
Diethylenetriamine-assisted hydrothermal synthesis of dodecahedral α-Fe2O3 nanocrystals with enhanced and stable photoelectrochemical activity†
Rui
Xu
,
You
Xu
,
Yi
Huang
,
Yanmei
Shi
and
Bin
Zhang
*
Department of Chemistry, School of Science, Tianjin University, and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China. E-mail: bzhang@tju.edu.cn
First published on 6th November 2014
Abstract
Dodecahedral α-Fe2O3 nanocrystals (D-hematite) were successfully synthesized through a hydrothermal method involving FeS-diethylenetriamine (FeS-DETA) hybrid nanosheets as starting materials. The photoanode made by assembling the as-obtained D-hematite on ITO substrates exhibited efficient photoelectrocatalytic water splitting activity and good stability in 1.0 M KOH aqueous solution under visible light irradiation.
1. Introduction
Photoelectrochemical (PEC) water splitting is an environmentally friendly and promising approach for converting solar energy into chemical energy.1–3 The pioneering work was published in 1972 by Fujishima and Honda, in which the “Honda–Fujishima effect” and photoelectrolysis using n-type TiO2 as the photoelectrode were first described.4 Many PEC materials, especially semiconductor oxides,5–10 have been investigated. As a typical example, hematite (α-Fe2O3) is considered as one of the most promising candidates for visible-light-driven PEC water splitting due to its abundance, low cost, non-toxicity, excellent stability and appropriate band positions for visible light absorption.11–15 However, the poor mobility of photogenerated carriers, short hole diffusion length and slow oxidation kinetics limit the performance of α-Fe2O3 in PEC water splitting.16–21 Therefore, it is necessary to develop facile and cost-effective methods to synthesize active α-Fe2O3 for PEC water splitting.Intensive research has been carried out to explore the synthesis of nanostructured α-Fe2O3,22–26 in forms such as nanoparticles,27–29 nanorods/nanowires,30–33 nanotubes,34 nanoplates/nanosheets,35–38 nanoflowers39,40 and nanocubes,41–45 owing to their larger active surface areas, better light absorption, and increased photocurrent compared with the corresponding bulk counterparts. However, the synthesis of nanostructured α-Fe2O3 polyhedra still remain challenging because polyhedral metal oxides have more complex crystal systems than metal nanocrystals.46–52 To date, only a few excellent reports have been produced on the preparation of polyhedral α-Fe2O3 nanocrystals. For example, the groups of Gao53 and Wang54 have established a solvothermal method to prepare tetrakaidecahedral and oblique parallelepiped hematite in the presence of carboxymethyl cellulose (CMC) and poly(vinylpyrrolidone) (PVP). Lv et al. reported an efficient fabrication of dodecahedral and octadecahedral α-Fe2O3 with the aid of fluoride anions.55 Meanwhile, the present applications of polyhedral α-Fe2O3 have mainly been focused on magnetic measurements56,57 and their gas-sensing properties.58,59 Thus, it is highly desirable to develop a facile strategy to fabricate single-crystalline polyhedral α-Fe2O3 for PEC water splitting.
Herein, we report the synthesis of single-crystalline dodecahedral α-Fe2O3 through a facile hydrothermal approach. The simple protocol involved the use of inorganic–organic hybrid FeS-amine nanosheets as the starting material, and a mixture of amine and water as the solvent and reaction agent (Scheme 1). It was found that the choice of amine was crucial to the formation of dodecahedral morphology; also, the dissolution of the amine facilitated the reaction process. Furthermore, the as-prepared dodecahedral α-Fe2O3 nanocrystals exhibited efficient PEC water-splitting activity and good stability under visible-light irradiation (λ > 420 nm), compared with commercial α-Fe2O3. Traditionally, to achieve efficient PEC water-splitting activity, α-Fe2O3 is mainly synthesized using a deposition technique at high temperature (spray pyrolysis and CVD, 400–500 °C) or solution-based methods with subsequent annealing steps (700–800 °C).12 However, in our experiments, photoactive dodecahedral α-Fe2O3 nanocrystals were prepared at a low temperature (200 °C) using a hydrothermal method. The photocurrent density of dodecahedral α-Fe2O3 nanocrystals is 3.06 mA cm−2 at the potential of 1.6 V versus RHE under visible-light illumination (λ > 420 nm). To the best of our knowledge, this value is comparable to or even higher than most of the reported undoped hematite films without any co-catalysts (Table S1†).
 | ||
| Scheme 1 Scheme illustrating the preparation of single-crystalline dodecahedral α-Fe2O3 through the anion-exchange reaction of the organic–inorganic FeS-DETA hybrid nanosheets with OH− at elevated temperature. | ||
2. Experimental section
2.1 Materials
All chemicals were of analytical grade and used as received without further purification.2.2 Synthesis of inorganic–organic hybrid FeS-DETA nanosheets
FeS-DETA was synthesized using a modified method reported by Yu's group.60,61 In a typical synthesis procedure, FeSO4·7H2O (0.2780 g), C2H5NS (0.1526 g), H2O (8 mL) and diethylenetriamine (DETA, 8 mL) were added to a Teflon-lined autoclave with a capacity of 20 mL. The mixture was stirred for 20 min to form a homogeneous suspension. The autoclave was then sealed and heated at 140 °C for 24 h. After the reaction, the autoclave was cooled down to room temperature naturally. In our manuscript, the hybrid nanosheets are named as FeS-DETA.2.3 Synthesis of dodecahedral α-Fe2O3
In a typical procedure, the as-obtained inorganic–organic hybrid FeS-DETA nanosheets were vigorously stirred for 30 min to form a homogeneous suspension. Then the as-obtained dispersions (0.8 mL) were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 3 min to remove the supernatant transparent solution which may contain redundant reagent. After centrifugation, the dispersions were diluted with 11.0 mL of H2O and 0.1 mL of DETA. Then the mixture was added to a Teflon-lined autoclave with a capacity of 20 mL and heated at 200 °C for 6 h. After cooling to room temperature, the precipitate was collected by centrifugation and washed with ethanol three times. The final product was dried in a vacuum oven at room temperature for 6 h.
000 rpm for 3 min to remove the supernatant transparent solution which may contain redundant reagent. After centrifugation, the dispersions were diluted with 11.0 mL of H2O and 0.1 mL of DETA. Then the mixture was added to a Teflon-lined autoclave with a capacity of 20 mL and heated at 200 °C for 6 h. After cooling to room temperature, the precipitate was collected by centrifugation and washed with ethanol three times. The final product was dried in a vacuum oven at room temperature for 6 h.
2.4 Photoelectrochemical measurements
Photoelectrochemical measurements were carried out using a standard three-compartment cell consisting of a working electrode, a Pt sheet counter electrode, and a Hg/HgO (1.0 M KOH) reference electrode performed using an electrochemical workstation (CHI 660D, CH Instruments, Austin, TX) under a 300 W Xe lamp equipped with a 420 nm cut-off filter. A solution of 1.0 M KOH (pH = 13.6) was employed as the electrolyte. Oxygen was purged from the electrolyte using bubbled nitrogen gas. An