamat, J. Phys. Chem. B, 2007, 111, 6839 CrossRef CAS PubMed.
- Y. Li, M. Hong, M. Miao, B. Qiu, Z. Lin, Z. Cai and G. Chen, J. Mater. Chem. B, 2013, 1, 1044–1051 RSC.
- E. Ozbay, Science, 2006, 311, 189 CrossRef CAS PubMed.
- E. Fort and S. Grésillon, J. Phys. D: Appl. Phys., 2008, 41, 013001 CrossRef.
- K. L. Gurunatha and E. Dujardin, J. Phys. Chem. C, 2013, 117, 3489 CAS.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171 CrossRef CAS PubMed.
- R. J. Umpleby, S. C. Baxter, Y. Chen, R. N. Shah and K. D. Shimizu, Anal. Chem., 2001, 73, 4584–4591 CrossRef CAS.
- Y. Miyake, H. Ishida, S. Tanaka and S. D. Kolev, Chem. Eng. J., 2013, 218, 350–357 CrossRef CAS PubMed.
- B. Pelaz, S. Jaber, D. J. de Aberasturi, V. Wulf, T. Aida, J. s. M. de la Fuente, J. Feldmann, H. E. Gaub, L. Josephson and C. R. Kagan, ACS Nano, 2012, 6, 8468 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. See DOI: 10.1039/c4cc05761a |
| This journal is © The Royal Society of Chemistry 2015 |