
Rhodium(III)-catalyzed C–H/C–C activation sequence: vinylcyclopropanes as versatile synthons in direct C–H allylation reactions†
Jia-Qiang
Wu‡
a,
Zhi-Ping
Qiu‡
b,
Shang-Shi
Zhang
a,
Jing-Gong
Liu
a,
Ye-Xing
Lao
a,
Lian-Quan
Gu
a,
Zhi-Shu
Huang
*a,
Juan
Li
*b and
Honggen
Wang
*a
aSchool of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China. E-mail: wanghg3@mail.sysu.edu.cn; ceshzs@mail.sysu.edu.cn
bDepartment of Chemistry, Jinan University, Huangpu Road West 601, Guangzhou, 510632, China. E-mail: tchjli@jnu.edu.cn
First published on 4th November 2014
Abstract
Succession of C–H activation and C–C activation was achieved by using a single rhodium(III) catalyst. Vinylcyclopropanes were used as versatile coupling partners. Mechanistic studies suggest that the olefin insertion step is rate-determining and a facile β-carbon elimination is involved, which represents a novel ring opening mode of vinylcyclopropanes.
Transition-metal-catalyzed C–H activation reactions have emerged as a fertile field for the construction of C–C and C–heteroatom bonds.1 Among the numerous transition metals, RhIII stands out for its advantageous features such as high efficiency, mild reaction conditions, and broad substrate scope.2 Due to their versatile reactivity towards transition-metal catalysis,3 alkenes are widely used as coupling partners in C–H activation reactions.4,5 Typically, after the C–H activation/alkene insertion steps, β-hydrogen elimination is a facile elementary step as demonstrated by the numerous examples of oxidative coupling of aryl C–H bonds with olefins in the presence of different metals (Fig. 1a). Recently, it was also disclosed that β-oxygen6 and β-nitrogen6d,7 elimination is feasible when RhIII was the catalyst, partly driven by the release of ring strain. Because of the chemical inertness of the C–C single bond, β-carbon elimination is generally unfavored and thus far less established,8 even though the selective C–C cleavage has attracted increasing interest in the synthetic community, as it offers a potential alternative for synthetic disconnection.9 Vinylcyclopropanes (VCPs), bearing an olefinic moiety and a cyclopropane ring, are useful organic synthons in synthetic chemistry.10 The catalytic ring-opening of vinylcyclopropanes can be achieved under the catalysis of Lewis acids (LA) via ionic reaction pathways,11 or by a radical pathway with an organic thiyl radical catalyst.12 Alternatively, the direct oxidative addition of cyclopropane to a low-valent nucleophilic transition metal to form a π-allyl metal complex as the key reaction intermediate is also a literature precedent (Fig. 1b).13 We reasoned that, by taking advantage of the multifold reactivities of vinylcyclopropanes, a C–H activation/alkene insertion sequence would generate a rhodacycle A. Thereafter, a subsequent β-carbon elimination would be thus feasible (Fig. 1b).14 Herein, we demonstrate that RhIII is an efficient catalyst for a sequential C–H activation and C–C activation15 (β-carbon elimination) reaction with vinylcyclopropanes as coupling partners (Fig. 1c).
