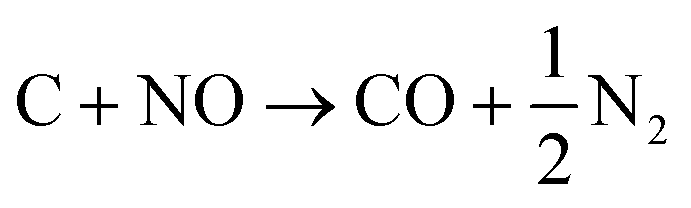Carbon-supported gas-cleaning catalysts enable syn gas methanation at atmospheric pressure†
Received
30th May 2014
, Accepted 20th September 2014
First published on 26th September 2014
Abstract
Though feasible for CO methanations in gas-cleaning applications ([CO] < 1%), carbon-nanotube-supported catalysts have never been implemented for the production of synthetic natural gas. To achieve sufficiently active catalysts, NiO particles were grafted onto multiwalled carbon nanotubes (MWNTs) using a modified incipient wetness impregnation technique that produced particles ranging from 3 to 18 nm in diameter. The reducibility of the deposited NiO particles was studied, as were the thermal stability and surface area of the composites. The materials were then applied as catalysts for CO methanation in syn gas, where CO concentration is greater than 9%, to synthesise synthetic natural gas (SNG) at atmospheric pressure. Ni/MWNTs containing only 13 wt% Ni catalysed the methanation of CO, achieving ~95% CO conversion and ~85% selectivity for CH4. The factors affecting the activity and stability of the composites were studied, and a method to regenerate the Ni/MWNTs catalyst was developed. Following regeneration, the developed catalysts could be reused with small losses in activity and selectivity.
Introduction
Synthetic (or substitute) natural gas (SNG), i.e. synthetic methane (CH4), is generally produced from synthesis gas (a mixture of CO and H2), which can be derived from renewable sources like biomass.1 The promise of the technology lies in the simple integration of SNG into established fuel (i.e. natural gas) distribution networks.1d The major reactions in the production of SNG are the methanation reactions (eqn (1)–(3)),2 with the methanation of CO (eqn (1)) being paramount. eqn (3) occurs primarily under H2-lean conditions.| | | CO + 3H2 ⇌ CH4 + H2O ΔH° = −206 kJ mol−1 | (1) |
| | | CO2 + 4H2 ⇌ CH4 + 2H2O ΔH° = −165 kJ mol−1 | (2) |
| | | 2CO + 2H2 ⇌ CH4 + CO2 ΔH° = −247 kJ mol−1 | (3) |
Since the discovery of methanation, extensive research has been carried out on various catalysts for the reaction, including those based upon Fe, Ru, Ni, Rh, Mo and Co.1d,2a,3 Of these, nickel has been extensively employed1d,2a,4 because of its high activity, high selectivity for methane and relatively low cost. Methanation catalysts have been applied in gas-cleaning processes to lower CO concentrations in H2 streams from ~1–2% to <100 ppm, preparing them for use in PEM (polymer electrolyte membrane) fuel cells and ammonia manufacture, and catalyst lifetimes are in years.2a Despite early efforts,2a,4a,5 significant challenges are still faced during the methanation of CO in synthesis gas, where relatively high CO concentrations (>12%) are common; these include the formation of hotspots within the reactor because of the highly exothermic reactions (eqn (1)–(3)), the sintering of the active metal particles (especially in the case of Ni) and carbon deposition on the catalysts (eqn (4)).2a,6
| | | 2CO ⇌ C + CO2 ΔH° = −172.5 kJ mol−1 | (4) |
Industrial methanations are commonly performed at high pressure (~60 bar) in order to reach a high yield of methane;7 however, the need to compress the gas prior to methanation is considered a major disadvantage of the process.8 Further, several researchers have designed systems that combine the production of syn gas from renewable biomass sources and the subsequent methanation of that syn gas, and these systems generally operate between 1 and 5 bar.9 Finally, the recently reported solar-driven reductions of CO2 to syn gas operate under atmospheric conditions.10 Thus a catalyst that offered both high CO conversion and high CH4 selectivity in methanation at low pressure would enable the more efficient operation of such cleaner-SNG products.
Multiwalled-carbon-nanotube-(MWNT)-supported11 and carbon-nanofibre-supported12 catalysts for CO methanation have been tested in the past. MWNTs are promising supports for methanation catalysts as they are mesoporous, and have high surface areas and good thermal stability.11,12 MWNT-supported Ni catalysts perform as well as those on traditional refractory supports (like Al2O3, SiO2, and MgO), but with lower metal loadings, because they do not form non-reducible mixed nickel oxides during synthesis,13 and because they have a large surface area available to stabilise crystallites. Additionally, their mesoporosity minimises mass-transfer resistance, allowing high gas velocities to be implemented even in reactors that require a low pressure drop across the catalytic bed; this is a definite advantage when highly exothermic reactions are occurring. The excellent thermal conductivity of MWNTs can aid the transfer of excess heat from the reaction bed. Moreover, metal can be recovered from spent catalysts simply by combusting the carbonaceous support in an oxygen-rich environment at moderate temperature (>700 °C). This is not possible with traditional refractory supports (vide supra), which generally require strong acids or bases to leach away the catalyst, thus necessitating post-leach processing operations.14
However, the nanocarbon-supported catalysts that have been tested for CO methanation to date,11,12 including a Ru-based catalyst11 that would be very expensive to implement for SNG synthesis, have only been tested under gas-cleaning conditions, i.e., for CO concentrations <2%. CO methanation at higher CO concentrations presents particular challenges (vide supra), and may pose additional stress on the catalyst. Of particular concern in the case of nanocarbon-based catalysts is the combination of hot-spot formation due the exothermicity of the catalyst, and the formation of stoichiometric amounts of H2O(g), which can etch carbon nanotubes.15 Further, Ni particles can sinter at high temperature.2a Thus these catalysts cannot necessarily be extended to the conditions of synthetic natural gas production, where the concentration of CO is an order of magnitude higher ([CO] > 12% in syn gas). On the other hand, these gas-cleaning catalysts effectively catalyse the methanation of dilute CO at atmospheric pressure. Therefore, if they could still be used at higher concentrations of CO, they would have the potential to produce SNG from uncompressed the syn gas. We therefore sought to determine whether CNT-supported Ni composites could be effective catalysts for SNG synthesis at atmospheric pressure and, if so, what the preferred and limiting process parameters would be. We investigated the feasibility of applying MWNT-supported Ni catalysts, which are active for CO methanation under gas-cleaning conditions, to the synthesis of SNG from syn gas at atmospheric pressure. Various catalysts and process metrics were investigated, as tolerance to feedstock deviations and catalyst durability is extremely important for widespread implementation, and have not yet been investigated for methanation catalysts supported on CNTs. The stability and potential for long-term implementation of the catalyst were also studied. To achieve the latter, the MWNT catalysts had to be regenerated occasionally by applying CO2, which is known to oxidise carbonaceous deposits6a,bvia the reverse of the Boudouard reaction (eqn (4)). Not only is this a relatively mild oxidation treatment, but it would also be convenient in the case of syn gas methanation coupled to a solar-driven CO2 reduction. Then, a simple bypass of the reduction system would provide the CO2 to remove C deposits. In any case, as regeneration with CO2 has not previously been applied to CNT-supported catalysts, the technique was studied in some detail in order to extend the catalyst lifetimes while maintaining performance.
