
Highly efficient deep-blue phosphorescence from heptafluoropropyl-substituted iridium complexes†
Jung-Bum
Kim‡
a,
Seung-Hoon
Han‡
b,
Kiyull
Yang
c,
Soon-Ki
Kwon
*d,
Jang-Joo
Kim
*a and
Yun-Hi
Kim
*b
aDepartment of Materials Science and Engineering and the Center for Organic Light Emitting Diodes, Seoul National University, Seoul 151-742, South Korea. E-mail: jjkim@snu.ac.kr
bDepartment of Chemistry and ERI, Gyeongsang National University, Jinju 66-701, South Korea. E-mail: ykim@gnu.ac.kr
cDepartment of Chemistry Education, Gyeongsang National University, Jinju 660-701, South Korea
dSchool of Materials Science and Engineering and ERI, Gyeongsang National University, Jinju 66-701, South Korea. E-mail: skwon@gnu.ac.kr
First published on 31st October 2014
Abstract
New deep-blue iridium complexes, consisting of a heptafluoropropyl (HFP) substituent at the 3′ position of 2′,4′′-difluorophenyl, have a deep HOMO level and decreased shoulder electronic transition and inhibit self-quenching due to the sterically hindered group without conjugation. An OLED using (HFP)2Ir(mpic) exhibited a maximum EQE of 21.4% with a CIE of (0.146, 0.165).
Red and green phosphorescent iridium complexes are used in organic light emitting diodes (OLEDs) for displays and solid state lighting systems. Recently, external quantum efficiencies (EQEs) of over 30% have been reported.1–5 However, development of highly efficient blue, especially deep blue emitting phosphorescent dyes still remains a challenge for high performance full color displays and high quality white OLEDs. Iridium(III)bis[(4,6-difluorophenyl)-pyridinato-N,C2′] picolinate (FIrpic) has been widely used as a dopant material for sky blue PHOLEDs with high external quantum efficiency.6–14 However, it is difficult to synthesize deep blue phosphorescent materials that exhibit high color purity as well as high photoluminescence quantum yields (PLQYs).15–17 Recently, a couple of strategies have been developed for deep-blue emitting iridium complexes, which are used in high-efficiency, deep blue phosphorescent OLEDs; however, the color purity was rather limited.18,19
One strategy involves controlling the energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) localized primarily on the phenyl and pyridyl moieties, respectively, of the cyclometalated ligands.20 The electron-withdrawing group in the phenyl part lowers the HOMO level while the electron-donating group in the pyridyl part increases the LUMO level. We had previously reported that the position of the substituent in dimethylated iridium complexes affects the color purity and efficiency of the corresponding OLEDs.21 We also recently reported that overly strong electron-withdrawing groups such as the heptafluorocarbonyl unit decreased both HOMO and LUMO levels.22 Therefore, appropriately strong electron-withdrawing groups are required to effectively decrease the HOMO level without lowering the LUMO level. A second strategy is to control the vibration shoulder intensities to have larger emission oscillator strength at the shortest wavelength than the oscillator strength of other longer wavelength shoulder peaks. Since most of the blue-emitting iridium complexes have vibrational shoulder peaks at a longer wavelength, larger oscillator strengths of the longer wavelength shoulder peaks have an effect of diminishing the color purity for deep blue.