indium–tin oxide (ITO) glass decorated with catalyst samples was used as the working electrode. For a typical procedure for fabricating the working electrode, 1 mg of α-Fe2O3 catalysts was dispersed in a solution containing water (0.5 mL), CH3CH2OH (0.5 mL) and 5 wt% Nafion solution (10 μL) before the mixture was ultrasonicated to generate a homogeneous ink. Then 160 μL of the catalyst ink was spread on an ITO glass substrate (loading amount: ~0.158 mg cm−2).3. Results and discussion
The FeS-DETA hybrid nanosheets were prepared by a facile hydrothermal reaction. Fig. 1a and 1b display the representative scanning electron microscopy (SEM) images of the FeS-DETA hybrid nanostructures, showing a typical 2D sheet-like morphology with a lateral size distribution of 200–1500 nm and a thickness of 50–100 nm. As shown in Fig. 1h, the powder X-ray diffraction (PXRD) pattern shows that the emergence of diffraction peaks is similar to the reported FeS-amine precursors.60,61 Fourier transform infrared (FTIR) spectra (Fig. 1g, cyan line) and energy-dispersive X-ray spectroscopy (EDS) analysis (Fig. S1, ESI†) further identify them as the inorganic–organic hybrid nanostructures.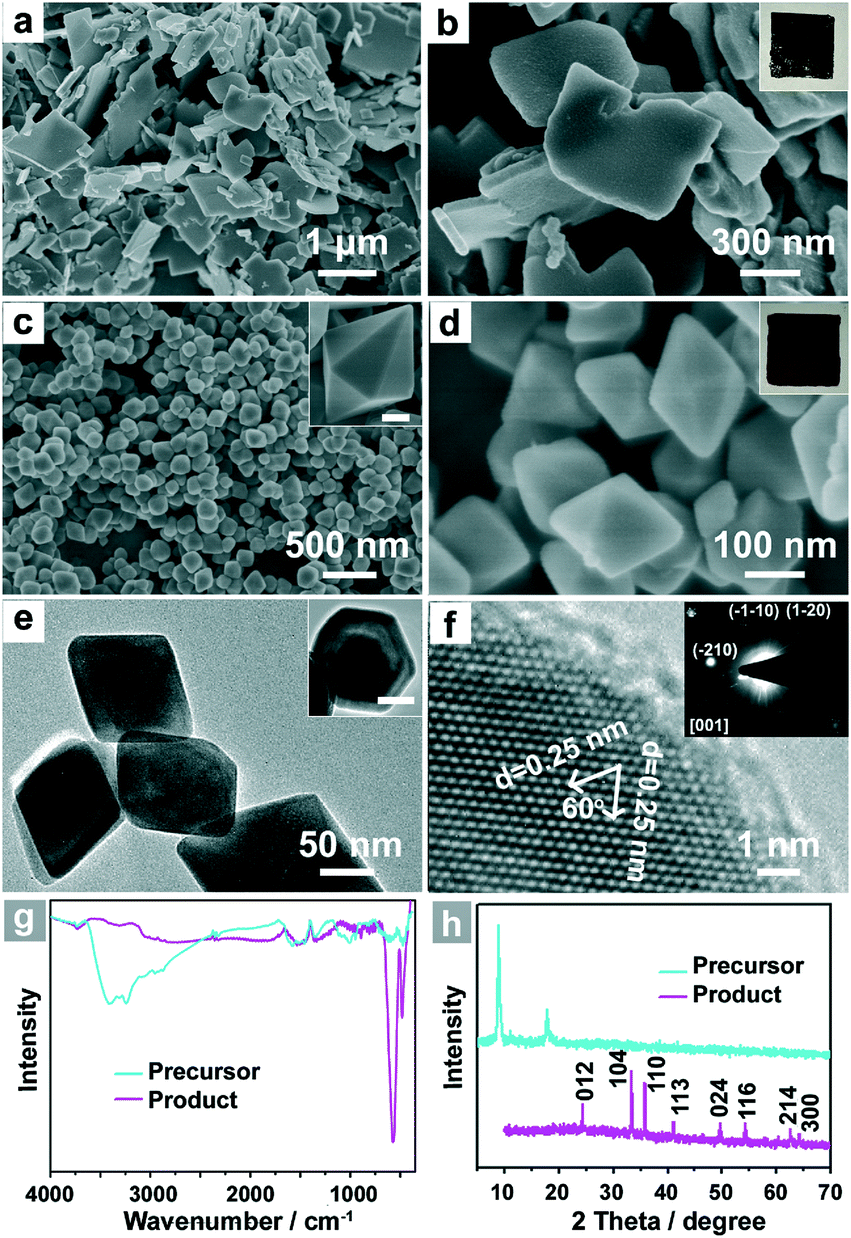 | ||
| Fig. 1 (a,b) SEM images and photograph (inset) of inorganic–organic hybrid FeS-DETA nanosheets. (c,d) SEM images and photograph (inset) of dodecahedral α-Fe2O3 (inset, scale bar is 50 nm). (e) Side view and top view (inset, scale bar is 50 nm) TEM image of the dodecahedral α-Fe2O3. (f) HRTEM image of dodecahedral α-Fe2O3 and the corresponding SAED pattern from the [001] zone axis (inset). (g,h) FTIR spectra (g) and XRD patterns (h) of the inorganic–organic hybrid FeS-DETA nanosheets (cyan line) and dodecahedral α-Fe2O3 (magenta line). | ||
The single-crystalline dodecahedral α-Fe2O3 was obtained through a simple hydrothermal treatment of the as-obtained FeS-DETA hybrid nanosheets. SEM images show their dodecahedral morphological features with a lateral size of 100–300 nm (Fig. 1c and 1d). The transmission electron microscopy (TEM) images of the products displayed in Fig. 1e clearly confirm the dodecahedral morphology from the side view and top view (inset). The representative high-resolution TEM (HRTEM) image and the associated selected area electron diffraction (SAED) pattern in the inset of Fig. 1f reveal the single crystalline structure of the dodecahedral α-Fe2O3. The HRTEM image shows that the crystal plane spacings are 0.25 nm, which can be index to the {110} or {1−20} lattice spacings of the rhombohedral hematite. As shown in Fig. 1h, all the diffraction peaks from the PXRD pattern can be attributed to the rhombohedral α-Fe2O3 (JCPDS 33-0664). No other peaks can be observed in XRD pattern, indicating the high purity of the as-obtained dodecahedral α-Fe2O3. The FTIR spectra further confirm the transformation from the inorganic–organic FeS-DETA hybrid nanosheets into the inorganic α-Fe2O3 (Fig. 1g). The photographs in the insets of Fig. 1b and 1d also clearly show the change from the precursor to the product. The EDS spectrum and thermogravimetric (TG) analysis demonstrate the composition and high purity of the α-Fe2O3 samples (Fig. S2 and S4 ESI†).
To clarify the formation mechanism of the dodecahedral α-Fe2O3, SEM, EDS and XRD were used to characterize the intermediates collected at different reaction stages (Fig. 2). At an early stage (Fig. 2a), the FeS-DETA hybrid nanosheets gradually became rough. When the anion-exchange reaction proceeded for another one hour (2 h, Fig. 2b), these small nanoparticles grew further and became small 2D nanosheets. The appearance of nanosheet intermediates could be attributed to the formation of layered FeOOH.62 The assumption was further confirmed by the analysis of XRD patterns. As shown in Fig. 2d, the peaks of the precursor became much weaker, and some peaks of FeOOH (JCPDS 18-0639) could be observed as the reaction proceeded for 2 h. When the reaction time was increased to 4 h, dodecahedral crystals with some nanosheets on the surface (Fig. 2c) appeared. The XRD pattern in Fig. 2d revealed that the intermediate was mostly α-Fe2O3 (JCPDS 33-0664). With increasing reaction time, dodecahedral α-Fe2O3 crystals with a smooth surface were eventually produced (Fig. 1c and 1d). Also, EDS analysis (Fig. 2a–c and S1–S3, ESI†) disclosed that the ratio of S:Fe experienced a decreasing and disappearing process. On the basis of these results, we propose the transformation mechanism of FeS-DETA sheets into dodecahedral α-Fe2O3, involving the anion-exchange reaction of inorganic–organic hybrid nanosheets and the dissolution of DETA, as displayed in Scheme 1. Initially, S2− ions are slowly exchanged by the OH− ions from water accompanied by the dissolution of DETA. Then, the Fe2+ ions are subsequently oxidized by the oxygen in the solution to form FeOOH. Finally, the dodecahedral α-Fe2O3 crystals appear and become smooth in the subsequent reaction process. The reactions involved are shown below:
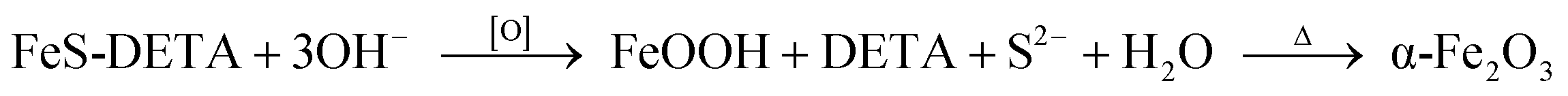
 | ||
| Fig. 2 (a,b,c) SEM image and (d) XRD spectra of the intermediates collected after the reaction proceeded for different times: (a) 1 h, (b) 2 h, (c) 4 h. | ||
With the stable dodecahedral α-Fe2O3 in hand, we tested their optical absorption properties through UV-vis spectroscopy and their PEC activity under visible-light illumination (λ > 420 nm) in an electrolyte of 1.0 M KOH (pH = 13.6). For comparison, commercial α-Fe2O3 nanoparticles were also evaluated. As presented in Fig. S12, ESI,† commercial α-Fe2O3 nanoparticles (C-hematite) also show a small size and uniform morphology. It can be seen from the UV-vis diffuse reflectance spectra that both C-hematite and D-hematite have a similar bandgap (Eg) of ca. 2.0 eV so that almost all UV light and a large part of the visible light can be employed, whereas D-hematite still shows a stronger absorption ability when compared with C-hematite in the visible region (Fig. 3a). To test their PEC performance, I–V curves have been measured under visible-light illumination (λ > 420 nm) and in the dark (Fig. 3b). In the polarization curves for the oxygen evolution reaction (OER), the photocurrent of D-hematite nanostructures (3.06 mA cm−2) is almost four times larger than that of C-hematite (0.77 mA cm−2) at the potential of 1.6 V versus a reversible hydrogen electrode (RHE). Fig. 3c displays that the photocurrent responses of D-hematite and C-hematite are prompt, steady and reproducible. The photocurrent–time course of D-hematite is presented in Fig. 3d, suggesting no obvious decrease of the photocurrent over a 10 h test. Electrochemical impedance spectroscopy (EIS) (Fig. S13, ESI†) shows that D-hematite exhibits a smaller impedance than C-hematite. The enhanced PEC activity of the D-hematite may be attributed to its unique morphological and structural features. To prove the advantage of the unique dodecahedral morphology, the I–V curves of the samples synthesized using FeS-TETA precursors were also examined. These polarization curves for the OER show that the PEC activities of the dodecahedral samples synthesized using FeS-DETA precursors and FeS-TETA precursors are better than commercial hematite and synthetic cubic hematite (Fig. S14, ESI†). Firstly, the D-hematite nanostructures harvest more visible light than C-hematite, as shown in Fig. 3a. The D-hematite from the FeS-DETA precursors may show better PEC activity than that from the FeS-TETA precursors considering their smaller size. What is more, the photocatalytic activity trend of the exposed facets is in the order of {110} > {012} ≫ {001}.35 The special dodecahedral nanostructures can offer a huge number of {110} facets according to the HRTEM image (Fig. 1f) for exhibiting better photoactivity whereas the dominant facet of cubic hematite is {012}.35,45 Secondly, the D-hematite with a large fraction of uncoordinated surface atoms can usually supply a high density of catalytic active sites.53 Thirdly, the good crystallinity of D-hematite is helpful for quick charge transfer of the photogenerated carriers.3 We expect that the PEC activity of such dodecahedral α-Fe2O3 can be further improved by surface treatment and through decoration with co-photocatalysts or dopants.
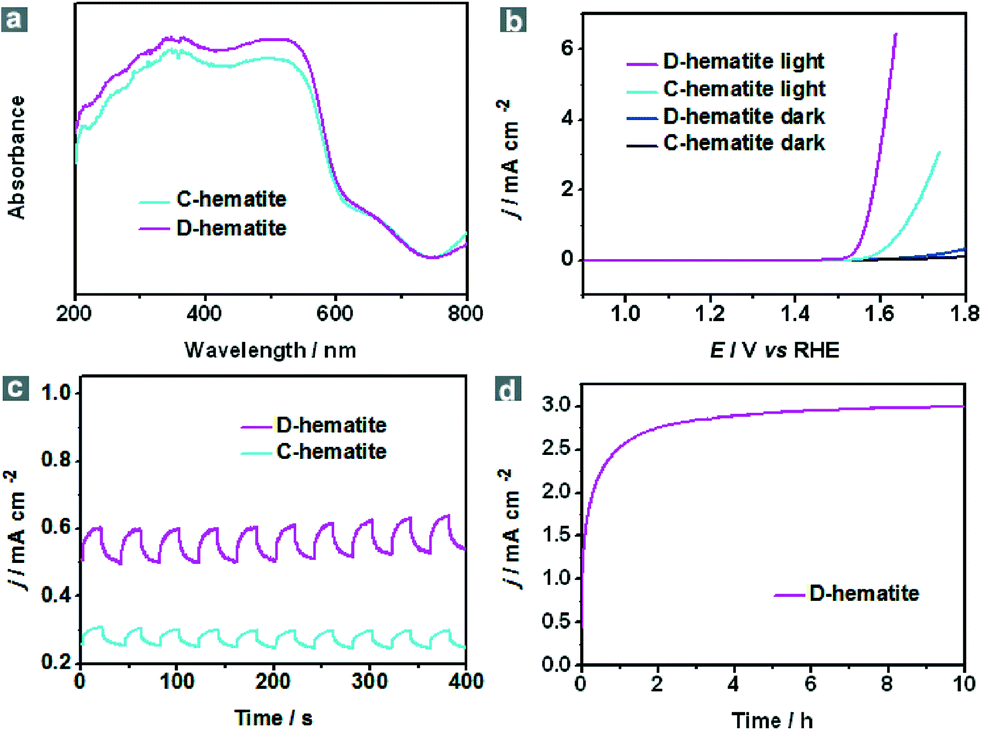 | ||
| Fig. 3 PEC performances of dodecahedral α-Fe2O3 (D-hematite, magenta line) and commercial α-Fe2O3 nanoparticles (C-hematite, cyan line): (a) UV-vis diffuse reflectance spectra; (b) iR-corrected polarization curves for the oxygen evolution reaction (OER) under visible-light illumination (λ > 420 nm) and in the dark (D-hematite, blue line. C-hematite, black line) with a scan speed of 10 mV s−1; (c) photocurrent response at an applied bias potential of 1.6 V vs. RHE in 1.0 M KOH aqueous solution under repeated on/off cycles of visible-light illumination (λ > 420 nm); (d) photocurrent–time curve at an applied bias potential of 1.6 V vs. RHE under visible-light illumination (λ > 420 nm) in 1.0 M KOH. | ||
4. Conclusion
In summary, dodecahedral α-Fe2O3 nanostructures have been successfully prepared via a simple and cost-effective DETA-assisted hydrothermal treatment of inorganic–organic hybrid FeS-DETA 2D nanosheet precursors. It is found that the presence of DETA is primarily responsible for the formation of the dodecahedral α-Fe2O3. These dodecahedral α-Fe2O3 nanostructures show enhanced PEC activity and good stability under visible-light irradiation (λ > 420 nm) in the presence of 1.0 M KOH as the electrolyte. In addition to their promising application in efficient PEC water splitting, dodecahedral α-Fe2O3 may find broad applications in the fields of organic catalysis,63 water treatment35 and dye-sensitized solar cells.64Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21422104 and no. 21373149) and the Innovation Foundation of Tianjin University.Notes and references
- M. Gratzel, Nature, 2001, 414, 338 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS.
- H. M. Chen, C. K. Chen, R. S. Liu, L. Zhang, J. Zhang and D. P. Wilkinson, Chem. Soc. Rev., 2012, 41, 5654 RSC.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294 RSC.
- L. Shang, T. Bian, B. Zhang, D. Zhang, L. Wu, C. H. Tung, Y. Yin and T. Zhang, Angew. Chem., Int. Ed., 2014, 53, 250 CrossRef CAS PubMed.
- C. Zhou, Y. Zhao, L. Shang, Y. Cao, L. Z. Wu, C. H. Tung and T. Zhang, Chem. Commun., 2014, 50, 9554 RSC.
- T. Bian, L. Shang, H. Yu, M. T. Perez, L. Z. Wu, C. H. Tung, Z. Nie, Z. Tang and T. Zhang, Adv. Mater., 2014, 26, 5613 CrossRef CAS PubMed.
- Y. Lin, G. Yuan, S. Sheehan, S. Zhou and D. Wang, Energy Environ. Sci., 2011, 4, 4862 CAS.
- K. Sivula, F. L. Formal and M. Gratzel, ChemSusChem, 2011, 4, 432 CrossRef CAS PubMed.
- R. H. Goncalves, B. H. R. Lima and E. R. Leite, J. Am. Chem. Soc., 2011, 133, 6012 CrossRef CAS PubMed.
- D. K. Zhong, M. Cornuz, K. Sivula, M. Gratzel and D. R. Gamelin, Energy Environ. Sci., 2011, 4, 1759 CAS.
- T. K. Townsend, E. M. Sabio, N. D. Browning and F. E. Osterloh, Energy Environ. Sci., 2011, 4, 4270 CAS.
- D. A. Wheeler, G. Wang, Y. Ling, Y. Li and J. Z. Zhang, Energy Environ. Sci., 2012, 5, 6682 CAS.
- N. J. Cherepy, D. B. Liston, J. A. Lovejoy, H. Deng and J. Z. Zhang, J. Phys. Chem. B, 1998, 102, 770 CrossRef CAS.
- A. G. Joly, J. R. Williams, S. A. Chambers, G. Xiong, W. P. Hess and D. M. Laman, J. Appl. Phys., 2006, 99, 053521 CrossRef PubMed.
- C. Y. Cummings, F. Marken, L. M. Peter, K. G. U. Wijayantha and A. A. Tahir, J. Am. Chem. Soc., 2012, 134, 1228 CrossRef CAS PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294 CrossRef CAS PubMed.
- G. Wang, Y. Ling and Y. Li, Nanoscale, 2012, 4, 6682 RSC.
- K. Sivula, R. Zboril, F. L. Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Gratzel, J. Am. Chem. Soc., 2010, 132, 7436 CrossRef CAS PubMed.
- A. Duret and M. Gratzel, J. Phys. Chem. B, 2005, 109, 17184 CrossRef CAS PubMed.
- N. T. Hahn, H. Ye, D. W. Flaherty, A. J. Bard and C. B. Mullins, ACS Nano, 2010, 4, 1977 CrossRef CAS PubMed.
- A. Kay, I. Cesar and M. Gratzel, J. Am. Chem. Soc., 2006, 128, 15714 CrossRef CAS PubMed.
- J. S. Xu and Y. J. Zhu, CrystEngComm, 2012, 14, 2702 RSC.
- U. Bjorksten, J. Moser and M. Gratzel, Chem. Mater., 1994, 6, 858 CrossRef.
- X. Mou, X. Wei, Y. Li and W. Shen, CrystEngComm, 2012, 14, 5107 RSC.
- N. K. Chaudhari, H. C. Kim, M. S. Kim, J. Park and J. S. Yu, CrystEngComm, 2012, 14, 2024 RSC.
- L. Vayssieres, C. Sathe, S. M. Butorin, D. K. Shuh, J. Nordgren and J. Guo, Adv. Mater., 2005, 17, 2320 CrossRef CAS.
- L. Vayssieres, N. Beermann, S. E. Lindquist and A. Hagfeldt, Chem. Mater., 2001, 13, 233 CrossRef CAS.
- M. Rachel, R. Mahfujur, J. M. D. MacElroy and C. A. Wolden, ChemSusChem, 2011, 4, 474 CrossRef PubMed.
- F. Meng, S. A. Morin and S. Jin, J. Am. Chem. Soc., 2011, 133, 8408 CrossRef CAS PubMed.
- S. K. Mohapatra, S. E. John, S. Banerjee and M. Misra, Chem. Mater., 2009, 21, 3048 CrossRef CAS.
- X. Zhou, J. Lan, G. Liu, K. Deng, Y. Yang, G. Nie, J. Yu and L. Zhi, Angew. Chem., Int. Ed., 2012, 51, 178 CrossRef CAS PubMed.
- S. Xiong, J. Xu, D. Chen, R. Wang, X. Hu, G. Shen and Z. L. Wang, CrystEngComm, 2011, 13, 7114 RSC.
- L. Li, H. B. Wu, L. Yu, S. Madhavi and X. W. Lou, Adv. Mater. Interfaces, 2014, 1, 1400050 Search PubMed.
- W. Cheng, J. He, T. Yao, Z. Sun, Y. Jiang, Q. Liu, S. Jiang, F. Hu, Z. Xie, B. He, W. Yan and S. Wei, J. Am. Chem. Soc., 2014, 136, 10393 CrossRef CAS PubMed.
- M. Lin, H. R. Tan, J. P. Y. Tan and S. Bai, J. Phys. Chem. C, 2013, 117, 11242 CAS.
- L. S. Zhong, J. S. Hu, H. P. Liang, A. M. Cao, W. G. Song and L. J. Wan, Adv. Mater., 2006, 18, 2426 CrossRef CAS.
- S. B. Wang, Y. L. Min and S. H. Yu, J. Phys. Chem. C, 2007, 111, 3551 CAS.
- X. Liang, X. Wang, J. Zhuang, Y. Chen, D. Wang and Y. Li, Adv. Funct. Mater., 2006, 16, 1805 CrossRef CAS.
- B. Jia and L. Gao, Cryst. Growth Des., 2008, 8, 1372 CAS.
- L. Zhang, H. B. Wu, S. Madhavi, H. H. Hng and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 17388 CrossRef CAS PubMed.
- W. Wu, R. Hao, F. Liu, X. Su and Y. Hou, J. Mater. Chem. A, 2013, 1, 6888 CAS.
- Y. Liang, L. Shang, T. Bian, C. Zhou, D. Zhang, H. Yu, H. Xu, Z. Shi, T. Zhang, L. Z. Wu and C. H. Tung, CrystEngComm, 2012, 14, 4431 RSC.
- J. Yang and T. Sasaki, Cryst. Growth Des., 2010, 10, 1233 CAS.
- Y. Xu, H. Wang, Y. Yu, L. Tian, W. Zhao and B. Zhang, J. Phys. Chem. C, 2011, 115, 15288 CAS.
- S. Inamdar, H. S. Choi, M. S. Kim, K. Chaudhari and J. S. Yu, CrystEngComm, 2012, 14, 7009 RSC.
- W. J. Li, E. W. Shi, W. Z. Zhong and Z. W. Yin, J. Cryst. Growth, 1999, 203, 186 CrossRef CAS.
- Y. Jun, J. Choi and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 3414 CrossRef CAS PubMed.
- Z. Zhang, B. Xu and X. Wang, Chem. Soc. Rev., 2014, 43, 7870 RSC.
- J. Yin, Z. Yu, F. Gao, J. Wang, H. Pang and Q. Lu, Angew. Chem., Int. Ed., 2010, 49, 6328 CrossRef CAS PubMed.
- Y. Yang, H. Ma, J. Zhuang and X. Wang, Inorg. Chem., 2011, 50, 10143 CrossRef CAS PubMed.
- B. Lv, Z. Liu, H. Tian, Y. Xu, D. Wu and Y. Sun, Adv. Funct. Mater., 2010, 20, 3987 CrossRef CAS.
- R. Liu, Y. Jiang, H. Fan, Q. Lu, W. Du and F. Gao, Chem. – Eur. J., 2012, 18, 8957 CrossRef CAS PubMed.
- T. K. Van, H. G. Cha, C. K. Nguyen, S. W. Kim, M. H. Jun and Y. S. Kang, Cryst. Growth Des., 2012, 12, 862 CAS.
- X. Li, W. Wei, S. Wang, L. Kuai and B. Geng, Nanoscale, 2011, 3, 718 RSC.
- X. Liu, J. Zhang, S. Wu, D. Yang, P. Liu, H. Zhang, S. Wang, X. Yao, G. Zhu and H. Zhao, RSC Adv., 2012, 2, 6178 RSC.
- Z. Zang, H. B. Yao, Y. X. Zhou, W. T. Yao and S. H. Yu, Chem. Mater., 2008, 20, 4749 CrossRef CAS.
- W. T. Yao, H. Z. Zhu, W. G. Li, H. B. Yao, Y. C. Wu and S. H. Yu, ChemPlusChem, 2013, 78, 723 CrossRef CAS.
- X. L. Fang, Y. Li, C. Chen, Q. Kuang, X. Z. Gao, Z. X. Xie, S. Y. Xie, R. B. Huang and L. S. Zheng, Langmuir, 2010, 26, 2745 CrossRef CAS PubMed.
- M. Shokouhimehr, Y. Piao, J. Kim, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 7039 CrossRef CAS PubMed.
- Y. Hou, D. Wang, X. H. Yang, W. Q. Fang, B. Zhang, H. F. Wang, G. Z. Lu, P. Hu, H. J. Zhao and H. G. Yang, Nat. Commun., 2013, 4, 1583 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental instruments and additional characterization results. See DOI: 10.1039/c4ce01872a |
| This journal is © The Royal Society of Chemistry 2015 |