Experimental section
Chemicals
Ethanol (EtOH, Absolute AR, Fronine), Ni(NO3)2·6H2O (>99 wt%, Sigma), H2SO4 (98 wt%, Sigma-Aldrich), and NaOH (98 wt%, Proanalys) were used as received. Gases (Coregas) NO (0.1% in 99.99% Ar), H2 (>99.999%), Ar (>99.999%), CO (25% in 99.995% Ar), He (internal standard, >99.999%), CH4 (>99.99%) and CO2 (>99.9%) were used as received.
Synthesis of MWNT composites
‘MWNTs’ were produced by chemical vapour deposition in a fluidised bed16 and purified using a microwave technique previously described (see ESI,† Section S1 and Fig. S1–S3).17 After purification, the Ni/MWNTs precatalysts, i.e. unreduced nickel-MWNT composites, were prepared by a previously described incipient wetness impregnation method.18 Typically, purified MWNTs (100–200 mg) were dispersed in EtOH (20–40 mL) and sonicated for 30 min using a Branson Sonifier 450 (maximum power output 400 W; output control at 20%) to obtain a homogeneous mixture. The required amount of Ni(NO3)2·6H2O was dissolved in ≤5 mL EtOH and the solution was added to the CNT dispersion, which was then stirred at 90 °C on a hotplate until dry. The resulting catalysts were further dried in a vacuum oven (120 °C, 24 h) and subsequently calcined (10 °C min−1 to 500 °C, hold 3 h) in NO (0.1% in Ar). The precatalysts were labelled 13Ni/MWNTs, where 13 is the theoretical Ni loading following reduction, in wt%. We also synthesised 8Ni/MWNTs precatalysts using this technique; these were intended to more closely mimic reported Ni/MWNTs gas-cleaning catalysts.12
Catalyst characterisation
Samples were examined using thermogravimetric analysis (TGA), transmission electron microscopy (TEM), X-ray diffraction (XRD), Raman spectroscopy (InVia, Renishaw), X-ray photoelectron spectroscopy (XPS), inductively coupled plasma-atomic emission spectrometry (ICP-AES, Vista AX, Varian), and temperature-programmed reduction with H2 (H2-TPR). Analytical methods are described in detail in the ESI† (Section S2).
Steady-state CO methanations
Methanations over the xNi/MWNTs catalysts (70 mg) were carried out in a microreactor (4 mm inner diameter, Fig. S4†). First, the catalysts were reduced in H2 (10% in Ar, 50 sccm (standard cubic centimetres per minute, where standard is defined as T = 25 °C, P = 1 atm), heat at 30 °C min−1 to 550 °C, hold 60 min) before being cooled to the reaction temperature (200 ≤ T ≤ 400 °C) under Ar (65 sccm). These catalysts were denoted xNi/MWNTs-r, where r indicates the application of a reductive treatment. Unless otherwise stated, the reactant stream for methanation was 5 sccm CO, 15 sccm H2, 2 sccm He and 15 sccm Ar, for a total flow of 37 sccm (thus [CO] = 14%; [H2] = 41%). For analysis, samples of the reactant and product gas streams (~13 sccm) were transferred via a heated capillary to the internally heated mass spectrometer. The conversion of CO (χ[CO]) and selectivity to CH4 (S[CH4]) are calculated as:| | 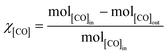 | (5) |
| | 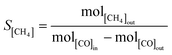 | (6) |
Unless otherwise mentioned, conversion and selectivity were measured after 5 h isothermal operation. Individual flow rates were computed by normalising the signal intensities of each gas (as recorded by the MS (mass units), i[gas]/i[He]) to known mass flow ratios (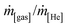 ), where He was used as an internal standard (see Section S3 and Fig. S5 and S6†).
), where He was used as an internal standard (see Section S3 and Fig. S5 and S6†).
Catalyst regeneration
The catalyst could be regenerated in two steps. It was first heated at 550 °C under 10% CO2 in Ar for 40 min, then at the same temperature in 10% H2 in Ar for 30 min. The development of this regeneration procedure is discussed in the section titled “Durability and Regeneration” (vide infra).
Results and discussion
Characteristics of Ni/MWNTs
The N2 adsorption–desorption isotherms of both the MWNTs and 13Ni/MWNTs were IUPAC Type-II (Fig. S7†),19 which demonstrated that the mesoporosity and high surface areas of the MWNTs were maintained throughout all treatments. When the MWNTs were calcined in NO, their surface area increased from 246 to 277 m2 g−1 (Table 1), likely caused by the oxidation of carbons by NO (eqn (7) and (8)),20 which is enhanced at elevated calcination temperatures (>400 °C) and by the presence of oxygenated surface groups in the purified MWNTs (visible by XPS; see Fig. S3†).21| | 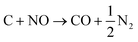 | (8) |
Table 1 Surface, porosity and crystallite properties of purified, NO-treated, and Ni-functionalised MWNTs
| Sample |
S
BET
[cm3 g−1] |
d
pore
[nm] |
V
pore
[cm3 g−1] |
Ni concentration [wt%] |
d
NiOx [nm] |
I
D/IGb |
| XPSc |
ICP |
TEM |
XRDd |
|
Determined from N2 adsorption–desorption isotherms recorded at −196 °C (Fig. S7†). BET surface area (SBET) was measured over P/P0 = 0.05–0.35 on the adsorption curve; average pore diameter (dpore) was calculated via BJH theory, and total pore volume (Vpore) was calculated for P/P0 = 0.995. Note that dpore refers to the diameter of interparticle pores, i.e. those between MWNTs.
I
D/IG was calculated from the Raman spectra (Fig. S8†).
Estimated from detection-sensitivity-adjusted peak areas.
Crystallite size (dNiOx) was calculated using the Scherrer equation (eqn S1†) applied to XRD patterns (Fig. 2). Errors represent the standard deviation of the calculated values for all peaks.
MWNTs were dispersed in EtOH then dried and calcined in NO (0.1% in Ar, 3 h, 500 °C).
Following a 150 h CO methanation at 350 °C in a fixed-bed CVD-type system (used to avoid contamination from quartz wool).