In this communication, we report three new deep-blue iridium complexes, which consist of a heptafluoropropyl (HFP) substituent at the 3′ position of 2′,4′′-difluorophenyl as the main ligand and a picolinate (pic), a picolinate with 3-methylpyridine (mpic) or a trifluoromethylated triazole (fptz) as the ancillary ligand. HFP was selected as an electron-withdrawing group that does not exhibit extended π-conjugation and a methyl group at the 4 position of the pyridine group for main ligand. Perfluoro alkyl groups have several effects in Ir(III) cyclometalated complexes. One is an appropriate electron-withdrawing effect without conjugation on the ligand π-system. The density functional theory (DFT) calculation showed that the heptafluoro alkyl group more strongly affects the HOMO level than the LUMO level, resulting in a large band gap and small oscillator strength of long wavelength vibrational shoulder peaks leading to deep-blue emission. Another effect is to provide steric hindrance around the metal to increase the PLQY.23 In addition, fluorinated substituents in the aromatic ligands of metal complexes make them highly volatile, enabling facile, high-yielding purification by sublimation, a feature beneficial for the mass production of displays. A trifluoromethyl-substituted triazole or a picolinate ancillary ligand is also utilized so that C-linked 2-pyridyl-azoles can form a stable chelate using two proximal nitrogen atoms. In addition, trifluoromethyl-substituted triazoles are strongly acidic, a feature reinforced by the incorporation of the electron-withdrawing heptafluoropropyl substituent.24
Fig. 1 illustrates the synthesis of the heptafluoropropyl substituted ligand and iridium complexes. The ligand, 2-(2,4-difluoro-3-(perfluoropropyl)phenyl)-4-methylpyridine, was obtained by Ulmann coupling of heptafluoropropyl iodide and 2-(2,4-difluoro-3-iodophenyl)-4-methylpyridine. 2-(2,4-Difluorophenyl)-4-methylpyridine was synthesized using iodine and under lithium diisopropylamide (LDA) base conditions. The cyclometalated iridium μ-chloro-bridged dimer was prepared by the reaction of the ligand and IrCl3·H2O in a mixture of 2-ethoxyethanol and water by the well-known Nonoyama reaction. The new iridium complexes were prepared by reacting the chloro-bridged dimer with the ancillary ligands in 2-ethoxyethanol with sodium bicarbonate. The new iridium complexes, (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz), were characterized by 1H-NMR and 19F-NMR spectroscopy and high-resolution mass spectrometry (HRMS) (Synthesis details in ESI†).
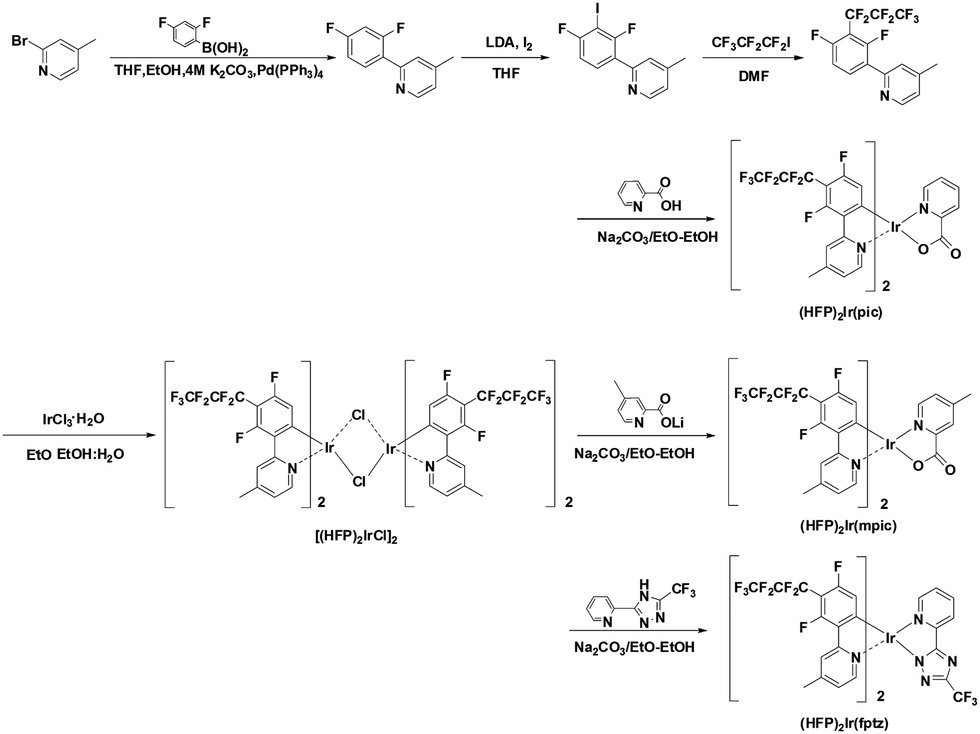 | ||
| Fig. 1 Synthetic schemes of the iridium complexes. | ||
DFT calculations of the iridium complexes were carried out by optimizing the geometries of the molecules using the B3LYP/6-31G basis set for the ligands, and the relativistic effective core potential of Los Alamos and Double-basis sets (LANL2DZ)25 for Ir (as implemented in Gaussian 03 package22). Time-dependent-DFT (TD-DFT)26 calculations were also performed to estimate the energy levels, orbital electron density distributions and singlet and triplet transition energies on the basis of the structures