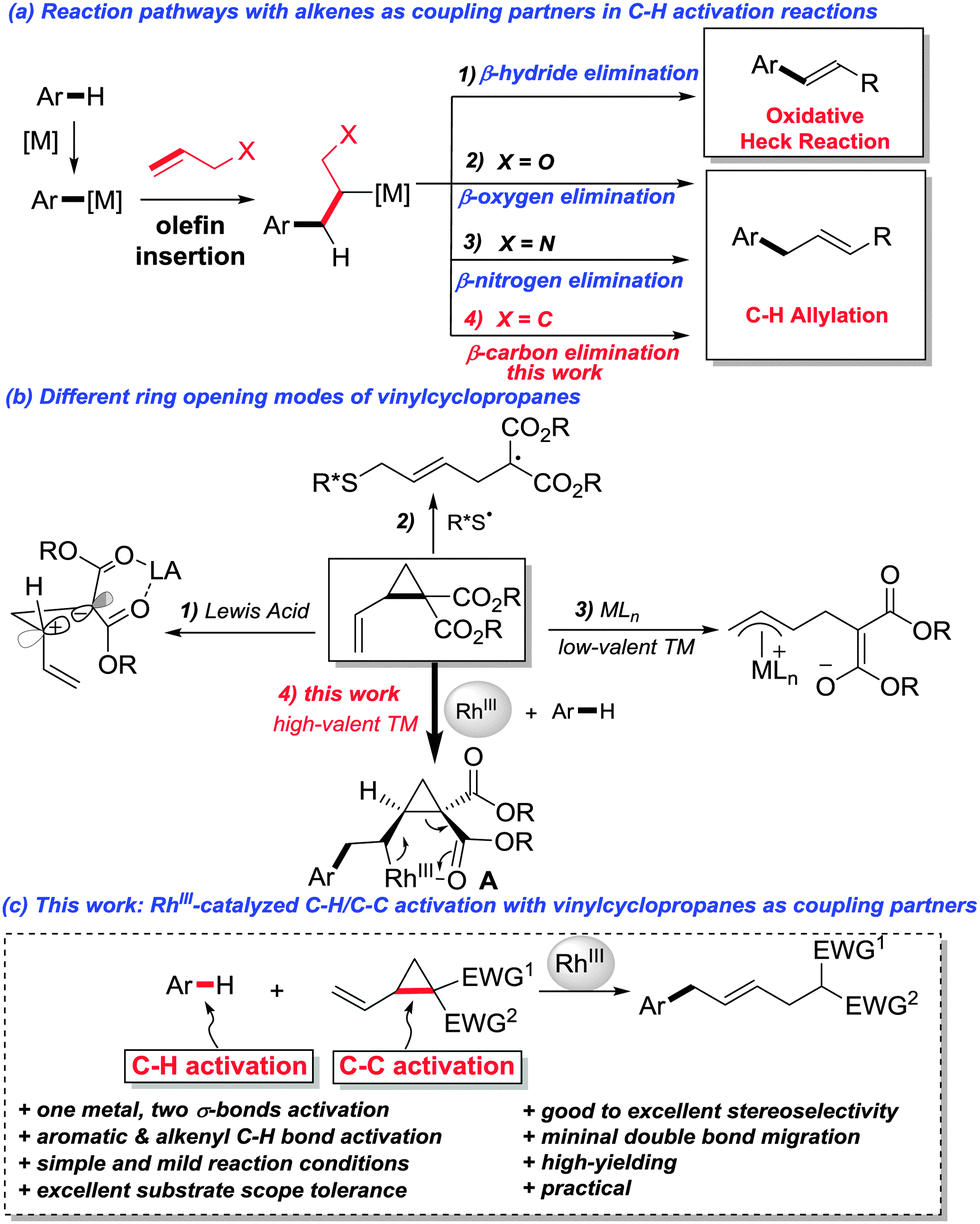 | ||
| Fig. 1 (a) Alkenes as coupling partners in C–H activation reactions; (b) ring opening modes of vinylcyclopropanes; (c) this work: RhIII-catalyzed C–H/C–C activation with vinylcyclopropanes as coupling partners. | ||
N-methoxybenzamides were chosen as substrates due to their high reactivities in C–H activation reactions. The reaction of 1a with dimethyl 2-vinylcyclopropane-1,1-dicarboxylate 2a in the presence of 5 mol% [RhCp*(CH3CN)3](SbF6)2 and 1 equivalent of CsOAc at 80 °C in MeOH delivered the desired product 3aa in 38% yield with an E/Z ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 1). It was found that CsOAc was crucial for the reaction as its omission or replacement with acid PivOH resulted in trivial reactivity (entries 2 and 3). Interestingly, water can also be used as solvent, affording 65% of the product, which demonstrates the great robustness of this novel transformation (entry 4). CF3CH2OH turned out to be a better solvent giving 90% yield (entry 5). Lowering the temperature from 80 °C to 30 °C gave a better stereoselectivity of 9:1 with a slight sacrifice in yield (78%, entry 6). A better E/Z ratio of 21:1 was achieved by switching the coupling partner from 2a to more sterically hindered diisopropyl 2-vinylcyclopropane-1,1-dicarboxylate 2b (entry 7). Notably, minimal migration of the newly formed double bond was observed under the optimized reaction conditions ([RhCp*(CH3CN)3](SbF6)2 (5 mol%), CsOAc (1.0 equiv.), CF3CH2OH (0.2 M), 30 °C).
