
A crucial process: organic matrix and magnesium ion control of amorphous calcium carbonate crystallization on β-chitin film
Yufei
Ma
ab and
Qingling
Feng
*cd
aMOE Key Laboratory of Biomedical Information Engineering, School of Life Science and Technology, Xi'an Jiaotong University, Xi'an, 710049, PR China
bBioinspired Engineering and Biomechanics Center, Xi'an Jiaotong University, Xi'an 710049, PR China
cState Key Laboratory of New Ceramics and Fine Processing, Department of Materials Science and Engineering, Tsinghua University, Beijing 100084, China
dLaboratory of Advanced Materials, Department of Materials Science and Engineering, Tsinghua University, Beijing 100084, China. E-mail: biomater@mail.tsinghua.edu.cn; Fax: +86 10 62771160; Tel: +86 10 62782770
First published on 8th October 2014
Abstract
Amorphous calcium carbonate (ACC) particles with a diameter of about 300 nm were synthesized first. Then the as-prepared products were used to investigate the ACC transformation processes occurring on chitin film under the control of a water soluble matrix (WSM, extracted from aragonite pearls, China) or magnesium ions in aqueous solution. Raman spectroscopy, transmission electron microscopy (TEM) together with selected area electron diffraction (SAED) and field-emission scanning electron microscopy (FE-SEM) equipped with energy-dispersive X-ray (EDX) were used to characterize the crystallized calcium carbonate from ACC. The results demonstrate that the existence of a WSM and chitin film offers the ability to control the pure aragonite phase, leading to the formation of rod-like aragonite crystal aggregates. Comparatively, the collaborative effect of magnesium ions and chitin film not only induces the formation of the aragonite crystal aggregates, but also inhibits the transformation from ACC to Mg-calcite. Moreover, a possible transformation mechanism has been proposed and discussed.
1. Introduction
Calcium carbonate (CaCO3) is one of the most important industrial and biological materials due to its abundance in nature and its wide applications in industry. In parallel, CaCO3 can be used as a model mineral for biomimetic research, in which biogenic control over mineral orientation, polymorph and morphology has been intensively studied.1 Additionally, many in vitro studies have focused on unraveling biomineralization mechanisms with the hope of mimicking the natural materials and controlling the material structures and properties. Calcium carbonate in nature has three anhydrous crystalline polymorphs (calcite, aragonite and vaterite) and two well-defined hydrous crystalline polymorphs, calcium carbonate monohydrate (CaCO3·H2O) and calcium carbonate hexahydrate (CaCO3·6H2O), as well as one amorphous form.2Amorphous calcium carbonate (ACC) is a hydrated, poorly ordered and metastable precursor to crystalline calcium carbonate. Many research results illustrate that ACC is a useful amorphous phase for many aspects and plays an important role in the biomineralization of CaCO3.3,4 ACC is found in various organisms such as mature sea urchin larvae4,5 and larvae of molluscan bivalves.6 On the other hand, ACC is used in temporary storage deposits and structural skeletal elements,7–9 and acts as a transient precursor to crystalline polymorphs.8,10,11 The rich variety of CaCO3 structures in nature may be due to the character of ACC, which is easily molded into many different shapes. Thus, it is of scientific and industrial interest to study the transformation mechanism from ACC to a crystalline structure. Despite its importance, however, there are few reports on ACC due to its unstable nature.
Previously, a simple route involving the use of low temperature with the presence of a high amount of magnesium was used to synthesize highly monodispersed spherules of ACC in the lab firstly.12 Additionally, two solutions, (I) an aqueous solution including Ca2+ ions and dimethyl carbonate (DMC) and (II) an aqueous NaOH solution, were required to prepare ACC particles.13 However, stabilization and crystallization of ACC are controlled by various factors, including temperature, pH, additive and solvent.14–17 Sawada has reported that ACC precipitated at lower temperatures is more stable, whereas increasing the precipitation temperature induces crystallization.14 Koga et al. found that increasing the alkalinity stabilizes the amorphous state and then this observation was quantified by determining the enthalpies for the transformation of ACC into calcite depending on the pH of the mother liquor.15 Recently, various additives have been used as stabilizers for ACC, such as magnesium ions,16,18,19 phosphate,14,20 polyglutamates and DNA,21–23 and the transformation of ACC has been investigated. Loste et al.24 reported that ACC was the first phase formed in aqueous solution containing magnesium ions. The magnesium content of the ACC was determined by the Mg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ca ratio in the solution, and increased systematically with increasing Mg:Ca ratio. Then the crystalline phase produced by the transformation of ACC depended on the magnesium content of ACC. Additionally, several research groups have studied the use of mixtures of different solvents to control the transformation of ACC or the crystallization of calcium carbonate.17,25–27 Chen et al.17,25 reported water-induced phase transformation of poly (4-sodium styrene sulfonate) or poly (acrylic acid) stabilized ACC in a water–ethanol solution at room temperature. Liu et al.28 described that the phase transformation of magnesium amorphous calcium carbonate could be easily realized in an ethanol–water mixed solution which is free from organic additives under mild conditions. Though each factor mentioned above has been investigated independently of its influence on ACC stabilization and transformation, their combined effect on ACC crystallization is more close to the complicated biomineralization process in nature, and the real ACC transformation detail is poorly understood at present.
:Ca ratio in the solution, and increased systematically with increasing Mg:Ca ratio. Then the crystalline phase produced by the transformation of ACC depended on the magnesium content of ACC. Additionally, several research groups have studied the use of mixtures of different solvents to control the transformation of ACC or the crystallization of calcium carbonate.17,25–27 Chen et al.17,25 reported water-induced phase transformation of poly (4-sodium styrene sulfonate) or poly (acrylic acid) stabilized ACC in a water–ethanol solution at room temperature. Liu et al.28 described that the phase transformation of magnesium amorphous calcium carbonate could be easily realized in an ethanol–water mixed solution which is free from organic additives under mild conditions. Though each factor mentioned above has been investigated independently of its influence on ACC stabilization and transformation, their combined effect on ACC crystallization is more close to the complicated biomineralization process in nature, and the real ACC transformation detail is poorly understood at present.
Recently, increasing evidence proves that biomineralization occurs through self-assembly and/or transformation of the amorphous precursor, such as in the case of sea urchin spines29,30 and oyster shells.31–33 Weiss et al.6 has reported that the initially deposited mineral phase of mollusc larval shells is predominantly amorphous calcium carbonate, which subsequently partially transforms into aragonite. Nassif et al.34 recently demonstrated that the nacreous tablets of adult mollusk shell nacre are coated by a thin surface layer of ACC. These studies suggest that the primary minerals are likely amorphous. The transient formation of ACC seems to be a general principle in biomineralization. In different invertebrates, chitin could be regarded as the substrate that binds other macromolecules, which in turn induce nucleation of the mineral phase. This is well described for mollusc shells, specifically for nacre, where chitin has been suggested as an important component of the molluscan organic matrix.35 Hence, chitin, as a common substrate, plays an important role in CaCO3 crystal growth. However, limited information is available on the effect of chitin on ACC transformation and crystallization.
In biological systems, both Mg and specialized organic macromolecules may be used to stabilize ACC.36 Previously, we reported that the water soluble matrix of aragonite pearls from Hyriopsis cumingii had the ability to induce the formation of aragonite crystals.37 In the present work, ACC particles are synthesized first. Then we perform a systematic study of the transformation process from ACC to crystalline phases on chitin film under the control of a WSM or magnesium ions in aqueous solution. The phase transformation of ACC is observed during the formation of the rod-like and the quasi-spherical aragonite aggregates, and a possible growth mechanism of the aragonite crystal aggregates with different morphologies has been proposed. Moreover, this study could also be used in the rational design of new materials with complex shapes assembled from precursors or stable amorphous structures.
2. Experimental
2.1 Materials
All chemicals are analytical grade. Calcium chloride dehydrate, magnesium chloride hexahydrate, sodium hydroxide, dimethyl carbonate, ethanol and acetone were obtained from Sinopharm Chemical Reagent Company and used without further purification. Chitin was purchased from Shanghai Chemical Reagent Company and cut into small pieces (area: ~1 cm2). Double-distilled water was used throughout the experimental process.2.2 Synthesis of Amorphous Calcium Carbonate (ACC)
0.147 g of CaCl2·2H2O and 421 μl of dimethyl carbonate dissolved in 80 ml of water were placed in a beaker at 4 °C. The reaction was started by adding 20 ml of 0.5 M sodium hydroxide to the reaction solution under stirring. After 2 min the precipitate was removed from the reaction mixture by centrifugation, then washed with acetone and ethanol and dried at 30 °C.38,392.3 Effects of the organic matrix and magnesium ions on ACC crystallization on chitin film
The WSM of aragonite pearls from Hyriopsis cumingii was extracted40 and prepared in the solution with a concentration of 100 ug ml−1. Magnesium chloride solutions (10 mM and 40 mM) were prepared and named as low Mg solution and high Mg solution, respectively. In the experiment group, 10 mg of ACC was immersed in 20 ml of WSM and Mg ion solutions. In the control group, 10 mg of ACC was immersed in 20 ml of water. The silicon slice and the prepared chitin film were used as the substrates for calcium carbonate crystal growth. The mineralization process lasted for 12 h at room temperature. Finally, the substrates with deposited CaCO3 crystals were taken from the solution carefully, washed with double-distilled water and dried for characterization. For the time-dependent crystallization study, the substrates were removed from the mineralization system at the designed time.2.4 Characterization