Following a 240 h CO methanation at 350 °C (Fig. 5).
|
| MWNTs |
246 |
16.2 |
2.02 |
— |
— |
— |
— |
0.78 |
| NO-MWNTe |
277 |
29.2 |
2.01 |
— |
— |
— |
— |
1.02 |
| 13Ni/MWNTs |
280 |
21.8 |
1.64 |
11.1 |
11.7 |
3–18 |
4.2 ± 0.5 (NiO) |
1.12 |
| 13Ni/MWNTs-r |
298 |
23.2 |
1.71 |
10.2 |
11.2 |
6–22 |
5.9 ± 0.1 (Ni) |
— |
| Used 13Ni/MWNTs-r |
326f |
21.6f |
1.53f |
9.1f |
10.3f |
7–28g |
10.7 ± 0.3g (Ni) |
1.18g |
The 13Ni/MWNTs had a larger average interparticle pore diameter (21.8 nm) and smaller pore volume (1.64 cm3 g−1) than the purified MWNTs (Table 1), indicating that NiO was deposited within the interparticle pores. The calcination parameters (500 °C in 0.1% NO in Ar) were chosen based upon the results of Sietsma et al.,18 who have shown that this treatment produces smaller metal–oxide nanoparticles than alternative treatments by controlling the decomposition rate of the metal salt. The effect of this atmosphere on the crystallinity of the MWNTs was investigated using Raman spectroscopy (Fig. S8†). All samples showed the two intense Raman peaks (D and G band peaks, Fig. S8†) that are typical of MWNT samples; the intensity ratio of these peaks, ID/IG, indicates the degree of disorder in the walls of the graphitic tubes within MWNT systems. ID/IG was 0.78 for the purified MWNTs, indicating good crystallinity, as expected given the synthesis and purification methods used.17 However, the NO-MWNTs and 13Ni/MWNTs had ID/IG of 1.02 and 1.12 respectively, implying that additional treatment increased the disorder and defects within the MWNT structure. Upon heating in air, the 13Ni/MWNTs decomposed at a lower temperature than the purified MWNTs (Fig. S9, cf. S2†); however, they remained stable to ~400 °C.
Consistent with the Raman spectroscopy results, the XPS spectrum of 13Ni/MWNTs catalysts (Fig. S10–S12†) confirmed the significant presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C sp2 bonds, whose binding energy (BE) of 284.5 eV was characteristic of graphitic carbon–carbon bonds in MWNTs.22 Deconvolution of the C1s core-level spectrum (Fig. S11†) also indicated the presence of sp3 C–C bonds (defects), as well as both sp3 and sp2 carbons bonded to oxygen, (
C sp2 bonds, whose binding energy (BE) of 284.5 eV was characteristic of graphitic carbon–carbon bonds in MWNTs.22 Deconvolution of the C1s core-level spectrum (Fig. S11†) also indicated the presence of sp3 C–C bonds (defects), as well as both sp3 and sp2 carbons bonded to oxygen, (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C*–O, BE = 286.5 eV; >C*O, BE = 287.7 eV). The sp3 carbons and C–O bonds were likely introduced during MWNT purification, as this is known to occur when MWNTs are treated with concentrated acid (5 M H2SO4) at high temperatures (>200 °C).23 These defect/oxygenated surface sites are desirable, as they increase the hydrophilicity of the MWNTs, thus facilitating their dispersion in solvents, and also provide preferential nucleation sites for the Ni ions.24 However, the high surface concentrations of these oxygenated groups that can be achieved by MWNT oxidation in extremely harsh conditions, like refluxing in conc. HNO3, are detrimental to structural stability,25 and were thus avoided. The high-temperature calcination of 13Ni/MWNTs in NO may also have contributed to their surface oxygen content, but did not introduce any nitrogen-based functionalities that were detectable by XPS (normally found around 400 eV, Fig. S10†). Transmission electron microscope (TEM) images of 13Ni/MWNTs showed a uniform coverage of discrete particles, presumably NiO, on the MWNT surface (Fig. 1a), despite the high target loadings of NiO. Some large particles and regions of agglomerations (Fig. 1b and S13a†) did exist, and likely reflected an incomplete/uneven dispersion of the MWNTs in the Ni(NO3)2 salt solution. On the 13Ni/MWNTs, from 200 randomly chosen particles in the TEM images (Fig. 1 and S13†), particle diameters ranged from 3 to 18 nm (Table 1); these small particles were likely formed due to the impregnation solvent (EtOH)26 and calcination parameters (0.1% NO, 500 °C)18 used. Despite that the 13Ni/MWNTs were vigorously tip-sonicated during TEM sample preparation, no free nanoparticles were observed, attesting to the strong attachment of the metal-oxide particles to the MWNTs.
C*–O, BE = 286.5 eV; >C*O, BE = 287.7 eV). The sp3 carbons and C–O bonds were likely introduced during MWNT purification, as this is known to occur when MWNTs are treated with concentrated acid (5 M H2SO4) at high temperatures (>200 °C).23 These defect/oxygenated surface sites are desirable, as they increase the hydrophilicity of the MWNTs, thus facilitating their dispersion in solvents, and also provide preferential nucleation sites for the Ni ions.24 However, the high surface concentrations of these oxygenated groups that can be achieved by MWNT oxidation in extremely harsh conditions, like refluxing in conc. HNO3, are detrimental to structural stability,25 and were thus avoided. The high-temperature calcination of 13Ni/MWNTs in NO may also have contributed to their surface oxygen content, but did not introduce any nitrogen-based functionalities that were detectable by XPS (normally found around 400 eV, Fig. S10†). Transmission electron microscope (TEM) images of 13Ni/MWNTs showed a uniform coverage of discrete particles, presumably NiO, on the MWNT surface (Fig. 1a), despite the high target loadings of NiO. Some large particles and regions of agglomerations (Fig. 1b and S13a†) did exist, and likely reflected an incomplete/uneven dispersion of the MWNTs in the Ni(NO3)2 salt solution. On the 13Ni/MWNTs, from 200 randomly chosen particles in the TEM images (Fig. 1 and S13†), particle diameters ranged from 3 to 18 nm (Table 1); these small particles were likely formed due to the impregnation solvent (EtOH)26 and calcination parameters (0.1% NO, 500 °C)18 used. Despite that the 13Ni/MWNTs were vigorously tip-sonicated during TEM sample preparation, no free nanoparticles were observed, attesting to the strong attachment of the metal-oxide particles to the MWNTs.