optimized in their ground states. The HOMO, HOMO − 1, LUMO, and LUMO + 1 for (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz) are shown in Fig. S1 (ESI†). The HOMO levels of (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz) were mostly distributed over the phenyl ring of the main ligand, with a large contribution from the d atomic orbital and a slight contribution from the ancillary ligand. For comparison, we calculated the HOMO and LUMO levels of (TFM)2Ir(pic), (TFM)2Ir(mpic), and (TFM)2Ir(fptz) with the already reported trifluoromethyl group.27 The HOMO levels of (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz) with the strongly electron withdrawing heptafluoropropyl group were calculated as −6.22, −6.17, and −6.49 eV, respectively, while the HOMO levels of (TFM)2Ir(pic), (TFM)2Ir(mpic), and (TFM)2Ir(fptz) with the trifluoromethyl group were calculated to be −6.17, −6.12, and −6.45 eV, respectively (see ESI,† Fig. S1, Tables S3–S6). These results suggest that the introduction of strongly electron-withdrawing heptafluoropropyl groups lowers the HOMO levels. The LUMO orbitals of the complexes were mostly distributed over one of the main ligands and the ancillary ligand. The LUMO levels of (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz) with the strongly electron withdrawing heptafluoropropyl group were calculated as −2.35, −2.25, and −2.46 eV, respectively, while those of (TFM)2Ir(pic), (TFM)2Ir(mpic), and (TFM)2Ir(fptz) with the trifluoromethyl group were calculated to be −2.33, −2.23, and −2.43 eV, respectively. Compared with the trifluoromethyl group, the strongly electron-withdrawing heptafluoropropyl group has a greater effect on the HOMO level than it does on the LUMO level. In addition, compared to the picolinate ancillary ligand, the strongly electron-withdrawing triazole ancillary ligand decreased the HOMO level while the electron-donating methylated picolinate ancillary ligand increased the LUMO level. As a result, the band gaps increase in the following order: (HFP)2Ir(pic) < (HFP)2Ir(mpic) < (HFP)2Ir(fptz). Moreover, the band gaps are larger than those of (TFM)2Ir(pic), (TFM)2Ir(mpic), and (TFM)2Ir(fptz), each of which contains the trifluoromethyl group. Recently, we reported that the strong heptafluorocarbonyl substituted iridium complexes, (HF)2Ir(pic) and (HF)2Ir(fptz), lowered the LUMO levels more than trifluorocarbonyl substituted iridium complexes, (TF)2Ir(pic) and (TF)2Ir(fptz), increasing the band gaps in the following order: (HF)2Ir(pic) < (TF)2Ir(pic) < (HF)2Ir(fptz) < (TF)2Ir(fptz).27 These results suggest that the strongly electron-withdrawing heptafluoropropyl group is an effective substituent for accessing the deepest blue emission.
Cyclic voltammetry (CV) was used to measure the HOMO levels of the iridium complexes (Fig. S2, ESI†). The HOMO levels of (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz) were found to be −5.94, −5.92, and −6.13 eV, respectively. The HOMO levels are in the same order as the DFT calculation. The LUMO levels were obtained from the optical band gaps and the HOMO levels. The HOMO and LUMO levels and the optical band gaps are summarized in Table S1 (ESI†).
Thermal properties were evaluated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) under a nitrogen atmosphere. The 5% weight loss temperatures of the iridium complexes were 366 °C for (HFP)2Ir(pic), 368 °C (HFP)2Ir(mpic), and 355 °C for (HFP)2Ir(fptz). Glass transition was not observed up to 250 °C. These results suggest that the new iridium complexes have good thermal stability (Fig. S3, Table S7, ESI†).
Fig. 2(a) shows the ultraviolet-visible (UV-vis) absorption and PL spectra of the iridium complexes in a CHCl3 solution (10−5 M) at room temperature. Each of the complexes, (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz), displayed strong absorption bands at around 255 nm, which can be attributed to intraligand π–π* transitions. The absorption bands at 277 nm for (HFP)2Ir(pic), 278 nm for (HFP)2Ir(mpic) and 272 nm for (HFP)2Ir(fptz) represent the spin-allowed 1π–π* transition (ligand-centered absorptions). The absorptions shown at 370 nm for (HFP)2Ir(pic), 372 nm for (HFP)2Ir(mpic), and 356 nm for (HFP)2Ir(fptz) can be assigned to the singlet metal to ligand charge-transfer (1MLCT) transitions. The spin-forbidden triplet 3MLCT or 3LC transitions (or both) appeared as low-energy absorption shoulders at approximately 410 nm for the complexes with the trifluoromethyl-substituted triazole and 420 nm for the complexes with the picolinate and the methylpicolinate ancillary ligand, respectively. The absorption maximum of the iridium complex with the methyl picolinate ancillary ligand was similar to that with the picolinate ancillary ligand, while the absorption maximum of the iridium complex with the trifluoromethyl-substituted triazole ancillary ligand was blue shifted by approximately 10 nm. The complexes (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz) showed intense blue phosphorescence in CHCl3 solution at room temperature, with the emission peaks at 451 and 479 nm for (HFP)2Ir(pic), 452 and 480 nm for (HFP)2Ir(mpic), and 446 and 475 nm for (HFP)2Ir(fptz).
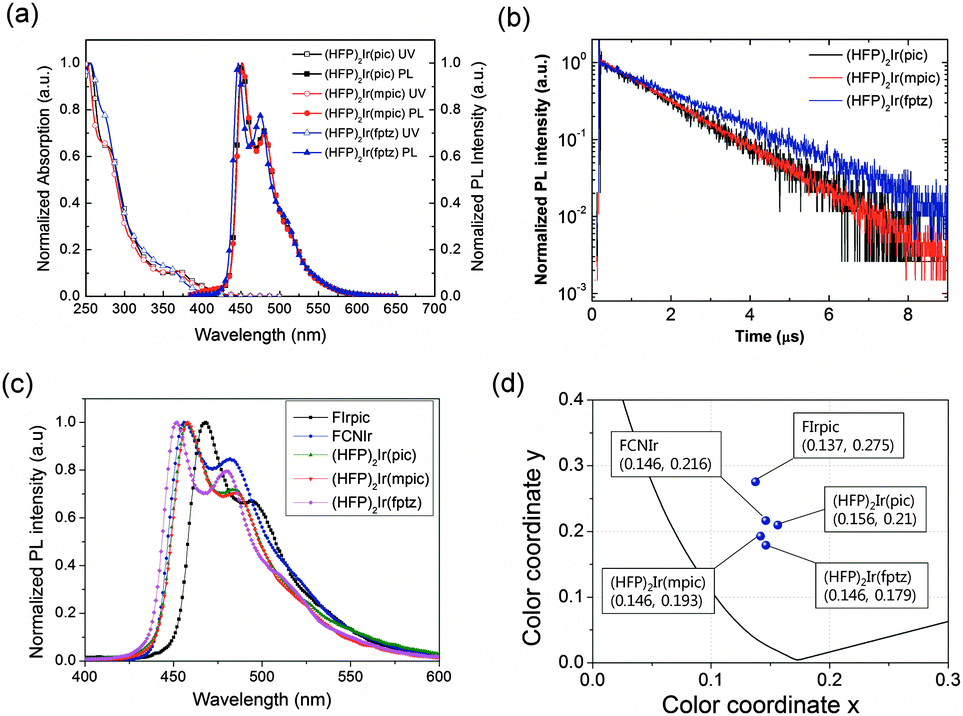 | ||
| Fig. 2 (a) UV-vis absorption and PL spectra of the iridium complexes in CHCl3. (b) Transient PL spectra of mCPPO1 films doped with 10 wt% iridium complexes. (c) PL spectra of mCPPO1 films doped with 10 wt% Firpic, FCNIr, (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz). (d) (CIE) coordinates calculated from PL spectra of Firpic, FCNIr, (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz). | ||
The PLQYs of the iridium complexes were measured using 9-(3-(9H-carbazole-9-yl)phenyl)-3-(dibromophenylphosphoryl)-9H-carbazole (mCPPO1) films doped with 10 wt% of the iridium complexes. An integrating sphere was used for the measurements. The PLQYs of (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz) were determined to be 81 ± 3%, 90 ± 3%, and 53 ± 3%, respectively. The heptafluoropropyl substitutents with a bulky ancillary ligand within the iridium complexes enhanced the steric hindrance of molecules to suppress the self-quenching due to reduction of bimolecular interactions, increasing the PLQYs.22,23
The transient PL of the doped films is shown in Fig. 2(b). The exciton lifetimes of the iridium complexes were 1.46, 1.47, and 1.99 for (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz), respectively. The iridium complexes with the picolinate-based ancillary ligand show relatively shorter lifetimes than does the iridium(III) complex with the trifluoromethyl-substituted triazole ligand.
Fig. 2(c) shows the normalized PL spectra of mCPPO1 films doped with 10 wt% Firpic, FCNIr, (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz). It is interesting to note that (HFP)2Ir(pic) and (HFP)2Ir(mpic) showed the lowest intensity of the 0–1 vibronic transitions among the complexes, resulting in the deepest blue Commission Internationale de l'Eclairage (CIE) coordinates calculated from the PL spectra of blue emitters. The color properties of materials are illustrated in Fig. 2(d), which show that our iridium complexes have the deepest CIE coordinates.
Phosphorescent blue OLEDs were fabricated using the iridium complexes as dopants within a phosphorescent emitting layer. The device structure of the phosphorescent blue OLEDs consisted of the following: glass/indium tin oxide (ITO, 70 nm)/ReO3 (4 wt%) doped N,N′-dicarbazolyl-3,5-benzene (mCP, 30 nm)/mCP (10 nm)/mCPPO1:iridium complexes (30 nm, 10 wt%)/diphenylphosphine oxide-4-(triphenylsilyl)phenyl (TSPO1, 10 nm)/Rb2CO3 (8 wt%)-doped mCP (30 nm)/Al (100 nm). The hole injection layer (HIL) was doped with a p-type dopant, ReO3 [rhenium(VI) oxide],28–30 and the electron injection layer (EIL) was doped with an n-type dopant, Rb2CO3,31–33 to facilitate efficient hole and electron injection from the electrodes. The mCP and TSPO1 layers functioned as the hole- and electron-transporting layers, respectively. Compound mCPPO1 was chosen as the host material owing to its high triplet energy and the balance of electron and hole mobilities that it offers.19 The LUMO level of mCPPO1 was 0.24 eV lower than that of mCP, and its HOMO level was 0.59 eV higher than that of TSPO1, leading to effective exciton blocking. Moreover, the triplet energy levels of all the constituent materials were higher than those of the iridium complexes. Therefore, it was expected that charge carrier and triplet exciton would be confined within the emitting material layer (EML).
The current density–voltage–luminance (J–V–L) characteristics of the OLEDs are shown in Fig. 3(b). The J–V characteristics of all the three devices were similar because of their similar structure, which varied only by the dopants used in each. In contrast, the devices based on (HFP)2Ir(pic) and (HFP)2Ir(mpic) showed higher maximum luminance than did (HFP)2Ir(fptz). The external quantum efficiencies (EQEs) of the OLEDs are shown in Fig. 3(c). The device with (HFP)2Ir(mpic) showed the highest EQE among the three devices followed by (HFP)2Ir(pic) and (HFP)2Ir(fptz), an observation consistent with the PLQYs of the dopants. The maximum EQEs were 19.7%, 21.4% and 14.2% for the devices containing (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz), respectively. The power efficiencies of the devices were 22.3, 25.9 and 14.5 lm W−1 for the devices containing (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz), respectively. The performances of the three OLEDs are summarized in Table S2 (ESI†). The electroluminescence (EL) spectra of the three devices at 5 mA cm−2 are shown in Fig. 3(d). The emission peaks (ELmax) of the OLEDs based on (HFP)2Ir(pic) and (HFP)2Ir(mpic) were located at 452 nm, while that of the device based on (HFP)2Ir(fptz) was located at 447 nm. The values of the full width at half maximum for the emission of all three devices were less than 50 nm. Owing to the spectral purities of the emissions from the OLEDs, the CIE coordinates (x,y) of the devices containing (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz) at 100 cd m−2 were (0.147, 0.164), (0.146, 0.165), and (0.152, 0.148), respectively.
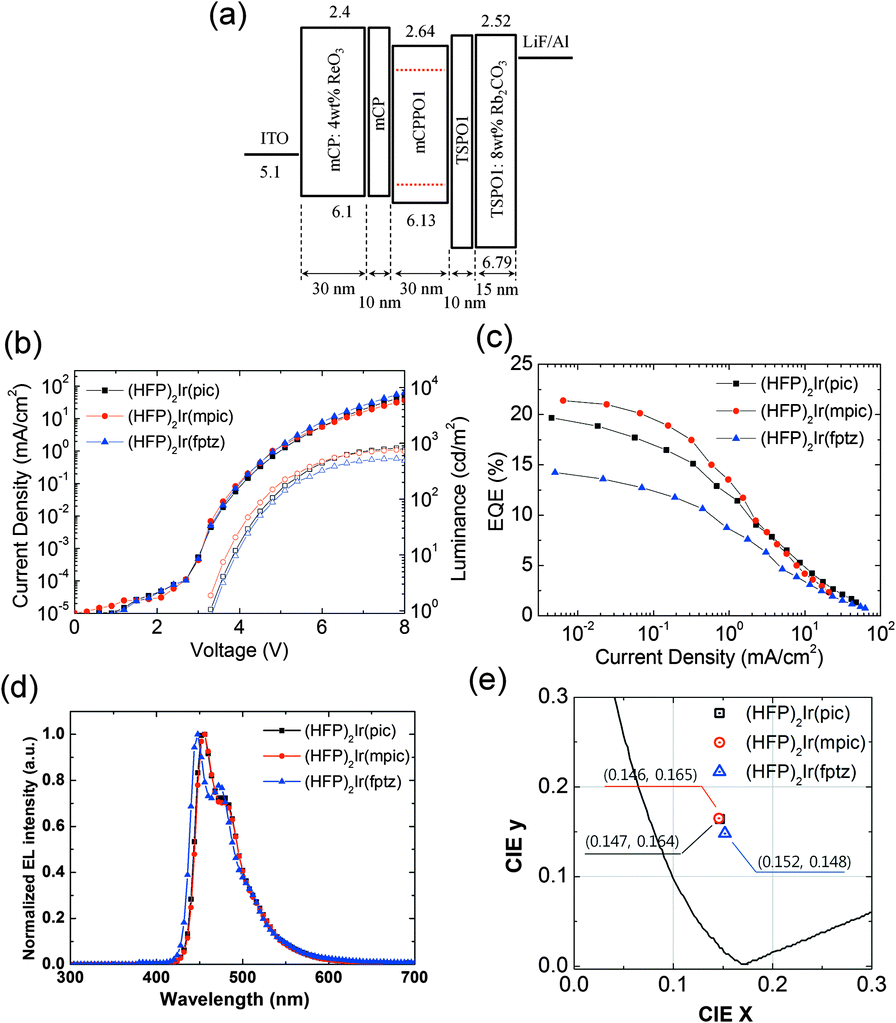 | ||
| Fig. 3 (a) Device structure and energy level diagrams of the OLEDs, (b) current density–voltage–luminance curves, (c) quantum efficiencies versus current density curves, and (d) normalized EL spectra of the different phosphorescent blue OLEDs at 5 mA cm−2. (e) Comparison of the CIE coordinates of the EL from the OLEDs. | ||
Three new deep-blue iridium(III) complexes, (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz), consisting of a heptafluoropropyl substituent at the 3′ position of 2′,4′′-difluorophenyl-3-methylpyridine as the main ligand and a picolinate, a picolinate with 3-methylpyridine or a trifluoromethylated triazole as the ancillary ligand, were successfully synthesized and characterized. We demonstrated that the strong electron-withdrawing heptafluoropropyl groups affect the HOMO level more than the LUMO level, as determined by TD-DFT calculations. As a result, these iridium complexes exhibited wide band gaps with high PL QYs. Phosphorescent OLEDs based on (HFP)2Ir(pic), (HFP)2Ir(mpic), and (HFP)2Ir(fptz) exhibited high maximum EQEs of 19.7%, 21.4% and 14.2% and CIE coordinates of (0.147, 0.164), (0.146, 0.165) and (0.152, 0.148), respectively. These CIE coordinates represent some of the deepest blue emissions ever achieved from phosphorescent OLEDs with considerably high EQEs.
This work was supported by the industry technology R&D program of MOITE/KEIT (10048317).
Notes and references
- D. Tanaka, H. Sasabe, Y.-J. Li, S.-J. Su, T. Takeda and J. Kido, Jpn. J. Appl. Phys., 2007, 46, 1 CrossRef.
- M. G. Helander, Z. B. Wang, J. Qiu, M. T. Greiner, D. P. P. Puzzo, Z. W. Liu and Z. H. Lu, Science, 2011, 332, 944 CrossRef CAS PubMed.
- Y.-S. Park, S. Lee, K.-H. Kim, S.-Y. Kim, J.-H. Lee and J.-J. Kim, Adv. Funct. Mater., 2013, 23, 4914 CrossRef CAS.
- S.-Y. Kim, W.-I. Jeong, C. Mayr, Y.-S. Park, K.-H. Kim, J.-H. Lee, C.-K. Moon, W. Brütting and J.-J. Kim, Adv. Funct. Mater., 2013, 23, 3896 CrossRef CAS.
- S. Lee, K.-H. Kim, D. Limbach, Y.-S. Park and J.-J. Kim, Adv. Funct. Mater., 2013, 23, 4105 CrossRef CAS.
- L. Xiao, S.-J. Su, Y. Agata, H. Lan and J. Kido, Adv. Mater., 2009, 21, 1271 CrossRef CAS.
- S.-J. Su, T. Chiba, T. Takeda and J. Kido, Adv. Mater., 2008, 20, 2125 CrossRef CAS.
- H. Sasabe, E. Gonmori, T. Chiba, Y.-J. Li, D. Tanaka, S.-J. Su, T. Takeda, Y.-J. Pu, K. Nakayama and J. Kido, Chem. Mater., 2008, 20, 5951 CrossRef CAS.
- N. Chopra, J. Lee, Y. Zheng, S.-H. Eom, J. Xue and F. So, Appl. Phys. Lett., 2008, 93, 143307 CrossRef PubMed.
- J.-K. Bin, N.-S. Cho and J.-I. Hong, Adv. Mater., 2012, 24, 2911 CrossRef CAS PubMed.
- C. Fan, Y. Li, C. Yang, H. Wu, J. Qin and Y. Cao, Chem. Mater., 2012, 24, 4581–4587 CrossRef CAS.
- C. Fan, L. Zhu, B. Jiang, D. Ma, J. Qin and C. Yang, Org. Electron., 2013, 14, 3163–3171 CrossRef CAS PubMed.
- C.-L. Ho and W.-Y. Wong, New J. Chem., 2013, 37, 1665–1683 RSC.
- C.-L. Ho, H. Li and W.-Y. Wong, New J. Organomet. Chem., 2014, 751, 261 CrossRef CAS PubMed.
- R. J. Holmes, S. R. Forrest, T. Sajoto, A. Tamayo, P. I. Djurovich, M. E. Thompson, J. Brooks, Y.-J. Tung, B. W. D'Andrade, M. S. Weaver, R. C. Kwong and J. J. Brown, Appl. Phys. Lett., 2005, 87, 243507 CrossRef PubMed.
- C.-H. Yang, Y.-M. Cheng, Y. Chi, C.-J. Hsu, F.-C. Fang, K.-T. Wong, P.-T. Chou, C.-H. Chang, M.-H. Tsai and C.-C. Wu, Angew. Chem., Int. Ed., 2007, 46, 2418 CrossRef CAS PubMed.
- Y.-C. Chiu, J.-Y. Hung, Y. Chi, C.-C. Chen, C.-H. Chang, C.-C. Wu, Y.-M. Cheng, Y.-C. Yu, G.-H. Lee and P.-T. Chou, Adv. Mater., 2009, 21, 2221 CrossRef CAS.
- S. O. Jeon, K. S. Yook, C. W. Joo and J. Y. Lee, Adv. Mater., 2010, 22, 1872 CrossRef CAS PubMed.
- S. O. Jeon, K. S. Yook, C. W. Joo and J. Y. Lee, Adv. Mater., 2011, 23, 1436 CrossRef CAS PubMed.
- P. J. J. Hay, Phys. Chem. A, 2002, 06, 1634 CrossRef.
- S. O. Jung, Y. Kang, H. S. Kim, Y. H. Kim, C. L. Lee, J.-J. Kim and S. K. Kwon, Eur. J. Inorg. Chem., 2004, 3415 CrossRef CAS.
- S. Lee, S.-O. Kim, H. Shin, H.-J. Yun, K. Yang, S.-K. Kwon, J.-J. Kim and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14321 CrossRef CAS PubMed.
- V. V. Grushin, N. Herron, D. D. LeCloux, W. J. Marshall, V. A. Petrov and Y. Wang, Chem. Commun., 2001, 1494, 10.1039/B103490C.
- S.-O. Kim, Q. Zhao, K. Thangaraju, J.-J. Kim, Y.-H. Kim and S.-K. Kwon, Dyes Pigm., 2011, 90, 139 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS PubMed.
- H.-J. Seo, K.-M. Yoo, M. Song, J.-S. Park, S.-H. Jin, Y.-I. Kim, Y.-I. Kim and J.-J. Kim, Org. Electron., 2010, 11, 564 CrossRef CAS PubMed.
- D.-S. Leem, H.-D. Park, J.-W. Kang, J.-H. Lee, J. W. Kim and J.-J. Kim, Appl. Phys. Lett., 2007, 91, 011113 CrossRef PubMed.
- J.-H. Lee, D.-S. Leem, H.-J. Kim and J.-J. Kim, Appl. Phys. Lett., 2009, 94, 123306 CrossRef PubMed.
- J.-H. Lee, D.-S. Leem and J.-J. Kim, Org. Electron., 2010, 11, 486 CrossRef CAS PubMed.
- D.-S. Leem, S.-Y. Kim, J.-J. Kim, M.-H. Chen and C.-I. Wu, Electrochem. Solid-State Lett., 2009, 12, J8 CrossRef CAS PubMed.
- M.-H. Chen, Y.-H. Chen, C.-T. Lin, G.-R. Lee, C.-I. Wu, D.-S. Leem, J.-J. Kim and T.-W. Pi, J. Appl. Phys., 2009, 105, 113714 CrossRef PubMed.
- S. Lee, J.-H. Lee, J.-H. Lee and J.-J. Kim, Adv. Funct. Mater., 2011, 21, 855 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, CV, TGA and DFT calculations. See DOI: 10.1039/c4cc07768g |
| ‡ Jung-Bum Kim and Seung-Hoon Han contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |