
Heat-treatment of metal–organic frameworks for green energy applications
Lacey
Lux†
,
Kia
Williams†
and
Shengqian
Ma
*
Department of Chemistry, University of South Florida, 4202 East, Fowler Avenue, Tampa, Florida 33620, USA. E-mail: sqma@usf.edu; Fax: +1 813 974 3203; Tel: +1 813 974 5217
First published on 9th September 2014
Abstract
Increasing population, global climate change, and dwindling fossil fuel reserves have led to a green energy revolution in the past few years, particularly in the fields of green energy generation and storage. While fuel cell, lithium-ion batteries, and supercapacitors are far from technical jargon in today's world, the sheer number of these devices is not accurately exhibited in the consumer market. Metal–organic frameworks (MOFs) are an emerging class of crystalline materials consisting of metal ion nodes and organic linkers that has shown promise in the fields of gas capture, gas storage, separations, catalysis, magnetism, fluorescence, and sensing to name only a few. The use of these materials as self-sacrificing templates, via calcination or pyrolysis, for the design of green energy generation and storage devices is the focus of this review.
1. Introduction
With increasing population and usage of fossil fuels, the need for alternative and sustainable energy technologies has become a necessity. Not only must such technology need to, at least, match the general activity of current energy storage conversion and storage devices, but there is also a need for feasible and affordable application and distribution. Interests such as fuel cells, batteries, capacitors, solar cells, etc. afford much opportunity for improvement and research focus.1 Metal–organic frameworks (MOFs) are a class of porous materials consisting metal ions or clusters coordinated by organic ligands. They are highly crystalline materials with a large structural diversity and permanent porosity. These features, along with the ability to fine tune the structure with various ligands and metal centers, lead to an array of applications including gas capture and storage,2 separations,3 catalysis,4 magnetism applications,5 fluorescence and sensing applications,6 and more in this rapidly developing field. The facile modification, as it relates to the tunability of the crystalline pore sizes and surface areas of MOFs, affords this class of compounds a high abundance of resultant structures in comparison to other classes of porous materials. Additionally, thermally activated MOFs impart distinct advantages to these materials when compared to their parent structures including electrolytic conductivity, a higher resistance to corrosion by the electrolyte, and a uniform distribution of catalytic centers affixed to a carbon support.MOFs have been studied for many energy related applications, particularly for gas storage and separations. What is less explored, however, is the use of MOFs as templates/precursors for energy conversion and storage devices. MOFs have shown early promise in different aspects of these technologies, and, being a relatively new field, there is still much more to be explored.7 In this review, we aim to narrow the focus on one very intriguing property of MOFs, the ability to serve as a template or self-sacrificing precursor for electrochemical applications after thermal treatment. This topic will cover the heat treatment of MOFs, including pyrolysis and calcination, and how it is applied in fuel cells, lithium-based batteries, and electric double-layer capacitors.
2. Fuel cells and MOFs as electrocatalyst precursors
Fuel cells comprise a widely studied group of energy conversion devices, offering efficient alternative fuel use and low emissions. According to the U.S. Department of Energy (DOE), over 200 light duty vehicles, more than 20 fuel cell buses, and approximately 60 fueling stations are currently in use in transportation applications in the U.S., and GM, Toyota, Honda, Hyundai, and other vehicle manufacturers intend to commercialize fuel cell vehicles by 2015.8 In order for fuel cells to be an adequate replacement for internal combustion engines (ICEs), there are key factors to be addressed and overcome. Herein, we will discuss different fuel cell types and the applications and challenges of heat-treated MOFs within them.Several types of fuel cells, including polymer electrolyte membrane fuel cells (PEMFCs), direct ethanol fuel cells (DEFCs), and direct borohydride fuel cells (DBFCs) have been targeted for MOF usage.8,9 It is widely accepted that platinum group metal (PGM) materials are the preferred catalyst at both the anode and the cathode. Despite this, the oxygen reduction reaction (ORR) at the cathode is typically significantly lower than the fuel oxidation at the anode (Fig. 1).10 Overcoming the slow kinetics requires increasing Pt content, which not only leads to higher fuel cell stack costs due to limited Pt reserves, but also lower durability, as Pt is subject to contamination.11 In addition to attempting to reduce Pt loading, increasing efforts have designing non-platinum group metal (non-PGM) and metal-free catalysts to replace PGM catalysts. The DOE target for 2020 is to develop a 60% peak-efficient, direct hydrogen fuel cell with 5000 hour durability at $30 kW−1.12
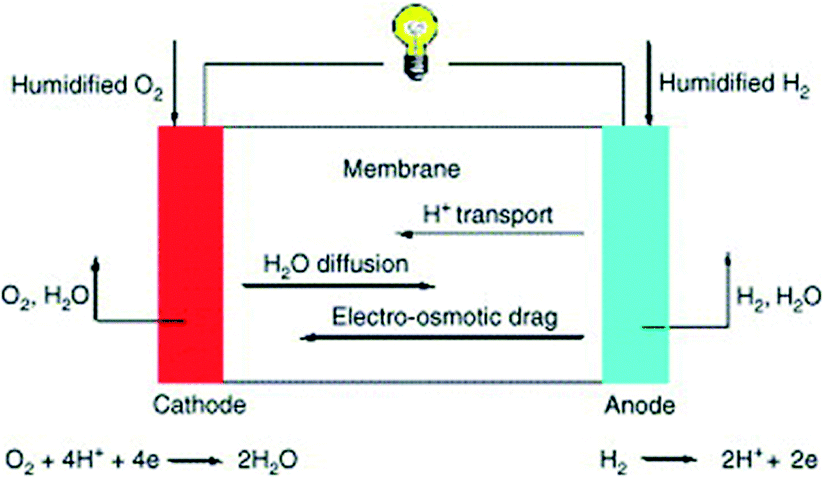 | ||
| Fig. 1 General schematic of a hydrogen fuel cell. Reprinted with permission from ref. 10. Copyright 2005, Elsevier B.V. | ||
The accessibility of metal-ion sites in MOFs, high volumetric density, and high micropore surface area give MOFs an ideal potential as catalyst precursors with no need of an additional carbon source prior to pyrolysis. To date, MOFs have been studied for use in fuel cells as both catalysts and as proton conductive membranes; however, only MOFs in use in catalyst design will be discussed in this work.
2.1 MOFs as precursors for precious metal electrocatalysts
DEFCs use ethanol as a liquid fuel source, and, thus, hold an advantage to PEMFCs in storage and transportation.13 They are not, however, immune to the challenges of current fuel cell technologies. Currently, platinum based catalysts are still accepted as most effective for the ethanol oxidation reaction (EOR) at the anode and ORR at the cathode. Nadeem et al. synthesized and studied the use of a MOF-5 (Zn4O(bdc)3, bdc = 1,4-benzenedicarboxylate) derived porous carbon (PC-900) as a support for PtFe nanoparticles.9a MOF-5 was carbonized at 900 °C, then doped with PtFe nanoparticles resulting in PtFe/PC-900. This EOR catalyst was compared to PtFe and Pt supported by Vulcan XC72 carbon catalysts, which were deemed PtFe/XC-72 and Pt/XC-72, respectively. The calculated electrochemically active surface area (ECSA) for PtFe/PC-900 was found to be 105.32 m2 g−1 compared to 73.31 m2 g−1 for PtFe/XC-72 and 44.11 m2 g−1 for Pt/XC-72. This was mainly attributed to the high dispersion of PtFe nanoparticles due to the larger Brunauer–Emmett–Teller (BET) surface area of PC-900 in comparison to XC-72. PtFe/PC-900 also shows high power density of 121 mW cm−2 and has a normalized activity per gram of Pt of 6.8 mA g−1 Pt.Further evidence for the advantage of MOF-5 as a precursor for carbon support comes from a similar work by Yi et al. wherein MOF-5 was carbonized with furfuryl alcohol (FA) for use in a Pt based catalyst for DBFCs, and deemed Pt/NPC.14 DBFCs are promising energy conversion alternatives in which aqueous borohydride solutions serve as fuel. Their fast anode kinetics, high theoretical open circuit voltage (OCV), and high energy density represent only a few of the many advantages that DBFCs provide in comparison to ICEs.15 As with most fuel cell types, the high cost and limited availability of the precious metals used at catalysts debilitate the commercialization potential of DBFCs. In Yi's work, Pt/NPC was also compared to Pt supported on XC-72 (Pt/XC-72). Pt/NPC not only shows a 36.38% higher current density than Pt/XC-72 for direct borohydride oxidation, but also has a power density of 54.34 mW cm−2 at 25 °C while Pt/XC-72 has a power density of 34.13 mW cm−2.
PEMFCs are the most widely studied fuel cells for mobile applications, owing to the use of hydrogen as a fuel and oxygen from the air as an oxidant to produce electricity, with water as the major byproduct. Kaliaguine et al. introduced Pt atoms into MOF-253 (Al(OH)(bpydc), bpydc = 2,2′ -bipyridine-5,5′-dicarboxylate) and pyrolyzed at temperatures ranging from 700–1050 °C, to reveal Pt nanoparticles (Pt-MOF) (Fig. 2).16 N2 isotherms of Pt-MOF show a decreased BET surface area and reveal mesopores, micropores, and a decrease in pore volume. After the removal of Al species, catalytic activity was tested at both the anode and the cathode for Pt-MOF treated at both 950 °C (C3) and 1050 °C (C4) and compared to the commercial electrode (BASF, 0.5 mgPt cm−2). The anode results of C3 proved more impressive, resulting in an OCV of ~970 mV, comparable to the ~960 mV of the commercially available electrocatalyst. Furthermore, the current densities at 0.6 V Pt-MOF and the commercial catalyst are 482 mA cm−2 and 537 mA cm−2, respectively, while the power densities are 0.58 W mgPt−1 and 0.64 W mgPt−1, respectively.
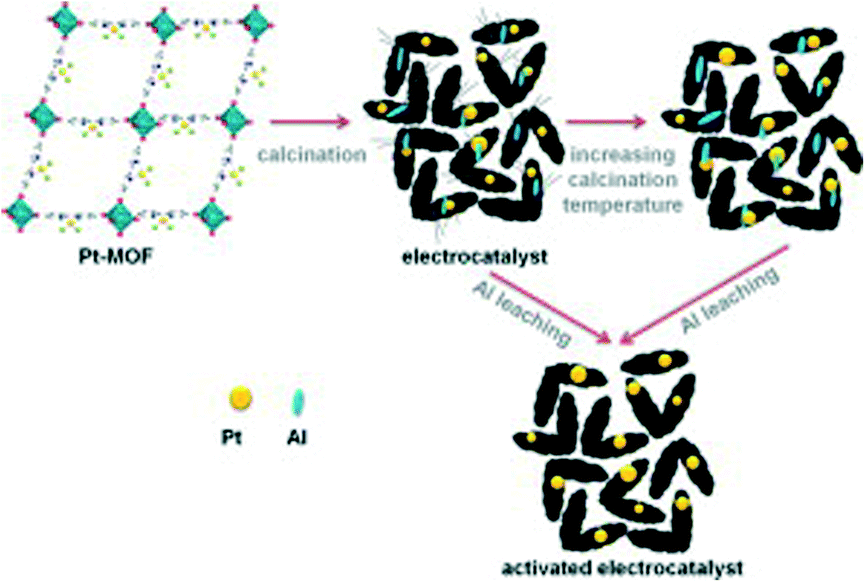 | ||
| Fig. 2 Synthesis schematic of Pt-MOF from MOF-253. Reprinted with permission from ref. 16. Copyright 2013, Elsevier B.V. | ||
2.2 MOFs as precursors for non-precious metal electrocatalysts
Previous studies have shown that macrocyclic catalysts with the Fe–N4 moiety likely lead to a 4-electron reduction pathway of oxygen to produce H2O as the desired product, which is one of the most important aspects of PEMFCs as clean energy sources.11 Binuclear Co–N4 moiety catalysts have also been shown to facilitate a 4-electron pathway reduction. Transition metal macrocyclic complexes generally do not have long-term stability, but thermal treatment after they have been absorbed on high surface area carbon support particles increases stability and catalytic activity.11While there are many different catalyst synthetic methods, M–N–C catalysts with metal–Nx (M–Nx, where M = Fe, Co, Ni, Mn, etc.) obtained through pyrolysis have shown activity in PEMFCs.17,18 The first example of a MOF as a precursor came in 2011 when Ma et al. synthesized a cobalt imidazole framework with regularly dispersed Co–N4 moieties using Co(NO3)2·6H2O and 3,5-imidazolate.17 Heat treatments were done between 500–900 °C. After pyrolysis, the catalysts were treated with sulfuric acid to remove metallic cobalt. The ORR activity of the catalyst treated at 750 °C was revealed to have an onset potential of 0.83 V. The mass activity at 0.8 V and 0.75 V are 0.14 Ag−1 and 1.34 Ag−1, respectively. This is an increase from the mass activity of the heat-treated sample before metallic cobalt removal. In addition, an increase in surface area was observed after acid sonication. It was proposed that increase in ORR activity for the sample at 750 °C can be ascribed to the retention of high surface area and micropores, and the formation of mesopores. Rotating ring disk electrode (RRDE) measurements were taken to study the electron transfer mechanism. The Koutecky–Levich equation was employed to find the number of electrons transferred, n. Calculations show that 3.3–3.6 electrons were transferred during the ORR catalysis.
To date the highest peak power density catalyst for M–N4 systems for ORR is 0.91 W cm−2, which was first reported by Dodelet et al. in 2011.19 ZIF-8, a zeolitic imidizolate framework based on 2-methylimidazole, was used as a carbon support for iron(II) acetate and 1,10-phenanthroline (phen), the nitrogen source. ZIF-8, iron(II) acetate, and phen were ballmilled together in various ratios, heated at various temperatures in Ar, and then subsequently heated in NH3. The best precursor ratio held a 20/80 mass ratio of phen to ZIF-8 and was heated at 1050 °C in Ar for 1 hour and 950 °C in NH3. The power density at 0.6 V was found to be 0.75 W cm−2, which is comparable to the commercial Pt catalyst. The volumetric activity of the Fe-based catalyst is increased by a factor of 2.3 in comparison to the Pt-based catalyst. This was attributed to the improved mass transport in the carbon support. This gives further evidence that the presence of both micropores and mesopores in the carbon support aid in improving activity, as Fe and N were observed to have no effect on mass transport. Since this study, many works using iron based catalysts with phen with MOF supports have been published.
Very recently, Dodolet et al. continued studies on this system to enhance performance and improve durability.20 The catalyst previously studied was subjected to a second heat treatment in NH3, but degrades more slowly if it only undergoes pyrolysis in Ar. Therefore, after only the initial heat treatment, the catalyst, thus named NC-Ar, was used to study activity and stability. To improve activity, the mass transport properties were targeted due to the thickness of non-PGM catalysts versus PGM catalysts. It was previously reported that graphitization is possible a source of stability.21 Adding 26 wt% carbon fibers with an average diameter of 150 nm to the catalyst precursor showed improvement in the catalyst performance; however, further heat treatments were needed to improve durability, which still remains and issue. One very important conclusion was drawn: degradation of good catalytic sites coincided with the activation of catalytic sites that were not originally active or had meager performance because of the improvement of mass transport.
ZIF-8 impregnated with furfuryl alcohol (FA) was both pyrolyzed, then doped with iron(II) acetate and phen, and doped with iron(II) acetate and phen, then pyrolyzed in Jaouen et al.'s recent work.22 The higher power performances observed in this work belong to those catalysts that contain not only micropores, but mesopores in the 2–4 nm range, related to the 1.8–2 nm range reported by Liu et al., again providing evidence that micropore and mesopore presence in a carbon support increases mass transport properties. Moreover, it is believed that, in this case, heat treatment in heat treatment in NH3 does not necessarily contribute to N content, but has a major function in creating micropores and small mesopores that connect already existing active sites to the surface.
In another work by Banerjee, Kurungot et al., to tune porosity of the carbon materials, Zn-based multi-ligand ZIFs using imidazole/2-nitroimidazole (ZIF-70), 2-nitroimidazole/benzimidazole (ZIF-68), and 2-nitroimidazole/5-chlobenzimidazole (ZIF-69) were embedded with iron phenanthroline complexes, [Fe(phen)3]2+, and heated at 900 °C in Ar (Fig. 3).23 It was found that the catalyst prepared from ZIF-70 (FeNC-70) has a larger surface area and is more porous, allowing for higher loading of [Fe(phen)3]2+. Rotating disk electrode (RDE) measurements reveal an onset potential of 0.80 V for FeNC-70, and RRDE results show n values of 3.3–3.8, favoring water production.
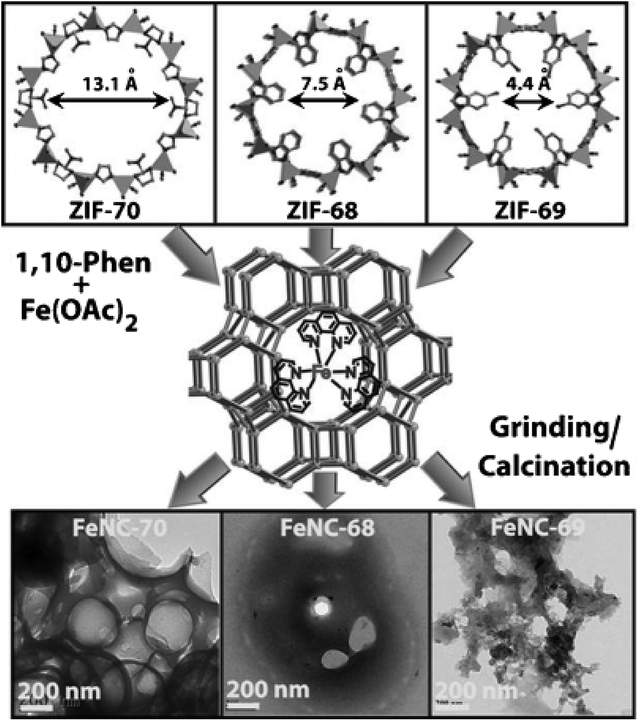 | ||
| Fig. 3 Synthetic pathway of FeNC-70, FeNC-68, and FeNC-69 from [Fe(phen)3]2+ embedded ZIF-70, ZIF-68, and ZIF-69, respectively. Reprinted with permission from ref. 23. Copyright 2013, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Jaouen and coworkers also investigated the replacement of phen with 2,46,-tris(2-pyridyl)-s-triazine (TPTZ) in the FeII/ligand/ZIF-8 precursor.24 The aim was to improve previous use of TPTZ on a high surface area carbon support by Zhang et al. by studying Fe-ligand coordination strength.25,26 It was found that it is imperative that Fe remain coordinated to the ligand during the impregnation of ZIF-8 so that the Fe-ligand complex remains on the surface of the ZIF-8 particles, ensuring catalytic activity.
Liu and coworkers' work continued with another replacement of phen on a ZIF-8 support.18 An iron imidazolate framework [Fe3(imid)6(imidH)2]x, coined, FeIM, was synthesized with ferrocene and imidazole. Metallic iron was removed after heat treatment at 700, 800, and 900 °C in Ar, yielding FeIM700, FeIM800, and FEIM900, respectively. FeIM was also ballmilled with ZIF-8 and pyrolyzed at 1050 °C in Ar, then 950 °C in NH3 for 15 minutes, giving FEIM/ZIF-8. RRDE measurements showed that FeIM700 has an onset potential of 0.855 °C and n values within the range of 3.70–3.81. FeIM/ZIF-8 showed the best activity overall with an onset potential of 0.915 V. Values of n lie within the range of 3.83–3.93, highly favoring the 4-electron transfer mechanism with a H2O2 yield <8.6%. In this case, it is believed that the ammonia treatment increases N content. For FeIM700, FeIM800, and FeIM900, activity decreases with decreasing N content, helping support importance of N content in ORR activity.
2.3 MOFs as precursors for metal-free electrocatalysts
High surface area carbons have been targets of studies for use in various energy related applications.27 Given that MOFs have already been used as precursors for high surface area carbon supports with metals, it is no surprise that they would be investigated for use with heteroatoms for applications such as fuel cells, lithium ion batteries, solar cells, etc. Kurungot et al., utilized in situ graphitic carbon nitride (g-C3N4) polymerization by (a) grinding MOF-5 and melamine together before heat treatment, and (b) soaking MOF-5 and melamine in an ethanol solution prior to heat treatment (Fig. 4).28 It is to be noted, unlike the ORR activities tested in the previous examples, which are typically done under acidic conditions (HClO4 or H2SO4), this example and the next are under alkaline conditions. MOFCN900, the catalyst prepared under the first synthetic method and heated at 900 °C shows the highest catalytic activity of all of the samples tested, and has a current density of 4.2 mA cm−2 with n value of 3.12.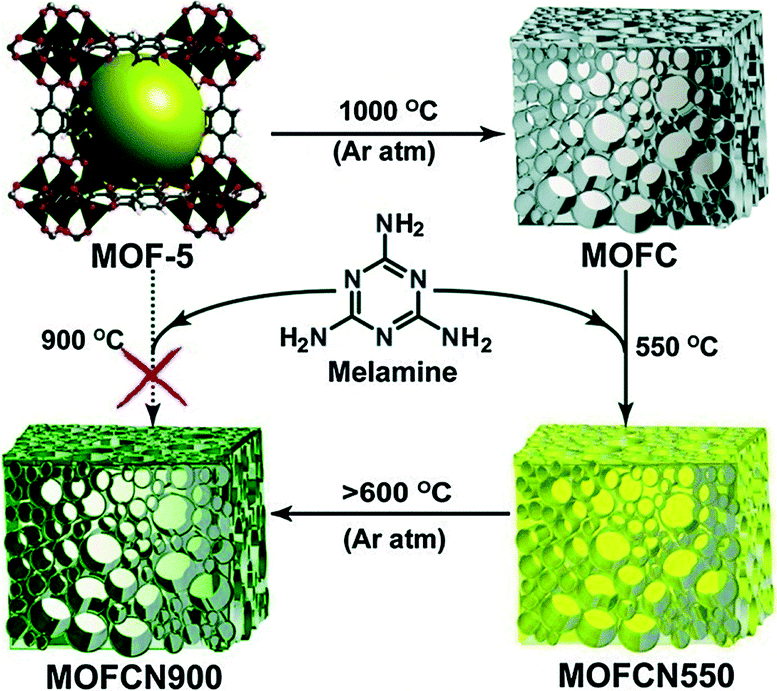 | ||
| Fig. 4 Construction of MOF-derived carbon via in situ g-C3N4 polymerization. Reprinted with permission from ref. 28. Copyright 2014, The Royal Society of Chemistry. | ||
In order to utilize an alternative green carbon precursor in comparison to previously used FA, Cao et al. (a) soaked ZIF-7 (Zn(NO3)2·(H2O)3) in an aqueous glucose solution prior to pyrolysis, and (b) ground ZIF-7 and glucose together prior to pyrolysis. The results were deemed carbon-L, for the method ‘a’ and carbon-S for method ‘b’.29 Carbon-L shows an onset potential of 0.85 V vs. RHE, and with n value 3.68 at 0.3 V, close to the commercial 20% Pt/C. It was also found to be tolerant to methanol whilst the Pt/C catalyst was poisoned. Studying the chronoamperometric response in 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s, carbon-L retained 75% of its relative current while 20% Pt/C only retained 53%.
000 s, carbon-L retained 75% of its relative current while 20% Pt/C only retained 53%.
2.4 Conclusions
The use of MOFs as self-sacrificing precursors for fuel cell electrocatalysts is an exceptionally fresh area of research. The first example was in 2011, and most of the works discussed in this section were published between 2013 and 2014. While strides have been made in recognizing design features of beneficial use, and some aspects of catalysts' activities have been akin to Pt based catalysts in similar conditions, major pitfalls arise in the durability of non-PGM catalysts based on MOFs. Also, although certain functionalities have been proven to have good catalytic activity, there is still a lack in understanding of the mechanisms in which ORR occurs in these catalysts, which necessitates further exploration in order to make the most efficient catalyst design.3. Green energy storage devices and MOFs as precursors to novel electrode materials
The current global energy consumption is predicted to double within the next few decades causing an increase in global air pollution and global warming. To ameliorate the resultant negative environmental impact, there has been an increased demand for the development of green energy resources. As a result of this and in conjunction with dwindling fossil fuel resources, there has been a considerable investigation into both green energy generation and storage systems. Devices that provide such energy storage include rechargeable batteries based on lithium ions or lithium–sulfur complexes along with electric double-layer capacitors or supercapacitors.Lithium-ion batteries (LIB) are the current industrial standard for portable power sources in our ever-expanding universe of smartphones, laptop computers, and other portable electronic devices. Additionally, there is scientific inquiry into their potential uses as a partial power source for electric vehicles in conjunction with primary renewable energy-based fuel cells. These batteries have a number of distinct advantages for the storage of green energy including a high energy density and relatively long life span.30 Batteries are the most recognizable of the green energy storage devices due to their deep integration into our everyday lives. The predominant anodic material used in today's LIBs is graphite with a theoretical capacity of 372 mA h g−1.31 As our society has progressed further into the information age, the electronic devices that we have come to depend upon are becoming more and more energetically demanding with future devices requiring a far greater capacity than even the highest theoretical value that graphite can provide. The current push in the development of new LIBs is to develop anodes that can out perform today's graphite standard.32 Metal oxides have long been investigated for this role as they have a propensity to adopt a number of structural geometries.33 As structural geometries necessarily impact the function of a material by tuning its chemical, physical, and electronic properties, metal oxides have the potential to provide a wide array of characteristics allowing for the optimization of energy storage in next-generation LIBs.
LIBs conventionally use a lithium metal oxide for the cathode and graphite as the anode. During charging cycles, electrons move from the cathode to the anode with the concomitant migration and intercalation of lithium ions from the cathode into the graphite anode.34 During the discharge cycle, an electric current is generated by the return of the lithium cations and subsequently, the electrons to the cathode and the recombination of the electrons with the lithium ions batteries (LSB) function via a similar mechanism but have a higher theoretical capacity due to sulfur's large specific capacity of 1675 mA h g−1.35
Supercapacitors (SCs) are another fundamental source of green energy storage. Rather than the electrochemical storage that occurs in batteries, electric double-layer capacitors store energy by predominantly storing charge electrostatically in a Helmholtz layer. Moreover, pseudocapacitors store charge by both electrostatic and electrochemical energy storage methods.36 Carbon electrodes, such as those used by the majority of SCs today, leave much to be desired. They often result in devices with a strong electric double-layer capacity but highly inhibited Faradaic charge storage.37 Ideally, both should be incorporated into a single device to optimize its theoretical energy storage capacitance. Advantages of SCs include a very high power density, rapid charge dispersion and response time, and exceptionally long cycling lives in comparison to the battery counterparts.38 However, they are handicapped by their low energy density. Pseudocapacitors allow for the realization of a higher energy density for devices by integrating both the electric double-layer and Faradaic charge storage where oxidation/reduction reactions that take place at the surface of the electrode result in chemical energy storage analogous to the mechanism of function for a battery.39
It is important to note that SCs vary fundamentally from their parent devices, capacitors.37 Traditional capacitors result in the transfer of energy by charge carriers migrating between electrodes. This movement of charge results in a charge separation between the electrodes and the necessary formation of a potentia between the electrodes that can be extricated and harvested via the application of an external circuit. On the other hand, SCs necessarily store their energy as a charge distribution in a Helmholtz double layer that interfaces with the electrolyte rather than as an electrochemical reaction. The energy density of these devices has been experimentally modulated by an increase in the electrode surface area to provide a drastic increase in the contacts that form the interface between the electrolyte and the electrode.40 Often time, nanoporous carbonaceous materials, such as activated carbons, are used to facilitate this increase in surface area.
One facet that both batteries and SCs share is that the surface area of their electrodes is crucial for the optimization of their functionality. For commercial alkaline batteries, metallic zinc comprising part of the anode is of a powdered morphology due to its increased surface area.41 The larger surface area facilitates increased specific interaction between the electrolyte and the electrode resulting in a lowering of the internal resistance and an increase in the power density of the battery. Superconductors benefit from an increase in electrode surface area for an analogous rationale.
3.1 MOFs as precursors to lithium-based secondary batteries
A wide variety of materials have been examined for their potential application in the development of electrode materials for secondary, or rechargeable, LIBs including transition metal chalcogenides and transition metal oxides.42 As transition metal chalcogenides suffer from a low aggregate operating voltage against metallic lithium of ca. 2 V,43 transition metal oxides have become have become a focus of the scientific community in the development of novel electrodes.44 The investigations of these oxides are necessarily biased toward the 3d transition metals as a result of their smaller mass and dimension leading to larger electric capacities in relation to their 4d and 5d counterparts.45Nanometric Co3O4 along with other nanoscale transition metal oxides of the formula MO (M = Co, Ni, Cu, Fe) has been established as an electrode material for LIBs since the publication of a letter in Nature by Poizot, et al. in 2000.46 Conventional synthetic strategies of nanometric Co3O4 preferentially result in the production of characteristic singular nanoparticles with a uniform size distribution, a trend that is ascribed to traditional homogeneous reaction conditions. Other morphologies of Co3O4 have also been produced including mesoporous single crystal nanoneedles47 comprised of nanocrystal building blocks and nanowires constructed via a virus biotemplating synthetic methodology.48 In comparison to the single crystal nanoparticles, both of these extended morphologies have proven to possess enhanced electrochemical performance in relation to specific capacitance, rate capability, and cycleability.
The first report of a material designed as a LIB electrode via a MOF-templating scheme was agglomerated Co3O4 by Liu, et al. in 2008.49 Cobalt-MOF, Co3(NDC)3(DMF)4 (NDC = 2,6-naphthalenedicarboxylate; DMF = N,N′-dimethylformamide) was synthesized by the solvothermal reaction of hexahydrate Co(NO3)2 and H2NDC in DMF resolving purple crystals. Calcination of the cobalt-MOF at 600 °C resulted in the formation of the agglomerated Co3O4 nanoparticles. The synthesis scheme is depicted in Fig. 5, where the cobalt metal nodes of the framework are converted to metal oxide subunits and further transformed into primary nanoparticles (ca. 25 nm). The primary nanoparticles subsequently aggregated into a densely packed secondary nanoparticulate structure (ca. 250 nm; BET surface area = 5.3 m2 g−1). A standard Co3O4 coin half-cell was constructed to determine the electrochemical capabilities of the as-prepared agglomerates at a current density of 50 mA g−1. The first discharge exhibited a high discharge capacity of 1118 mA h g−1 and capacity retention was ca. 75% for the first cycle. The capacity retention reached a maximum of 965 mA h g−1 (86% of initial capacity) after 50 cycles. The resultant high capacity of the as-prepared Co3O4 agglomerate is attributed to its porous primary–secondary structural organization that provides a greater global electrode accessibility facilitating increased efficiency in electrolyte transportation and lithium-ion diffusion. The porous structure also serves to minimize the structural damage caused by lithium ion intercalation and release.
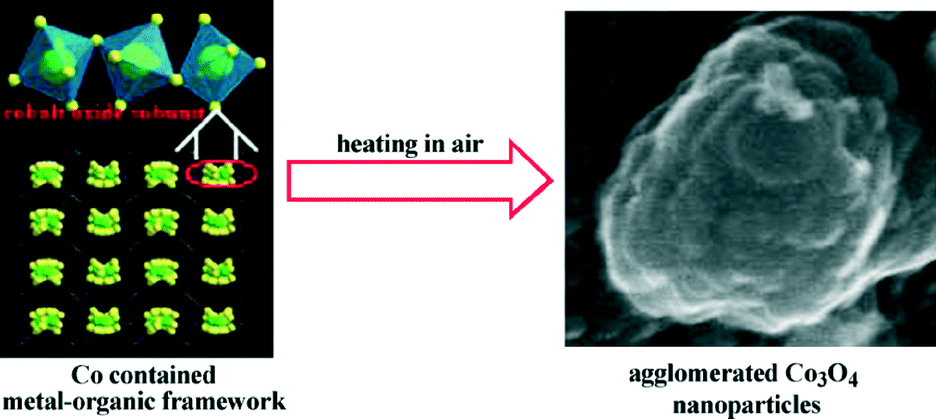 | ||
| Fig. 5 Synthetic scheme for the production of agglomerated Co3O4 nanoparticles via calcination of a MOF carbon source. Reprinted with permission from ref. 49. Copyright 2010, Elsevier B.V. | ||
These phenomena can be confirmed within other porous morphologies of Co3O4 such as nanowires. Porous Co3O4 nanowires have been synthesized from porous coordination polymers (PCPs).50 Hydrothermal synthesis of cobalt and nitrilotriacetic acid (NA) forms a Co–NA nanowire template. Calcination of the template material at 450 °C facilitates the oxidation of cobalt and the concomitant decomposition of the template giving porous Co3O4 nanowires. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) determined an average nanowire length on an order of magnitude of 10 s of nm and a uniform diameter of 150 nm. The as-prepared nanowires were found to have a BET surface area of ca. 26 m2 g−1 by N2 sorption isotherms. Electrochemical characterization of the as-prepared Co3O4 nanowires proceeded by the usage of the as-prepared nanowires as the positive electrode of a standard Co3O4 half-cell. After the first discharge, the cathodic potential peak shows a marked decrease in the second discharge but an approximate stable discharge in cycles 2 through 10. Cyclic voltammetry experiments confirmed the initial capacity of the Co3O4 nanowires as 1366 mA h g−1 at a current density of 300 mA g−1 and the retention of a reversible capacity of 810 mA h g−1 at 30 cycles, or ca. 56% of its initial capacity. Synthetic reaction conditions were also investigated for significant electrochemical effect. Solvent and metal counterion composition were found to effect the nanowire morphology with homogeneous nanowire diameters resulting from water-rich phases and regular, smooth nanowires when chloride ions (Cl−) were included as a counterion to the cobalt. The effect of Cl− in a synthetic route employing CoCl2 as the metallic reagent was ascribed to its hydrogen bond stabilization of the nanowires in a water-rich local environment. The decrease in the electrochemical performance from the agglomerated Co3O4 nanoparticles to that of the weaker as-prepared nanowire morphology is consistent with materials having higher BET surface areas resulting in decreased electrochemical performance.
Another transition metal oxide that has been studied for use in LIB electrodes is Fe2O3 as it has a theoretical capacity of ca. 1000 mA h g−1,33 a value far greater than the theoretical capacity of conventional anodic graphite. However, it is well-known that nanometric Fe2O3 suffers from poor cycleability due to large volume changes upon lithium ion insertion and extraction.51 Fe2O3 is also known to have a low conductivity with respect to other transition metal oxides consistent with hindrance of electrochemical performance, particularly during the charge/discharge process at high current densities.52 Conventional methodologies have focused on altering the size and morphology of Fe2O3 structures via traditional synthetic methods. However, such techniques have produced limited candidates of which many are further impeded by production costs and sub-optimal specific capacities and rate capability.53 MOF-templating has been used as an alternate method for producing spindle-like mesoporous α-Fe2O3via the solvothermal synthesis of MIL-88(Fe) subsequently followed by a two-step calcination process conducted at 500 °C and 380 °C sequentially.51 The former temperature under an inert atmosphere produces the FeOx–C composite; the latter under ambient gases facilitates the removal of carbonaceous residues and the formation of α-Fe2O3 with a spindle-like morphology with a BET surface area of 75 m2 g−1. The synthesis scheme is shown in Fig. 6. While such a high surface area could be considered to be a detriment in some electrode materials, the surface of the as-prepared α-Fe2O3 is considered to be beneficial to the two-step calcination due to the carbon formed during the first step. The formed carbonaceous residue transiently buffers the interior of the material against further structural contraction during the final heat treatment of the calcination process. The electrochemical performance of the spindle-like α-Fe2O3 was considered and found to be an improvement over other α-Fe2O3 morphologies. The as-prepared sample presented a Coulombic efficiency of 69% at a current density of 200 mA g−1 (0.2 C) after one cycle by retained a high capacity of ca. 97% (911 mA h g−1; discharge rate fixed at 0.1 C) from cycles 3–100 at a current density of 100 mA h g−1. The relatively poor efficiency associated with the higher current density is common amongst α-Fe2O3 morphologies and is attributed to the formation of a solid electrolyte interface and electrolyte decomposition causing irreversible capacity loss. The high specific capacity is attributed to its porous structure and small nanometric particles that allow for electrolyte migration and lithium-ion diffusion while remaining a buffer against volume changes inherent in lithium-ion intercalation and extraction, analogous to the rationale of the Co3O4 agglomerated nanoparticles. Other MOF-templated morphologies of α-Fe2O3 have been produced including a nanocube structure based on MOF-templated Prussian blue nanocubes,54 although the electrochemical properties of the agglomerated nanoparticles outweigh those of the nanocube morphology in respect to novel electrode design. Other oxides, such as cuprous and cupric, have also been used in the development of anodic materials for LIBs as metal deficient p-type semiconductor materials although they have been outshone by other materials developed in the field.55
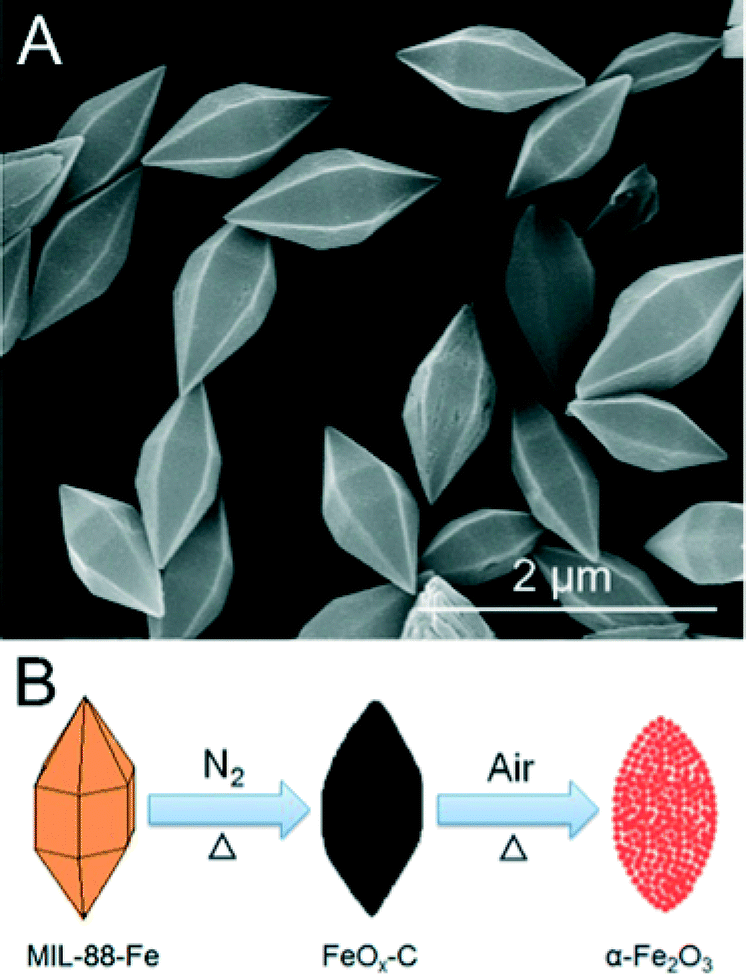 | ||
| Fig. 6 (a) SEM images of the as-prepared MIL-88(Fe) (b) Synthetic scheme for the production of the spindle-like α-Fe2O3 morphology via the calcination of a MOF carbon source.55 Reprinted with permission from ref. 51. Copyright 2012, American Chemical Society. | ||
LSBs have an advantage over their LIB counterparts due to their higher energy density and reduced cost.56 Importantly, LSBs have a very high theoretical energy density of 2500 Wh kg−1, which is significantly higher than the 500 Wh kg−1 associated with traditional LIBs.57 Inhibitory constraints to the practical application of LSBs stem from the solubility of polysulfides in organic electrolytes and the formation of intermediates (formula: Li2Sx (1 ≤ x ≥ 8)) during both the charging and discharging processes.58
The high solubility of the polysulfides results in a dynamic loss of mass in the sulfur cathode leading to a drastic reduction in capacitance upon recycling. The polysulfides can also migrate through the electrolyte to the metallic lithium anode causing the formation of Li2S, an insoluble that can result in a further decrease of long term function.59
Examples of LSBs derived via the calcination or carbonization of MOF precursors include microporous carbon polyhedron (MPCPs),60 but their performance is still unsuitable as a viable competitor of LIBs.
3.2 MOFs as precursors to novel supercapacitor electrode materials
SC electrolytes can be delineated into aqueous or organic moieties. Aqueous electrolytes, most commonly based on sulfuric acid (H2SO4) or potassium hydroxide (KOH), offer low internal resistance but are necessarily limited to a comparatively low operational voltage of ca. one volt per cell.61 Organic electrolytes, such as those based on acetonitrile or propylene carbonate (PC), allow for a higher cellular voltage due to the cost of increased internal resistance.Liu, et al. produced the first MOF-templated material formulated for use in superconductors in 2008. Furfuryl alcohol (FA) was introduced into a sample of MOF-5 via vapor deposition resulting in the polymerization of FA within the pores of the MOF-5 framework (FA@MOF-5) upon heat treatment.49 Pyrolysis of FA@MOF-5 at multiple temperatures resulted in distinct structures with variable surface areas. When pyrolysis was conducted at 1000 °C, the BET surface area reached a maximum among their samples of 2872 m2 g−1. At this temperature, pyrolysis of the FA@MOF-5 resulted in global framework collapse where the framework acted as a self-sacrificial scaffold for the formation of a nanoporous carbon structure. The resultant nanoporous carbon was examined as a potential novel material for superconductor electrodes in a 1.0 M H2SO4 electrolyte.
Cyclic voltammetry experiments gave a value for the capacitance of 204 F g−1 when considered at a sweep rate of 5 mV s−1. The specific capacitance of the material was found to be as high as 258 F g−1via galvanostatic charge/discharge experiments.
To the best of our knowledge, MOF-templated electrode materials targeted for SC using aqueous electrolytes have far out performed their organic-based counterparts. The preeminent examples of the electric double-layer capacitor anodic material produced from a MOF-templating procedure is HCPs-0.2, also denoted as a worm-like mesoporous carbon (WMC) in a previous publication.66
Deng, et al. first synthesized a series of hierarchical porous carbons in 2010 from a rationally designed nonporous MOF, or a metal–organic coordination polymer (MOCP) as it is referred to in the Materials Letters publication.72 The template for the HCPs series later resulted from a synthetic deviation from the conventional MOF-5 protocol.66 Reaction of terephthalic acid, zinc nitrate hexahydrate, and bismuth nitrate pentahydrate in DMF resulted in a crystalline product. The particular synthesis of HPCs-0.2 required 0.2 mmol of the bismuth salt. After drying and the addition of glycerol as a carbon source, the product was pyrolized at 1000 °C for 8 h. In total, there were four derivatives of the parent hierarchical porous carbon produced labeled by the concentration of bismuth added for each particular trial, 0, 0.1, 0.2, and 0.4 mmol of bismuth in the precursor, respectively. The electrochemical parameters of the HPCs were examined via galvanostatic charge/discharge experiments. For the particular example of HCPs-0.2, a combination of the HCPs-0.2 product, carbon black, and polytetrafluoroethylene (PTFE, 5%) was used as a working electrode in a conventional three-electrode experimental setup with 6 M KOH as the electrolyte. The counter and reference electrodes were nickel foil and Hg/HgO, respectively. Routine characterization confirmed a mesoporous carbon prior to calcination with pore sizes centered on 2.6 and 6.3 nm and a BET surface area of 2587 m2 g−1. The resultant morphology was not one of nanoparticles or nanowires, but of structures denoted as a “flower-like” structure consisting of nanosheets. The large gaps left by the aggregation of these sheets facilitates a faster transport of electrolytes through the system in comparison with traditional electrode materials or materials with a lower porosity. Galvanostatic charge/discharge experiments confirmed the functionality of the resultant product as a novel electrode material for electric double-layer capacitors with a capacitance of 344 F g−1. The deviation in the synthetic route between the second paper where the resultant structure was denoted as HCPs-0.2 and the first in which is was denoted as WMC is slight. The only differing experimental parameter was the calcination temperature at 950 °C and 1000 °C, respectively. However, since the eutectic zinc–bismuth alloy produced during the synthesis of the HPCs vaporizes above 950 °C, the pertinent experimental details can be considered to be equivalent.
To the best of our knowledge, WMC/HCPs-0.2 is the best performing novel material synthesized from a MOF template or precursor for the application of SC electrodes. The second-best material formed via a calcination process is activated MAC (MAC-A) with a capacitance of 274 F g−1, a 70 F deviation from WMC/HCPs-0.2. A selection of SC electrode materials formed by the carbonization or calcination of MOFs can be seen in Table 1 which are delineated by activity in alkaline, acidic, and ionic/organic electrolytes, respectively.
| Sample | S BET (m2 g−1) | Capacitance (F g−1) | Current density (mA g−1) | Sweep rate (mV s−1) | Temperature (°C) | Electrolyte |
|---|---|---|---|---|---|---|
| Acidic electrolytes | ||||||
| NPC49 | 2872 | 258 | 250 | — | 1000 | 1.0 M H2SO4 |
| NPC650 (ref. 62) | 1521 | 222 | 50 | — | 650 | 1.0 M H2SO4 |
| AS-ZC-800 (ref. 63) | 2972 | 211 | 10 | 800 | 1.0 M H2SO4 | |
| C800 (ref. 27b) | 2169 | 188 | 250 | — | 800 | 1.0 M H2SO4 |
| A-ZC-800 (ref. 63) | 1341 | 170 | — | 10 | 800 | 1.0 M H2SO4 |
| C1000 (ref. 27b) | 3405 | 161 | 250 | — | 1000 | 1.0 M H2SO4 |
| S-ZC-800 (ref. 63) | 1955 | 158 | — | 10 | 800 | 1.0 M H2SO4 |
| NPC530 (ref. 62) | 3040 | 158 | 50 | — | 530 | 1.0 M H2SO4 |
| NPC800 (ref. 62) | 1141 | 151 | 50 | — | 800 | 1.0 M H2SO4 |
| NPC1000 (ref. 62) | 2524 | 149 | 50 | — | 1000 | 1.0 M H2SO4 |
| NPC900 (ref. 62) | 1647 | 148 | 50 | — | 900 | 1.0 M H2SO4 |
| Z-800 (ref. 64) | 720 | 130 | — | 50 | 800 | 1.0 M H2SO4 |
| Z-900 (ref. 64) | 1075 | 128 | — | 50 | 900 | 1.0 M H2SO4 |
| Z-1000 (ref. 64) | 1110 | 112 | — | 50 | 1000 | 1.0 M H2SO4 |
| ZC-800 (ref. 63) | 1051 | 104 | — | 10 | 800 | 1.0 M H2SO4 |
| Basic electrolytes | ||||||
| HPCs-0.2 (ref. 65) | 2587 | 344 | 50 | — | 1000 | 6 M KOH |
| WMC66 | 2587 | 344 | 50 | — | 950 | 6 M KOH |
| MAC-A67 | 2222 | 274 | 250 | — | MAC activated at 700 | 6 M KOH |
| HPCs-0.4 (ref. 65) | 2857 | 241 | 100 | — | 1000 | 6 M KOH |
| MC-A67 | 1673 | 222 | 250 | — | MC activated at 700 | 6 M KOH |
| HPs-0.1 (ref. 65) | 2137 | 215 | 100 | — | 1000 | 6 M KOH |
| MPC-A67 | 1271 | 187 | 250 | — | MPC activated at 700 | 6 M KOH |
| HPCs-0 (ref. 65) | 1796 | 185 | 100 | — | 1000 | 6 M KOH |
| C-S700 (ref. 68) | 817 | 182 | — | 100 | 700 | 6 M KOH |
| MgC69 | 2322 | 173 | 500 | — | 700 | 30% KOH |
| MPC67 | 1543 | 170 | 250 | — | 900 | 6 M KOH |
| BaC69 | 955 | 169 | 500 | — | 700 | 30% KOH |
| HPC67 | 1391 | 166 | 100 | — | 1000 | 6 M KOH |
| C-S900 (ref. 68) | 704 | 156 | — | 100 | 900 | 6 M KOH |
| MC67 | 1812 | 149 | 250 | — | 900 | 6 M KOH |
| C-C1700 (ref. 68) | 311 | 117 | — | 100 | 700 | 6 M KOH |
| MAC67 | 384 | 72 | 250 | — | 900 | 6 M KOH |
| Basic electrolytes | ||||||
| C-C1900 (ref. 68) | 199 | 70 | — | 100 | 900 | 6 M KOH |
| Organic/neutral salt electrolytes | ||||||
| MgC69 | 2322 | 180 | 500 | — | 700 | EMImBF4 |
| 3D HPC70 | 1224 | 175 | 600 | — | 950 | 1 M NEt4BF4 in PC |
| BaC69 | 955 | 171 | 500 | — | 700 | EMImBF4 |
| MAC-A67 | 2222 | 168 | 250 | — | MAC activated at 700 | 1 M NEt4BF4 acetonitrile |
| MC-A67 | 1673 | 126 | 250 | — | MC activated at 700 | 1 M NEt4BF4 acetonitrile |
| MDC-D71 | 3526 | Ca. 120 | — | 50 | 900 | 1 M NaCl |
| MC67 | 1812 | 113 | 250 | — | 900 | 1.5 M NEt4BF4 acetonitrile |
| MPC67 | 1543 | 111 | 250 | — | 900 | 1 M NEt4BF4 acetonitrile |
| MPC-A67 | 1271 | 110 | 250 | — | MPC activated at 700 | 1 M NEt4BF4 acetonitrile |
| MAC67 | 384 | 4 | 250 | — | 900 | 1 M NEt4BF4 acetonitrile |
Zhou, et al. prepared two mesoporous carbons via the simple and direct pyrolysis of magnesium citrate and barium citrate.69 The resultant materials, MgC and BaC, were further examined for their potential electrochemical efficacy in organic electrolytes. MgC proved to be the most efficient material of the two, and of all MOF-derived materials for electrode function in ionic/organic electrolytes, with a capacitance of 180 F g−1 in EMImBF4 (1-ethyl-3-methylimidazolium tetrafluoroborate). BaC synthesized from an analogous synthetic scheme was found to have a capacitance of 171 F g−1 using the same electrolyte system.
Presently, it can easily be seen that novel MOF-derived electrodes formulated for use in alkaline electrolytes, primarily KOH, are superior to those functional in acidic or organic electrolytes, i.e. H2SO4 and EMImBF3, respectively. This can be attributed to the complete ionization of KOH in solution, in comparison to sulfuric acid and organic electrolytes which undergo only partial ionization. The ionization of H2SO4 is only considered to be sufficiently strong during its first dissociation process leaving H2SO4 as a relatively electrolyte. Organic electrolytes necessarily cannot be classified as strong electrolytes but do have their advantages in the development of electrodes for SCs due to allowing for a higher operational voltage per cell.73
3.3 Conclusion
While not quite as new as the use of MOFs as self-sacrificing precursors for fuel cell electrocatalysts, the use of MOFs as precursors or templates for the development of novel electrode materials for lithium-based batteries (LIB, LSB) and SCs can still be classified recently introduced area of research. In the years since the initial publications describing MOF-derived novel electrodes for both LIBs and SCs, the field has undoubtedly made significant strides in the formulation of new devices such as LSBs. While LIBs are pervasive, the field still faces a number of challenges. Some of the most dominant of these is the need for more increased structural stability that can withstand the volume changes associated lithium-ion intercalation and extraction over longer cycling lives,74 and needs for improved capacity, overall increased cycling lives, the development of perfectly lithium-compatible electrolytes, and batteries made of more environmentally sustainable materials.75 While MOFs have made an impact on the field of lithium-based batteries, it is our opinion that their use in this field is only just being explored and that the innate diversity and porosity of MOFs will continue to contribute to the development of these materials. We feel that the same sentiments can be extrapolated to the use of MOFs in the development of electrodes for SCs. Shortcomings of the published MOF-derived electrodes still remain particularly the low energetics of the current devices, i.e. low capacitance and low energy density. The current literature on this topic has revealed that high specific surface areas, tailored pores in terms of both size and functionality, low internal electrical resistances, increased cycling performance and lowered cost for development will be beneficial to the development of novel electrodes for this application. It is our belief that MOFs can be prominent contributors as potential platforms for the continuing development of electrodes as they relate to renewable energy storage devices, namely lithium-based batteries and SCs.4. Conclusions and future perspective
Worldwide climate change and the imminent decline of nonrenewable energy sources such as fossil fuels have culminated in a global search for green energy generation and storage alternatives. MOF-derived green energy alternatives show great promise for both energy generation, via FCs, and energy storage, via LIBs, LSBs, and SCs. However, as with all work in this emerging field, there still is room for further improvement before these devices can be effectively transplanted from the laboratory to the consumer market.Acknowledgements
The authors acknowledge the University of South Florida for the financial support of this work. Kia Williams is funded by NSF Florida-Georgia LSAMP HRD #1139850.References
- (a) A. Choudhury, H. Chandra and A. Arora, Application of solid oxide fuel cell technology for power generation—A review, Renewable Sustainable Energy Rev., 2013, 20, 430–442 CrossRef CAS PubMed; (b) X. Zhou, J. Qiao, L. Yang and J. Zhang, A Review of Graphene-Based Nanostructural Materials for Both Catalyst Supports and Metal-Free Catalysts in PEM Fuel Cell Oxygen Reduction Reactions, Adv. Energy Mater., 2014, 4, 1301523 Search PubMed; (c) M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanostructured carbon-metal oxide composite electrodes for supercapacitors: a review, Nanoscale, 2013, 5(1), 72–88 RSC; (d) H. Choi, C. Nahm, J. Kim, C. Kim, S. Kang, T. Hwang and B. Park, Review paper: Toward highly efficient quantum-dot- and dye-sensitized solar cells, Curr. Appl. Phys., 2013, 13, S2–S13 CrossRef PubMed; (e) L. Lu, X. Han, J. Li, J. Hua and M. Ouyang, A review on the key issues for lithium-ion battery management in electric vehicles, J. Power Sources, 2013, 226, 272–288 CrossRef CAS PubMed; (f) A. J. Heeger, 25th anniversary article: bulk heterojunction solar cells: understanding the mechanism of operation, Adv. Mater., 2014, 26(1), 10–28 CrossRef CAS PubMed; (g) M. Beidaghi and Y. Gogotsi, Capacitive energy storage in micro-scale devices: recent advances in design and fabrication of micro-supercapacitors, Energy Environ. Sci., 2014, 7(3), 867 RSC.
- (a) M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Hydrogen storage in metal-organic frameworks, Chem. Rev., 2012, 112(2), 782–835 CrossRef CAS PubMed; (b) Y. He, W. Zhou, G. Qian and B. Chen, Methane storage in metal–organic frameworks, Chem. Soc. Rev., 2014, 43, 5657–5668 RSC.
- J. R. Li, J. Sculley and H. C. Zhou, Metal-organic frameworks for separations, Chem. Rev., 2012, 112(2), 869–932 CrossRef CAS PubMed.
- (a) K. Leus, Y.-Y. Liu and P. Van Der Voort, Metal-Organic Frameworks as Selective or Chiral Oxidation Catalysts, Catal. Rev.: Sci. Eng., 2014, 56(1), 1–56 CrossRef CAS; (b) V. Lykourinou, Y. Chen, X. S. Wang, L. Meng, T. Hoang, L. J. Ming, R. L. Musselman and S. Ma, Immobilization of MP-11 into a mesoporous metal-organic framework, MP-11@mesoMOF: a new platform for enzymatic catalysis, J. Am. Chem. Soc., 2011, 133(27), 10382–10385 CrossRef CAS PubMed.
- (a) P. Canepa, Y. Chabal and T. Thonhauser, When metal organic frameworks turn into linear magnets, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 094407 CrossRef; (b) Q. Zhang, B. Li and L. Chen, First-principles study of microporous magnets M-MOF-74 (M = Ni, Co, Fe, Mn): the role of metal centers, Inorg. Chem., 2013, 52(16), 9356–9362 CrossRef CAS PubMed.
- (a) M. J. Dong, M. Zhao, S. Ou, C. Zou and C. D. Wu, A luminescent dye@MOF platform: emission fingerprint relationships of volatile organic molecules, Angew. Chem., Int. Ed., 2014, 53(6), 1575–1579 CrossRef CAS PubMed; (b) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Metal-organic framework materials as chemical sensors, Chem. Rev., 2012, 112(2), 1105–1125 CrossRef CAS PubMed.
- (a) A. Morozan and F. Jaouen, Metal organic frameworks for electrochemical applications, Energy Environ. Sci., 2012, 5(11), 9269 RSC; (b) S.-L. Li and Q. Xu, Metal–organic frameworks as platforms for clean energy, Energy Environ. Sci., 2013, 6(6), 1656 RSC.
- Multi-Year Research, Development and Demonstration Plan, 2012.
- (a) I. A. Khan, A. Badshah, N. Haider, S. Ullah, D. H. Anjum and M. A. Nadeem, Porous carbon as electrode material indirect ethanol fuel cells (DEFCs) synthesized by the direct carbonization of MOF-5, J. Solid State Electrochem., 2014, 18, 1545–1555 CrossRef CAS PubMed; (b) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The chemistry and applications of metal-organic frameworks, Science, 2013, 341(6149), 1230444 CrossRef PubMed.
- B. Wang, Recent development of non-platinum catalysts for oxygen reduction reaction, J. Power Sources, 2005, 152, 1–15 CrossRef CAS PubMed.
- J. Zhang, PEM Fuel Cell Electrocatalysts and Catalyst Layers, 2008 Search PubMed.
- D. Papageorgopoulos, FY 2013 Annual Progress Report, DOE Hydrogen and Fuel Cells Program, 2013 Search PubMed.
- (a) M. Zhiani, H. A. Gasteiger, M. Piana and S. Catanorchi, Comparative study between platinum supported on carbon and non-noble metal cathode catalyst in alkaline direct ethanol fuel cell (ADEFC), Int. J. Hydrogen Energy, 2011, 36(8), 5110–5116 CrossRef CAS PubMed; (b) M. Z. F. Kamarudin, S. K. Kamarudin, M. S. Masdar and W. R. W. Daud, Review: Direct ethanol fuel cells, Int. J. Hydrogen Energy, 2013, 38(22), 9438–9453 CrossRef CAS PubMed.
- J. Liu, H. Wang, C. Wu, Q. Zhao, X. Wang and L. Yi, Preparation and characterization of nanoporous carbon-supported platinum as anode electrocatalyst for direct borohydride fuel cell, Int. J. Hydrogen Energy, 2014, 39, 6729–6736 CrossRef CAS PubMed.
- J. Ma, N. A. Choudhury and Y. Sahai, A comprehensive review of direct borohydride fuel cells, Renewable Sustainable Energy Rev., 2010, 14(1), 183–199 CrossRef CAS PubMed.
- F. Afsahi, H. Vinh-Thang, S. Mikhailenko and S. Kaliaguine, Electrocatalyst synthesized from metal organic frameworks, J. Power Sources, 2013, 239, 415–423 CrossRef CAS PubMed.
- S. Ma, G. A. Goenaga, A. V. Call and D. J. Liu, Cobalt imidazolate framework as precursor for oxygen reduction reaction electrocatalysts, Chem. – Eur. J., 2011, 17(7), 2063–2067 CrossRef CAS PubMed.
- D. Zhao, J.-L. Shui, C. Chen, X. Chen, B. M. Reprogle, D. Wang and D.-J. Liu, Iron imidazolate framework as precursor for electrocatalysts in polymer electrolyte membrane fuel cells, Chem. Sci., 2012, 3(11), 3200 RSC.
- E. Proietti, F. Jaouen, M. Lefevre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- N. Larouche, R. Chenitz, M. Lefèvre, E. Proietti and J.-P. Dodelet, Activity and stability in proton exchange membrane fuel cells of iron-based cathode catalysts synthesized with addition of carbon fibers, Electrochim. Acta, 2014, 115, 170–182 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt, Science, 2011, 332(6028), 443–447 CrossRef CAS PubMed.
- A. Morozan, M. T. Sougrati, V. Goellner, D. Jones, L. Stievano and F. Jaouen, Effect of Furfuryl Alcohol on Metal Organic Framework-based Fe/N/C Electrocatalysts for Polymer Electrolyte Membrane Fuel Cells, Electrochim. Acta, 2014, 119, 192–205 CrossRef CAS PubMed.
- T. Palaniselvam, B. P. Biswal, R. Banerjee and S. Kurungot, Zeolitic imidazolate framework (ZIF)-derived, hollow-core, nitrogen-doped carbon nanostructures for oxygen-reduction reactions in PEFCs, Chem. – Eur. J., 2013, 19(28), 9335–9342 CrossRef CAS PubMed.
- J. Tian, A. Morozan, M. T. Sougrati, M. Lefevre, R. Chenitz, J. P. Dodelet, D. Jones and F. Jaouen, Optimized synthesis of Fe/N/C cathode catalysts for PEM fuel cells: a matter of iron-ligand coordination strength, Angew. Chem., Int. Ed., 2013, 52(27), 6867–6870 CrossRef CAS PubMed.
- L. Zhang, K. Lee, C. W. B. Bezerra, J. Zhang and J. Zhang, Fe loading of a carbon-supported Fe–N electrocatalyst and its effect on the oxygen reduction reaction, Electrochim. Acta, 2009, 54(26), 6631–6636 CrossRef CAS PubMed.
- A. Velázquez-Palenzuela, L. Zhang, L. Wang, P. L. Cabot, E. Brillas, K. Tsay and J. Zhang, Fe–Nx/C electrocatalysts synthesized by pyrolysis of Fe(II)–2,3,5,6-tetra(2-pyridyl)pyrazine complex for PEM fuel cell oxygen reduction reaction, Electrochim. Acta, 2011, 56(13), 4744–4752 CrossRef PubMed.
- (a) M. Pumera, Graphene-based nanomaterials for energy storage, Energy Environ. Sci., 2011, 4(3), 668 RSC; (b) H. L. Jiang, B. Liu, Y. Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, From metal-organic framework to nanoporous carbon: toward a very high surface area and hydrogen uptake, J. Am. Chem. Soc., 2011, 133(31), 11854–11857 CrossRef CAS PubMed.
- S. Pandiaraj, H. B. Aiyappa, R. Banerjee and S. Kurungot, Post modification of MOF derived carbon via g-C3N4 entrapment for an efficient metal-free oxygen reduction reaction, Chem. Commun., 2014, 50(25), 3363–3366 RSC.
- P. Zhang, F. Sun, Z. Xiang, Z. Shen, J. Yun and D. Cao, ZIF-derived in situ nitrogen-doped porous carbons as efficient metal-free electrocatalysts for oxygen reduction reaction, Energy Environ. Sci., 2014, 7(1), 442 CAS.
- J.-M. Tarascon and M. Armand, Issues and challenges facing rechargeable lithium batteries, Nature, 2001, 414(6861), 359–367 CrossRef CAS PubMed.
- B. Gao, A. Kleinhammes, X. P. Tang, C. Bower, L. Fleming, Y. Wu and O. Zhou, Electrochemical intercalation of single-walled carbon nanotubes with lithium, Chem. Phys. Lett., 1999, 307(3–4), 153–157 CrossRef CAS.
- M. Armand and J.-M. Tarascon, Building better batteries, Nature, 2008, 451(7179), 652–657 CrossRef CAS PubMed.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanostructured metal oxide-based materials as advanced anodes for lithium-ion batteries, Nanoscale, 2012, 4(8), 2526–2542 RSC.
- J.-M. Tarascon and D. Guyomard, Li metal-free rechargeable batteries based on Li1+xMn2O4 cathodes (o<x<1) and carbon anodes, J. Electrochem. Soc., 1991, 138(10), 2864–2868 CrossRef CAS PubMed.
- R. Elazari, G. Salitra, A. Garsuch, A. Panchenko and D. Aurbach, Sulfur-impregnated activated carbon fiber cloth as a binder-free cathode for rechargeable Li-S batteries, Adv. Mater., 2011, 23(47), 5641–5644 CrossRef CAS PubMed.
- J. Schiffer, D. Linzen and D. U. Sauer, Heat generation in double layer capacitors, J. Power Sources, 2006, 160(1), 765–772 CrossRef CAS PubMed.
- B. E. Conway, Transition from "supercapacitor" to "battery" behavior in electrochemical energy storage, J. Electrochem. Soc., 1991, 138(6), 1539–1548 CrossRef CAS PubMed.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, Supercapacitor devices based on graphene materials, J. Phys. Chem. C, 2009, 113(30), 13103–13107 CAS.
- E. Frackowiak and F. Begiun, Carbon materials for the electrochemical storage of energy in capacitors, Carbon, 2001, 39(6), 937–950 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Graphene-based ultracapacitors, Nano Lett., 2008, 8(10), 3498–3502 CrossRef PubMed.
- A. M. Bernardes, D. C. R. Espinosa and J. A. S. Tenório, Recycling of batteries: a review of current processes and technologies, J. Power Sources, 2004, 130(1–2), 291–298 CrossRef CAS PubMed.
- A. Morozan, B. Jousselme and S. Palacin, Low-platinum and platinum-free catalysts for the oxygen reduction reaction at fuel cell cathodes, Energy Environ. Sci., 2011, 4(4), 1238 CAS.
- L. Heredy and L. R. McCoy, Lithium-molten Salt Cell with Transition Metal Chalcogenide Positive Electrode, US Pat. 3,898,096, Aug 5, 1975, 1975 Search PubMed.
- A. Lueking and R. T. Yang, Hydrogen Spillover from a Metal Oxide Catalyst onto Carbon Nanotubes—Implications for Hydrogen Storage, J. Catal., 2002, 206(1), 165–168 CrossRef CAS.
- M. Dekkers, G. Rijnders and D. H. A. Blank, ZnIr[sub 2]O[sub 4], a p-type transparent oxide semiconductor in the class of spinel zinc-d[sup 6]-transition metal oxide, Appl. Phys. Lett., 2007, 90(2), 021903 CrossRef PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nano-sized transition-metal oxides as negative-electrode materials for lithium-ion batteries, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- X. Y. Xue, S. Yuan, L. L. Xing, Z. H. Chen, B. He and Y. J. Chen, Porous Co3O4 nanoneedle arrays growing directly on copper foils and their ultrafast charging/discharging as lithium-ion battery anodes, Chem. Commun., 2011, 47(16), 4718–4720 RSC.
- K. T. Nam, D. W. Kim, P. J. Yoo, C. Y. Chiang, N. Meethong, P. T. Hammond, Y. M. Chiang and A. M. Belcher, Virus-enabled synthesis and assembly of nanowires for lithium ion battery electrodes, Science, 2006, 312(5775), 885–888 CrossRef PubMed.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, Metal-organic framework as a template for porous carbon synthesis, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef PubMed.
- C. Li, X. Yin, L. Chen, Q. Li and T. Wang, Synthesis of cobalt ion-based coordination polymer nanowires and their conversion into porous Co3O4 nanowires with good lithium storage properties, Chemistry, 2010, 16(17), 5215–5221 CrossRef PubMed.
- X. Xu, R. Cao, S. Jeong and J. Cho, Spindle-like mesoporous alpha-Fe(2)O(3) anode material prepared from MOF template for high-rate lithium batteries, Nano Lett., 2012, 12(9), 4988–4991 CrossRef CAS PubMed.
- Y. C. Lee, Y. L. Chueh, C. H. Hsieh, M. T. Chang, L. J. Chou, Z. L. Wang, Y. W. Lan, C. D. Chen, H. Kurata and S. Isoda, p-Type alpha-Fe2O3 nanowires and their n-type transition in a reductive ambient, Small, 2007, 3(8), 1356–1361 CrossRef CAS PubMed.
- (a) W. Cho, Y. H. Lee, H. J. Lee and M. Oh, Systematic transformation of coordination polymer particles to hollow and non-hollow In(2)O(3) with pre-defined morphology, Chem. Commun., 2009,(31), 4756–4758 RSC; (b) W. Cho, Y. H. Lee, H. J. Lee and M. Oh, Multi ball-in-ball hybrid metal oxides, Adv. Mater., 2011, 23(15), 1720–1723 CrossRef PubMed; (c) W. Cho, S. Park and M. Oh, Coordination polymer nanorods of Fe-MIL-88B and their utilization for selective preparation of hematite and magnetite nanorods, Chem. Commun., 2011, 47(14), 4138–4140 RSC.
- L. Zhang, H. B. Wu, R. Xu and X. W. Lou, Porous Fe2O3 nanocubes derived from MOFs for highly reversible lithium storage, CrystEngComm, 2013, 15(45), 9332 RSC.
- (a) J. Zhang, J. Liu, Q. Peng, X. Wang and Y. Li, Nearly monodisperse Cu2O and CuO nanospheres: preparation and applications for sensitive gas sensors, Chem. Mater., 2006, 18(4), 867–871 CrossRef CAS; (b) M. Izaki, T. Shinagawa, K.-T. Mizuno, Y. Ida, M. Inaba and A. Tasaka, Electrochemically constructed p-Cu2O/n-ZnO heterojunction diode for photovoltaic device, J. Phys. D: Appl. Phys., 2007, 40(11), 3326–3329 CrossRef CAS; (c) B. Skarman, D. Grandjean, R. Benfield, A. Hinz, A. Andersson and L. Wallenberg, Carbon Monoxide Oxidation on Nanostructured CuO/CeO Composite Particles Characterized by HREM, XPS, XAS, and High-Energy Diffraction, J. Catal., 2002, 211(1), 119–133 CAS; (d) B. White, M. Yin, A. Hall, D. Le, S. Stolbov, T. Rahman, N. Turro and S. O'Brien, Complete CO oxidation over Cu2O nanoparticles supported on silica gel, Nano Lett., 2006, 6(9), 2095–2098 CrossRef CAS PubMed; (e) X. Wang, J. A. Rodriguez, J. C. Hanson, D. Gamarra, A. Martinez-Arias and M. Fernandez-Garcia, In situ studies of the active sites for the water gas shift reaction over Cu-CeO2 catalysts: complex interaction between metallic copper and oxygen vacancies of ceria, J. Phys. Chem. B, 2006, 110(1), 428–434 CrossRef CAS PubMed; (f) Z. Zhong, V. Ng, J. Luo, S.-P. Teh, J. Teo and A. Gedanken, Manipulating the self-assembling process to obtain control over the morphologies of copper oxide in hydrothermal synthesis and creating pores in the oxide architecture, Langmuir, 2007, 23(11), 5971–5977 CrossRef CAS PubMed; (g) X. Zheng, X. Zhang, X. Wang, S. Wang and S. Wu, Preparation and characterization of CuO/CeO2 catalysts and their applications in low-temperature CO oxidation, Appl. Catal., A, 2005, 295(2), 142–149 CrossRef PubMed; (h) M.-F. Luo, Y.-P. Song, J.-Q. Lu, X.-Y. Wang and Z.-Y. Pu, Identification of CuO species in high surface area CuO-CeO2 catalysts and their catalytic activities for CO oxidation, J. Phys. Chem. C, 2007, 111(34), 12686–12692 CrossRef CAS; (i) P. G. Harrison, I. K. Ball, W. Azelee, W. Daniell and D. Goldfarb, Nature and surface redox properties of copper(II)-promoted cerium(IV) oxide CO-oxidation catalysts, Chem. Mater., 2000, 12(12), 3715–3725 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, A highly ordered nanostructured carbon-sulphur cathode for lithium-sulphur batteries, Nat. Mater., 2009, 8(6), 500–506 CrossRef CAS PubMed.
- H.-J. Ahn, K.-W. Kim, J.-H. Ahn and G. Cheruvally, Encyclopedia of Electrochemical Power Sources, Elsevier, 2010, vol. V, p. 155 Search PubMed.
- Z. Wang, X. Li, Y. Cui, Y. Yang, H. Pan, Z. Wang, C. Wu, B. Chen and G. Qian, A Metal–Organic Framework with Open Metal Sites for Enhanced Confinement of Sulfur and Lithium–Sulfur Battery of Long Cycling Life, Cryst. Growth Des., 2013, 13(11), 5116–5120 Search PubMed.
- S.-E. Cheon, K.-S. Ko, J.-H. Cho, S.-W. Kim, E.-Y. Chin and H.-T. Kim, Rechargeable Lithium Sulfur Battery, J. Electrochem. Soc., 2003, 150(6), A800 CrossRef CAS PubMed.
- H. B. Wu, S. Wei, L. Zhang, R. Xu, H. H. Hng and X. W. Lou, Embedding sulfur in MOF-derived microporous carbon polyhedrons for lithium-sulfur batteries, Chemistry, 2013, 19(33), 10804–10808 CrossRef CAS PubMed.
- A. Burke, Ultracapacitors: why, how, and where is the technology, J. Power Sources, 2000, 91(1), 37–50 CrossRef CAS.
- B. Liu, H. Shioyama, H. Jiang, X. Zhang and Q. Xu, Metal–organic framework (MOF) as a template for syntheses of nanoporous carbons as electrode materials for supercapacitor, Carbon, 2010, 48(2), 456–463 CrossRef CAS PubMed.
- A. J. Amali, J. K. Sun and Q. Xu, From assembled metal-organic framework nanoparticles to hierarchically porous carbon for electrochemical energy storage, Chem. Commun., 2014, 50(13), 1519–1522 RSC.
- W. Chaikittisilp, M. Hu, H. Wang, H. S. Huang, T. Fujita, K. C. Wu, L. C. Chen, Y. Yamauchi and K. Ariga, Nanoporous carbons through direct carbonization of a zeolitic imidazolate framework for supercapacitor electrodes, Chem. Commun., 2012, 48(58), 7259–7261 RSC.
- S. Mo, Z. Sun, X. Huang, W. Zou, J. Chen and D. Yuan, Synthesis, characterization and supercapacitive properties of hierarchical porous carbons, Synth. Met., 2012, 162(1–2), 85–88 CrossRef CAS PubMed.
- D. Yuan, J. Chen, S. Tan, N. Xia and Y. Liu, Worm-like mesoporous carbon synthesized from metal–organic coordination polymers for supercapacitors, Electrochem. Commun., 2009, 11(6), 1191–1194 CrossRef CAS PubMed.
- J. Hu, H. Wang, Q. Gao and H. Guo, Porous carbons prepared by using metal–organic framework as the precursor for supercapacitors, Carbon, 2010, 48(12), 3599–3606 CrossRef CAS PubMed.
- P. Su, L. Jiang, J. Zhao, J. Yan, C. Li and Q. Yang, Mesoporous graphitic carbon nanodisks fabricated via catalytic carbonization of coordination polymers, Chem. Commun., 2012, 48(70), 8769–8771 RSC.
- J. Zhou, X. Yuan, W. Xing, W.-J. Si and S.-P. Zhuo, Mesoporous carbons derived from citrates for use in electrochemical capacitors, New Carbon Materials, 2010, 25(5), 370–375 CrossRef.
- S.-L. Jin, H.-G. Deng, L. Zhan, W.-M. Qiao and L.-C. Ling, Synthesis of 3D hierarchical porous carbon as electrode material for electric double layer capacitors, New Carbon Materials, 2012, 27(2), 87–92 CrossRef CAS.
- S. J. Yang, T. Kim, K. Lee, Y. S. Kim, J. Yoon and C. R. Park, Solvent evaporation mediated preparation of hierarchically porous metal organic framework-derived carbon with controllable and accessible large-scale porosity, Carbon, 2014, 71, 294–302 CrossRef CAS PubMed.
- H. Deng, S. Jin, L. Zhan, Y. Wang, S. Qiao, L. Tang, X. Liang, W. Qiao and L. Ling, Synthesis and electrochemical performance of a laminated hollow porous carbon, Mater. Lett., 2010, 64(10), 1187–1189 CrossRef CAS PubMed.
- R. Kotz and M. Carlen, Principles and applications of electrochemical capacitors, Electrochim. Acta, 1999, 45(15–16), 2483–2498 Search PubMed.
- J. O. Besenhard, J. Yang and M. Winter, Will advanced lithium-alloy anodes have a chance in lithium-ion batteries?, J. Power Sources, 1997, 68(1), 87–90 CrossRef CAS.
- M. S. Whittingham, Lithium batteries and cathode materials, Chem. Rev., 2004, 104(10), 4271–4302 CrossRef CAS.
Footnote |
| † These two authors provided equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2015 |