:1 (Table 1, entry 1). It was found that CsOAc was crucial for the reaction as its omission or replacement with acid PivOH resulted in trivial reactivity (entries 2 and 3). Interestingly, water can also be used as solvent, affording 65% of the product, which demonstrates the great robustness of this novel transformation (entry 4). CF3CH2OH turned out to be a better solvent giving 90% yield (entry 5). Lowering the temperature from 80 °C to 30 °C gave a better stereoselectivity of 9:1 with a slight sacrifice in yield (78%, entry 6). A better E/Z ratio of 21:1 was achieved by switching the coupling partner from 2a to more sterically hindered diisopropyl 2-vinylcyclopropane-1,1-dicarboxylate 2b (entry 7). Notably, minimal migration of the newly formed double bond was observed under the optimized reaction conditions ([RhCp*(CH3CN)3](SbF6)2 (5 mol%), CsOAc (1.0 equiv.), CF3CH2OH (0.2 M), 30 °C).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Solvent | Additive | T [°C] | Yield [%] | E/Zb |
| a 1a (0.2 mmol), 2 (0.24 mmol), [RhCp*(CH3CN)3](SbF6)2 (5 mol%), additive (1.0 equiv.), solvent (1 mL), 24 h, isolated yield. b Determined by 1H NMR. c With additional tween 20 (30 mol%). | ||||||
| 1 | Me (2a) | MeOH | CsOAc | 80 | 38 | 9:1 |
| 2 | Me (2a) | MeOH | — | 80 | <5 | — |
| 3 | Me (2a) | MeOH | PivOH | 80 | <5 | — |
| 4 | Me (2a) | H2Oc | CsOAc | 30 | 65 | 8:1 |
| 5 | Me (2a) | CF3CH2OH | CsOAc | 80 | 90 | 6:1 |
| 6 | Me (2a) | CF3CH2OH | CsOAc | 30 | 78 | 9:1 |
| 7 | iPr ( 2b ) | CF3CH2OH | CsOAc | 30 | 79 | 21:1 |
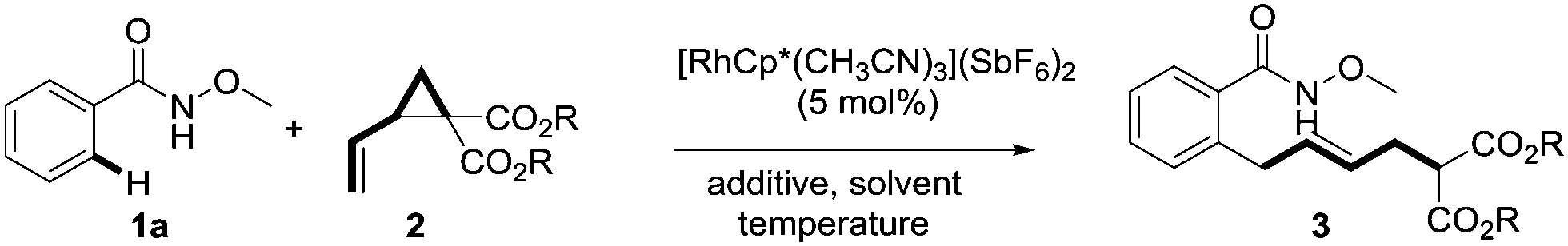
With the optimized reaction conditions in hand, we next explored the generality of this reaction by variation of N-methoxybenzamide 1 (Scheme 1). To our delight, the reaction is compatible with a variety of functionalities such as bromo (3bb), iodo (3cb), methoxy (3eb), trifluoromethyl (3fb), cyano (3hb), nitro (3ib), ester (3jb), acetyl (3kb), and even chloromethyl (3lb), providing ample opportunity for further derivatization of the products. Ortho-substituents did not hamper the reactivity (3mb–3pb). When meta-substituted substrates were used, good regioselectivities favouring the less hindered position were observed (3qb–3sb). N-Methoxy-2-naphthamide 1t provided the allylation product at the C3 position exclusively. Oxime represents another type of applicable substrate, giving the desired product in reasonable yield (3ub). The selective C2-allylation of indoles also worked well under the assistance of a pyrimidyl directing group, albeit with moderate E/Z ratios (3vb and 3wb).