The morphology and elemental composition of calcium carbonate crystals were observed using a field-emission scanning electron microscope (FESEM, JSM-7001F) with energy-dispersive X-ray spectroscopy (EDX, Oxford Instruments). A transmission electron microscope (TEM, JEOL-2011) operated at 200 kV together with selected area electron diffraction (SAED) were used to determine the polymorph. The amorphism of calcium carbonate was identified by X-ray powder diffraction (XRD, D8 ADVANCE) with Cu Kα radiation (40 kV, 40 mA) and a Fourier transform infrared spectrometer (FT-IR, Nicolet 6700) from 4000 to 400 cm−1 at room temperature by the KBr pellet method. Thermogravimetric analysis-mass spectrometry (TGA-MS) curves were measured under argon using a Mettler Toledo ThermoSTAR TGA/SDTA 851. Raman spectroscopy (RM 2000) was used to confirm the polymorph of the deposited CaCO3. Some typical crystals that could represent the overall situation were selected to analyze separately by a Raman spectrometer with the Raman shifts ranging from 100 to 1200 cm−1, and an argon ion laser with a wavelength of 613 nm was used.3. Results and discussion
3.1 Characterization of ACC particles
The synthesized calcium carbonate was characterized by SEM, X-ray diffraction, FTIR and TGA-MS. Calcium carbonate synthesized using CaCl2·2H2O, DMC and NaOH completely consisted of spherical particles with a diameter of about 300 nm and the surface of particles looked smooth (Fig. 1a). The X-ray diffraction pattern reveals some broad bumps located at 2θ of ~30° and ~45° (Fig. 1b), which reflect the poorly ordered nature of the solid. In addition, there are also two little peaks at 2θ of 29.5° and 47.7°. This indicates that small amounts of calcite are present in these poorly ordered solids (ICDD PDF 05-586). Furthermore, there is a broad and strong absorption peak at 866 cm−1, accompanied by a split peak at 1420 and 1475 cm−1 in the FT-IR spectra (Fig. 1c). ACC has a characteristic broad ν2 absorption band at ~866 cm−1, and a split peak at ~1418 and 1475 cm−1.8 Hence, it also shows that the overwhelming majority of calcium carbonate particles are amorphous. Actually, Ajikumar et al.12 first described a simple and efficient strategy for the synthesis of monodispersed microspheres of ACC by using a low-temperature precipitation of calcium carbonate without any organic additives. The endothermic processes at 89 °C and 140 °C are proved to be loss of water, identified by thermogravimetric analysis-mass spectrometry (Fig. 1d). Water is lost in two distinct steps. From 65 to 120 °C, the first step is assigned to loosely bound water. In the second step from 120 to 250 °C, structural water is lost with concurrent formation of calcite. Water stabilizes the amorphous phase, and if water is completely removed, crystallization occurs.8,41 Finally, at around 500 °C, the sample decomposes to calcium oxide and carbon dioxide.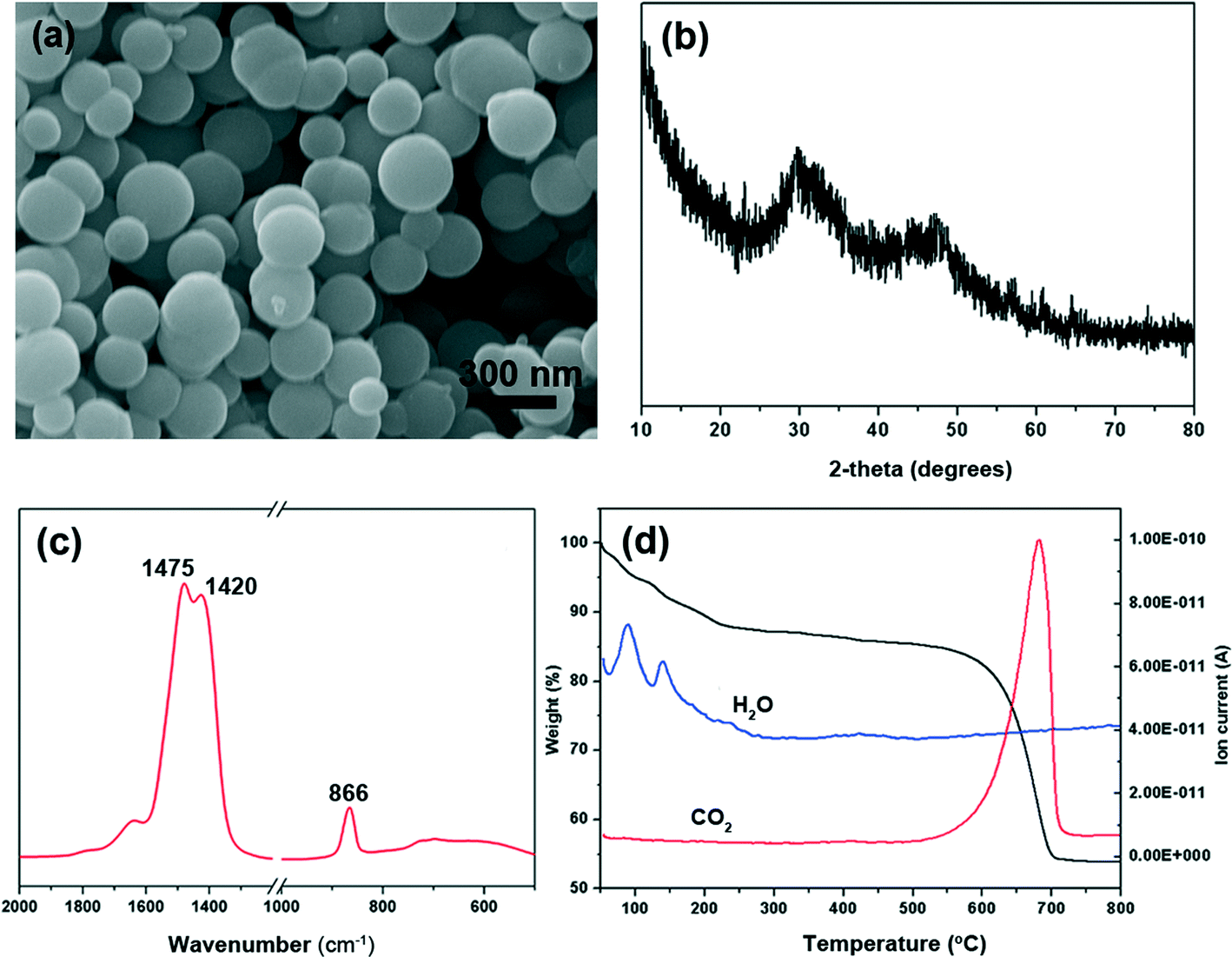 | ||
| Fig. 1 Characterization of the prepared amorphous calcium carbonate. (a) SEM image; (b) X-ray pattern; (c) FT-IR spectra; (d) TGA-MS curve. | ||
3.2 Effect of the WSM from freshwater pearls on ACC crystallization on chitin film
ACC was crystallized by soaking ACC powders into the solution with or without a WSM on silicon slice and chitin film. The precipitated CaCO3 crystals, leading to the formation of calcite and aragonite, were determined by Raman spectroscopy (Fig. 2). The inset in each figure shows the morphology of the analyzed crystal which represents the overall crystals. The peaks at 283 cm−1, 713 cm−1 and 1087 cm−1 are characteristic peaks of calcite. While the peaks at 154 cm−1 and 206 cm−1 are due to the typical Raman spectral lines of aragonite.42 Furthermore, according to a previous study, a pair of peaks at 700 cm−1 and 704 cm−1 is characteristic of aragonite,43,44 which is also observed in our Raman data (Fig. 2e and f). For aragonite, the site symmetry of the CO32− ion causes the two doubly degenerate ν4 in-plane bending modes to split into a pair of non-degenerate modes.45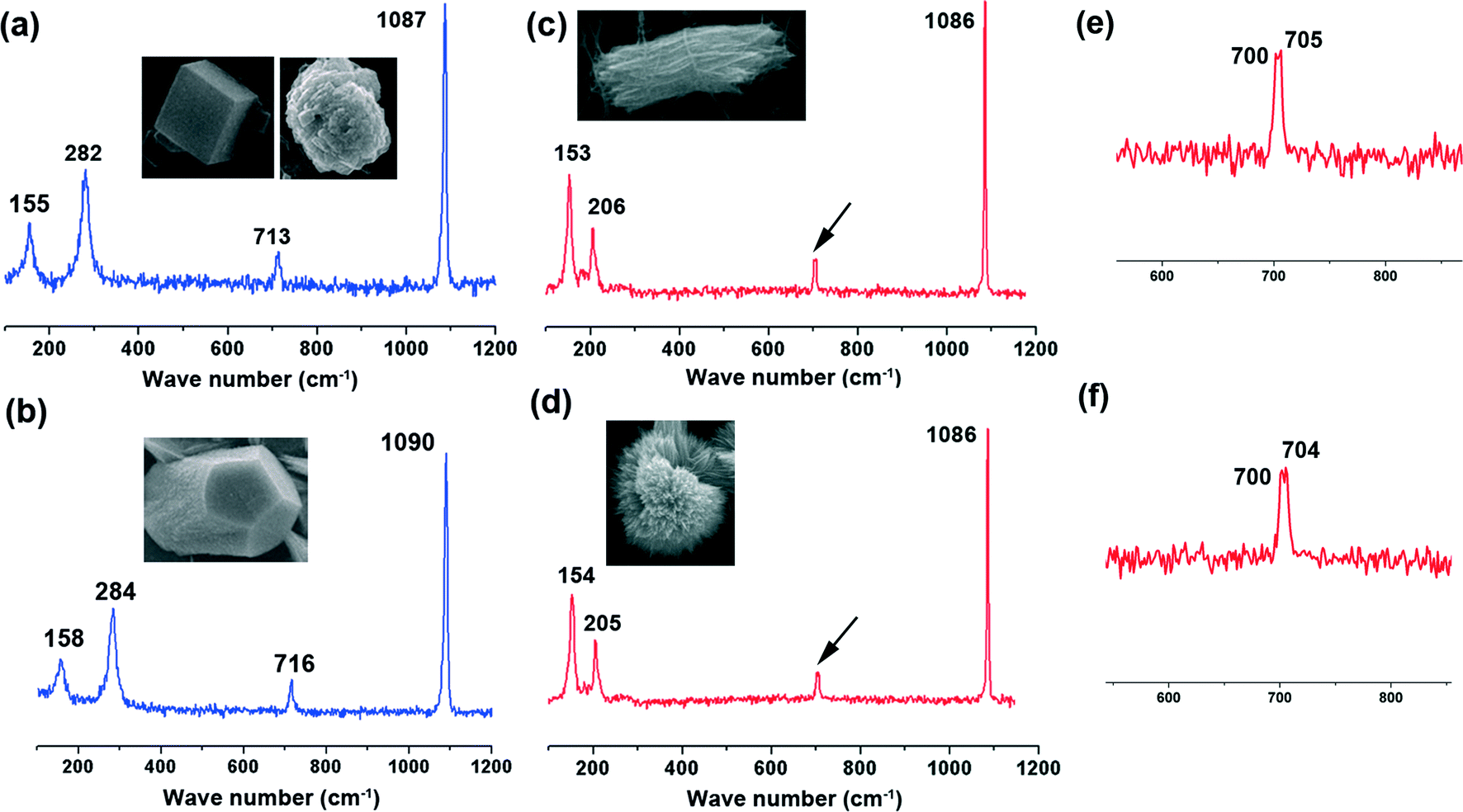 | ||
| Fig. 2 Raman spectrograms of CaCO3 crystals with different morphologies transformed from ACC particles under the control of a water soluble matrix or magnesium ions on silicon slice or chitin film in aqueous solution. (a) Calcite; (b) Mg-calcite; (c) aragonite; (d) aragonite; (e) and (f) are the magnified peaks indicated by the black arrows in (c) and (d), respectively. The inset in each figure indicates the typical crystal morphology. | ||
ACC mineralization occurs on the surface of the silicon slice and chitin film. In the absence of a WSM, only calcite is formed either on the silicon slice or on the chitin film despite the difference in the crystal morphology. Calcite crystals with exposed (104) crystalline faces grow on the silicon slice (Fig. 3a), and irregular spherical calcite crystals are observed on the chitin film (Fig. 3b). This is attributed to the different substrates and there exists an interaction between CaCO3 crystals and the chitin film. Some amino groups exist in chitin macromolecules.46 Hence, the interaction between –NH3+ in chitin macromolecules and CO32− could affect calcium carbonate crystallization, which leads to the irregular calcite formation. Liu et al.47 grafted different kinds of functional groups on monocrystalline silicon chips to study their effects on calcium carbonate crystallization and they found that the amount of the typical calcite with (104) crystalline faces was dramatically decreased on –NH2 modified substrates. This could be due to the inhibiting effect of amino groups on (104) crystalline faces. Consequently, the chitin film only has an effect on the CaCO3 morphology rather than on the polymorph. Incorporating a WSM into the crystallization procedure leads to a significant change in the crystal polymorph. Introducing a WSM into the mineralization solution results in the formation of aragonite accompanied by calcite on the silicon slice (Fig. 3c). In contrast, the rod-like aragonite crystals predominantly grow when the template is chitin film (Fig. 3d). The trigger for inducing the formation of aragonite, the polymorph in nacre, is therefore the addition of the WSM from freshwater pearls. The WSM possesses many carboxylic groups.48 The interfacial interaction between the functional groups of the WSM and the ions in the solution can reduce the nucleation energy of aragonite, which is responsible for the formation of aragonite crystals.37 Although the WSM additive alone has the capacity to induce aragonite, the combination of chitin film and WSM enhances significantly the amount of aragonite. Hence, the combination of WSM with chitin film can offer the ability to synthetically control the pure aragonite phase.
 | ||
| Fig. 3 SEM images of CaCO3 crystals transformed from ACC particles in aqueous solution with or without a water soluble matrix at room temperature for 12 h. (a) Crystal growth without a WSM on silicon slice, calcite precipitated; (b) crystal growth without a WSM on chitin film, calcite precipitated; (c) crystal growth with a WSM on silicon slice, calcite and aragonite precipitated; (d) crystal growth with a WSM on chitin film, aragonite precipitated. The scale bars in all the micrographs are 3 μm. | ||
3.3 Effect of magnesium ions on ACC crystallization on chitin film
To investigate the effect of magnesium ions on ACC mineralization, ACC powder was immersed in low Mg solution (10 mM) and high Mg solution (40 mM). ACC crystallization took place on silicon slice or chitin film. When ACC crystalized in low Mg solution on silicon slice, a majority of spindle-like aragonite aggregates composed of little needle-like crystals were observed (Fig. 4a), accompanied by some irregular Mg-calcite determined by Raman spectroscopy (Fig. 2b). The spectrum exhibits that the band characteristics [two lattice modes at 158 and 284 cm−1, ν1 (CO32−) symmetric stretching at 1090 cm−1 and ν4 (CO32−) antisymmetric bending at 716 cm−1] are similar to the respective values of Mg-calcite reported in the previous work.38 The observed shift in the positions of carbonate anions towards higher wavenumbers results from the higher polarizing power of Mg2+ in comparison to that of Ca2+ in pure calcite.49 In contrast, ACC crystallization on chitin film leads to the formation of the rod-like aragonite aggregates only (Fig. 4b). The crystalized calcium carbonate obtained in high Mg solution on silicon slice is shown in Fig. 4c. Some quasi-spherical aragonite aggregates appear, coexisting with a few Mg-calcites (Fig. 4c). Each quasi-spherical aragonite aggregate is also made up of the needle-like crystals. Comparatively, only quasi-spherical aragonite aggregates are obtained on chitin film without other calcium carbonate polymorphs (Fig. 4d). Hence, it is concluded that the formation of the spindle-like aragonite aggregates, the rod-like aragonite aggregates, the quasi-spherical aragonite superstructures and the irregular Mg-calcite all come from the transformation of the ACC precursors. | ||
| Fig. 4 SEM images of CaCO3 crystals transformed from ACC particles under the control of magnesium ions at room temperature for 12 h. (a) [Mg2+] = 10 mM, silicon slice; Mg-calcite and aragonite precipitated; (b) [Mg2+] = 10 mM, chitin film; aragonite precipitated; (c) [Mg2+] = 40 mM, silicon slice; Mg-calcite and aragonite precipitated; (d) [Mg2+] = 40 mM, chitin film; aragonite precipitated. The scale bars in all the micrographs are 3 μm. | ||
It is well known that ACC is easier to dissolve in solution and thereby rapidly transforms into calcium carbonate crystals under ambient conditions in the absence of any additives.4,50 Magnesium ions, as cosolutes of calcium, have higher hydration energy, and dehydration of the magnesium ions prior to incorporation in the calcite lattice creates a barrier to the growth of calcite nuclei.24 This is the reason why aragonite crystals can be obtained more easily in the presence of sufficient magnesium. It is worth noting that magnesium ions just induce the formation of aragonite in which magnesium is not incorporated.51 The previous report also has demonstrated that via the control of polymers or biomolecules, a mixture of aragonite with Mg-calcite was obtained by ACC crystallization in aqueous solution.22
Mg-ACC nanoparticles serve as the intermediate precursors for the formation of Mg-calcite crystals, which is also a very popular transformation process for biogenic calcite minerals.34,52,53 For instance, Long et al.54 synthesized high-magnesium calcite via polymer-stabilized amorphous calcium magnesium carbonate precursors under mild conditions. This in vitro fabrication of high-magnesium calcite provided some indirect clues to the formation mechanism of biogenic Mg-calcite. Mg2+ ions can incorporate into the structures of calcite. Additionally, Mg2+ ions are more strongly hydrated than Ca2+ ions and are specifically adsorbed on the crystal faces of calcite, producing crystals elongated along the c axis,55 which results in a pronounced morphology change compared with the typical rhombohedral morphology of calcite. A trend was found that the aragonite morphology changed from rod-like aggregates to quasi-spherical aggregates with an increase in Mg2+ ion concentration, which indicated that magnesium ions acted as shape modifiers in our experiment. The existence of Mg-calcite was determined by the substrate on which ACC crystallization happened. On chitin film, no Mg-calcite crystals grew in low Mg or high Mg solutions. Therefore, we believe that the combination of magnesium ions and the chitin substrate not only induces the growth of aragonite crystals but also inhibits the transformation from ACC to Mg-calcite.
3.4 Process of ACC crystallization under the control of an organic matrix or magnesium ions on chitin film
The time-dependent morphology evolution process was carefully examined to understand the formation process of ACC crystallization under the control of a WSM or magnesium ions on chitin film (Fig. 5). Initially, ACC precursors with a diameter of ~300 nm are obtained (Fig. 5a) by separating them from the mother solution and washing with ethanol and acetone, because the amorphous phase is very unstable in aqueous solution. The sample precipitated on chitin film after 6 h under the control of the WSM is shown in Fig. 5b. There exist some aragonite crystal aggregates with a rod-like shape and some other ACC particles. The existence of ACC indicates that the WSM has the capacity to stabilize ACC in aqueous solution. The diameter and length of aragonite crystal aggregates are about 1 μm and 3 μm, respectively. Each rod-like aragonite crystal aggregate is composed of needle-like crystals. After 12 h, all ACCs crystalize to form aragonite crystals and the pre-existing aragonite crystal aggregates grow bigger with a length of ~5 μm and a diameter of ~1.8 μm (Fig. 5c). When magnesium ions as a kind of additive were added into the solution, the transformation process from ACC into aragonite crystal aggregates differed from that with the addition of the WSM. Magnesium ions diffused and were present within the structure of ACC and thus changed the morphology from the spherical shape into the ellipsoid-like morphology after 2 h (Fig. 5d). It led to the formation of Mg-ACC which was proved by energy-dispersive X-ray spectroscopy (Fig. 5g) and TEM with selected area electron diffraction (Fig. 5h). The SAED pattern shows that the sample is still in the amorphous state and the EDX analysis result shows the co-existence of calcium, magnesium, oxygen and carbon (Fig. 5g), which indicates that the ACC contains magnesium. With prolonged time, this ellipsoid-like Mg-ACC disappeared and many needle-like aragonite primary crystals with a length of ~750 nm grew on chitin film after mineralization for 6 h (Fig. 5e). Previous work reported that the growth of aragonite was preferred along the c axis.56 As a result of that, under normal temperature and pressure, aragonite crystals are constructed like fine needles. When the mineralization time reached 12 h, the rod-like aragonite crystal aggregates (length: ~5 μm) appeared on chitin film in 10 mM magnesium ion solution (Fig. 5f) while the quasi-spherical aragonite aggregates (diameter: ~6 μm) grew in 40 mM magnesium ion solution (Fig. 5i). | ||
| Fig. 5 SEM images of the ACC crystallization process occurring on chitin film under the control of a water soluble matrix or magnesium ions. (a) The original ACC particles; the crystallization process with a WSM at (b) 6 h #ACC and aragonite# and (c) 12 h #aragonite#; the crystallization process with magnesium ions at (d) 2 h #Mg-ACC#, (e) 6 h #aragonite#, (f) 12 h [Mg2+ ion concentration: 10 mM] #aragonite# and (i) 12 h [Mg2+ ion concentration: 40 mM] #aragonite#; (g) EDX results of the sample. The square area in (d) indicates the analysis location; (h) SAED pattern of the precipitate in (d). ## indicates the crystal polymorph of the precipitated CaCO3. | ||
On the basis of the above results and analyses, the possible mechanism of ACC crystallization on chitin film under the control of the WSM (Fig. 6a) or magnesium ions (Fig. 6b) is proposed and briefly discussed. When the WSM was added in the solution, firstly, the WSM adsorbed on the surface of ACC particles to form the WSM stabilized ACC. In this case, some ACC particles with the original size were still observed after 6 h in the solution (Fig. 5b). Hence, the WSM inhibited the dissolution of ACC so that the dissolution–recrystallization process rarely took place. It is concluded that a solid–solid phase transformation is the primary mechanism for ACC crystallization under the control of the WSM. The previous literature reported that ACC had a short-range order structure.8 This short-range order structure might function as the basic unit that is reconstructed into the corresponding crystalline form. The local centers of the ordered structure aligned and coalesced cooperatively to form crystalline microdomains. As the mineralization time went on, these crystalline microdomains grew and coalesced continually, leading to the formation of needle-like primary crystals. Subsequently, new ACC particles are deposited on the surface of the needle-like primary crystals and then are transformed into crystal forms. Tang et al.57 believed that this surface nucleation and subsequent growth, and transformation of ACC happened at the same time. The nucleation and growth of new primary crystals on the surface of the pre-formed crystal resulted in the merging of adjoining primary crystals and eventually the rod-like crystal aggregates are formed.
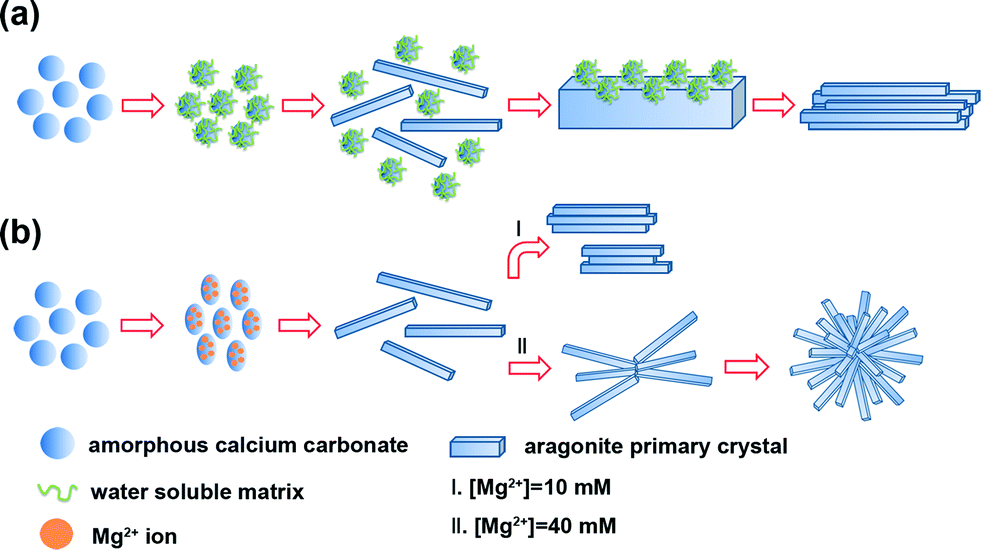 | ||
| Fig. 6 Schematic illustration of the ACC crystallization process occurring on chitin film under the control of (a) a water soluble matrix or (b) magnesium ions. | ||
In contrast, the process of ACC crystallization on chitin film under the control of magnesium ions is quite different (Fig. 6b). When Mg2+ ions were introduced in the mineralization system, Mg-ACC with the ellipsoid-like morphology appeared (Fig. 5d). In the previous studies, some Mg-ACC with imperfect spherical morphologies was reported but the specific reason was still unclear.20,54,58,59 Hence, we suppose that Mg2+ ions adsorbed on ACC initially and then entered into the structure of ACC, which led to the ellipsoid-like morphology. The role that magnesium may play in the control of ACC stabilization should be considered. Recently, the high dehydration energy of magnesium ions has been considered to be responsible for the stabilizing effect of magnesium ions on ACC.39 Magnesium present within the structure of ACC is likely to prevent its dehydration, hence reducing the rate of dissolution and decreasing its overall solubility.60 This decrease in solubility reduces the supersaturation in the solution, which is the possible reason for Mg-ACC stabilization. Additionally, another factor contributes to explain how magnesium ions stabilize the amorphous state. Bond lengths around Mg are expected to be shorter relative to Ca–O bond lengths.61 Some literature reported that the Mg–O bond lengths measured in ACC were even shorter compared to those of crystalline Mg carbonate minerals.62This indicates that magnesium ions in ACC are not influenced by the host ACC phase. Thus, the presence of magnesium ions in ACC causes significant distortion of the local atomic structure, favoring a disordered atomic structure of the bulk and consequently stabilizing the amorphous phase. Therefore, in the present work, Mg-ACC disappeared after 6 h and the needle-like aragonite primary crystals formed (Fig. 5e). With prolonged time, the needle-like primary crystals joined together by aggregation to become larger rod-like crystal aggregates in the solution with low Mg ion concentration (Fig. 5f). Comparatively, in high Mg ion concentration solution, this kind of needle-like primary crystal first integrated into cross-linked crystal aggregates. Then, the needle-like primary crystals continued to be attracted, joined and attached onto the cross-linked crystal aggregates to produce uniform quasi-spherical crystal aggregates (Fig. 5i). The growth process of CaCO3 crystals along with the morphological change has been observed for other similar structures such as “dumbbell”63 and “peanut”57,64 morphologies. Despite all this, it must be pointed out that the exact mechanism for ACC crystallization with different morphologies is extremely complicated and needs to be further studied.
4. Conclusions
In summary, ACC particles with a diameter of about 300 nm are synthesized first. Then we have investigated the phase transition process from ACC to the crystalline phase on chitin film by adding a WSM or magnesium ions in aqueous solution. The WSM may stabilize ACC, and the existence of the WSM and chitin film leads to the formation of rod-like aragonite crystal aggregates. Comparatively, the collaborative effect of magnesium ions and chitin film not only induces the formation of aragonite crystal aggregates, but also inhibits the transformation from ACC to Mg-calcite. A possible growth mechanism of the aragonite crystal aggregates with different morphologies is thereby proposed. This study may give some useful clues for understanding the biomineralization process of CaCO3 in nature. Furthermore, the present work provides some new insights into the preparation of CaCO3 aggregates with complex shape and fine structure assembled from amorphous precursors.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (51361130032, 51472139) and the Doctor Subject Foundation of the Ministry of Education of China (20120002130002).References
- F. C. Meldrum and H. Coelfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed.
- F. C. Meldrum, Int. Mater. Rev., 2003, 48, 187–224 CrossRef CAS PubMed.
- J. Aizenberg, D. A. Muller, J. L. Grazul and D. R. Hamann, Science, 2003, 299, 1205–1208 CrossRef CAS PubMed.
- S. Raz, P. C. Hamilton, F. H. Wilt, S. Weiner and L. Addadi, Adv. Funct. Mater., 2003, 13, 480–486 CrossRef CAS.
- E. Beniash, J. Aizenberg, L. Addadi and S. Weiner, Proc. R. Soc. London, Ser. B, 1997, 264, 461–465 CrossRef CAS.
- I. M. Weiss, N. Tuross, L. Addadi and S. Weiner, J. Exp. Zool., 2002, 293, 478–491 CrossRef CAS PubMed.
- J. Aizenberg, G. Lambert, L. Addadi and S. Weiner, Adv. Mater., 1996, 8, 222–226 CrossRef CAS.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959–970 CrossRef CAS.
- J. Aizenberg, S. Weiner and L. Addadi, Connect. Tissue Res., 2003, 44, 20–25 CrossRef CAS.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161–1164 CrossRef CAS PubMed.
- Y. Politi, R. A. Metzler, M. Abrecht, B. Gilbert, F. H. Wilt, I. Sagi, L. Addadi, S. Weiner and P. U. P. A. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20045–20045 CrossRef PubMed.
- P. K. Ajikumar, L. G. Wong, G. Subramanyam, R. Lakshminarayanan and S. Valiyaveettil, Cryst. Growth Des., 2005, 5, 1129–1134 CAS.
- J. Liu, S. Pancera, V. Boyko, J. Gummel, R. Nayuk and K. Huber, Langmuir, 2012, 28, 3593–3605 CrossRef CAS PubMed.
- K. Sawada, Pure Appl. Chem., 1997, 69, 921–928 CrossRef CAS.
- N. Koga, Y. Z. Nakagoe and H. Tanaka, Thermochim. Acta, 1998, 318, 239–244 CrossRef CAS.
- R. S. K. Lam, J. M. Charnock, A. Lennie and F. C. Meldrum, CrystEngComm, 2007, 9, 1226–1236 RSC.
- S. F. Chen, J. H. Zhu, J. Jiang, G. B. Cai and S. H. Yu, Adv. Mater., 2010, 22, 540–545 CrossRef CAS PubMed.
- Y. Nishino, Y. Oaki and H. Imai, Cryst. Growth Des., 2009, 9, 223–226 CAS.
- J. Jiang, M. R. Gao, Y. H. Qiu and S. H. Yu, Nanoscale, 2010, 2, 2358–2361 RSC.
- C. Gunther, A. Becker, G. Wolf and M. Epple, Z. Anorg. Allg. Chem., 2005, 631, 2830–2835 CrossRef.
- T. Kato, Adv. Mater., 2000, 12, 1543–1546 CrossRef CAS.
- S. Raz, S. Weiner and L. Addadi, Adv. Mater., 2000, 12, 38–42 CrossRef CAS.
- N. A. J. M. Sommerdijk, E. N. M. van Leeuwen, M. R. J. Vos and J. A. Jansen, CrystEngComm, 2007, 9, 1209–1214 RSC.
- E. Loste, R. M. Wilson, R. Seshadri and F. C. Meldrum, J. Cryst. Growth, 2003, 254, 206–218 CrossRef CAS.
- X. R. Xu, A. H. Cai, R. Liu, H. H. Pan, R. K. Tang and K. W. Cho, J. Cryst. Growth, 2008, 310, 3779–3787 CrossRef CAS PubMed.
- L. Liu, B. Hu, S. F. Chen, S. J. Liu, J. Jiang, G. B. Cai and S. H. Yu, CrystEngComm, 2010, 12, 3593–3598 RSC.
- X. Geng, L. Liu, J. Jiang and S. H. Yu, Cryst. Growth Des., 2010, 10, 3448–3453 CAS.
- Y. Y. Liu, J. Jiang, M. R. Gao, B. Yu, L. B. Mao and S. H. Yu, Cryst. Growth Des., 2013, 13, 59–65 CAS.
- S. Albeck, S. Weiner and L. Addadi, Chem. – Eur. J., 1996, 2, 278–284 CrossRef CAS.
- Y. Ma, S. R. Cohen, L. Addadi and S. Weiner, Adv. Mater., 2008, 20, 1555–1559 CrossRef CAS.
- Z. J. Ma, J. Huang, J. Sun, G. N. Wang, C. Z. Li, L. P. Xie and R. Q. Zhang, J. Biol. Chem., 2007, 282, 23253–23263 CrossRef CAS PubMed.
- A. P. Wheeler, K. W. Rusenko, D. M. Swift and C. S. Sikes, Mar. Biol., 1988, 98, 71–80 CrossRef CAS.
- Y. Zhang, L. P. Xie, Q. X. Meng, T. M. Jiang, R. L. Pu, L. Chen and R. Q. Zhang, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol, 2003, 135, 565–573 CrossRef.
- N. Nassif, N. Pinna, N. Gehrke, M. Antonietti, C. Jager and H. Colfen, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12653–12655 CrossRef CAS PubMed.
- I. M. Weiss and V. Schonitzer, J. Struct. Biol., 2006, 153, 264–277 CrossRef CAS PubMed.
- J. Aizenberg, G. Lambert, S. Weiner and L. Addadi, J. Am. Chem. Soc., 2002, 124, 32–39 CrossRef CAS PubMed.
- Y. F. Ma, L. Qiao and Q. L. Feng, Mater. Sci. Eng., C, 2012, 32, 1963–1970 CrossRef CAS PubMed.
- B. Borzecka-Prokop, A. Weselucha-Birczynska and E. Koszowska, J. Mol. Struct., 2007, 828, 80–90 CrossRef CAS PubMed.
- D. Di Tommaso and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2010, 12, 894–901 RSC.
- Y. F. Ma, Y. H. Gao and Q. L. Feng, Mater. Sci. Eng., C, 2011, 31, 1338–1342 CrossRef CAS PubMed.
- G. Wolf and C. Gunther, J. Therm. Anal. Calorim., 2001, 65, 687–698 CrossRef CAS.
- N. V. Vagenas and C. G. Kontoyannis, Vib. Spectrosc., 2003, 32, 261–264 CrossRef CAS.
- G. Behrens, L. T. Kuhn, R. Ubic and A. H. Heuer, Spectrosc. Lett., 1995, 28, 983–995 CrossRef CAS.
- W. Z. Wang, G. H. Wang, Y. K. Liu, C. L. Zheng and Y. J. Zhan, J. Mater. Chem., 2001, 11, 1752–1754 RSC.
- A. Anderson, Spectrosc. Lett., 1996, 29, 819–825 CrossRef CAS.
- I.-Y. Kim, S.-J. Seo, H.-S. Moon, M.-K. Yoo, I.-Y. Park, B.-C. Kim and C.-S. Cho, Biotechnol. Adv., 2008, 26, 1–21 CrossRef CAS PubMed.
- X. J. Liu, Y. H. Gao and Q. L. Feng, Acta Mater. Compos. Sinica, 2012, 29, 21–27 CAS.
- F. Nudelman, B. A. Gotliv, L. Addadi and S. Weiner, J. Struct. Biol., 2006, 153, 176–187 CrossRef CAS PubMed.
- J. M. Alia, Y. D. de Mera, H. G. M. Edwards, P. G. Martin and S. L. Andres, Spectrochim. Acta, Part A, 1997, 53, 2347–2362 CrossRef.
- M. Faatz, F. Grohn and G. Wegner, Adv. Mater., 2004, 16, 996–1000 CrossRef CAS.
- A. Mucci, Am. J. Sci., 1983, 283, 780–799 CrossRef CAS.
- D. Gebauer, A. Volkel and H. Colfen, Science, 2008, 322, 1819–1822 CrossRef CAS PubMed.
- J. Mahamid, A. Sharir, L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12748–12753 CrossRef CAS PubMed.
- X. Long, Y. Ma and L. Qi, Cryst. Growth Des., 2011, 11, 2866–2873 CAS.
- G. Falini, S. Fermani, M. Gazzano and A. Ripamonti, J. Mater. Chem., 1998, 8, 1061–1065 RSC.
- S. Weiner and L. Addadi, J. Mater. Chem., 1997, 7, 689–702 RSC.
- H. Tang, J. G. Yu and X. F. Zhao, Mater. Res. Bull., 2009, 44, 831–835 CrossRef CAS PubMed.
- H. Nebel and M. Epple, Z. Anorg. Allg. Chem., 2008, 634, 1439–1443 CrossRef CAS.
- N. Xu, Y. Li, L. Zheng, Y. Gao, H. Yin, J. Zhao, Z. Chen, J. Chen and M. Chen, Chem. Eng. J., 2014, 251, 102–110 CrossRef CAS PubMed.
- J. D. Rodriguez-Blanco, S. Shaw, P. Bots, T. Roncal-Herrero and L. G. Benning, J. Alloys Compd., 2012, 536, S477–S479 CrossRef CAS PubMed.
- M. Faatz, F. Grohn and G. Wegner, Mater. Sci. Eng., C, 2005, 25, 153–159 CrossRef PubMed.
- Y. Politi, D. R. Batchelor, P. Zaslansky, B. F. Chmelka, J. C. Weaver, I. Sagi, S. Weiner and L. Addadi, Chem. Mater., 2010, 22, 161–166 CrossRef CAS.
- S. H. Yu, H. Colfen and M. Antonietti, J. Phys. Chem. B, 2003, 107, 7396–7405 CrossRef CAS.
- X. H. Jiang, M. B. Zheng, H. Q. Chen, L. J. Pan, J. Tao and J. M. Cao, Wuji Cailiao Xuebao, 2008, 23, 1283–1286 CAS.
| This journal is © The Royal Society of Chemistry 2015 |