
Fluoride, bifluoride and trifluoromethyl complexes of iridium(I) and rhodium(I)†
Byron J.
Truscott
,
Fady
Nahra
,
Alexandra M. Z.
Slawin
,
David B.
Cordes
and
Steven P.
Nolan
*
EaStCHEM, School of Chemistry, University of St. Andrews, North Haugh, St. Andrews, Fife KY16 9ST, UK. E-mail: snolan@st-andrews.ac.uk
First published on 27th October 2014
Abstract
Herein we report robust methods for the preparation and full characterisation of a range of Ir(I) and Rh(I) fluoride and bifluoride complexes using N-heterocyclic carbenes (NHCs) as ancillary ligands. The processes that link the fluoride and the bifluoride species are investigated and reports of the first Ir–bifluoride and Ir(I)–NHC and Rh(I)–NHC trifluoromethyl complexes are revealed.
Fluorinated organometallic complexes are highly interesting species. Fluorides are known to confer unique properties to the metal and their coordination can lead to fascinating reactivity. Nevertheless, metal fluorides remain the least understood member of the metal-halide family.1
Additionally, the catalytic preparation of fluorinated organic compounds is an area of great importance, particularly for the radio therapeutic, pharmaceutical and agrochemical sectors.2 The isolation of fluorinated organometallic complexes permits us to study the behaviour of potential catalysts and catalytic intermediates under homogeneous conditions.3 This knowledge is of paramount importance in designing new catalytic cycles and improving existing fluorination methods.2b,4
The interaction between a hard base such as fluoride and a low-oxidation state metal (behaving as a soft Lewis acid) should lead to weak metal–fluoride bonds.1a,5 Stabilisation of M–F bonds and isolation of LTM–fluorides has mostly been reported using phosphine, N-donor or σ-aryl ancillary ligands.1,6 Some of the most extensively investigated fluoride complexes are analogues of Vaska's complex ([IrCl(CO)(PPh3)2]) and related rhodium complexes.1a,6c,7 In contrast, fluoride complexes bearing olefinic ligands remain scarce,8 and even more scarce are those containing N-heterocyclic carbenes (NHCs).6a,9 NHC ligands have featured extensively in the literature as electron-rich donor ligands, and have proven particularly useful in stabilising hard/soft metal–ligand mismatches such as metal hydroxides.10
Bifluoride complexes [M(FHF)] are very interesting, and since the first report by Coulson11 on Pt, several examples of metal bifluorides have been reported, including Mn,12 W,13 Pd,14 Ru,15 Mo,16 Ni,17 and Rh,7a,8b,18 mostly bearing phosphine or amine based supporting ligands. The only examples of LTM–HF2 complexes bearing NHC ligands are those reported by the groups of Leyssens on Cu;19 and by Whittlesey on Rh (bearing ring expanded six-membered NHC ligands).18c,20 Furthermore, reports of LTM(NHC) complexes bearing trifluoromethyl (CF3) groups are exclusive to Cu.21 In order to build on the reactivity of Ir(I) and Rh(I) compounds, we aimed to establish robust methods for the preparation of fluorinated Ir(I)- and Rh(I)-complexes bearing F, HF2 and CF3 moieties.
Based on previous success in stabilising M–OH bonds on Rh(I) and Ir(I), our strategy was to use the [M(cod)(NHC)(X)] scaffold (M = Rh, Ir; cod = 1,5-cyclooctadiene; X = Cl, OH) to gain access to M(I)–F bonds.10 Gratifyingly, a transmetallation/halide substitution reaction between [Ir(cod)(NHC)Cl] and AgF6b delivered [Ir(cod)(NHC)F] complexes 1–4 as yellow solids in excellent yields (Scheme 1). Multinuclear NMR spectroscopy and elemental analysis were used to unambiguously confirm the identity of the products with the 19F NMR signals corresponding to the Ir(I)–F moiety resonating between −221 and −227 ppm (see ESI†).
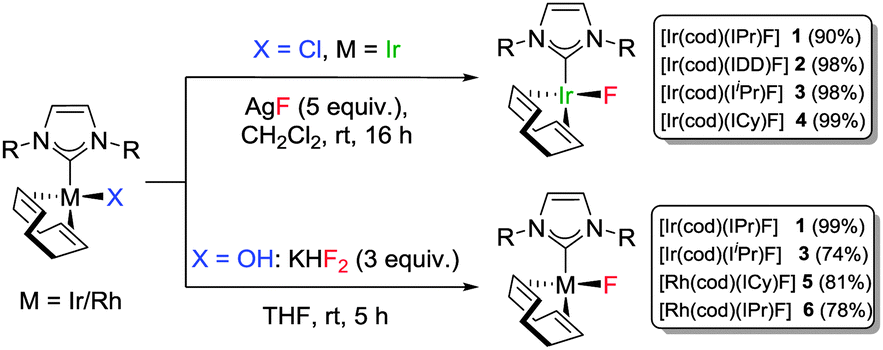 | ||
| Scheme 1 Preparation of Ir(I) and Rh(I)–fluoride complexes. | ||
It was clear that the three species were intrinsically linked and we were able to convert the fluoride complex to either the Ir(I)–OH (via reaction with CsOH or KOH), or to the Ir(I)–Cl (via reaction with NaCl). However, when we attempted to extend the methodology to rhodium, the reactions were at best very slow (65% conversion after 60 h for [Rh(cod)(IPr)Cl]). Treating either of the metal chlorides or hydroxides with fluoride salts such as KF or CsF was also unsuccessful and a new approach had to be devised. To our delight, the Rh(I)–OH reacted with cheap and readily available KHF2 to deliver the desired Rh(I)–F in high yield in just 5 h. This method proved general for both metals (Scheme 1) and analysis by 19F NMR indicated that Rh(I)–F signals resonated at a higher field than the analogous Ir(I)–F complexes (δ −258, br (5); δ −253, d, 1JRhF = 77 Hz (6)).
X-ray analysis of complexes 1, 3, 5 and 6 (Fig. 1 and Fig. S56 and S57, ESI†) indicated 16e−, square planar geometries. Compared to the parent complexes, the M(I)–F bonds (1.991–2.032 Å for Ir and 2.016–2.092 Å for Rh) are shorter than M(I)–Cl bonds (2.334–2.382 Å)22 and closer in magnitude to the corresponding M(I)–hydroxides (2.014–2.038 Å).10,23
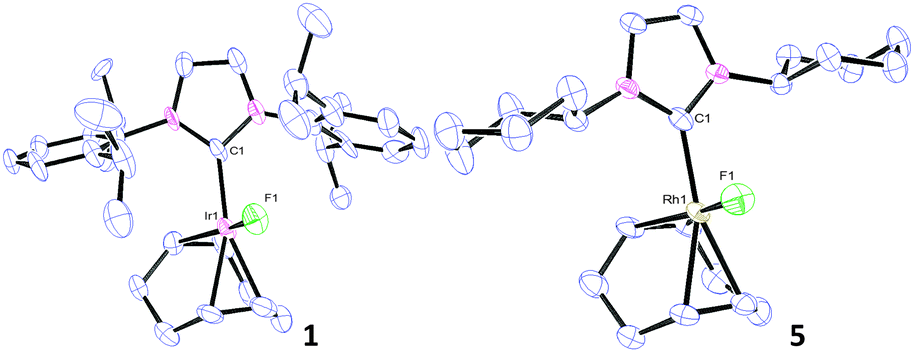 | ||
| Fig. 1 Thermal ellipsoid representations (50%) of 1 and 5. H atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (°), numbers in parentheses are for second independent molecule: 1: Ir(1)–C(1) 2.014(16), [2.029(17)]; Ir(1)–F(1) 2.023(9), [2.002(11)]; C(1)–Ir(1)–F(1): 87.5(5), [88.2(6)]. 5: Rh(1)–C(1) 2.022(3); Rh(1)–F(1) 2.018(2); C(1)–Rh(1)–F(1) 88.20(11). | ||
As a third method, we examined the reaction between the Rh(I) and Ir(I)–hydroxides and Et3N·3HF. Ir(I)–OH and Rh(I)–OH complexes were reacted with Et3N·3HF (0.33 equiv.) in benzene to successfully deliver the Rh(I) and Ir(I)–fluorides in good to excellent yields (Scheme 2).
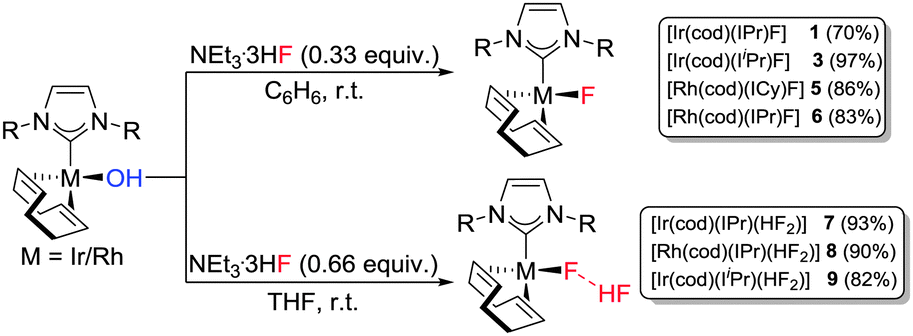 | ||
| Scheme 2 Reaction of Rh(I)- and Ir(I)- hydroxides with Et3N·3HF. | ||
As described by Riant and Leyssens,19b changing the solvent to THF and increasing the relative concentration of Et3N·3HF (0.66 equiv.) afforded the expected bifluoride species, [Ir(cod)(IPr)(HF2)] (7) and [Rh(cod)(IPr)(HF2)] (8) from the corresponding M–hydroxides (Scheme 2). NMR spectroscopic analyses of these new complexes showed subtle differences to the analogous fluoride complexes (1 and 5). At room temperature the 1H NMR spectrum for each compound contained a low-field broad doublet (δ 11.4, 1JFH = 395 Hz (7); δ 12.3, 1JFH = 350 (8)). Upon cooling (200 K, CD2Cl2) the signals partially resolved into doublets of doublets in each case with a spin–spin coupling of 395 and 40 Hz for 7 and 350 and 25 Hz for 8 (see ESI†). These 1H NMR signals were assigned to the acidic proton of the bifluoride moiety; with a strong bond to one fluoride and a weaker bond to a second fluoride. The assignment was confirmed by the identification of two fluoride groups by 19F NMR analysis of 7 and 8.
In each case, a 19F NMR signal was observed in the same region as that found for 1 and 6, corresponding to the metal-bound (proximal) fluoride. In addition, a doublet resonated further downfield with a spin–spin coupling constant of 395 Hz on 7 and 350 Hz on 8. These signals were assigned to the distal fluoride, showing a strong bond to the acidic proton. At 200 K, all 19F NMR signals resolved into distinct doublets of doublets, e.g. for 7: δ −178 (dd, 1JHF = 395, 2JFF = 120, Ir–F⋯HF), −239 (dd, 2JFF = 120, 1JHF = 40, Ir–F⋯HF, see ESI†). Based on these results, we proposed the composition of 7 and 8 as [Ir(cod)(IPr)(FHF)] and [Rh(cod)(IPr)(FHF)], respectively (Scheme 2) with the HF2 moieties comprising a strong bond between the metal and the proximal fluoride, and a relatively weak hydrogen bond to the HF moiety (i.e. [M]–F⋯HF as opposed to [M]⋯F–HF), in agreement with literature reports.15b,18a,b,19b,20
X-ray crystallographic analysis of 7 and 8 (Fig. 2) showed square planar complexes, similar to the mono-fluorides. The X-ray structure for 7 shows a significantly elongated M–F bond. This weakening of the M–F bond is a consequence of the hydrogen bond to HF.15b The F–F separation on 7 is in the range of 2.35–2.39 Å and 2.32–2.33 Å for complex 8. These values are considerably less than the sum of the van der Waals radii of two F atoms (2.94 Å)24 and are indicative of strong hydrogen bonds to the two fluorides, in agreement with reports for other metal bifluorides including those of Mo and Rh.13,14c,15b,c,16–18,19b,20
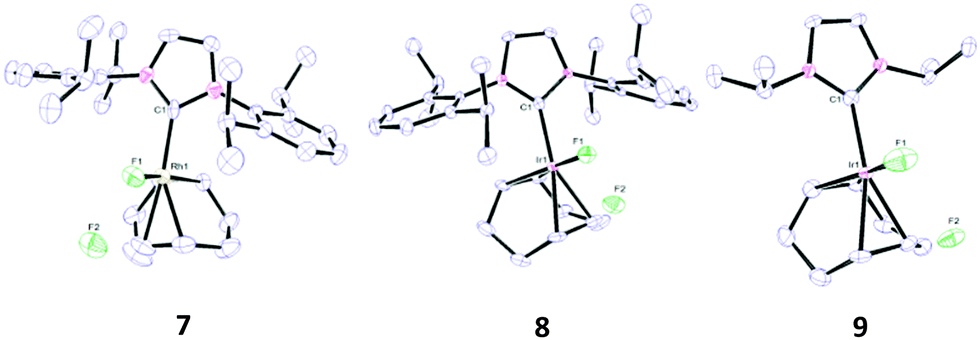 | ||
| Fig. 2 Thermal ellipsoid representations of 7, 8 and 9 showing 50% probability. H atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (°) 7: Ir(1)–C(1) 2.033(4), [2.049(4)]; Ir(1)–F(1) 2.057(3), [2.062(2)]; C(1)–Ir(1)–F(1) 88.92(13), [92.59(12)]. 8: Rh(1)–C(1) 2.047(5), [2.035(6)]; Rh(1)–F(1) 2.089(3), [2.069(3)]; C(1)–Rh(1)–F(1) 93.09(16), [89.14(18)]. 9: Ir(1)–C(1) 2.045(6), [2.053(6)]; Ir(1)–F(1) 2.064(5), [2.082(4)]; C(1)–Ir(1)–F(1) 91.0(2), [87.5(2)]. | ||
The unsymmetrical nature of the bifluoride ligand is further supported by FTIR analyses of 7 and 8, which showed two broad bands each (7: ν = 2623 and 1807 cm−1; 8: ν = 2530 and 1890 cm−1) corresponding to the H–F modes of the bifluoride (absent from FTIR of 1 or 6, see ESI†).14b,16,20 Compared to the IR bands expected for free HF (ν = 3896 and 3895 cm−1 in an Ar matrix),25 it is clear that there is weakened hydrogen bonding within the HF moiety as a result of the H-bond to the proximal fluoride, causing a shift to lower wavenumber.
Compared to symmetrical inorganic bifluoride salts, for example Cs+, K+ or Na+ HF2− (1372–1284 cm−1),26 the IR bands of 7 and 8 are considerably higher in wavenumber, indicating that the HF2 moiety in these complexes is highly unsymmetrical, with a stronger hydrogen bond to one fluorine atom than the other.
In addition to 7 and 8, an alkyl-substituted NHC complex of Ir(I)–(HF2) was prepared in the form of [Ir(cod)(IiPr)(HF2)] (9) (Scheme 2). The less sterically encumbered 9 exhibited different structural characteristics to 7 and 8, both in the solid-state and in solution. For example, at room temperature the 1H NMR signal corresponding to the acidic proton resonates as a broad unresolved peak, as do both signals on the 19F NMR spectrum (see ESI†). Upon cooling to 200 K, the signals resolved into doublets of doublets in the same way as 7 and 8.
From the X-ray structure for 9 (Fig. 2), it is apparent that while the Ir–F bond is comparable in length to 7, the F–F separation (2.457(6), [2.587(6)] Å) is considerably larger, even compared to the values for most metal bifluorides reported to date.8b,13–16,18a–c,19b,20 On closer inspection of the X-ray structures of 7 and 8, it is clear that F(1) and the isopropyl proton on each side of the IPr ligand are in very close proximity to each other (ca. 2.33–2.49 Å in 7; 2.32–2.43 Å in 8). This is considerably less than the sum of the van der Waals radii for H and F (2.67 Å)24 Such a close contact is not possible in 9 and the added flexibility may account for the fluxional behaviour in solution that is associated with 9 (this close contact is not observed on the monofluoride complexes).
Next we studied the link between the fluoride and bifluoride complexes. The ease with which the bifluoride can be generated from the monofluoride and the lability of the HF molecule is especially important for catalytic fluorination and fluorination/ring-opening processes,27 where catalysts, particularly metal fluorides are coupled with an HF source. In order to examine the persistence of the bifluoride moiety in the presence of a base, both the Rh(I) and Ir(I)–bifluorides (7 and 8) were reacted with a stoichiometric amount of KOH. In both cases, full conversion to the corresponding M–F (1 and 6) was seen within 2 h. Interestingly, the addition of a second equivalent of KOH yielded the corresponding M(I)–OH; whilst addition of Et3N·3HF (0.33 equiv.) to 1 or 6 regenerated M–HF2 (7 and 8). Hence, the three species are inherently linked (Scheme 3) and in the presence of an excess of Et3N·3HF, a metal fluoride or hydroxide will more than likely deliver the metal bifluoride.
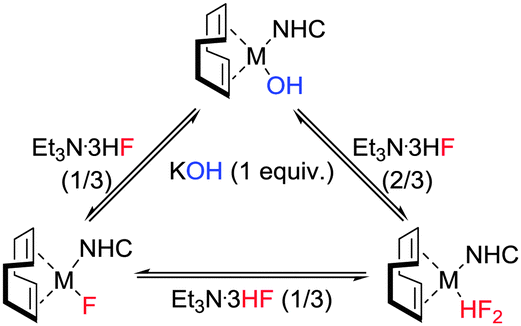 | ||
| Scheme 3 Processes linking M(I)–OH, M(I)–F and M(I)–HF2. | ||
Having established reliable routes to mono and bifluoride complexes of Ir(I) and Rh(I), our final goal was to access the first examples of Ir(I)– and Rh(I)–NHC trifluoromethyl complexes. Our initial attempts were conducted using Ruppert–Prakash's reagent, trifluoromethyl(trimethyl)silane (TMSCF3). Unfortunately all attempts to transfer the CF3 moiety from Si to Ir or Rh failed. The only complex that reacted with TMSCF3 was [Ir(cod)(IiPr)(OH)], delivering [Ir(cod)(IiPr)(OSiMe3)] (10) (96%).10b Reactions between TMSCF3 and the other Ir(I)– or Rh(I)–hydroxides, –chlorides, or –bifluorides were all unsuccessful and clearly a new approach was necessary. Roper has previously prepared Ir(I)–CF3 complexes from [Ir(CO)2(PPh3)2H] using Hg(CF3)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 and similar strategies have been employed on other metals using Cd(CF3)2. In an attempt to avoid such toxic reagents, we considered the use of Ag to transfer the CF3. Hence AgCF3 was prepared in situ from AgF and TMSCF3 at −40 °C29 and reacted with [Ir(cod)(IiPr)Cl] to give Ir(I)–CF3 (11) as a bright red solid (Scheme 4). All attempts at isolating the Ir–IPr and Rh–IPr analogues using this procedure failed. In order to rationalise these results we studied the structure of the isolated compound, 11. Hence, single crystals of 11 were grown and analysed by X-ray diffraction (Fig. 3).
28 and similar strategies have been employed on other metals using Cd(CF3)2. In an attempt to avoid such toxic reagents, we considered the use of Ag to transfer the CF3. Hence AgCF3 was prepared in situ from AgF and TMSCF3 at −40 °C29 and reacted with [Ir(cod)(IiPr)Cl] to give Ir(I)–CF3 (11) as a bright red solid (Scheme 4). All attempts at isolating the Ir–IPr and Rh–IPr analogues using this procedure failed. In order to rationalise these results we studied the structure of the isolated compound, 11. Hence, single crystals of 11 were grown and analysed by X-ray diffraction (Fig. 3).
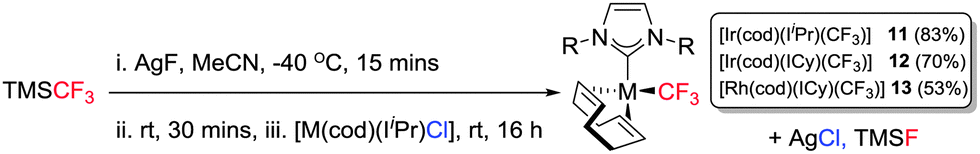 | ||
| Scheme 4 Preparation of [M(cod)(NHC)(CF3)] complexes (11–13). | ||
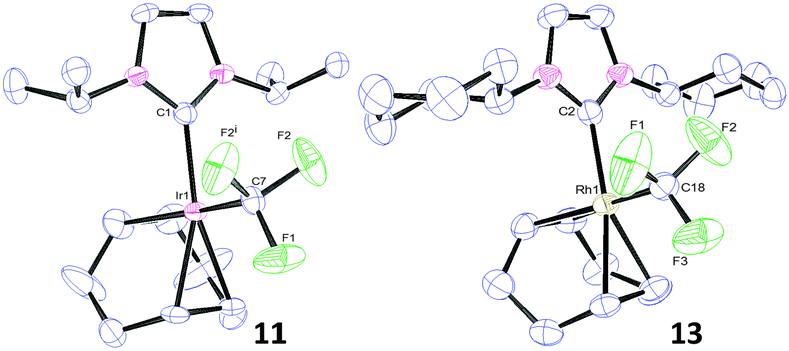 | ||
| Fig. 3 Thermal ellipsoid representations of 11 and 13 showing 50% thermal ellipsoid probability. H atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (°): 11: Ir(1)–C(1) 2.065(8); Ir(1)–C(7) 2.086(8); C(7)–F(1) 1.357(10); C(1)–Ir(1)–C(7) 88.3(3). 13: Rh(1)–C(2) 2.042(5); Rh(1)–C(18) 2.067(5); C(18)–F(1) 1.366(6); C(8)–Rh(1)–C(29) 87.3(2). | ||
In the solid-state it was apparent that two of the fluorine atoms were in very close proximity to a methine proton on each side of the NHC ligand (ca. 2.45 Å). This distance is considerably less than the sum of the Van der Waals radii of H and F (2.67 Å)24 and may indicate an interaction between the two groups. Furthermore, in the 13C{1H} NMR spectrum of 11, a spin–spin coupling interaction causes one of the methyl signals to split into a quartet (δ 22.8 ppm, JFC = 1 Hz). Since the methyl group is separated from the fluorine atoms by six bonds, the interaction must be a “through-space” coupling.30 This evidence indicates that there are considerable steric factors to take into account and that the presence of bulky NHC ligand substituents such as diisopropyl(phenyl) might hinder the coordination of a CF3 group to the metal centre.
To test this hypothesis, we investigated the reaction between [M(cod)(ICy)Cl] and AgCF3, which delivered [M(cod)(ICy)(CF3)] 12 and 13 (Scheme 4). The same structural features were evident on the 13C{1H} NMR spectrum, showing a spin–spin coupling interaction between a methylene on the cyclohexyl ring and the fluoride groups of the CF3 on 12. Furthermore, an X-ray crystal structure of 13 (Fig. 3) indicated a similar close proximity between the fluorides and a proton on each of the cyclohexyl rings (2.51–2.52 Å). Hence, this synthetic method is somewhat limited by the steric bulk of the NHC ligands and therefore, independent of the metal used. These structural investigations have given us further insights into the stability of this scaffold and the viability of the method.
In conclusion, we have demonstrated three methods for accessing Ir(I)–NHC fluoride compounds, two of which were effective on rhodium. The first examples of Ir(I)–bifluorides are reported in high yields, via a general method. Access to Ir(I) and Rh(I)–F and –HF2 complexes enabled examination of the link between the two species. This knowledge could prove paramount in developing new fluorination strategies since the bifluoride can be quantitatively generated from the monofluoride in solution and vice versa. A new method was also established to access the first examples of Ir(I)–NHC trifluoromethyl complexes. This method was also applied to access the first 5-membered NHC-based Rh(I)–CF3 complex. Consequently, we have assembled a toolbox for the preparation of fluorinated complexes to take advantage of the reactivity of already successful Ir(I)– and Rh(I)–NHC scaffolds. This will enable us to pursue fluorination strategies and may well provide active catalytic species or catalytic intermediates in the future.
The ERC (Advanced Researcher Award FUNCAT to SPN), the EPSRC and Sasol (Stipend to BJT) are gratefully acknowledged for support. Umicore AG are thanked for their gift of materials. We thank Prof. Andrew Smith for access to FTIR spectrometry. Thank you to Dr A. Collado and S. R. Patrick for helpful discussions. SPN is a Royal Society Wolfson Award holder.
Notes and references
- For reviews on fluorination see: (a) N. M. Doherty and N. W. Hoffmann, Chem. Rev., 1991, 91, 553–573 CrossRef CAS; (b) E. F. Murphy, R. Murugavel and H. W. Roesky, Chem. Rev., 1997, 97, 3425–3468 CrossRef CAS PubMed.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (c) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS PubMed.
- M. G. Campbell and T. Ritter, Org. Process Res. Dev., 2014, 18, 474–480 CrossRef CAS.
- Articles demonstrating fluorination strategies: (a) E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679–1681 CrossRef CAS PubMed; (b) H. G. Lee, P. J. Milner and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 3792–3795 CrossRef CAS PubMed; (c) M.-G. Braun and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 12990–12993 CrossRef CAS PubMed; (d) M. H. Katcher and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 17402–17404 CrossRef CAS PubMed; (e) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 10795–10798 CrossRef CAS PubMed; (f) K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134–7135 CrossRef CAS PubMed; (g) J. R. Brandt, E. Lee, G. B. Boursalian and T. Ritter, Chem. Sci., 2014, 5, 169–179 RSC; (h) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- (a) K. Fagnou and M. Lautens, Angew. Chem., Int. Ed., 2002, 41, 26–47 CrossRef CAS; (b) M. Gorol, N. C. Mösch-Zanetti, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Eur. J. Inorg. Chem., 2004, 2678–2682 CrossRef CAS; (c) R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS; (d) C. Becker, I. Kieltsch, D. Broggini and A. Mezzetti, Inorg. Chem., 2003, 42, 8417–8429 CrossRef CAS PubMed.
- (a) J. Fawcett, D. A. J. Harding, E. G. Hope, K. Singh and G. A. Solan, Dalton Trans., 2010, 39, 10781–10789 RSC; (b) C. J. Bourgeois, S. A. Garratt, R. P. Hughes, R. B. Larichev, J. M. Smith, A. J. Ward, S. Willemsen, D. Zhang, A. G. DiPasquale, L. N. Zakharov and A. L. Rheingold, Organometallics, 2006, 25, 3474–3480 CrossRef CAS; (c) V. V. Grushin, Acc. Chem. Res., 2009, 43, 160–171 CrossRef PubMed.
- (a) S. A. Macgregor, D. C. Roe, W. J. Marshall, K. M. Bloch, V. I. Bakhmutov and V. V. Grushin, J. Am. Chem. Soc., 2005, 127, 15304–15321 CrossRef CAS PubMed; (b) V. V. Grushin and W. J. Marshall, J. Am. Chem. Soc., 2004, 126, 3068–3069 CrossRef CAS PubMed.
- (a) P. Kläring, A.-K. Jungton, T. Braun and C. Müller, Eur. J. Inorg. Chem., 2012, 1430–1436 CrossRef; (b) J. Vicente, J. Gil-Rubio and D. Bautista, Inorg. Chem., 2001, 40, 2636–2637 CrossRef CAS.
- Examples of TM–NHC fluoride complexes (a) J. R. Herron and Z. T. Ball, J. Am. Chem. Soc., 2008, 130, 16486–16487 CrossRef CAS PubMed; (b) D. S. Laitar, PhD thesis, Massachusetts Institute of Technology, 2006; (c) D. S. Laitar, P. Müller, T. G. Gray and J. P. Sadighi, Organometallics, 2005, 24, 4503–4505 CrossRef CAS; (d) T. Schaub, M. Backes and U. Radius, J. Am. Chem. Soc., 2006, 128, 15964–15965 CrossRef CAS PubMed; (e) T. Schaub, P. Fischer, A. Steffen, T. Braun, U. Radius and A. Mix, J. Am. Chem. Soc., 2008, 130, 9304–9317 CrossRef CAS PubMed; (f) S. L. Chatwin, M. G. Davidson, C. Doherty, S. M. Donald, R. F. R. Jazzar, S. A. Macgregor, G. J. McIntyre, M. F. Mahon and M. K. Whittlesey, Organometallics, 2005, 25, 99–110 CrossRef; (g) S. P. Reade, D. Nama, M. F. Mahon, P. S. Pregosin and M. K. Whittlesey, Organometallics, 2007, 26, 3484–3491 CrossRef CAS; (h) J. Fawcett, D. A. J. Harding, E. G. Hope, K. Singh and G. A. Solan, Dalton Trans., 2009, 6861–6870 RSC.
- (a) B. J. Truscott, G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Org. Biomol. Chem., 2011, 9, 7038–7041 RSC; (b) B. J. Truscott, D. J. Nelson, C. Lujan, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2013, 19, 7904–7916 CrossRef CAS PubMed.
- D. R. Coulson, J. Am. Chem. Soc., 1976, 98, 3111–3119 CrossRef CAS.
- U. Bentrup, K. Harms, W. Massa and J. Pebler, Solid State Sci., 2000, 2, 373–377 CrossRef CAS.
- V. J. Murphy, D. Rabinovich, T. Hascall, W. T. Klooster, T. F. Koetzle and G. Parkin, J. Am. Chem. Soc., 1998, 120, 4372–4387 CrossRef CAS.
- (a) N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796–3797 CrossRef CAS PubMed; (b) N. A. Jasim and R. N. Perutz, J. Am. Chem. Soc., 2000, 122, 8685–8693 CrossRef CAS; (c) D. C. Roe, W. J. Marshall, F. Davidson, P. D. Soper and V. V. Grushin, Organometallics, 2000, 19, 4575–4582 CrossRef CAS.
- (a) M. K. Whittlesey, R. N. Perutz, B. Greener and M. H. Moore, Chem. Commun., 1997, 187–188 RSC; (b) N. A. Jasim, R. N. Perutz, S. P. Foxon and P. H. Walton, Dalton Trans., 2001, 1676–1685 RSC; (c) M. S. Kirkham, M. F. Mahon and M. K. Whittlesey, Chem. Commun., 2001, 813–814 RSC.
- V. J. Murphy, T. Hascall, J. Y. Chen and G. Parkin, J. Am. Chem. Soc., 1996, 118, 7428–7429 CrossRef CAS.
- T. Braun, S. P. Foxon, R. N. Perutz and P. H. Walton, Angew. Chem., Int. Ed., 1999, 38, 3326–3329 CrossRef CAS.
- (a) J. Vicente, J. Gil-Rubio, D. Bautista, A. Sironi and N. Masciocchi, Inorg. Chem., 2004, 43, 5665–5675 CrossRef CAS PubMed; (b) W. J. Marshall and V. V. Grushin, Organometallics, 2004, 23, 3343–3347 CrossRef CAS; (c) C. Segarra, E. Mas-Marzá, J. P. Lowe, M. F. Mahon, R. C. Poulten and M. K. Whittlesey, Organometallics, 2012, 31, 8584–8590 CrossRef CAS; (d) D. Noveski, T. Braun and S. Krückemeier, J. Fluorine Chem., 2004, 125, 959–966 CrossRef CAS PubMed.
- (a) T. Vergote, F. Nahra, D. Peeters, O. Riant and T. Leyssens, J. Organomet. Chem., 2013, 730, 95–103 CrossRef CAS PubMed; (b) T. Vergote, F. Nahra, A. Welle, M. Luhmer, J. Wouters, N. Mager, O. Riant and T. Leyssens, Chem. – Eur. J., 2012, 18, 793–798 CrossRef CAS PubMed; (c) T. Vergote, F. Nahra, A. Merschaert, O. Riant, D. Peeters and T. Leyssens, Organometallics, 2014, 33, 1953–1963 CrossRef CAS.
- N. Bramananthan, M. Carmona, J. P. Lowe, M. F. Mahon, R. C. Poulten and M. K. Whittlesey, Organometallics, 2014, 33, 1986–1995 CrossRef CAS.
- (a) G. G. Dubinina, H. Furutachi and D. A. Vicic, J. Am. Chem. Soc., 2008, 130, 8600–8601 CrossRef CAS PubMed; (b) A. Zanardi, M. A. Novikov, E. Martin, J. Benet-Buchholz and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 20901–20913 CrossRef CAS PubMed.
- (a) W. A. Herrmann, C. Köcher, L. J. Gooßen and G. R. J. Artus, Chem. – Eur. J., 1996, 2, 1627–1636 CrossRef CAS; (b) M. Ahmed, C. Buch, L. Routaboul, R. Jackstell, H. Klein, A. Spannenberg and M. Beller, Chem. – Eur. J., 2007, 13, 1594–1601 CrossRef CAS PubMed; (c) R. A. Kelly III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef.
- B. J. Truscott, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2013, 42, 270–276 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- L. Andrews and G. L. Johnson, J. Phys. Chem., 1984, 88, 425–432 CrossRef CAS.
- B. S. Ault, Acc. Chem. Res., 1982, 15, 103–109 CrossRef CAS.
- (a) J. J. Topczewski, T. J. Tewson and H. M. Nguyen, J. Am. Chem. Soc., 2011, 133, 19318–19321 CrossRef CAS PubMed; (b) J. Zhu, G. C. Tsui and M. Lautens, Angew. Chem., Int. Ed., 2012, 12353–12356 CrossRef CAS PubMed; (c) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC; (d) J. A. Akana, K. X. Bhattacharyya, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 7736–7737 CrossRef CAS PubMed; (e) J. A. Kalow and A. G. Doyle, Tetrahedron, 2013, 69, 5702–5709 CrossRef CAS PubMed.
- P. J. Brothers, A. K. Burrell, G. R. Clark, C. E. F. Rickard and W. R. Roper, J. Organomet. Chem., 1990, 394, 615–642 CrossRef CAS.
- Y. Zeng, L. Zhang, Y. Zhao, C. Ni, J. Zhao and J. Hu, J. Am. Chem. Soc., 2013, 135, 2955–2958 CrossRef CAS PubMed.
- J.-C. Hierso, Chem. Rev., 2014, 114, 4838–4867 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and full characterisation details for new complexes. CCDC 1000750 (1), 1000751 (3), 1000752 (5), 1001354 (6), 1000753 (7), 1000754 (8), 1000755 (9), 1000756 (11), 1000757 (13). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc07772e |
| This journal is © The Royal Society of Chemistry 2015 |