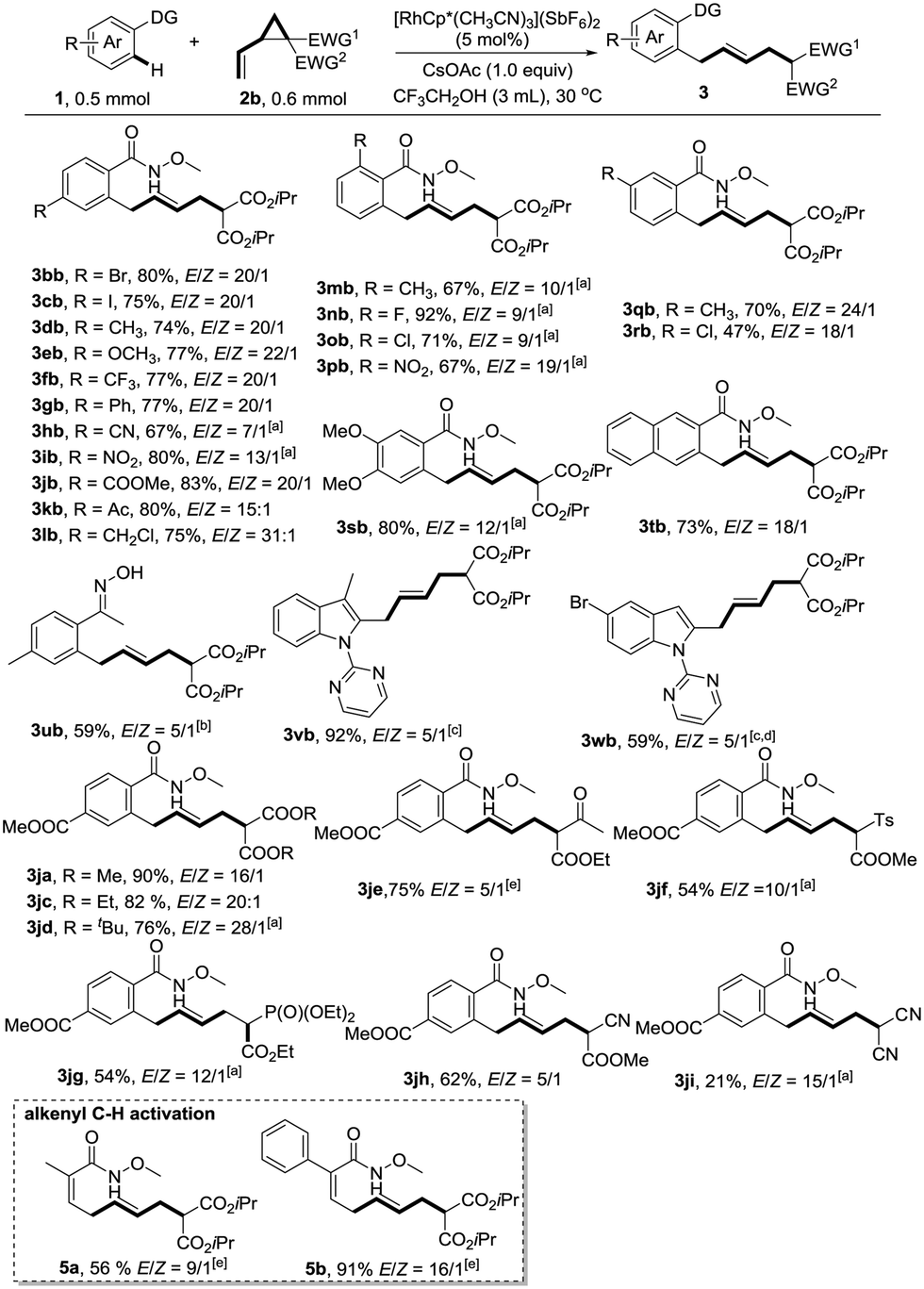 | ||
| Scheme 1 RhIII-catalyzed coupling reaction of vinylcyclopropanes 2b with various substrates. a50 °C; bMeOH was used as solvent, rt; cDCE was used as solvent, rt; dadditional 5 mol% RhIII was added after 24 h; e0 °C. | ||
The substrate scope of vinylcyclopropane 2 is also remarkable. VCPs 2 could be readily prepared from the corresponding activated methylene compounds and (E)-1,4-dibromobut-2-ene. As mentioned before, the introduction of a more sterically hindered group gave a better E/Z ratio, but at the same time retarded the reaction (3javs.3jbvs.3jd). Thus, di-tert-butyl 2-vinylcyclopropane-1,1-dicarboxylate 2d gave the highest E/Z ratio of 28:1. A slight increase of temperature to 50 °C was necessary to maintain the good yield (76%). A variety of other electron-withdrawing functional groups such as ketone (3je), sulfone (3jf), phosphonate (3jg), and cyano (3jh) were successfully employed in this reaction, giving the corresponding products in reasonable yields. However, the use of dicyano vinylcyclopropane provided the desired product 3ji in low yield (21%) due to the low conversion. The reaction was also applicable to alkenyl C–H activation reactions, giving skipped dienes with valuable handles for further transformations (5a and 5b). In general, good to excellent (5:1–31:1) E/Z ratios were observed. It should be mentioned that mixtures of diastereomers of 2e–h were used in this transformation.
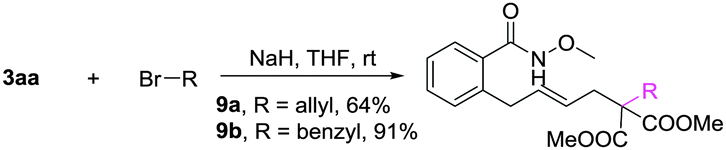
A gram-scale synthesis was performed using 2 mol% of catalyst and no decrease in efficiency was observed (eqn (1)). To document the potential utility of 3 in synthesis, the derivatization of 3 was conducted. Firstly, a Krapcho decarbalkoxylation of 3aa in the presence of NaCN in wet DMSO afforded the monoester 6 in 70% yield (eqn (2)).16 Secondly, a palladium(II)-catalyzed aerobic oxidative cyclization and a subsequent isomerization of the double bond gave isoquinolin-1(2H)-one 7 in 84% yield (eqn (3)). Furthermore, epoxidation of the double bond with 3-chloroperbenzoic acid (m-CPBA) followed by an intramolecular epoxide ring-opening delivered 1,2-amino alcohol 8 in 48% yield (eqn (4)). Finally, the elongations of the side chain were also successful using allyl bromide and benzyl bromide as alkylation reagents, forming the corresponding products 9a and 9b in 64% and 91% yield, respectively (eqn (5)).
To gain insight into the mechanism, a stoichiometric amount of TEMPO was subjected to the reaction to probe the possibility of a radical initiated ring-opening of cyclopropane.17 Comparable yield was obtained, indicating that a radical pathway is not likely.12,18 A small kinetic isotope effect value of 1.7 was obtained from a parallel experiment,17 suggesting that the C–H bond cleavage is not involved in the turn-over limiting step.19
To better understand the reaction mechanism, DFT computations17 were carried out (Fig. 2). The deprotonation of the amino group takes place first, which is followed by concerted metalation–deprotonation (CMD) to form a rhodacycle C.20 The free energy of the CMD transition state is 21.4 kcal mol−1. The removal of a neutral acetic acid from C is followed by the olefin coordination, generating (E)-D with an energy of 9.7 kcal mol−1. Olefin insertion into the Rh–C bond via a transition state (E)-D-TS gives the intermediate (E)-E. After that, a β-carbon elimination event takes place to cleave the carbon–carbon single bond of cyclopropane in (E)-F to form the intermediate (E)-G. This step was found to be energetically favored. The protonation of (E)-G under the assistance of AcOH via(E)-G-TS furnishes (E)-H. A proto-demetallation leads to the final product.
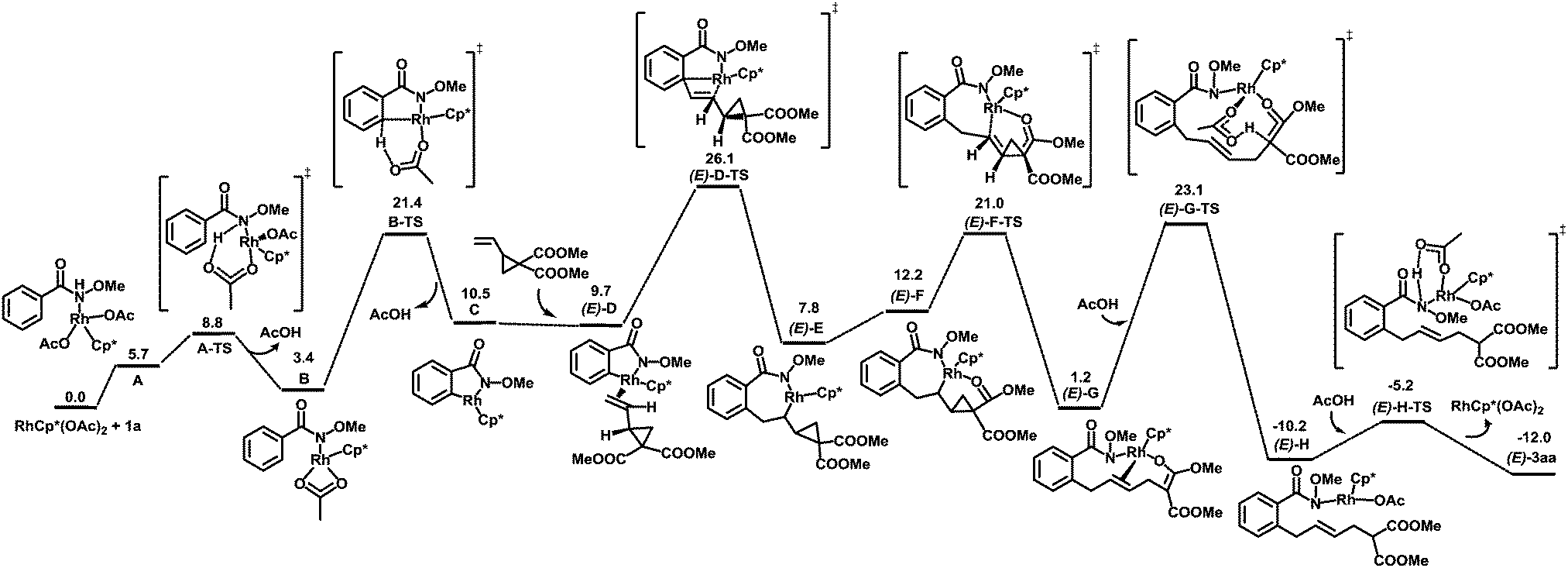 | ||
| Fig. 2 Calculated energy profiles for the RhIII-catalyzed sequential C–H/C–C activation reaction. | ||
Gram-scale synthesis:
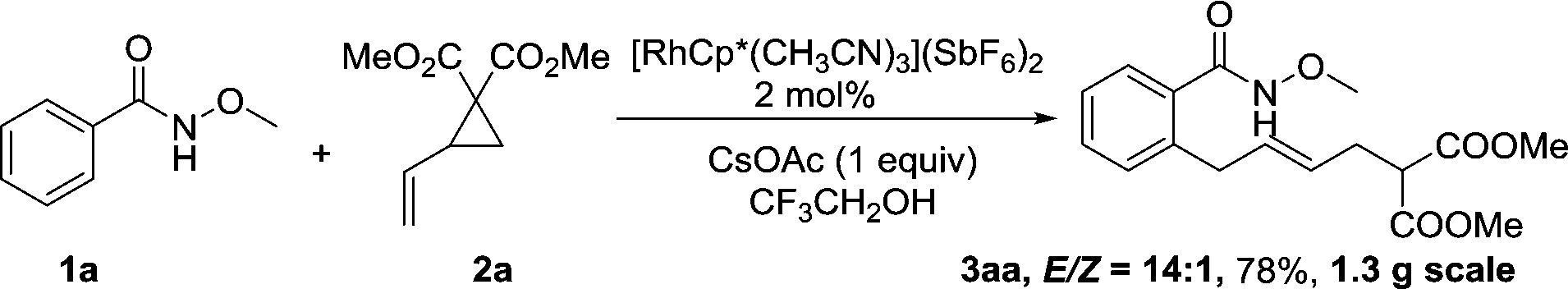 | (1) |
Krapcho decarbalkoxylation:
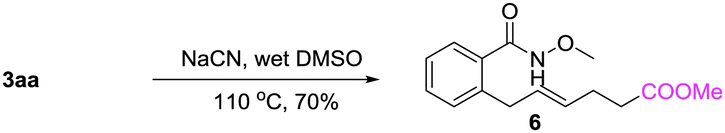 | (2) |
Oxidative cyclization:
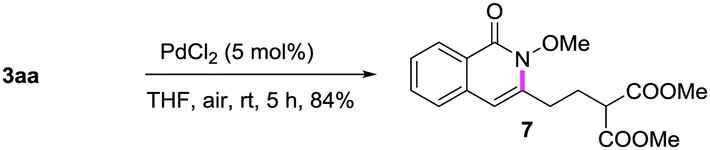 | (3) |
Epoxidation and subsequent cyclization:
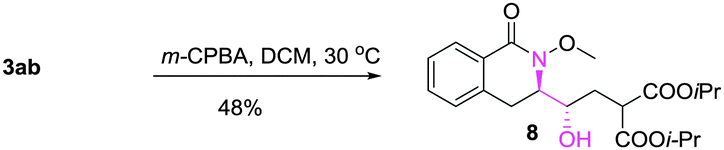 | (4) |
Side chain elongation:
 | (5) |
In summary, we have developed a RhIII-catalyzed sequential C–H/C–C activation reaction, by taking advantage of the multifold reactivity of vinylcyclopropanes. The reaction offers a simple and practical route for the synthesis of allylated arenes and skipped dienes with valuable handles. Besides, the reaction features excellent substrate scope tolerance, good stereoselectivity and is able to produce a high yield. Valuable building blocks were synthesized from the generated products. Mechanistic studies suggest that a formal β-carbon elimination is involved in the reaction mechanism. The reaction represents a rare example of unifying C–H and C–C cleavage into a single approach to synthesize complex molecules.
We are grateful for the support of this work through a Start-up Grant from Sun Yat-sen University and the National Natural Science Foundation of China (Grant No. 81330077 and 21103072).
Notes and references
- For recent reviews on C–H activation, see: (a) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (e) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (f) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (g) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (h) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (i) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (j) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef PubMed; (k) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (l) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (m) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (n) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- For recent reviews on RhIII-catalyzed C–H activations: (a) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (b) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2011, 44, 814 Search PubMed; (c) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (d) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS.
- (a) M. C. Willis, Chem. Rev., 2010, 110, 725 CrossRef CAS PubMed; (b) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed.
- For recent examples of RhIII-catalyzed C–H activation using alkenes as coupling partner, see: (a) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS PubMed; (b) S. H. Park, J. Y. Kim and S. Chang, Org. Lett., 2011, 13, 2372 CrossRef CAS PubMed; (c) A. S. Tsai, M. Brasse, R. G. Bergman and J. A. Ellman, Org. Lett., 2011, 13, 540 CrossRef CAS PubMed; (d) T. K. Hyster, L. Knorr, T. R. Ward and T. Rovis, Science, 2012, 388, 500 CrossRef PubMed; (e) B. Ye and N. Cramer, Science, 2012, 338, 504 CrossRef CAS PubMed; (f) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2013, 135, 66 CrossRef CAS PubMed; (g) Y. Unoh, Y. Hashimoto, D. Takeda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2013, 15, 3258 CrossRef CAS PubMed; (h) T. Zhang, L. Wu and X. Li, Org. Lett., 2013, 15, 6294 CrossRef CAS PubMed; (i) X.-S. Zhang, Q.-L. Zhu, Y.-F. Zhang, Y.-B. Li and Z.-J. Shi, Chem. – Eur. J., 2013, 19, 11898 CrossRef CAS PubMed; (j) S. C. Chuang, P. Gandeepan and C. H. Cheng, Org. Lett., 2013, 15, 5750 CrossRef CAS PubMed; (k) L. Huang, Q. Wang, J. Qi, X. Wu, K. Huang and H. Jiang, Chem. Sci., 2013, 4, 2665 RSC; (l) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188 CrossRef CAS PubMed; (m) Z. Shi, M. Boultadakis-Arapinis, D. C. Koester and F. Glorius, Chem. Commun., 2014, 50, 2650 RSC; (n) P. Becker, D. L. Priebbenow, R. Pirwerdjan and C. Bolm, Angew. Chem., Int. Ed., 2014, 53, 269 CrossRef CAS PubMed; (o) B. Li, J. Ma, W. Xie, H. Song, S. Xu and B. Wang, Chem. – Eur. J., 2013, 19, 11863 CrossRef CAS PubMed.
- Examples using palladium or ruthenium as catalyst, palladium: (a) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS; (b) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed; (c) R. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215 CrossRef CAS PubMed; (d) X.-S. Zhang, Z.-W. Li and Z.-J. Shi, Org. Chem. Front., 2014, 1, 44 RSC; (e) Y. Zhao, G. He, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2948 CrossRef CAS PubMed; (f) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586 CrossRef CAS PubMed; (g) K. J. Stowers, K. C. Fortner and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 6541 CrossRef CAS PubMed; (h) N. P. Grimster, C. Gauntlett, C. R. A. Godfrey and M. J. Gaunt, Angew. Chem., Int. Ed., 2005, 44, 3125 CrossRef CAS PubMed ; Ruthenium: ; (i) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS; (j) M. Schinkel, I. Marek and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 3977 CrossRef CAS PubMed.
- (a) H. Wang, N. Schroder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386 CrossRef CAS PubMed; (b) H. Wang, B. Beiring, D.-G. Yu, K. Collins and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12430 CrossRef CAS PubMed; (c) C. Feng, D. Feng and T.-P. Loh, Org. Lett., 2013, 15, 3670 CrossRef CAS PubMed; (d) S. Yu and X. Li, Org. Lett., 2014, 16, 1200 CrossRef CAS PubMed; (e) Z. Qi and X. Li, Angew. Chem., Int. Ed., 2013, 52, 8995 CrossRef CAS PubMed.
- (a) Y. Zhang, Q. Wu and S. Cui, Chem. Sci., 2014, 5, 297 RSC; (b) E. Jijy, P. Prakash, M. Shimi, P. M. Pihko, N. Josepha and K. V. Radhakrishnan, Chem. Commun., 2013, 49, 7349 RSC.
- (a) T. Satoh and M. Miura, Top. Organomet. Chem., 2005, 14, 1 CAS; (b) M. Murakami, M. Makino, S. Ashida and T. Matsuda, Bull. Chem. Soc. Jpn., 2006, 79, 1315 CrossRef CAS.
- (a) M. Gozin, A. Weisman, Y. Ben-David and D. Milstein, Nature, 1993, 364, 699 CrossRef CAS; (b) A. Masarwa and I. Marek, Chem. – Eur. J., 2010, 16, 9712 CrossRef CAS PubMed; (c) K. Chen, H. Li, Z.-Q. Lei, Y. Li, W.-H. Ye, L.-S. Zhang, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 9851 CrossRef CAS PubMed.
- Selected Reviews: (a) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (b) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed.
- (a) G. Sartori, F. Bigi, G. Casiraghi and G. Casnati, Tetrahedron, 1983, 39, 1761 CrossRef CAS; (b) D. B. England, T. D. O. Kuss, R. G. Keddy and M. A. Kerr, J. Org. Chem., 2001, 66, 4704 CrossRef CAS PubMed; (c) P. D. Pohlhaus, S. D. Sanders, A. T. Parsons, W. Li and J. S. Johnson, J. Am. Chem. Soc., 2008, 130, 8642 CrossRef CAS PubMed; (d) A. T. Parsons, A. G. Smith, A. J. Neel and J. S. Johnson, J. Am. Chem. Soc., 2010, 132, 9688 CrossRef CAS PubMed.
- T. Hashimoto, Y. Kawamata and K. Maruoka, Nat. Chem., 2014, 6, 702 CAS.
- Selected examples: (a) J. Moran, A. G. Smith, R. M. Carris, J. S. Johnson and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 18618 CrossRef CAS PubMed; (b) S. Sebelius, V. J. Olsson and K. J. Szabó, J. Am. Chem. Soc., 2005, 127, 10478 CrossRef CAS PubMed; (c) S. Sebelius, V. J. Olsson, O. A. Wallner and K. J. Szabó, J. Am. Chem. Soc., 2006, 128, 8150 CrossRef CAS PubMed; (d) Y. Sumida, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 4677 CrossRef CAS PubMed; (e) A. P. Dieskau, M. S. Holzwarth and A. Plietker, J. Am. Chem. Soc., 2012, 134, 5048 CrossRef CAS PubMed and references cited therein.
- For a recent example involving a C–H activation/nucleophilic addition to a ketone/β-carbon elimination mechanism, see: S. Yu and X. Li, Org. Lett., 2014, 16, 1220 CrossRef CAS PubMed.
- (a) A. Masarwa, D. Didier, T. Zabrodski, M. Schinkel, L. Ackermann and I. Marek, Nature, 2014, 505, 199 CrossRef CAS PubMed; (b) T. Seiser, O. A. Roth and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 6320 CrossRef CAS PubMed; (c) C. Aïssa and A. Fürstner, J. Am. Chem. Soc., 2007, 129, 14836 CrossRef PubMed; (d) S. I. Kozhushkov, D. S. Yufit and L. Ackermann, Org. Lett., 2008, 10, 3409 CrossRef CAS PubMed; (e) L. Ackermann, S. I. Kozhushkov and D. S. Yufit, Chem. – Eur. J., 2012, 18, 12068 CrossRef CAS PubMed.
- A. P. Krapcho, J. F. Weimaster, J. M. Eldridge, E. G. E. Jahngen, A. J. Lovey and W. P. Stephens, J. Org. Chem., 1978, 43, 138 CrossRef CAS.
- See ESI† for detail.
- J. E. Baldwin, Chem. Rev., 2003, 103, 1197 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, structural proofs, spectral data, mechanism study are provided. See DOI: 10.1039/c4cc07839j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |