
Air stable NHCs: a study of stereoelectronics and metallorganic catalytic activity†
Samantha K.
Furfari
,
Matthew R.
Gyton
,
Daniel
Twycross
and
Marcus L.
Cole
*
School of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: m.cole@unsw.edu.au
First published on 8th October 2014
Abstract
The air stable NHC IPrBr is reported. A stereoelectronic study of IPrBr and its similarly stable relative IMesBr demonstrates metal complex specific changes in NHC donicity versus the ubiquitous IPr and IMes. Application to a Suzuki coupling and an iridium transfer hydrogenation gives superior outcomes using IPrBr and IMesBr.
The increased catalytic activity and scope of N-heterocyclic carbene (NHC) complexes frequently surpasses those of their phosphine counterparts1 and is often attributed to the increased σ-donor nature of NHCs and thus greater catalyst stability.2 We have an ongoing interest in the chemistry of 4,5-dibromoimidazol-2-ylidenes (Scheme 1),3 especially the impact of halogenation on their catalytic activities in organocatalysis and those of their metal complexes in metallorganic catalysis. Unlike their 4,5-dichloro analogues, which are an expanding class,4 there is a only single example of a 4,5-dibromoimidazol-2-ylidene; IMesBr (Scheme 1)3c and as yet no examples of 4,5-diiodo species.
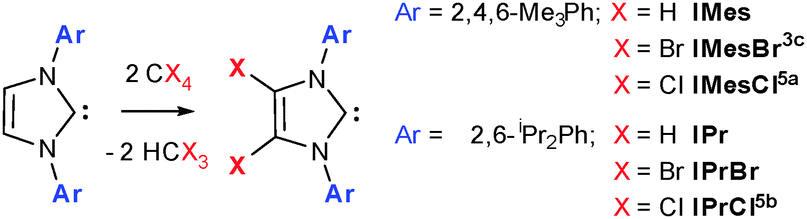 | ||
| Scheme 1 The NHCs IMes and IPr (X = H) and the preparation of their 4,5-dichloro and 4,5-dibromo analogues. | ||
IMesBr is a rare example of an air stable free NHC. Despite this attractive feature there is only a single report of its application to homogeneous catalysis6 and, perhaps more surprisingly, none to organocatalysis. This contrasts the ubiquity of the ‘parent’ NHC IMes and its bulkier relative IPr (Scheme 1) in the same catalytic arenas.1,2 Many have attributed the poor uptake in IMesCl, IMesBr and IPrCl to a predicted decrease in catalytic activity borne out of lower relative sigma donicity arising from 4,5-substituent withdrawal.1,2 Although the decrease in ligand donation has been established for IPrCl versus IPr,7 recent studies focusing on catalyses with mechanistic steps that benefit from improved metal electrophilicity,4ae.g. hydrocupration and -amination,4b have showcased the superior utility of 4,5-dichloroimidazol-2-ylidenes in some transformations.
Herein we report the air stable NHC IPrBr (Scheme 1, Fig. 1) and a unique stereoelectronic and catalytic study of IPrBr and IMesBr.† This comprises the application and comparison of a suite of structural and spectroscopic techniques (IR, 13C NMR spectroscopy, XPS and XRD, see Scheme 1, Table 1) and correlation with the activities of IPrBr and IMesBr metal complexes in two catalytic transformations with comparison to IPr and IMes analogues. These data indicate metal complex specific changes in NHC donicity and support the premise that 4,5-dibromination can generate superior catalysts relative to those coordinated by IMes and IPr.†
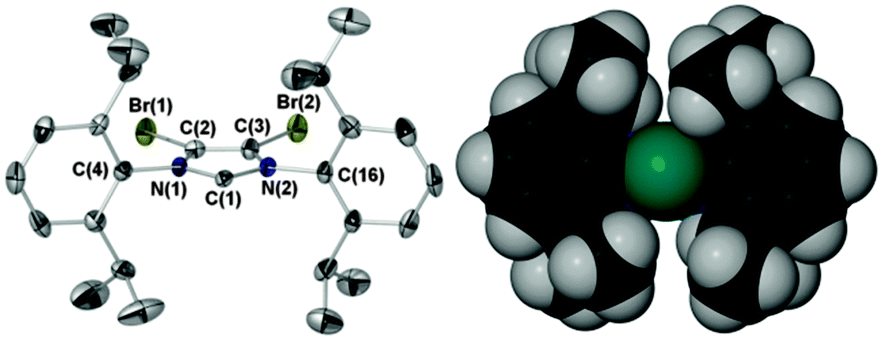 | ||
| Fig. 1 Ellipsoid plot of air stable IPrBr (50% probability) and space fill image with C2 in teal. Selected bond lengths (Å) and angles (°) with IPr values5b in parentheses: C(1)–N(1) 1.372(5) [1.364(3)], C(1)–N(2) 1.379(5) [1.369(3)], C(2)–C(3) 1.328(6) [1.336(3)], Br(1)–C(2) 1.862(4), Br(2)–C(3) 1.858(4), N(1)–C(1)–N(2) 101.8(3) [101.5(2)], Σ N(1) angles 359.9(9) [359.9(6)], Σ N(2) angles 360.1(9) [360.0(6)], C(4)aryl:heterocycle 89.7 [78.1], C(16)aryl:heterocycle 86.2 [65.4], %VBur 41.5 [41.0].8 | ||
| Ligand | IPrBr | IPr | IMesBr | IMes |
|---|---|---|---|---|
| 1L Ir–CNHC | 2.035(8) Å | 2.054(5) Å | 2.042(3) Å | 2.055(5) Å |
| 1L L solid G%12 | 38.49% | 38.89% | 33.33% | 32.83% |
| 1L Ir 4f7/2 BE | 61.01 eV | 61.10 eV | 61.10 eV | 61.25 eV |
| 2L IR ave CO ν | 2027.9 cm−1 | 2023.9 cm−1 | 2026.3 cm−1 | 2023.1 cm−1 |
| 3L 13C NMR | δ 174.7 | δ 177.5 | δ 175.0 | δ 177.2 |
| Catalysis I | 97% | 95% | 42% | 32% |
| Catalysis II | 38% | 36% | 30% | 22% |
IPrBr is readily prepared via the treatment of IPr with two equivalents of CBr4 (Scheme 1).† Similar reactions using the parent imidazolium salt or silver halide NHC complexes proved unsuccessful thereby confirming the requirement for a free NHC precursor. Moreover, attempts to dihalogenate IMes using N-chloro- or N-bromosuccinimide failed to yield 4,5-halogenated species even in the presence of suitable halide nucleophiles. Such reactions afforded the known carbene adducts of Cl2 and Br2.3b,c IPrBr and IMesBr do not react further with CBr4. This contrasts the reactivity of IMesCl, which generates electron poor olefins and chloronium salts upon reaction with CCl45a and thereby serves to eliminate contamination of IPrBr with similar co-products.
Crystalline samples of IPrBr tolerate air for several weeks when stored with a dessicant. Storage in the absence of a dessicant leads to decomposition over a period of one week affording an intractable mixture of non-imidazolium species. IPrBr deteriorates in toluene solution over a 24 h period to afford imidazolium salts that retain 4,5-dibromo substitution. As the solid-state structure of IPrBr closely resembles that of air sensitive IPr (Fig. 1)5b the origin of its air stability is unclear. Like IPrCl,5b the 4,5-halogens of IPrBr promote near orthogonal orientation of the N-arenes to the heterocycle plane.† This leads to a slight increase in steric bulk vis-à-vis IPr but lower bulk than IPrCl (%VBur 41.5, IPr 41.0, IPrCl 42.25b).8 In view of the lower stability of IPrBr in solution we suggest that the increased hydrophobicity of bromine, and indeed chlorine,9 may be the true origin of the observed air stabilities of IPrBr and IMesBr, and the noted increased stabilities of IPrCl and IMesCl.5
Aside from the absence of a 4,5-H2 resonance in its 1H NMR spectrum (IPr 6.62 ppm, C6D6)5b the most significant difference in the 1H and 13C NMR spectra of IPrBr with respect to those of IPr is the substantial spin–orbit-induced upfield shift of its 4,5-C2 resonance (IPrBr δ 107.56, IPr 121.5, C6D6).5b This is consistent with data for IMes and IMesBr (4,5-C213C NMR IMesBr δ 107.0, IMes 121.5, C6D6)3c and occurs without dramatic shifting of the NCN resonance (IPrBr δ 222.36, IPr 220.6, C6D6).†
Several researchers have put forward IR spectroscopy methods for the measurement of ligand donation using probe metal carbonyl stretching frequencies.10 A number of alternative probes have been developed. Of note are Huynh's application of 13C NMR spectroscopy with probe trans-[PdBr2(iPrBzim)(L)] species (iPrBzim = 1,3-iPr2benzimidazol-2-ylidene),11a Albrecht11b and Fukuzawa's11c use of X-ray photoelectron spectroscopy (XPS) to compare relative core 3d-electron binding energies at palladium, and Plenio's application of Lever electrochemical methods.11d These methods offer ready access to highly sensitive measures of ligand donicity and circumvent the use of hazardous metal carbonyls.
Reaction of IPrBr and IMesBr with [{Ir(μ-Cl)(cod)}2] (cod = 1,5-cyclooctadiene), [{Pd(μ-Br)Br(iPrBzim)}2] and [{Pd(μ-Cl)(Cin)}2] (Cin = η3-cinnamyl anion) affords [IrCl(cod)(NHC)] (1L, X = NHC), trans-[PdBr2(iPrBzim)(NHC)] (3L) and [PdCl(Cin)(NHC)] (4L) respectively (Fig. 2).† Placement of 1L under a carbon monoxide atmosphere gives ready access to the cis-[IrCl(CO)2(NHC)] (2L) complexes (Fig. 2).† Salient structural and spectroscopic data for the free NHCs and 1L, 2L and 3L (L = IPrBr, IPr, IMesBr, IMes) are listed in Table 1.† It is noteworthy that chloride–bromide exchange between the palladium or iridium chlorides of 1L, 2L and 4L (L = IPrBr and IMesBr) and the NHC 4,5-bromines was not detected during their synthesis or catalytic application. Indeed, attempts to activate the C–Br bonds of IPrBr and IMesBr under a variety of harsh reaction conditions including reaction with [Ni(cod)2], heating with cuprous cyanide in DMF at reflux and reaction with iPrMgCl† met with retention of the C–Br functions. Activation was only observed upon extended treatment of IPrBr with magnesium metal in tetrahydrofuran at reflux. This afforded the parent NHC IPr.
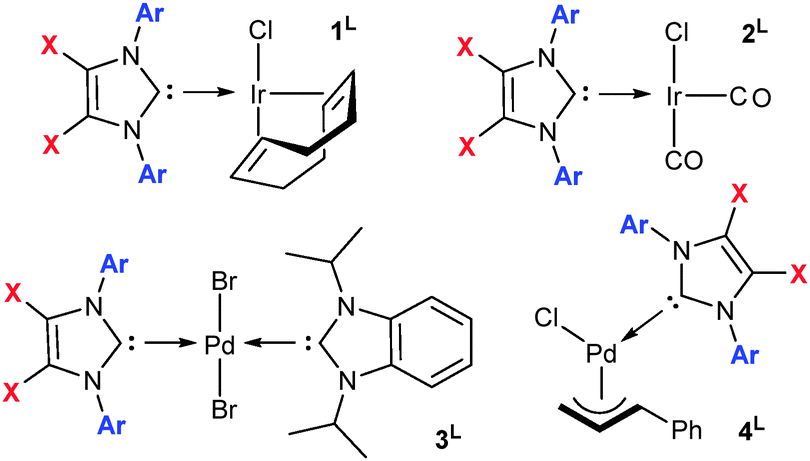 | ||
| Fig. 2 Complexes used herein. Selected data presented in Table 1. Ar = 2,6-iPr2Ph: X = Br L = IPrBr, X = H L = IPr; Ar = 2,4,6-Me3Ph: X = Br L = IMesBr, X = H L = IMes. | ||
The Ir–CNHC contacts of 1IMesBr (2.042(2) Å) and 1IPrBr (2.035(8) Å)† are shorter than those of 1IMes and 1IPr (2.055(5) and 2.054(2) Å).7 For IMesBr this occurs despite an increase in NHC bulk at the iridium (solid G% IMesBr 33.33, IMes 32.83; IPrBr 38.49, IPr 38.89%) thereby endorsing a notion of superior donation for IMesBr in this instance.12 The X-ray photoelectron spectra of 1IPrBr and 1IMesBr, which represents the first time XPS has been applied to [IrCl(cod)(NHC)] complexes, exhibit reduced (−0.09 eV, −0.15 eV) iridium 4f-electron binding energies relative to those of 1IPr and 1IMes (Table 1). This is also indicative of superior NHC donicity. The average IR C–O stretch of complexes 2L and the iPrBzim NCN resonances of complexes 3L (Table 1) are consistent with a decrease in sigma donation upon bromination (∼2 cm−1 higher and ∼2 ppm upfield respectively). The IR outcomes for 2L oppose those predicted based on 1L data (XPS, XRD) despite both containing iridium in the same oxidation state and geometry. Given the number of stereoelectronic measures used herein it is noteworthy that the relative donation of the 4,5-dibrominated and the non-brominated NHCs is not clearly differentiated. As such these data are suggestive of metal complex specific relative donation for 4,5-dibromo and 4,5-dihydro NHCs, and indicate air stability is achieved herein without a major loss of donor character.
Two relatively slow catalytic processes were employed to investigate the influence of 4,5-dibromination on the catalytic activities of IPrBr and IMesBr metal complexes: (I) the 4L catalyzed room temperature Suzuki coupling of bromomesitylene with naphthalene-1-boronic acid to afford a sterically encumbered biaryl,13 and (II) the 1L catalyzed transfer hydrogenation of acetophenone in 2-propanol using a low catalyst loading (Scheme 2).14
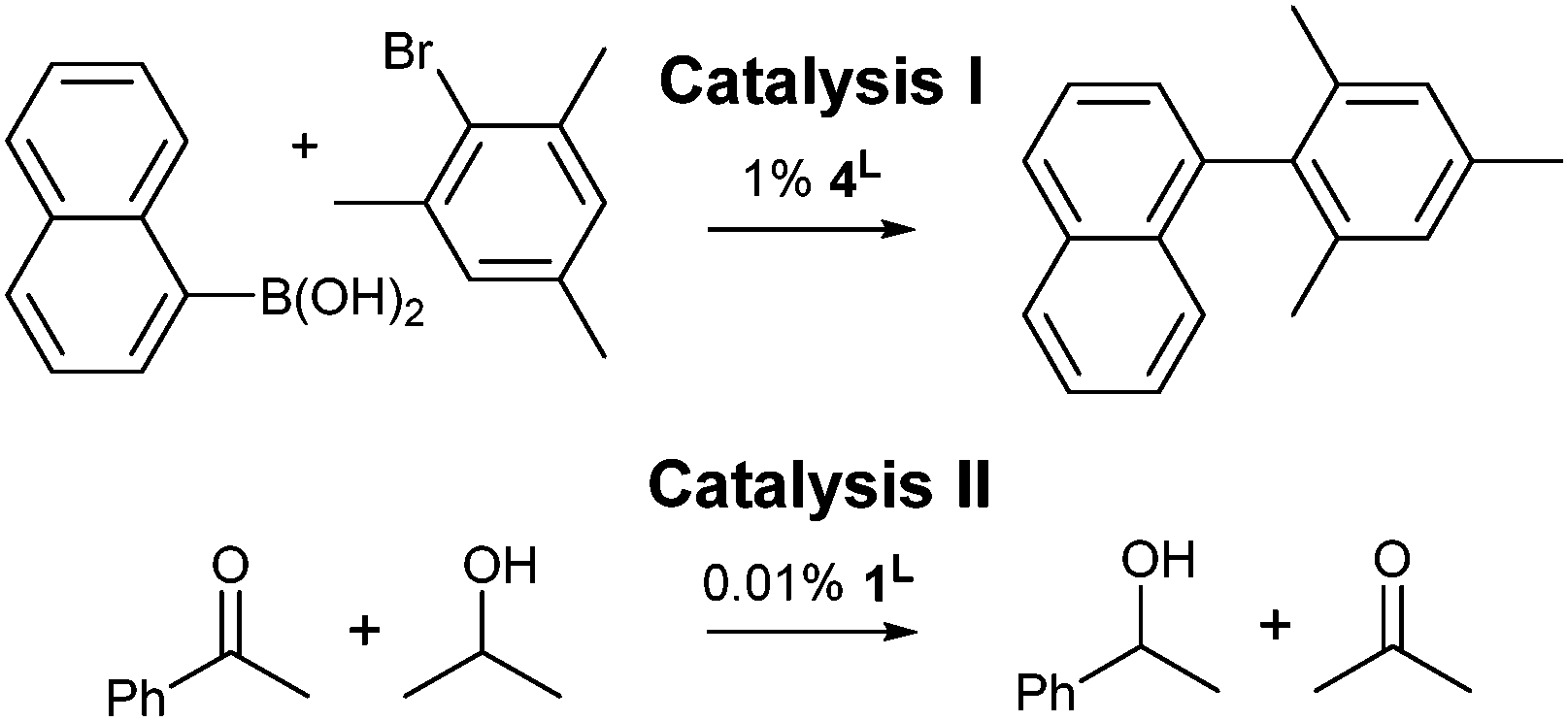 | ||
| Scheme 2 Synthetic transformations chosen to correlate NHC donor character with catalytic activity. See Table 1 for outcomes and Fig. 2 for 1L and 4L. | ||
The extents of conversion for catalyses I and II were assessed upon quenching of the reaction mixtures at 2 and 42 hours respectively (Table 1).† Palladium precatalysts 4IPrBr and 4IPr afford exceptional outcomes (97 and 95% respectively) in catalysis I. Catalysts 4IMesBr and 4IMes exhibit distinct but much lower activities that favour the IMesBr (42 versus 32%).† A control experiment using solely phenylboronic acid confirms that the C–Br functions of 4IPrBr are retained during catalysis and not arylated. In combination with 13C NMR data collected on 3L systems11a that suggest poorer donation to palladium(II) by IPrBr and IMesBr, these outcomes may indicate that room temperature Suzuki coupling benefits from moderate electron withdrawal at the NHC 4,5-positions (Table 1).†
Catalysis II demonstrates a trend that is consistent with XPS and XRD data for 1L, wherein analysis of the quenched reaction mixtures after 42 hours reveals that 1IPrBr and 1IMesBr (38 and 30% conversion) outperform their 4,5-dihydro counterparts (36 and 22% respectively). As the structural data for 1IMesBr and 1IPrBr identify an increase in ligand bulk for IMesBr versus IMes and a decrease for IPrBr versus IPr (Table 1) these outcomes do not simply follow the trend in NHC steric bulk and are consistent with the proposal that 4,5-dibromination of IMes and IPr not only generates an air stable NHC but also leads to an enhancement in some organometallic catalyses.
In summary, using a wide range of structural and spectroscopic probes we have established that the donor strengths of IPrBr and IMesBr are comparable to or surpass those of IPr and IMes. Preliminary catalytic studies indicate that IPrBr and IMesBr can generate more active organometallic species than those of IPr and IMes. We propose that IPrBr and IMesBr merit further investigation in catalysis, especially organocatalysis where their stability will be an asset to the synthetic chemist.
The authors would like to acknowledge the Australian Research Council for partial funding of this research (DP110104759) and the Australian Commonwealth for the provision of Australian Postgraduate Awards (SKF, MRG and DT).
Notes and references
- N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, Netherlands, 2011 Search PubMed.
- (a) S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef CAS PubMed; (b) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- (a) S. G. Alexander, M. L. Cole, S. K. Furfari and M. Kloth, Dalton Trans., 2009, 2909–2911 RSC; (b) M. L. Cole, C. Jones and P. C. Junk, New J. Chem., 2002, 26, 1296–1303 RSC; (c) M. L. Cole, A. J. Davies and C. Jones, J. Chem. Soc., Dalton Trans., 2001, 2451–2452 RSC.
- See for example: (a) M. Pompeo, J. L. Farmer, R. D. J. Froese and M. G. Organ, Angew. Chem., Int. Ed., 2014, 53, 3223–3226 CrossRef CAS PubMed; (b) K. Semba, M. Shinomiya, T. Fujihara, J. Terao and Y. Tsuji, Chem. – Eur. J., 2013, 19, 7125–7132 CrossRef CAS PubMed; (c) L. H. Peeck and H. Plenio, Organometallics, 2010, 29, 2761–2766 CrossRef CAS.
- (a) A. J. Arduengo, F. Davidson, H. V. R. Dias, J. R. Goerlich, D. Khasnis, W. J. Marshall and T. K. Prakasha, J. Am. Chem. Soc., 1997, 119, 12742–12749 CrossRef CAS; (b) A. J. Arduengo, R. Krafczyk, R. Schmutzler, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523–14534 CrossRef CAS.
- C. A. Urbina-Blanco, X. Bantriel, H. Clavier, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2010, 6, 1120–1126 CrossRef CAS PubMed.
- R. A. Kelly, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef CAS.
- Standard SambVca settings (3.5 Å sphere, 2.10 Å M–C bond, 0.05 Å mesh spacing, H atoms omitted, 1.17 Bondi radii scaling) using a .cif of the free NHC: A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS.
- Based on log PC6H5X − log
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PC6H6 values in A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525–616 CrossRef CAS ; H = 0.00, Cl = 0.71, Br = 0.86.
PC6H6 values in A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525–616 CrossRef CAS ; H = 0.00, Cl = 0.71, Br = 0.86. - (a) C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2593–2596 CrossRef; (b) A. R. Chianese, X. Li, M. C. Janzen, J. W. Faller and R. H. Crabtree, Organometallics, 2003, 22, 1663–1667 CrossRef CAS; (c) S. Wolf and H. Plenio, J. Organomet. Chem., 2009, 694, 1487–1492 CrossRef CAS.
- (a) H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395–5404 CrossRef CAS; (b) M. Heckenroth, A. Neels, M. G. Garnier, P. Aebi, A. W. Ehlers and M. Albrecht, Chem. – Eur. J., 2009, 15, 9375–9386 CrossRef CAS PubMed; (c) T. Terashima, S. Inomata, K. Ogata and S.-i. Fukuzawa, Eur. J. Inorg. Chem., 2012, 1387–1393 CrossRef CAS; (d) S. Leuthäußer, D. Schwarz and H. Plenio, Chem. – Eur. J., 2007, 13, 7195–7203 CrossRef PubMed.
- Standard solid G settings (normalisation distance 2.28 Å): I. A. Guzei and M. Wendt, Dalton Trans., 2006, 3991–3999 RSC.
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS PubMed.
- J. W. Ogle and S. A. Miller, Chem. Commun., 2009, 5728–5730 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses, catalyses, X-ray crystal structures for IPrBr, 1IPrBr and 1IMesBr, steric calculations and XPS data. CCDC 831264–831266. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc06809b |
| This journal is © The Royal Society of Chemistry 2015 |