DOI:
10.1039/C4CE01795A
(Paper)
CrystEngComm, 2015,
17, 98-106
Co-crystals and co-crystal hydrates of vanillic acid†
Received
1st September 2014
, Accepted 1st November 2014
First published on 18th November 2014
Abstract
A series of co-crystals of vanillic acid (VA) with caffeine (CAF), theophylline (THP), theobromine (THB), nicotinamide (NAM), isonicotinamide (INM), acridine (ACR) and urea (U) have successfully been prepared. The co-crystals involving the methylated xanthines indicate different host: guest ratios with co-crystals VA·2CAF, 2VA·THP and the co-crystal hydrate VA·THB·2H2O. The nicotinamide and acridine co-crystals (VA·NAM and VA·ACR) displayed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratios in contrast to the isonicotinamide co-crystal hydrate 2VA·2INM·2H2O. Co-crystallisation of vanillic acid with urea gave 2VA·2U. The co-crystals and co-crystal hydrates were characterised by single crystal X-ray diffraction and thermal analysis. Grinding and slurry experiments were also performed.
:1 molar ratios in contrast to the isonicotinamide co-crystal hydrate 2VA·2INM·2H2O. Co-crystallisation of vanillic acid with urea gave 2VA·2U. The co-crystals and co-crystal hydrates were characterised by single crystal X-ray diffraction and thermal analysis. Grinding and slurry experiments were also performed.
Introduction
Crystal engineering plays an important role in the preparation of multicomponent crystals, the main objective being to obtain compounds with enhanced physicochemical properties.1–3 The supramolecular synthon approach where complementary functional groups pair up is key in the design of new crystal structures.4–6 In particular the sythons involving carboxylic acid and nitrogen-containing functionalities have received much attention. The acid-pyridine heterosynthon was recently used as a tool in a study of the 1:1 co-crystals of isonicotinamide with benzoic acid and fluorobenzoic acids.7 Other examples of structures containing this synthon include the substituted 2-anilinonicotinic acids,8 co-crystals of picolinamide9 and 4-hydroxybenzoic acid.10,11 The acid–amide heterosynthon also features prominently as was observed in a series of co-crystals involving substituted benzamides,12–14 with dicarboxylic acids. In co-crystal systems involving the methylated xanthines the acid-imidazole heterosynthon is commonly featured.15–17 In this study a series of co-crystals of vanillic acid (VA) were prepared and characterised. VA was chosen due to its hydrogen bonding potential in the form of three different functional groups. The strongest hydrogen bond donor is the carboxylic acid group followed by the hydroxyl group. It also possesses a hydrogen bond acceptor (methoxy group). VA is an oxidised form of vanillin and is found in vanilla extract.18 VA is used as a flavouring agent and a food additive.19 The co-crystals of VA with pyrazinecarboxamide,20 ethenzamide,21 4-chloro-3,5-dimethyl-1H-pyrazole22 and andrographolide23 have been reported. The crystal structure of VA itself is also known and displays the R22(8) acid–acid homosynthon.24 For the co-crystal of VA with pyrazinecarboxamide, the acid–acid and amide–amide homosynthons are maintained compared to the co-crystal of VA and ethenzamide which displays the acid–amide heterosynthon. For the present study we have focused on a series of co-crystal formers which include the methylated xanthines (caffeine, theophylline and theobromine), nicotinamide, isonicotinamide, acridine and urea. The structure of vanillic acid and the co-crystal formers are given in Scheme 1.
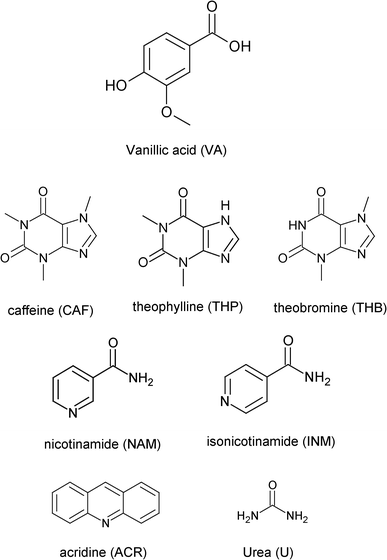 |
| | Scheme 1 Vanillic acid and co-crystal formers used in this study. | |
Experimental section
Crystal growth
1, VA·2CAF.
Vanillic acid (40 mg, 0.238 mmol) and caffeine (46.2 mg, 0.238 mmol) were dissolved in ethylmethyl ketone to form a dilute solution. Crystals were obtained via slow evaporation under ambient conditions after 1 week. Similar crystals were obtained using a 50:50 mixture of chloroform and methanol as a solvent.
2, 2VA·THP.
Vanillic acid (40 mg, 0.238 mmol) and theophylline (42.9 mg, 0.238 mmol) were dissolved in water. The dilute solution was left to evaporate and crystals were obtained after 4 weeks.
3, VA·THB·2H2O.
Vanillic acid (40 mg, 0.238 mmol) and theobromine (42.9 mg, 0.238 mmol) were dissolved in water. The dilute solution was left to evaporate and crystals were obtained after 4 d.
4, VA·ACR.
Vanillic acid (40 mg, 0.238 mmol) and acridine (42.6 mg, 0.238 mmol) were dissolved in diethyl ether. Crystals were obtained after 2 d.
5, VA·NAM.
Vanillic acid (40 mg, 0.238 mmol) and nicotinamide (29.1 mg, 0.238 mmol) were dissolved in ethyl methyl ketone. Crystals were obtained after 1 week. Similar crystals were obtained with the solvents acetone, isopropanol, ethanol and methanol.
6, 2VA·2INM·2H2O.
Vanillic acid (40 mg, 0.238 mmol) and isonicotinamide (29.1 mg, 0.238 mmol) were dissolved in methanol. Slow evaporation yielded crystals after 2 weeks. Similar crystals were obtained with isopropanol and tetrahydrofuran.
7, 2VA·2U.
Vanillic acid (40 mg, 0.238 mmol) and urea (28.6 mg, 0.476 mmol) were dissolved in ethanol with crystals obtained after 2 weeks. Similar crystals were obtained with solvents acetone, isopropanol and tetrahydrofuran.
Structure analysis
Cell dimensions were established from intensity data measured on a Nonius Kappa CCD25 and a Bruker DUO APEX II26 diffractometer using graphite-monochromated Mo-Kα radiation. The intensity data were collected by the standard phi scan and omega scan techniques, scaled and reduced using DENZO-SMN27 or SAINT-Plus.28 All of the structures were solved using direct methods and refined by full-matrix least squares with SHELX-97,29 refining on F2. The program X-Seed30 was used as a graphical interface. For all the structures the non-hydrogen atoms were located in the difference electron density map and refined anisotropically. All hydrogens not involved in hydrogen bonding were placed with geometric constraints and allowed to refine isotropically. The hydroxyl and carboxylic hydrogens of the vanillic acid and the hydrogens attached to nitrogen were found in the difference electron density map and allowed to refine isotropically.
Thermal analysis
Differential scanning calorimetry (DSC) was performed for all crystals. Crushed crystals were placed in crimped and vented pans and analysed using a Perkin Elmer 6 series system with a purge gas of nitrogen at 20 ml min−1. Samples were analysed between 303–600 K at a heating rate of 10 K min−1. For the co-crystal hydrates, 3 and 6, thermogravimetric analysis was performed on the crushed crystals.
Results and discussion
Structural analysis
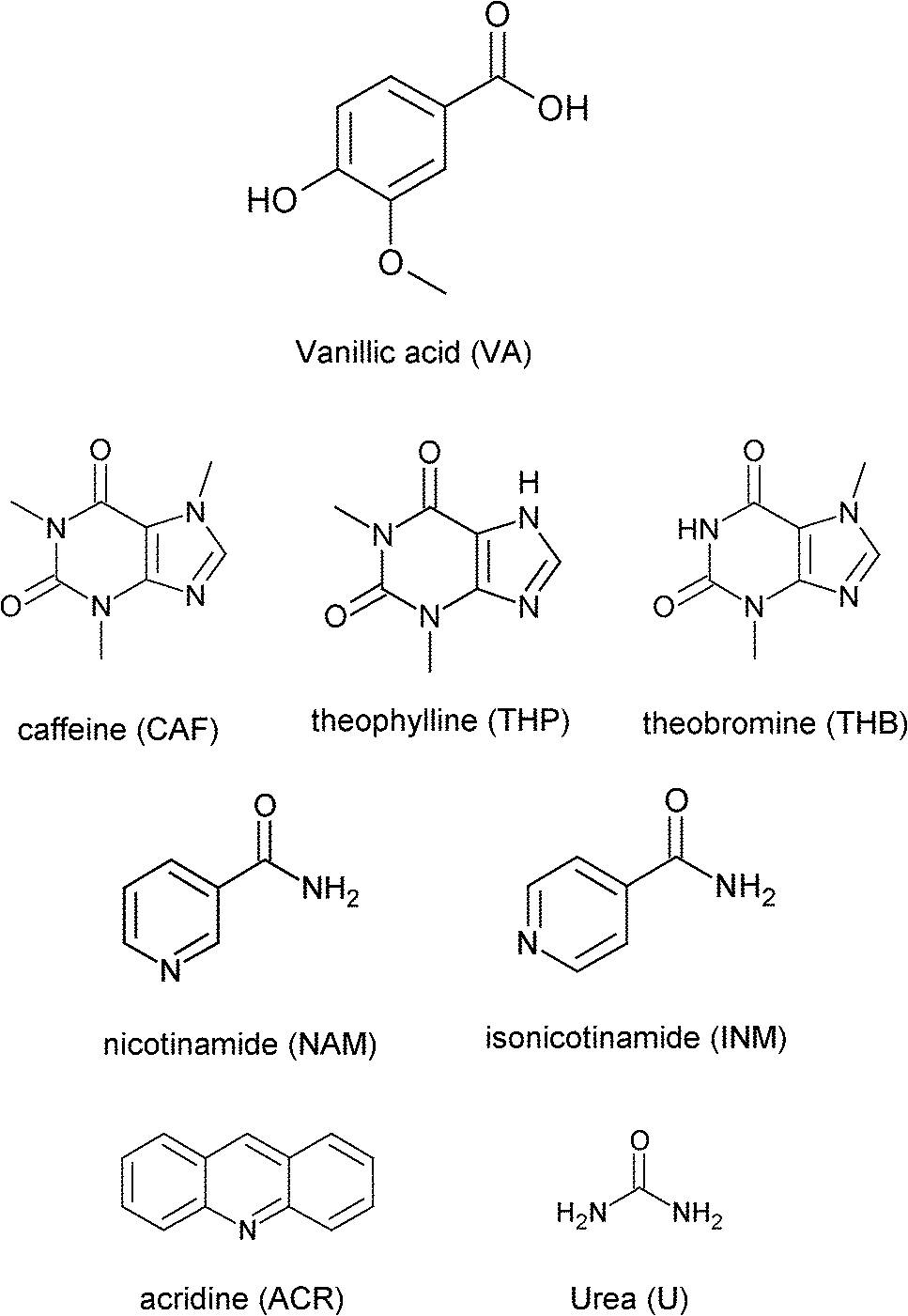
The crystal data for structures 1–4 are summarised in Table 1. The co-crystal of vanillic acid with caffeine, VA·2CAF (1), solved successfully in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit contains one VA molecule and two CAF molecules (Z = 2) which form a hydrogen bonded unit (Fig. 1). The carboxylic acid hydrogen of VA is linked to the imidazole nitrogen of CAF and the hydroxyl hydrogen of the same VA forms hydrogen bonds with the carbonyl oxygen of the second CAF molecule. The units are linked via weak CH⋯N and CH⋯O interactions. Adjacent CAF molecules utilise π⋯π stacking (3.420 Å) and the VA also interacts with CAF molecules via π⋯π stacking (3.546 Å).
. The asymmetric unit contains one VA molecule and two CAF molecules (Z = 2) which form a hydrogen bonded unit (Fig. 1). The carboxylic acid hydrogen of VA is linked to the imidazole nitrogen of CAF and the hydroxyl hydrogen of the same VA forms hydrogen bonds with the carbonyl oxygen of the second CAF molecule. The units are linked via weak CH⋯N and CH⋯O interactions. Adjacent CAF molecules utilise π⋯π stacking (3.420 Å) and the VA also interacts with CAF molecules via π⋯π stacking (3.546 Å).
Table 1 Crystal data table for the co-crystals of VA with the methylated xanthines and acridine
| Compound |
1
|
2
|
3
|
4
|
| Structural formula |
C8H8O4·2C8H10N4O2 |
2C8H8O4·C7H8N4O2 |
C8H8O4·C7H8N4O2·2H2O |
C8H8O4·C13H9N |
| Molecular mass (g mol−1) |
556.54 |
515.45 |
384.35 |
347.36 |
| Data collection temp. (K) |
173(2) |
173(2) |
173(2) |
173(2) |
| Crystal system |
Triclinic |
Monoclinic |
Triclinic |
Monoclinic |
| Space group |
P |
P21/c |
P |
P21/n |
|
a (Å) |
10.110(2) |
11.2741(6) |
8.2148(16) |
8.4690(17) |
|
b (Å) |
10.525(2) |
15.7712(10) |
8.2504(17) |
22.621(5) |
|
c (Å) |
12.221(2) |
13.3746(8) |
13.132(3) |
8.9534(18) |
|
α (°) |
77.69(3) |
90 |
102.82(3) |
90 |
|
β (°) |
81.26(3) |
90.572(2) |
97.46(3) |
90.68(3) |
|
γ (°) |
82.69(3) |
90 |
93.98(3) |
90 |
| Volume (Å3) |
1249.7(4) |
2378.0(2) |
856.1(3) |
1715.1(6) |
|
Z
|
2 |
4 |
2 |
4 |
|
D
c, calc density (g cm−3) |
1.479 |
1.440 |
1.491 |
1.345 |
| Absorption coefficient (mm−1) |
0.114 |
0.115 |
0.122 |
0.094 |
|
F(000) |
584 |
1076 |
404 |
728 |
|
θ range |
1.72–28.34 |
1.81–28.33 |
1.61–27.13 |
1.80–27.53 |
| Limiting indices |
−13,9; ±14; −16,15 |
±14; ±21; ±17 |
±10; ±10; ±16 |
−11,10; −25, 29; −11,5 |
| Reflections collected/unique |
12163/6186 |
29262/5876 |
7070/3767 |
8132/3933 |
| Goodness-of-fit on F2 |
1.038 |
1.047 |
1.027 |
0.965 |
| Final R indices [I > 2sigma(I)] |
R
1 = 0.0542; wR2= 0.1448 |
R
1 = 0.0470; wR2 = 0.1195 |
R
1 = 0.0415; wR2 = 0.1045 |
R
1 = 0.0511; wR2 = 0.1057 |
|
R indices (all data) |
R
1 = 0.0782; wR2 = 0.1605 |
R
1 = 0.0653; wR2 = 0.1299 |
R
1 = 0.0609; wR2 = 0.1156 |
R
1 = 0.1092; wR2 = 0.1276 |
| Largest diff. peak and hole (e Å−3) |
0.885; −0.264 |
0.316; −0.242 |
0.260; −0.236 |
0.235; −0.234 |
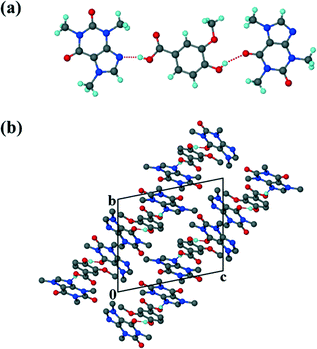 |
| | Fig. 1 (a) Hydrogen bonding in 1. (b) Packing diagram of 1 along [100]. Hydrogen bonding is indicated with dashed lines. Hydrogens not involved in hydrogen bonding have been omitted. | |
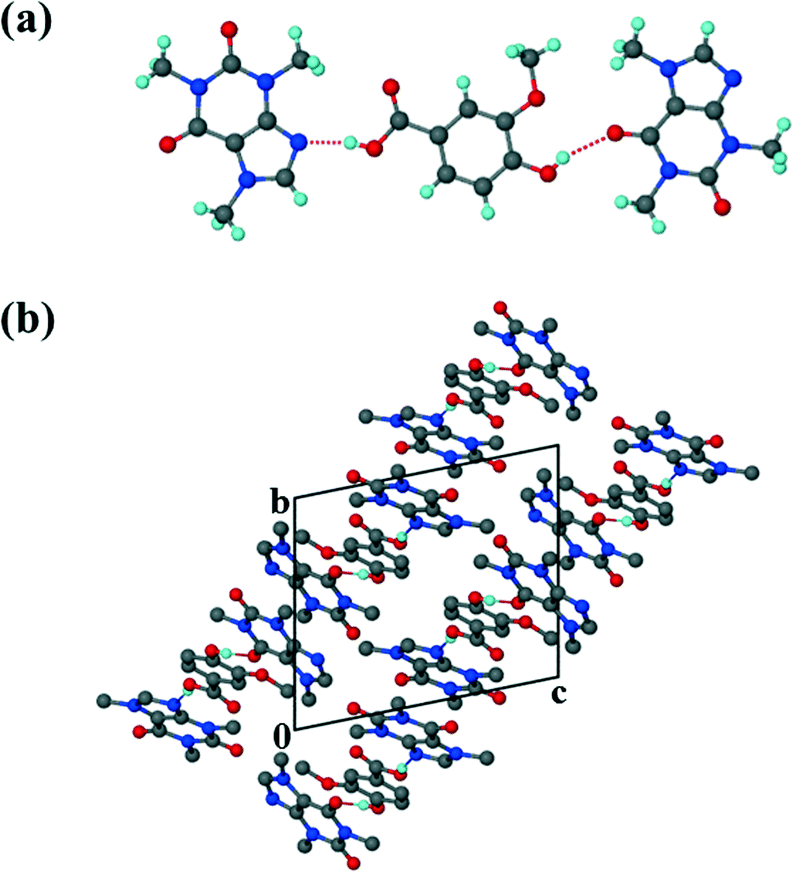
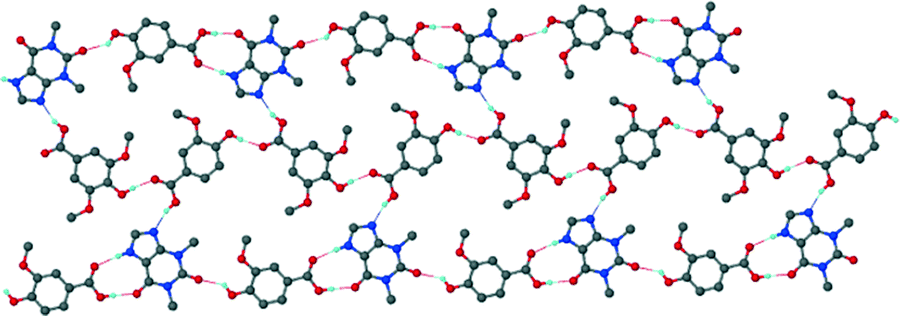
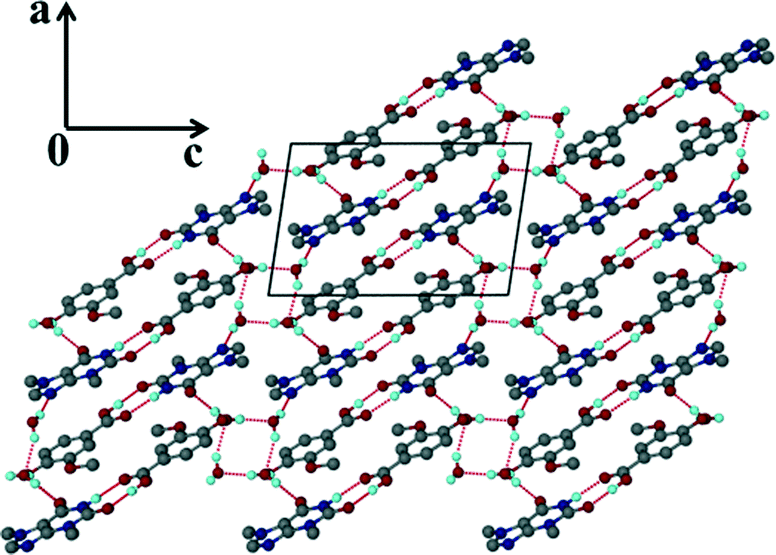
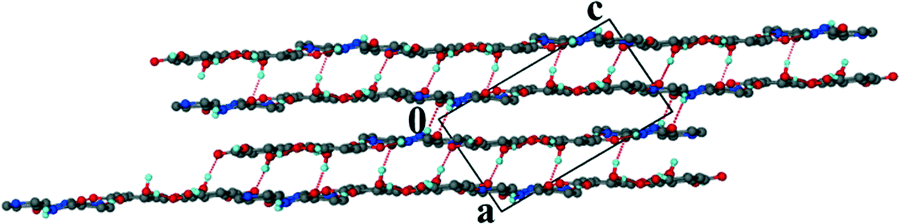
For the theophylline co-crystal, 2VA·THP (2), the structure solved in P21/c with two VA molecules and one THP molecule in the asymmetric unit. One of the VA molecules has a disordered methoxy group which refined with site occupancy factors of 0.812 and 0.188 respectively. The hydrogen bond network is more extensive due to the additional N–H donor group of the THP (Fig. 2). A VA molecule and a THP molecule form a hydrogen bonded ring which can be described as R22(9) using graphset notation,31 in addition to the R33(9) ring linking two VA and one THP molecule. This ring also utilises a weak C–H⋯O interaction (d(CO) = 3.031 Å). The hydrogen bonded layers in 2VA·THP stack in sheets parallel to [30![[2 with combining overline]](https://www.rsc.org/images/entities/char_0032_0305.gif) ] as shown in Fig. 3. The distance between the layers is approximately 3.750 Å with π⋯π stacking between VA and THP molecules.
] as shown in Fig. 3. The distance between the layers is approximately 3.750 Å with π⋯π stacking between VA and THP molecules.
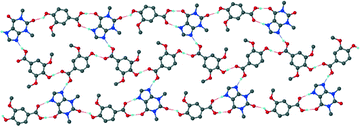 |
| | Fig. 2 Hydrogen bonding in 2. Only the major component of the disordered VA has been shown for clarity. | |
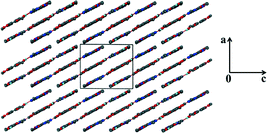 |
| | Fig. 3 Hydrogen bonded layers found in 2 parallel to [30]. | |
It is interesting to note the differences in crystal packing in the absence of the methoxy group of VA i.e. comparison of structures involving 4-hydroxybenzoic acid. Co-crystallisation of THP and 4-hydroxybenzoic acid yielded a 1:1 co-crystal.32 The R22(9) ring network persists, however the para-hydroxyl group forms a hydrogen bond with another imidazole nitrogen and not with a carbonyl group as was observed in 2VA·THP. Three co-crystals of CAF and 4-hydroxybenzoic acid have also been reported in the CSD;33 caffeine 4-hydroxybenzoic acid monohydrate,34 caffeine bis(4-hydroxybenzoic acid)35 and bis(caffeine) 4-hydroxybenzoic acid.35,36 The latter structure has units composed of one molecule of 4-hydroxybenzoic acid hydrogen bonded to two caffeine molecules. The Nimidazole⋯(COOH) hydrogen bond is maintained but the hydroxyl group forms a hydrogen bond with the urea carbonyl moiety and not the amide carbonyl as was observed in VA·2CAF. Zhang et al.35 described the structures of eight co-crystals of caffeine with hydroxybenzoic acids. The Nimidazole⋯(COOH) hydrogen bond was found in all eight co-crystals and follows the best acceptor/best donor model37,38 with the carboxylic acid as the best donor and the imidazole nitrogen as the best acceptor.34 However the hydrogen bonds involving the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of caffeine were less predictable. Of the eight co-crystals, three structures contained both OH(hydroxy)⋯O(amide) and OH(hydroxy)⋯O(urea) hydrogen bonds, while two contained only OH(hydroxy)⋯O(amide), another two only OH(hydroxy)⋯O(urea) and one contained neither interaction.
O groups of caffeine were less predictable. Of the eight co-crystals, three structures contained both OH(hydroxy)⋯O(amide) and OH(hydroxy)⋯O(urea) hydrogen bonds, while two contained only OH(hydroxy)⋯O(amide), another two only OH(hydroxy)⋯O(urea) and one contained neither interaction.
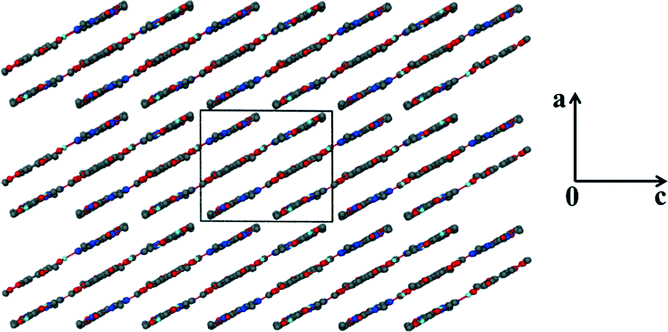
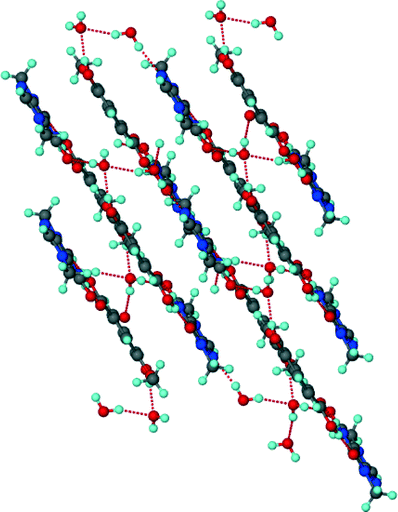
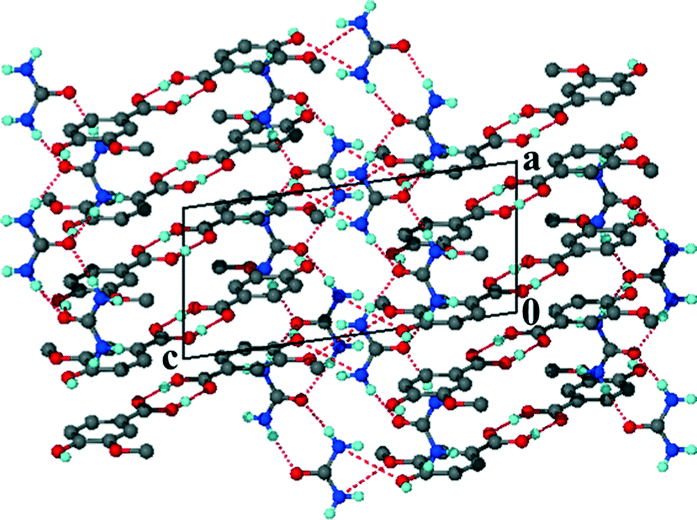
Unlike the previous two structures, theobromine formed a co-crystal hydrate with VA. Structure 3 (VA·THB·2H2O) solved successfully in the triclinic space group P with one THB, one VA and two water molecules in the asymmetric unit (Z = 2). The presence of water molecules disrupts the expected Nimidazole⋯(COOH) interaction and instead the VA and THB molecules form acid–imide heterosynthons which can be described as R22(8) rings. The VA-THB dimers pack in columns along [100] with the water molecules sandwiched between the columns (Fig. 4). The water molecules act as bridges linking the VA-THB dimers, with a water molecule acting as hydrogen bond donor to the imidazole nitrogen. A search of the CSD (May 2014 update) of THB co-crystals with carboxylic acids revealed five co-crystals39–42 and one co-crystal hydrate.43 All five co-crystals exhibited the Nimidazole⋯(COOH) heterosynthon. The co-crystal hydrate43 (gallic acid theobromine dihydrate) displayed the acid–imide heterosynthon with a water molecule donating a hydrogen bond to the imidazole nitrogen, which is consistent with our observation. For structure 3, water molecules occupy cavities and form tetramers which can be described as R44(8) rings. The VA and THB molecules stack in layers which utilise the aromatic rings of both molecules in the form of π⋯π interactions, the distance between the layers is 3.740 Å, with water molecules bridging the layers (Fig. 5).
 |
| | Fig. 4 Packing diagram of 3 along [010] illustrating the hydrogen bonded ring networks. | |
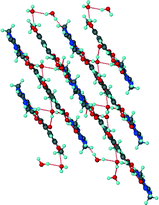 |
| | Fig. 5 Hydrogen bonded layers found in 3. | |
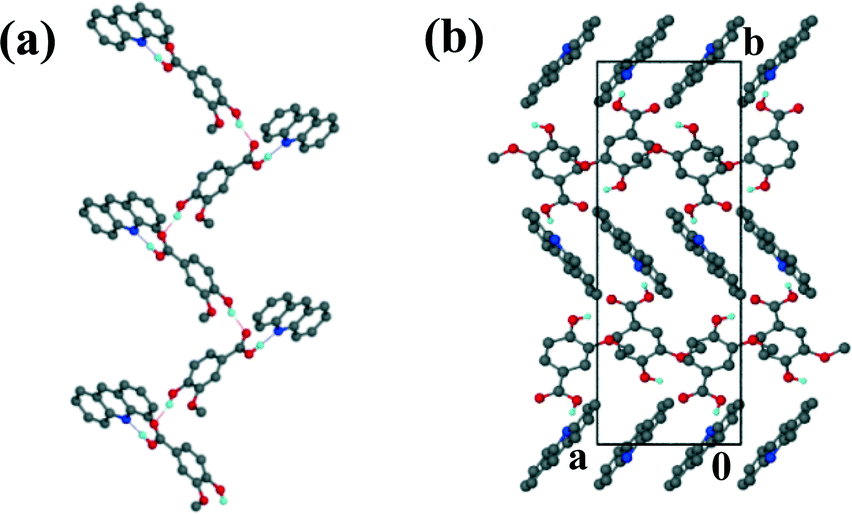
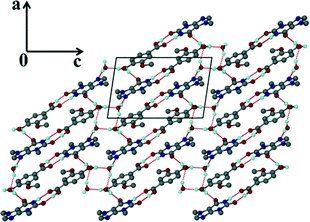
The acridine co-crystal, 4, solved in the monoclinic space group P21/n, with one ACR and one VA molecule in the asymmetric unit (Z = 4). The infinite hydrogen bonded chains (Fig. 6a) form a zig-zag motif with each VA molecule hydrogen bonded to one ACR molecule via (VA)COOH⋯N(ACR) hydrogen bonds and the hydroxyl group of VA linked to the carbonyl of another VA molecule.
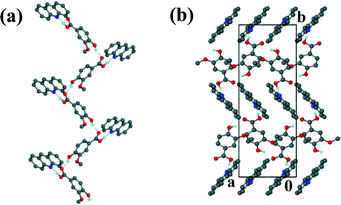 |
| | Fig. 6 Structure 4: (a) hydrogen bonding and (b) packing diagram along [001]. | |
Adjacent ACR molecules interact via π⋯π stacking with a minimum distance of 3.733 Å. The packing diagram along [001] is shown in Fig. 6b.
Table 2 summarises the crystal data for the NAM, INM and urea co-crystals. The NAM co-crystal, 5 and the INM co-crystal hydrate, 6, were both solved in P. For 5, the two VA molecules form a R22(8) dimer with the hydroxyl groups of VA linked to NAM molecules via one N–H bond, Fig. 7. The other N–H bond is linked to a neighbouring NAM molecule via the carbonyl group to form chains along [100]. In addition, two VA molecules and two NAM molecules are connected to form R44(16) hydrogen bonded rings. Very weak π⋯π interactions (4.470 Å) present between VA and NAM molecules. Comparison with the 1:1 4-hydroxybenzoic acid NAM co-crystal44,45 shows the acid–amide heterosynthon forming R22(8) rings with neighbouring acid–amide pairs connected to form R24(8) rings, in contrast to the acid homosynthon found in 5. The acid–amide heterosynthon is also found in the 1:1 4-hydroxybenzoic acid INM co-crystal.46 The asymmetric unit for structure 6 contains two VA, two INM and two water molecules.
Table 2 Crystal data table for the co-crystals of VA with nicotinamide, isonicotinamide and urea
| Compound |
5
|
6
|
7
|
| Structural formula |
C8H8O4·C6H6N2O |
2C8H8O4·2C6H6N2O·2H2O |
2C8H8O4·2CH4N2O |
| Molecular mass (g mol−1) |
290.27 |
616.58 |
456.41 |
| Data collection temp. (K) |
173(2) |
173(2) |
173(2) |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P |
P |
P |
|
a (Å) |
4.8723(10) |
7.9000(16) |
6.9821(14) |
|
b (Å) |
8.0058(16) |
13.196(3) |
10.232(2) |
|
c (Å) |
18.018(4) |
13.735(3) |
15.388(3) |
|
α (°) |
78.85(3) |
85.02(3) |
99.68(3) |
|
β (°) |
84.50(3) |
85.32(3) |
95.37(3) |
|
γ (°) |
75.62(3) |
78.68(3) |
103.46(3) |
| Volume (Å3) |
667.1(2) |
1395.6(5) |
1043.8(4) |
|
Z
|
2 |
2 |
2 |
|
D
c, calc density (g cm−3) |
1.445 |
1.467 |
1.452 |
| Absorption coefficient (mm−1) |
0.111 |
0.116 |
0.120 |
|
F(000) |
304 |
648 |
480 |
|
θ range |
2.31–28.33 |
2.94–27.71 |
1.36–26.74 |
| Limiting indices |
h: −6, 3; k: −10, 9; l: −23, 24 |
h: ±10; k: ±17; l: ±17 |
h: ±8; ±12; l: ±19 |
| Reflections collected/unique |
5518/3283 |
12277/6451 |
11587/4411 |
| Goodness-of-fit on F2 |
1.004 |
1.021 |
0.969 |
| Final R indices [I > 2sigma(I)] |
R
1 = 0.0442; wR2 = 0.1042 |
R
1 = 0.0511; wR2 = 0.1183 |
R
1 = 0.0474; wR2 = 0.1024 |
|
R indices (all data) |
R
1 = 0.0678; wR2 = 0.1164 |
R
1 = 0.0967; wR2 = 0.1414 |
R
1 = 0.0845; wR2 = 0.1191 |
| Largest diff. peak and hole (e Å−3) |
0.255; −0.313 |
0.215; −0.264 |
0.215; −0.265 |
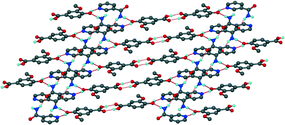 |
| | Fig. 7 Packing diagram of 5 illustrating the hydrogen bonding. | |
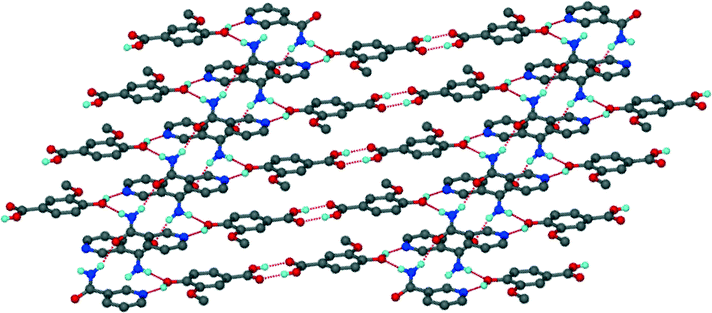
Unlike structure 5 which demonstrates the carboxylic acid homosynthon, the INM co-crystal hydrate displays amide homosynthons, R22(8), connected via R24(8) rings. Two VA and two water molecules are linked to form R44(20) rings. In addition, the –COOH group of VA is involved in –OH⋯N and –CO⋯HN hydrogen bonds to two INM molecules. The water bridged layers are illlustrated in Fig. 8. The layers associate via π⋯π interactions with a minimum distance of 3.669 Å between the centroids of INM and VA molecules.
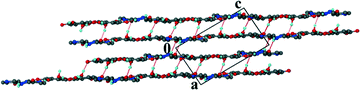 |
| | Fig. 8 Packing diagram of 6 along [010]. | |
The urea co-crystal, 7, also solved in P with two VA and two urea molecules in the asymmetric unit. The structure exhibits the carboxylic acid homosynthon and the amide homosynthon, both rings can be described as R22(8).
The VA dimers stack in layers parallel to [100] displaying π⋯π stacking with the distance between the centroids of VA molecules approximately 3.606 Å. The urea molecules form ribbons along [100], illustrated in Fig. 9 with the carbonyl group of the urea hydrogen bonded to the hydroxyl of a VA molecule. Thus the urea oxygen acts as a trifurcated acceptor to two urea molecules and a VA molecule.
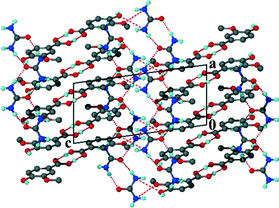 |
| | Fig. 9 Packing diagram of 7 along [010]. | |
The packing for most of the co-crystals and co-crystal hydrates in this study is characterised by hydrogen bonded rings, the exceptions are 1, where there are units comprising one VA and two CAF molecules and chains of VA and ACR molecules as seen in 4. In the two cases where water is included in the structure (3 and 6), the water molecules utilise all of their donor and acceptor sites.
For 3, four water molecules form tetramers, R44(8) rings which in turn act as either a hydrogen bond donor (via (water)OH⋯O(THB)/(water)OH⋯Namide(THB)) or acceptor group (via (water)O⋯HO(VA)) to the acid–amide heterosynthon and for 6, two VA and two water molecules form R44(20) rings. Comparison of the utilisation of the hydrogen bond donor and acceptor groups of the VA molecule revealed that the INM cocrystal hydrate, 6, utilises all of the H-bonding sites for intermolecular interactions. For this structure both VA molecules in the asymmetric unit participate fully in hydrogen bonding, with the methoxy groups in particular acting as H-bond acceptors to water molecules. In the case of the urea co-crystal, 7, there are weak interactions between the N–H bonds of urea and the –OCH3 group with (d(N⋯O) = 3.168 Å, 3.194 Å, 3.376 Å and 3.225 Å). The –OCH3 group also acts as an intramolecular H-bond acceptor to the adjacent –OH of the VA. The geometries of this intramolecular bond, d(O⋯O)/Å, <OH⋯O/° are as follows: 1: 2.691 Å, 109.0°; 2: 2.695 Å, 108.7°; 2.765 Å, 107.3°; 3: 2.682 Å, 108.3°; 4: 2.677 Å, 107.9° and 5: 2.688 Å, 106.2°, 6: none and 7: 2.702 Å, 111.2°. This correlates well with previous studies where it was found that six membered rings had the largest propensity of being involved in intramolecular hydrogen bonding.47 Not surprisingly π⋯π stacking also features prominently due to the aromaticity of VA and the co-crystal formers. There is also variation in the strength of the π⋯π interactions found for the structures, with a distance of 3.546 Å between VA and CAF molecules (1) and 4.470 Å between VA and NAM molecules (5). The hydrogen bond data for all structures is summarised in Table 3.
Table 3 Hydrogen bonding data for all the co-crystals and co-crystal hydrates
| Compound |
Donor (D) |
Acceptor (A) |
D⋯A/Å |
D–H/Å |
H⋯A/Å |
D–H⋯A/° |
|
x, y − 1, z.
1 − x, 0.5 + y, −z − 0.5.
1 − x, y − 0.5, −z − 0.5.
−x, −y − 1, −z + 1.
−x, −y, −z + 2.
−x + 1, −y + 1, −z + 1.
x, y + 1, z.
x − 0.5, 0.5 − y, 0.5 + z.
x − 1, y, z.
1 + x, y, z.
3 − x, 1 − y, −z.
−x, −y + 1, −z.
−x, −y, −z + 1.
−x + 1, −y + 1, −z + 2.
x + 1, y, z + 1.
−x + 1, −y + 2, −z.
x − 1, y, z.
|
|
1
|
O3 |
O7 |
2.802(2) |
0.86(1) |
1.99(2) |
157(1) |
| O2 |
N14 |
2.684(2) |
0.88(2) |
1.83(2) |
163(I) |
|
2
|
O2 |
O5 |
2.644(2) |
0.94(3) |
1.71(3) |
175(2) |
| N16 |
H16 |
2.770(2) |
0.93(1) |
1.85(1) |
172(1) |
| O2A |
N14 |
2.679(2) |
0.93(3) |
1.78(3) |
162(2) |
| O3 |
O6a |
2.679(2) |
0.92(1) |
1.92(1) |
140(1) |
| O3A |
O1Ab |
2.616(2) |
0.92(3) |
1.71(3) |
170(3) |
| C15 |
O3Ac |
3.031(2) |
0.95 |
2.18 |
148 |
|
3
|
O1 |
O8 |
2.652(2) |
0.94(3) |
1.72(3) |
172(2) |
| N17 |
O2 |
2.825(2) |
0.92(2) |
1.91(2) |
172(2) |
| O3 |
O5 |
2.719(2) |
0.88(3) |
1.86(3) |
165(2) |
| O5 |
O7d |
2.802(2) |
0.86(3) |
1.94(3) |
179(3) |
| O5 |
O6e |
2.763(2) |
0.84(3) |
1.93(3) |
172(2) |
| O6 |
N13f |
2.901(2) |
0.90(1) |
2.01(3) |
169(2) |
| O6 |
O5g |
2.868(2) |
0.85(3) |
2.06(3) |
157(3) |
|
4
|
O1 |
N9 |
2.562(2) |
0.97(1) |
1.60(1) |
174(3) |
| O3 |
O2h |
2.647(2) |
0.92(3) |
1.78(3) |
157(3) |
|
5
|
O2 |
O1f |
2.606(2) |
0.84 |
1.78 |
168 |
| N6 |
O3i |
2.973(2) |
0.92(1) |
2.06(1) |
170(1) |
| N6 |
O5j |
2.847(2) |
0.88(1) |
2.00(1) |
162(1) |
| O3 |
N10k |
2.675(2) |
0.91(1) |
1.86(1) |
148(1) |
|
6
|
O1 |
N9 |
2.629(2) |
1.04(1) |
1.60(1) |
169(1) |
| O3 |
O6g |
2.657(2) |
1.00(1) |
1.67(1) |
168(1) |
| O5 |
O4 |
3.051(2) |
0.89(1) |
2.19(1) |
163(1) |
| N16 |
O17Aa |
2.899(2) |
1.00(1) |
1.91(1) |
173(1) |
| N16A |
O17g |
2.931(2) |
0.98(1) |
1.97(1) |
169(1) |
| N16 |
O17Al |
3.000(2) |
0.97(1) |
2.21(1) |
138(1) |
| N16A |
O2 |
3.043(2) |
0.96(1) |
2.09(1) |
171(1) |
| O6 |
O17m |
2.825(2) |
0.99(1) |
1.85(1) |
169(1) |
| O6 |
O4A |
2.982(2) |
0.90(1) |
2.12(1) |
161(1) |
| O5 |
O2An |
2.833(2) |
0.95(1) |
1.89(1) |
175(1) |
| O1A |
N9Ao |
2.604(2) |
1.04(1) |
1.57(1) |
179(1) |
| O3A |
O5a |
2.644(1) |
1.03(1) |
1.65(1) |
159(1) |
|
7
|
O1 |
O2A |
2.615(2) |
0.99(1) |
1.63(1) |
177(1) |
| O1A |
O2 |
2.664(2) |
0.95(1) |
1.72(1) |
174(1) |
| O3 |
O5p |
2.747(2) |
0.83(1) |
1.97(1) |
156(1) |
| O3A |
O6q |
2.619(2) |
0.79(1) |
1.86(1) |
162(1) |
| N9 |
O6 |
2.864(2) |
0.85(1) |
2.02(1) |
172(1) |
| N9 |
O2Aa |
3.192(3) |
0.90(1) |
2.37(1) |
152(1) |
| N11 |
O2Aa |
3.273(3) |
0.86(1) |
2.48(1) |
153(1) |
| N11 |
O6i |
2.871(2) |
0.87(1) |
2.01(1) |
171(1) |
| N12 |
O5j |
2.963(2) |
0.88(1) |
2.10(1) |
169(1) |
| N12 |
O3Af |
3.036(2) |
0.88(1) |
2.24(1) |
150(1) |
| N14 |
O5 |
2.976(2) |
0.90(1) |
2.08(1) |
172(1) |
| N14 |
O3Af |
3.273(3) |
0.88(1) |
2.59(1) |
136(1) |
Thermal analysis
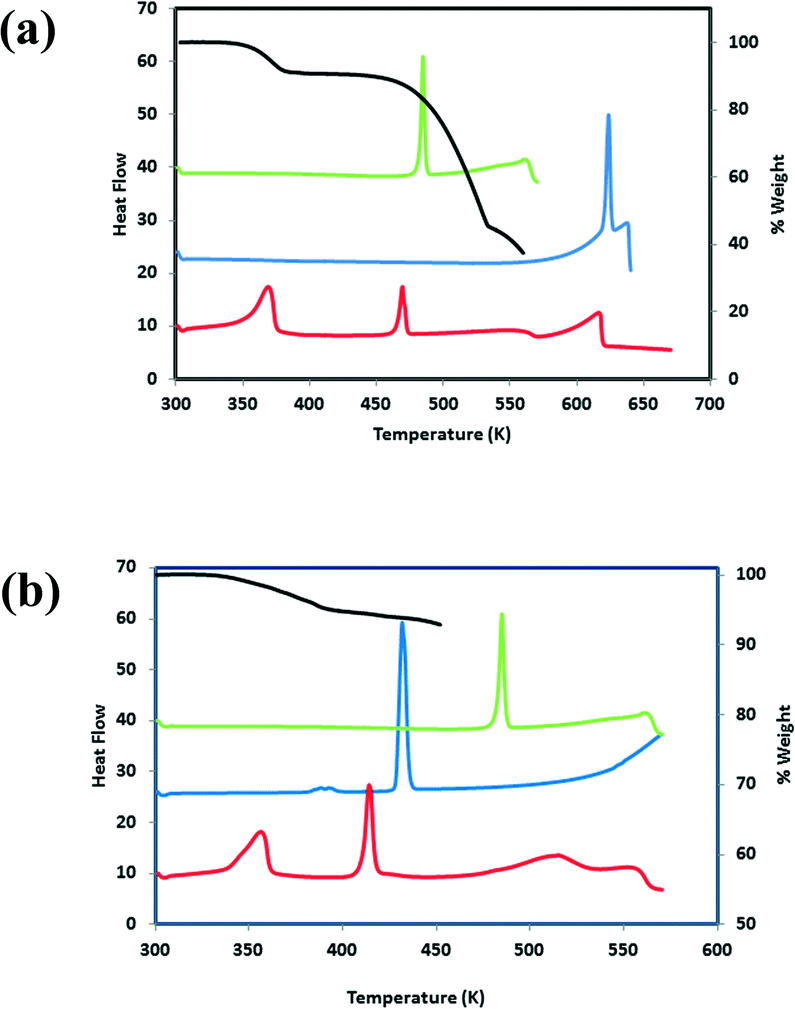
DSC results for VA and the co-crystal formers are summarized in Table 4. All of the co-crystals gave one peak in the DSC corresponding to the melting point (Table 5). The DSC analysis of the co-crystal hydrates showed two peaks, the first corresponding to the loss of water and the second due to the melt.
Table 4 DSC results for vanillic acid and the co-crystal formers
| VA and co-crystal formers |
Endotherm 1 (Tonset, K) |
Endotherm 2 (Tonset, K) |
| VA |
482.3 |
|
| CAF |
419.6 (broad) |
508.4 |
| THP |
547.7 |
|
| THB |
623.7 |
|
| ACR |
363.5 |
375.8 |
| NAM |
401.6 |
|
| INM |
382.1 |
428.4 |
| U |
410.0 |
|
Table 5 DSC results for the co-crystals and co-crystal hydrates
| Compound |
Endotherm 1 (Tonset, K) |
Endotherm 2 (Tonset, K) |
|
1
|
437.7 |
|
|
2
|
457.9 |
|
|
3
|
360.3 |
465.2 |
|
4
|
438.7 |
|
|
5
|
407.6 |
|
|
6
|
348.9 |
410.2 |
|
7
|
435.6 |
|
We note that the melting points for compounds 4–7 fall in-between that of the starting components compared to the melting points for compounds 1–3 which are all lower than that of the starting components. TG analyses for the hydrates were correlated to the stoichiometric ratios found in the asymmetric unit. For 3, the experimental mass loss was 9.3% (calculated mass loss 10.3%) and for 6, the experimental mass loss was 5.0% (calculated mass loss 5.9%). For 3, one of the water molecules participates in three hydrogen bonds, as a hydrogen bond donor to the imidazole nitrogen and a water molecule. It also acts as a hydrogen bond acceptor to another water molecule. The second water molecule is involved in four hydrogen bonds; as a hydrogen bond donor to a carbonyl group of a THB molecule and a water molecule. It also acts as a hydrogen bond acceptor to the hydroxyl group of VA and another water molecule. The TG and DSC curves for 3 and 6 are shown in Fig. 10. We note that even though the water molecules in 3 occupy cavities and are involved in hydrogen bonding, the loss of water (Tonset = 360.3 K, Tpeak = 367.8 K) is observed below 373 K. A similar result was found for 6 where both water molecules participate in hydrogen bonding. For 6, one of the water molecules are linked to three VA molecules whereas the other water molecule connects two VA molecules and one INM molecule, however the loss of water is observed at Tonset = 348.9 K (Tpeak = 354.3 K). Zaworotko et al.43 described the structure and thermal stability of eleven co-crystal hydrates. The study highlighted the general unpredictability of correlating the structure to the thermal stability of hydrates with the observation that channel hydrates have a tendancy to demonstrate low thermal stability.
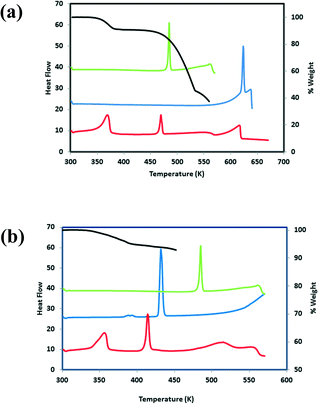 |
| | Fig. 10 (a) DSC plots for 3 (red), VA (green) and THB (blue). TG plot for 3. (b) DSC plots for 6 (red), VA (green) and INM (blue). TG plot for 6 (black). | |
Grinding and slurry experiments
Grinding and slurry experiments were conducted on mixtures of VA and each of the co-crystal formers. For the grinding experiments, molar ratios of VA and each of the co-crystal formers were manually ground using a mortar and pestle. Slurry experiments were conducted using 5–10 ml of solvent added to the known mixture of acid and the co-crystal former. The suspensions were stirred at room temperature overnight. For both types of experiments the powders were analysed using powder X-ray diffraction (PXRD). The results are summarised in Table 6. Co-crystal 1 was prepared after grinding for 45 min and it was also prepared from the slurry experiment. The PXRD spectra for the slurry and the grinding experiments of 2:1 mixtures of VA:THP did not match that of the physical mixture and were also different from the calculated PXRD pattern obtained from LAZYPULVERIX.48 TG and DSC analysis of the powders obtained from the grinding and the slurry experiments revealed that water was present in the structure, thus a hydrate was prepared. The DSC of the hydrate showed two peaks; Ton = 336.3 K due to the loss of water and Ton = 457.6 K due to the co-crystal melt. For 2 the DSC gave one peak (Ton = 457.9 K). The TG showed a mass loss of 6.6% (calc. 5.8%) corresponding to the loss of 2 moles of water. Thus the thermal analysis results confirmed the preparation of the co-crystal dihydrate 2VA·THP·2H2O. Attempts at obtaining single crystals of the dihydrate were unsuccessful. The slurry and neat grinding experiments involving 1:1 mixtures of VA:THB were unsuccessful with the PXRD's for both experiments matching that of the physical mixture. However addition of a few drops of water with grinding resulted in 3. Co-crystal 4 was prepared from the slurry experiment, no reaction occurred in the grinding experiment. Neat grinding of 1:1 molar ratios of VA and INM did not result in reaction but addition of a few drops of water gave the product 5. A slurry experiment involving a 1:1 ratio of VA:ACR in diethyl ether resulted in 6. Co-crystal 7 was successfully prepared from a slurry conversion experiment and from liquid assisted grinding; neat grinding resulted in the physical mixture. In summary we note that neat grinding was not successful for most of the co-crystal preparations. Only co-crystal 1 and the co-crystal dihydrate 2 were prepared using this method. Liquid assisted grinding however did result in co-crystal formation and this is not surprising as the addition of a few drops of solvent with grinding is known to increase the kinetics of a reaction.49 The slurry experiments were mostly successful, however compounds 3 and 5 could not be prepared via this method.
Table 6 Slurry, neat grinding and liquid assisted grinding experimentsa
| Compound |
Slurry |
Neat grinding |
Liquid-assisted grinding |
|
X: the physical mixture/unidentified phase formed, √: same phase as co-crystallisation, −: experiment not performed.
|
|
1
|
√ |
√ |
— |
|
2
|
Co-crystal hydrate |
Co-crystal dihydrate |
— |
|
3
|
X |
X |
√ |
|
4
|
√ |
X |
√ |
|
5
|
X |
X |
√ |
|
6
|
√ |
X |
√ |
|
7
|
√ |
X |
√ |
Conclusion
Vanillic acid displays diversity in its packing arrangements by forming co-crystals with different stoichiometries with the methylated xanthines. VA·2CAF exhibits units with one VA molecule hydrogen bonded to two CAF molecules; weak CH⋯N and CH⋯O contacts exist between individual units. This is in contrast to 2VA·THP and VA·THB·2H2O where the hydrogen bonded ring networks pack to form layers with π⋯π interactions between the layers. The packing for VA·ACR is characterised by continuous chains of VA and ACR molecules with π⋯π interactions between ACR molecules. Differences in stoichiometry and packing are also found when comparing VA·NAM, where the acid homosynthon prevails to 2VA·2INM·2H2O, where the amide homosynthon is present. Also where very weak π⋯π interactions (4.470 Å) are present between VA and NAM molecules there are shorter π⋯π interactions (3.669 Å) between VA and INM molecules. For both hydrates (VA·THB·2H2O and 2VA·2INM·2H2O) it was found that water molecules are incorporated into the hydrogen bonding networks resulting in more complex systems. For 2VA·2U the acid homosynthons stack in columns with the amide homosynthons, forming continuous chains. Thus VA utilises all of the functional groups (carboxylic acid, hydroxyl, methoxy and aromatic) in the different packing arrangements with the methylated xanthines, acridine and the selected amides. This demonstrates the versatility of vanillic acid in forming co-crystals with compounds that contain appropriate donor and acceptor groups. Thus vanillic acid provides an interesting system for co-crystallisation studies due to its range of functional groups.
Acknowledgements
We thank the National Research Foundation (South Africa) and the Cape Peninsula University of Technology for funding.
Notes and references
-
C. B. Aakeröy and P. D. Chopade, Co-crystals: Synthesis, Structure and Applications, Supramolecular Chemistry: From Molecules to Nanomaterials, 2012 Search PubMed.
- A. J. Smith, P. Kavura, K. K. Arora, S. Kesani, J. Tan, M. J. Zaworotko and R. D. Shytle, Mol. Pharmacol., 2013, 10, 2948–2961 CrossRef CAS PubMed.
- K. Biradha, C.-Y. Su and J. J. Vittal, Cryst. Growth Des., 2011, 11, 875–886 CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- C. B. Aakeröy, J. Desper and J. F. Urbina, Chem. Commun., 2005, 2820–2822 RSC.
- R. Dubey and G. R. Desiraju, Chem. Commun., 2014, 50, 1181–1184 RSC.
- S. Long and T. Li, Cryst. Growth Des., 2009, 9, 4993–4997 CAS.
- H. C. S. Chan, G. R. Woollam, T. Wagner, M. U. Schmidt and R. A. Lewis, CrystEngComm, 2014, 16, 4365–4368 RSC.
- B. R. Sreekanth, P. Vishweshwar and K. Vyas, Chem. Commun., 2007, 2375–2377 RSC.
- S. Aitipamula, A. B. H. Wong, P. S. Chow and R. B. H. Tan, CrystEngComm, 2013, 15, 5877–5887 RSC.
- S. Tothadi and G. R. Desiraju, Cryst. Growth Des., 2012, 12, 6188–6198 CAS.
- S. Tothadi and G. R. Desiraju, Chem. Commun., 2013, 49, 7791–7793 RSC.
- S. Tothadi, S. Joseph and G. R. Desiraju, Cryst. Growth Des., 2013, 13, 3242–3254 CAS.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, T. B. Borchardt, L. R. MacGillivray and G. G. Z. Zhang, Mol. Pharmaceutics, 2007, 4, 339–346 CrossRef PubMed.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, L. R. MacGillivray and G. G. Z. Zhang, Cryst. Growth Des., 2009, 9, 1932–1943 Search PubMed.
- N. Schultheiss, M. Roe and S. X. M. Boerrigter, CrystEngComm, 2011, 13, 611–619 RSC.
- A. K. Sinha, U. K. Sharma and N. Sharma, Int. J. Food Sci. Nutr., 2008, 59, 299–326 CrossRef CAS PubMed.
-
http://apps.who.int/food-additives-contaminants-jecfa-database/chemical.aspx?chemID=3298
.
- T. K. Adalder, R. Sankolli and P. Dastidar, Cryst. Growth Des., 2012, 12, 2533–2542 CAS.
- S. Aitipamula, A. B. H. Wong, P. S. Chow and R. B. H. Tan, CrystEngComm, 2012, 14, 8515–8524 RSC.
- C. B. Aakeröy, E. P. Hurley and J. Desper, Cryst. Growth Des., 2012, 12, 5806–5814 Search PubMed.
- K. Suresh, N. R. Goud and A. Nangia, Chem. – Asian J., 2013, 8, 3032–3041 CrossRef CAS PubMed.
- B. Kozlevcar, D. Odlazek, A. Golobic, A. Pevec, P. Strauch and P. Segedin, Polyhedron, 2006, 25, 1161–1166 CrossRef CAS.
-
COLLECT, data collection software, Nonius, Delft, The Netherlands, 1998 Search PubMed.
-
APEX 2, Version 1.0-27, Bruker AXS Inc, Madison, Wisconsin, USA, 2005 Search PubMed.
-
Z. Otwinowski and W. Minor in Methods in Enzymology, Macromolecular Crystallography, ed. C. W. Carter Jr and R. M. Sweet, Academic Press, 1997, part A, vol. 276, p. 307 Search PubMed.
-
SAINT-Plus, Version 7.12, Bruker AXS Inc., Madison, Wisconsin, USA, 2004 Search PubMed.
-
G. M. Sheldrick, SHELX-97: Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Barbour, X-Seed - A Software Tool for Supramolecular Crystallography, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323–338 CrossRef CAS PubMed.
-
Cambridge Structural Database, ConQuest, version 5.35, November 2013 Search PubMed.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2012, 14, 2381–2385 RSC.
- D.-K. Bucar, R. F. Henry, X. Lou, R. W. Duerst, L. R. MacGillivray and G. G. Z. Zhang, Cryst. Growth Des., 2009, 9, 1932–1943 CAS.
- A. K. Mahapatra, P. Sahoo, H. K. Fun and S. Goswami, Asian J. Chem., 2008, 20, 1761–1765 CAS.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, Angew. Chem., Int. Ed., 2001, 40, 3240–3242 CrossRef.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- E. Shefter, T. F. Brennan and P. Sackman, Chem. Pharm. Bull., 1971, 19, 746–752 CrossRef CAS.
- S. Karki, L. Fabian, T. Friščić and W. Jones, Org. Lett., 2007, 9, 3133–3136 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, S. Karki, L. Fabian, T. Friščić, G. M. Day and W. Jones, Chem. Commun., 2010, 46, 2224–2226 RSC.
- N. Madusanka, M. D. Eddleston, M. Arhangelskis and W. Jones, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 72–80 CAS.
- H. D. Clarke, K. K. Arora, H. Bass, P. Kavuru, T. Teng Ong, T. Pujari, L. Wojtas and M. J. Zaworotko, Cryst. Growth Des., 2010, 10, 2152–2167 CAS.
- J. I. Arenas-Garcia, D. Herrera-Ruiz, K. Mondragon-Vasquez, H. Morales-Rojas and H. Hopfl, Cryst. Growth Des., 2010, 10, 3732–3742 CAS.
- A. Lemmerer, D. A. Adsmond and J. Bernstein, Cryst. Growth Des., 2011, 11, 2011–2019 CAS.
- P. Vishweshwar, A. Nangia and V. M. Lynch, CrystEngComm, 2003, 5, 164–168 RSC.
- B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601–2611 CrossRef CAS PubMed.
- K. Yvon, W. Jeitschko and E. Parthé, LAZYPULVERIX, A Computer Program for calculating X-ray and neutron diffraction powder patterns, J. Appl. Crystallogr., 1977, 10, 73–74 CrossRef.
- A. Delori, T. Friščić and W. Jones, CrystEngComm, 2012, 14, 2350–2362 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The thermal analysis results and PXRD results are available in the ESI. CCDC 1022106–1022112 contains the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce01795a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.