 |
| | Fig. 1 (a, b) Transmission electron micrographs (TEM) of 13Ni/MWNTs. | |
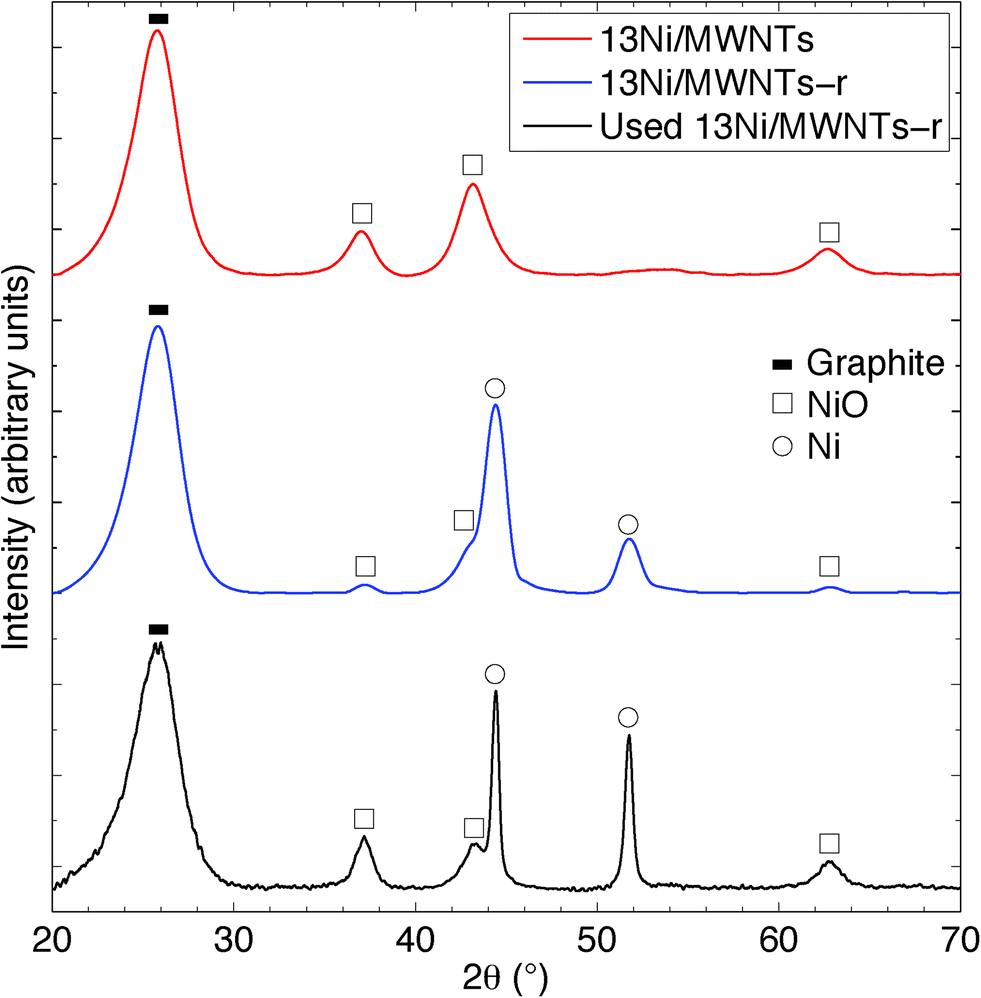
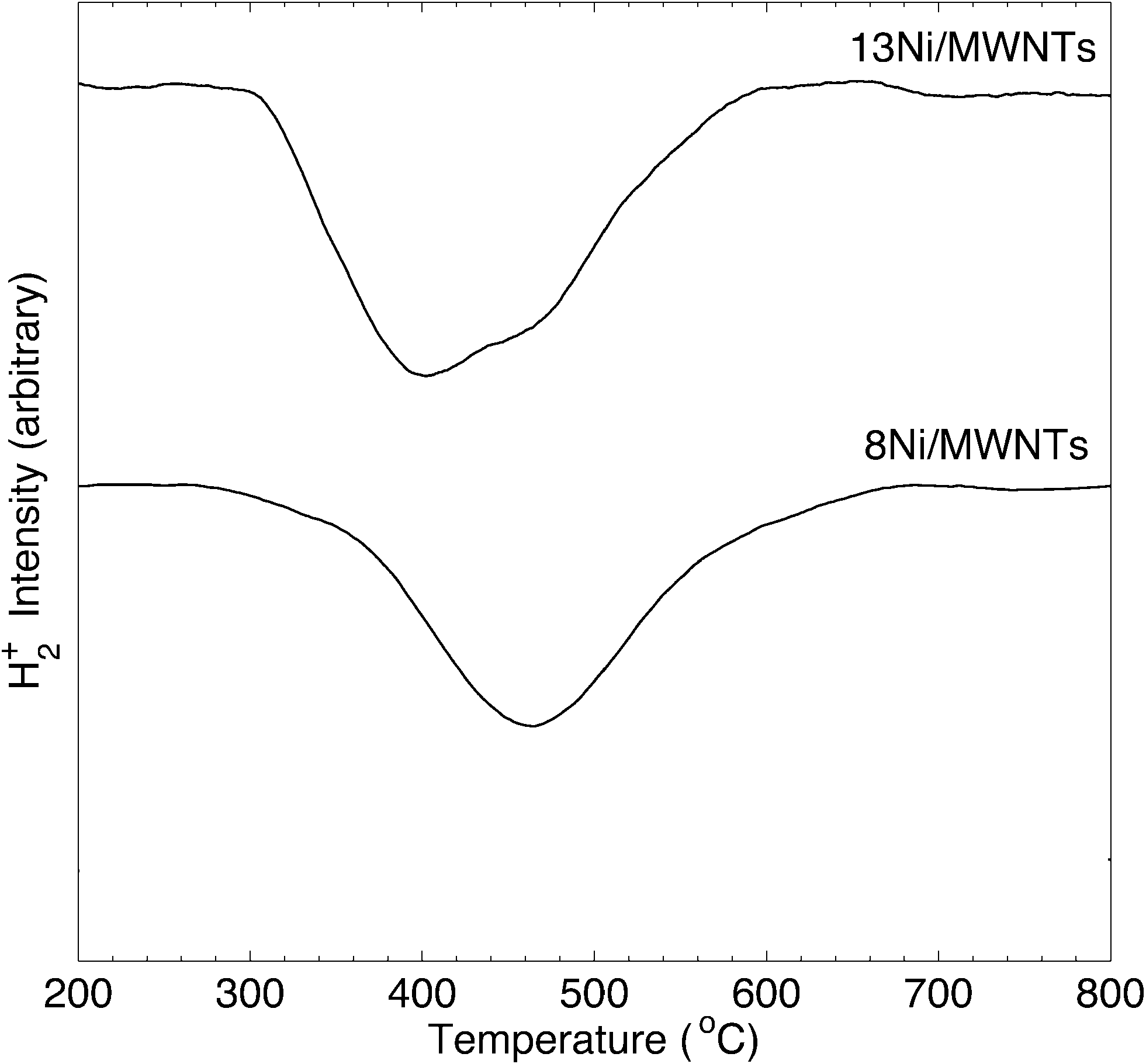
The crystallite structure of the deposited particles was investigated using X-ray diffraction (XRD, see Fig. 2, S14, and S15†). The XRD pattern (Fig. 2) of as-synthesised 13Ni/MWNTs confirmed the presence of NiO (JCPDS 47-1049), with diffraction peaks at 37.1, 43.5 and 62.9° assigned to the (111), (200), and (001) diffraction planes, respectively. The average crystallite size, calculated using the Scherrer equation (eqn S1†), was 4.2 ± 0.5 nm (Table 1), within the range of sizes measured under the TEM (Fig. 1 and S13†). We note, however, that intensity contributions from the MWNT (100) plane (at 43.5°, Fig. S15†) could broaden the peak at 43.5° and contribute to errors in calculated crystallite sizes. X-ray photoelectron spectra (XPS) of fresh 13Ni/MWNTs (Fig. S12†) showed a peak at 856.5 eV, corresponding to the 2p3/2 peak of Ni2+, similar to the value reported for NiO.27 No peak for Ni0 (BE (Ni 2p3/2) = 852.8 eV27) was observed on the as-synthesised 13Ni/MWNTs. The reduction of this precatalyst in H2 (3% in Ar, dT/dt = 10 °C min−1) was studied using mass spectrometry (Fig. 3); for these experiments, H2 concentration had to be limited to 3% (in Ar) so that changes in the H2+ signal could be clearly observed. MWNT-supported NiO samples typically show a single reduction peak, which is assigned to the reduction of Ni2+ in NiO to Ni0.26,28
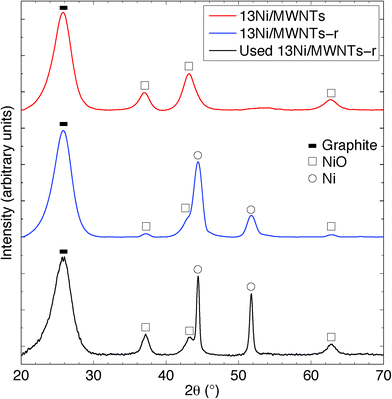 |
| | Fig. 2 X-ray diffraction patterns of 13Ni/MWNTs, 13Ni/MWNTs-r, and 13Ni/MWNTs-r after use (350 °C, reaction time = 240 h). Indicated phases are: (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) the graphite structure (002) plane of MWNTs at 26.5°; (□) NiO (JCPDS 47-1049) at 37.1° (111), 43.5° (200) and 62.9° (001); and (○) Ni (JCPDS 45-1027) at 44.8° (111) and 51.8° (200). ) the graphite structure (002) plane of MWNTs at 26.5°; (□) NiO (JCPDS 47-1049) at 37.1° (111), 43.5° (200) and 62.9° (001); and (○) Ni (JCPDS 45-1027) at 44.8° (111) and 51.8° (200). | |
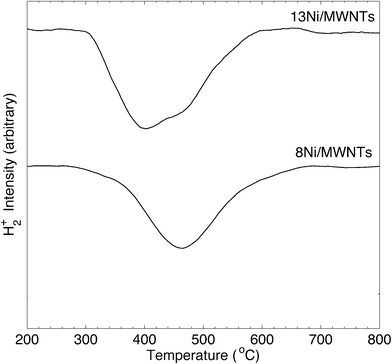 |
| | Fig. 3 H2-TPR of xNi/MWNTs. Analysis conditions: ~50 mg sample, 3% H2 in Ar, 60 sccm, heating at 10 °C min−1 over 200–800 °C, MS sampling at 15 scans min−1. | |
The reduction of NiO on 13Ni/MWNTs started at ~300 °C and peaked at ~400 °C. A shoulder was visible at ~460 °C, and the H2 consumption tailed off at ~600 °C. However, the precatalysts were fully reduced under the conditions used prior to catalysis (60 sccm, 10% H2 in Ar, 550 °C, 1 h), as confirmed by H2-TPR of the reduced catalyst (Fig. S16†). Further, the reduction of NiO to Ni was validated by XRD analysis (Fig. 2). The XRD pattern of the reduced catalyst 13Ni/MWNTs-r contained distinct peaks at 44.8 and 51.8°, which were assigned to the (111) and (200) diffraction planes of Ni0 (JCPDS 47-1049), respectively; still, small peaks at 37.1, 43.5 and 62.9° (attributed to NiO and possibly MWNTs, vide supra) were present. This could indicate incomplete reduction; however, given that the reduced 13Ni/MWNTs-r sample could not be further reduced (Fig. S16†), any unreduced NiO was inaccessible. On the other hand, we cannot discount the formation of NiO on exposure to atmosphere prior to and during XRD analysis. Reduction of the Ni/MWNTs produced Ni particles larger than the initial NiO particles, as evidenced by the sharper Ni peaks in the XRD patterns (Fig. 2). Previous reports have suggested that Ni particles can sinter due to the migration and coalescence of any weakly bound particles during reduction,27 and this may also have occurred in our system. Finally, consistent with the XRD results, the XPS spectrum of the 13Ni/MWNTs-r (Fig. S12†) showed a strong Ni0 2p3/2 peak at 852.8 eV,27 corresponding to the peak of Ni0, as well as a weaker peak for Ni2+ at 856.2 eV.
Methanation
The xNi/MWNTs composites were tested as catalysts for CO methanation using a plug-flow microreactor and a range of temperatures and conditions. Control experiments revealed that none of the MWNTs, NO-MWNTs or H2-treated NO-MWNTs catalysed significant CO methanation (Fig. S18†). Residual impurities in the MWNTs (Fig. S2†), derived from the Fe/Al2O3 catalyst used to produce them, may be weak methanation catalysts, and likely contributed to the small background reaction rate (Fig. S18†). Additionally, unreduced 13Ni/MWNTs (i.e. prior to reduction) also showed some methanation activity (Fig. S18†); H2 from the reactant stream likely reduced small amounts of NiO to Ni at the methanation temperatures, allowing it to catalyse CO methanation.
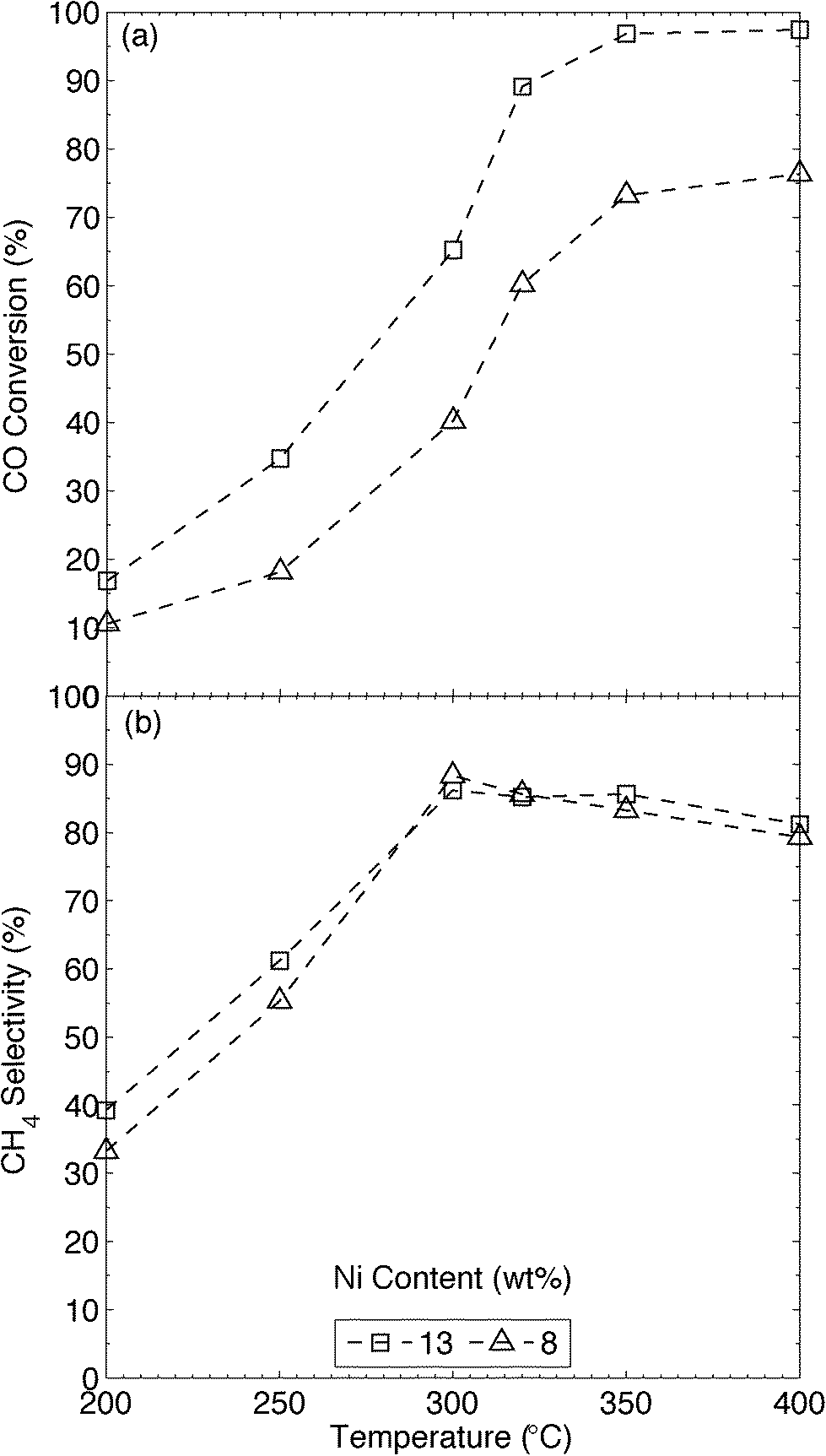
Temperature is critical in the methanation reaction, and we thus tested the MWNT-supported catalysts at 50 °C intervals over the applicable range (200–400 °C).1d,6c,11,29 All catalytic tests were carried out above 200 °C in order to avoid the generation of tetracarbonylnickel (Ni(CO)4),29e which is formed from CO and metallic Ni but decomposes above 160 °C, even in the presence of CO.30 The upper CO methanation temperature tested was limited by the supports themselves, as the graphitic and amorphous carbons of the MWNTs are hydrogenated, albeit slowly, by H2 at temperatures above 450 °C (Fig. S16† and ref. 31). In any case, a low reaction temperature is desirable for catalytic CO methanation; the high reaction rates generated over catalysts containing Ni and Ru produce large amounts of heat, which can cause hotspots to form in the reactor; these can cause thermal stresses and sintering in the catalyst bed.29e,32 To minimise these issues, and to reduce the need for complex heat-transfer systems (e.g. heat exchangers) in an industrial setting, the reaction temperature must be minimised, with H2/CO ratios as high as possible. Further, equilibrium methane yields are severely affected by reaction temperature (see Fig. S19† and ref. 29b and c); thus, theoretical CH4 yields are highest at lower operating temperatures.29e,32 An optimum temperature of 350 °C has been reported for the methanation of dilute CO ([CO] = 1.25 vol%)12 over Ni-CNT (7.8 wt%) catalysts. Thus first we compared our catalysts (8Ni/MWNTs), which produced CO conversions (Fig. 4) similar to those observed by Zhang et al.12 over Ni-CNT, despite that we used [CO] ~ 14 vol%. In comparison, CO methanation over 13Ni/MWNTs-r (Fig. 4) substantially increased at 300 °C and maximum CO conversion (~95%) was reached at 350 °C, with high CH4 selectivities (~85%) being obtained at or above 300 °C. These CO conversions, and the selectivity to CH4 measured over the 13Ni/MWNTs-r, were close to the expected equilibrium values reported by Gao and co-workers;29c the inclusion of the Ar carrier gas (keeping the total pressure at 1 atm) in the thermodynamic calculations did not significantly change the predicted equilibrium CO conversions or CH4 selectivities in the temperature range studied here (200–400 °C, Fig. S19†). No higher Ni loadings were tested, as the reaction was very near thermal equilibrium over 13Ni/MWNTs-r at 350 °C. Additionally, increasing the Ni content on the MWNTs from 8 to 13 wt% did not cause CO conversion to peak at a lower temperature (within the examined range), contrary to the case for aluminate-supported catalysts.29b,33 The main byproduct detected from CO methanation over 13Ni/MWNTs-r was CO2, with negligible amounts of higher-order hydrocarbons being detected, in agreement with previous reports.29b,34 At lower temperatures (<300 °C), less CO was consumed (CO conversion ≤~35%), and selectivity to CH4 suffered (~60%). The reaction products under these conditions were not investigated.
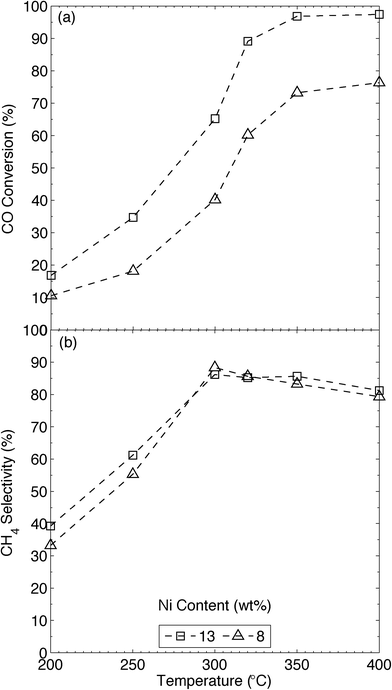 |
| | Fig. 4 (a) CO conversion and (b) CH4 selectivity of CO methanation over xNi/MWNTs-r, as functions of temperature. Experimental details: catalysts (70 mg) were reduced (550 °C, 10% H2 in Ar, 50 sccm, 60 min) and cooled (Ar, 65 sccm) to reaction temperature prior to introducing the reactants (13.5% CO, 41% H2, 2 sccm He and balance Ar, total flow = 37 sccm). Values reported are after 5 h isothermal operation. | |
For comparison, we synthesised 13Ni/γ-Al2O3 catalysts, and tested them under the same conditions used for the Ni/MWNT catalysts (Fig. S18†). The highest CO conversion over these catalysts was only 60%, and occurred at the highest temperature tested (400 °C); no maximum in CO conversion occurred over the 13Ni/γ-Al2O3 catalysts below 400 °C (recall that 13Ni/MWNTs yielded ~95% conversion at 350 °C). The greater activity of the Ni/MWNT catalysts was likely due to a higher proportion of available Ni0 particles at the same total nickel loading; in comparison, Al2O3-supported Ni catalysts contain significant amounts of less-reducible nickel aluminates that do not catalyse CO methanation.29b,33 In addition to the optimum methanation temperature, system metrics like weight-hourly space velocities (WHSVs, in standard cubic centimetres per gram of catalyst per hour) and H2/CO ratios (HCOR) were investigated to ascertain the boundary operating conditions for the 13Ni/MWNTs-r catalysts (Section S14†). These metrics can easily be altered in an industrial setting by increasing the amount of catalyst used and by applying a water-gas shift reactor, respectively; nevertheless, in our system, acceptable rates of CO methanation were found at WHSVs ≤ 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scc gcat−1 h−1 (Fig. S20†) and HCOR ≥ 3 (Fig. S21†).
000 scc gcat−1 h−1 (Fig. S20†) and HCOR ≥ 3 (Fig. S21†).
Durability and regeneration
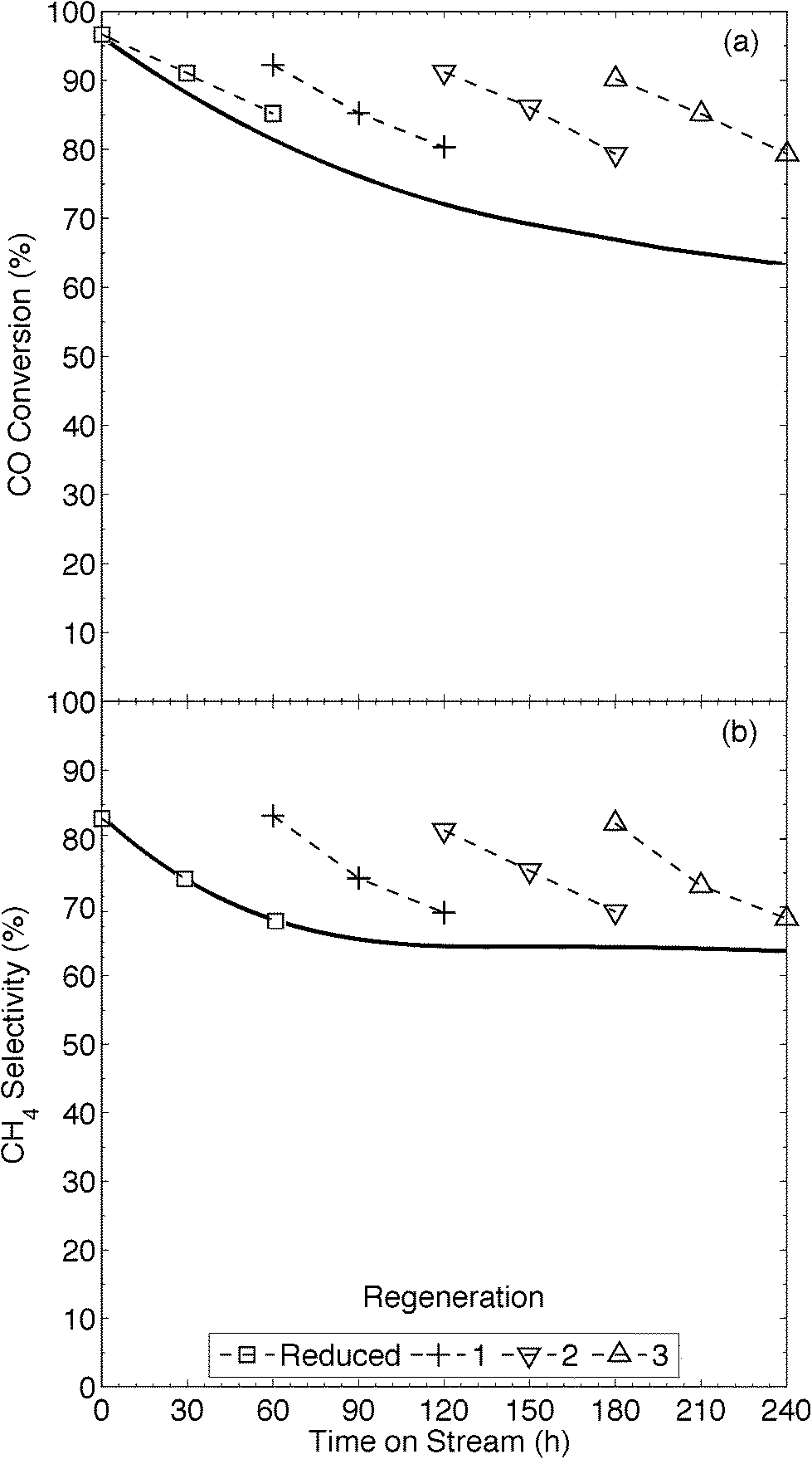
The XRD pattern (Fig. 2) of our 13Ni/MWNTs-r after use (350 °C, 240 h) showed no evolved phases, although the peaks at 37.1, 43.5, and 62.9° did narrow, indicating an increase in the average Ni crystallite size from 5.9 nm for fresh catalysts to 10.7 nm for used catalysts. Although the increase in particle size after a 240 h use pointed to sintering and thus a decrease in the number of active sites, the used catalysts were still active (Fig. 5). Over the 240 h run, the pressure measured upstream of the catalyst bed increased from 15.1 to 21.8 psi, and this change can only be attributed to morphological changes in the catalysts. Two types of changes were possible: (1) Ni particle agglomeration, which indeed occurred (see above), and (2) carbon deposition (eqn (4), (9), and (10)).
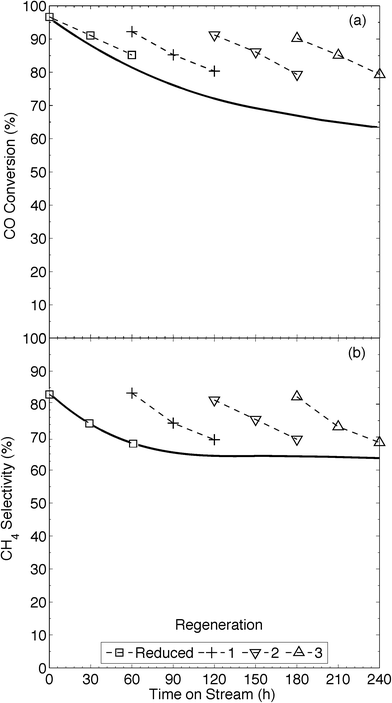 |
| | Fig. 5 Impact of time on stream during continuous operation (solid line, —) and operation with periodic regeneration (dashed lines, – – –) on (a) conversion and (b) CH4 selectivity of CO methanation over 13Ni/MWNTs-r at 350 °C. Experimental conditions: catalysts (70 mg) were reduced (550 °C, 10% H2 in Ar, 50 sccm, 60 min) and cooled (Ar, 65 sccm) to 350 °C prior to introducing the reactants (13.5% CO, 41% H2, 2 sccm He and balance Ar, total flow = 37 sccm). In situ regeneration: step 1: 10% CO2 in Ar, 40 min, 550 °C; step 2: 10% H2 in Ar, 0.5 h, 550 °C, at 60 h intervals. | |
On Ni-based catalysts, carbon deposition occurs as adsorbed atomic carbon (Cα), polymeric carbon films (Cβ), and NixCy,5,6,35 and is mostly formed via the Boudouard reaction (eqn (4)). In fact, Bartholomew et al.6a reported that carbon deposition happened more readily on smaller Ni particles; thus, a balance must be struck between the high catalytic activity of small, dispersed Ni nanoparticles and their consequent susceptibility to coking. Coking lowers on-stream times, negating gains in CO conversion and CH4 selectivity. At the temperatures used here, coking would be expected to form reactive, amorphous carbon forms, like adsorbed (Cα) and polymeric (Cβ) carbon.6a,36 Filamentous carbon formation has been shown to occur at temperatures >450 °C, and its formation can deactivate a catalyst by encapsulating Ni and by plugging the pores of a support, restricting access of the reactants to catalyst crystallites within the pores.6a,37 Further, previous studies have reported the distinct formation of filamentous C, platelet graphite and disordered amorphous C during the dissociation of CO on Ni crystals.6a,38 To investigate the morphological changes in our catalysts, we required a large, quartz-free sample of the catalyst after use. This was not available from the flow-through reactor, in which the quartz wool used to hold the sample in place became entrained in the catalyst, so the 13Ni/MWNTs-r were applied in a fixed-bed reactor having a conformation akin to a chemical vapour deposition (CVD) apparatus and a ceramic plate to hold the catalyst. After a 150 h use in this reactor, the catalysts had SBET = 326 m2 g−1, significantly higher than that of the reduced catalysts, with little change in the average pore size and pore volume (Table 1). The rise in specific surface area is indicative of carbon deposition. Also, Raman analysis of the used catalysts pointed to a possible increase in disordered carbon, seen as a slight increase in the ID/IG (Table 1). However, the slight increase may not be representative, as the significant presence of graphitic carbon could mask the extent of amorphous carbon deposited. Neither post-reaction TGA (Fig. S22†) nor XRD (Fig. 2) permitted the type of deposited C to be identified either. Further, when the reactor and catalyst were weighed, used to catalyse CO methanation for 48 h at 350 °C, and then dried under flowing Ar (40 sccm, 300 °C, 1 h) and weighed again, the weight of the catalyst bed had increased by ~5%. Whereas the mesoporous nature of the catalysts was not significantly impacted by the deposition of carbon, CO methanation performance was affected. Based upon the weight gain, increased surface area, and slight change in MWNT crystallinity (Raman) that occur during methanation over 13/Ni/MWNTs-r, the carbon balance of the reaction was <1; that is, some carbon from CO was deposited on the catalyst rather than converted to CH4.
The industrial application of these 13Ni/MWNTs-r catalysts will depend on their potential for high activity over long times on-stream; their deactivation by carbon deposition is thus problematic. Although steam and air are generally preferred for the oxidation of deposited carbon,5,6 such treatments would severely affect these MWNT-supported Ni catalysts, severely oxidising Ni and possibly the MWNTs. The oxidation of carbon supports have caused Ni crystallites to be lost.6a CO2 oxidises carbonaceous deposits via the reverse of the Boudouard reaction (eqn (4))6a,b and, although this reaction occurs more slowly than oxidation by H2O(g) or O2, it permits the gasification of more reactive (amorphous) carbon deposits without destroying the graphitic supports, and with minimal oxidation of the metallic Ni crystallites.39 We therefore explored regeneration with CO2. TGA of the 13Ni/MWNTs under CO2 showed them to be stable up to ~620 °C (Fig. S23†), lower than the ~780 °C value for NO-treated MWNTs (Fig. S23†); this difference was likely due to catalytic contributions from the deposited Ni nanoparticles. After ‘regeneration’ with CO2 (10% in Ar for 40 min at 550 °C), the 13Ni/MWNTs-r proved less active for CO methanation (χCO = 70%, Fig. S24†) than the fresh 13Ni/MWNTs-r, suggesting that some Ni was oxidised by CO2 (eqn (11)).35b However, the CO2-treated catalysts retained significantly higher CO methanation activity than the unreduced 13Ni/MWNTs (Fig. S18†).
Thus, although CO2 treatment is not as severe as air treatment, and is thus less prone to oxidise Ni0, a short reduction step was still required to completely restore CO methanation activity of the CO2-treated catalysts. These were subjected to H2 (10% in Ar, 0.5 h, 550 °C) prior to reuse as CO-methanation catalysts. In addition to reducing nickel oxides, H2 can, at temperatures >420 °C, hydrogenate carbons that have been deposited by the Boudouard reaction, at least on Al2O3-supported Ni,6b,38 so we also tested H2 as a sole regeneration agent. H2 reacts with carbon deposits on Ni/Al2O3 more slowly than steam does,40 so longer exposures to H2 are needed to gasify C deposits. We found that extended exposure of 13Ni/MWNTs to H2 (10% H2 in Ar, 5 h, 550 °C) caused significant coalescence of the Ni0 nanoparticles, increasing the overall crystallite size (Fig. S25†), and we therefore selected the tandem CO2/H2 regeneration procedure, in which the H2 treatment is brief because it is only designed to reduce Ni, but not to gasify C, for further study. 13Ni/MWNTs-r catalysts that had been used for catalysis, then reactivated in CO2 and then H2 were only slightly less active and selective catalysts in subsequent CO methanations (Fig. 5). Thus, it is likely that the decrease in activity observed during the extended methanation was caused entirely by carbonaceous intermediates and byproducts, and could therefore be reversed by the treatment used. The regeneration methodology increased the on-stream time, with χCO = 80%, from 60 to 240+ h (Fig. 5).
Conclusions
Ni-functionalised MWNTs had been used as CO-methanation catalysts under gas-cleaning conditions, but had not been tested in syn gas, where [CO] is much higher. Thus, Ni/MWNTs were produced using an incipient wetness impregnation technique with Ni(NO3)2·4H2O as the Ni precursor and dilute NO (in Ar) as the calcination atmosphere; this treatment gives smaller NiO nanoparticles on MWNTs than traditional impregnation techniques. The Ni/MWNT composites bore well-dispersed NiO nanoparticles 3–18 nm in diameter.
A detailed study of the applicability of these Ni/MWNTs to CO methanation in syn gas at atmospheric pressure was carried out, with a focus on long-term stability and reusability. The reduced Ni/MWNT composites catalysed CO methanation to give high conversion (~95%) and good selectivity to CH4 (~80%) over a long reaction period (>24 h). Clearly, the exothermic reactions being catalysed did not induce catalyst deactivation; it is likely that the high surface area and mesoporous structure of the MWNTs, which would have reduced mass-transfer resistance, protected against catalyst damage due to hot-spot formation. Further, the thermal stability of the MWNTs may have minimised the sintering of the small active Ni nanoparticles. However, carbon deposition did occur under the investigated conditions, and significantly deactivated the catalyst. Thus, a CO2-based treatment, not previously reported for MWNT-supported methanation catalysts, was developed to gasify the deposited carbon and regenerate the catalysts. Not surprisingly, the CO2 gasification oxidised some of the Ni0 to NiO, which necessitated a short reduction with H2 to complete the catalyst regeneration. Notably, the byproducts of the CO2-based treatment were mostly small amounts of unreacted CO2 with CO, which could be recycled into the feed stream. The catalytic activity, stability and reusability of the Ni-loaded heterogeneous catalysts are promising for use in SNG synthesis from syn gas under milder conditions, including at atmospheric pressure. This syn gas can be generated from biomass, and has recently been generated from the solar-driven reduction of CO2.10a–c The catalysts were applicable at high CO concentrations (~14%) and WHSVs (32000 scc gcat−1 h−1). Future work on this system will endeavour to lower the amount of Ni required for a successful reaction, and to test selective CO methanation in the significant presence of CO2. Also, the losses of catalytic activity after multiple regenerations, likely caused by carbon deposition and sintering, will need to be studied in some detail in order to ascertain the recyclability and possible long-term implementation of the catalyst.
Acknowledgements
M.F.V is grateful to the University of Sydney and the Australian Research Council for funding this research. N. N. is grateful for the financial support by CSIRO-National Research Flagships Program for providing the Postgraduate Scholarship (Future Manufacturing Flagship). The authors thank the Australian Centre for Microscopy & Microanalysis (ACMM), University of Sydney for TEM facilities and to Mr. V. Lo for his assistance with TEM analysis, to Dr. L. Carter from the School of Chemistry, University of Sydney, for her assistance with Raman spectroscopy analysis, to Dr. B. Gong from the University of New South Wales Surface Analytical Centre for his assistance with XPS analysis, to S. Billington from MKS Spectra for his assistance with MS set-up, and to Dr. J. Shi from the School of Chemical and Biomolecular Engineering, University of Sydney, for assistance with N2 adsorption measurements and ICP analysis. The authors are also grateful to Dr. A. Abbas for his assistance in thermodynamic equilibrium simulations.
Notes and references
-
(a) P. Jaramillo, W. M. Griffin and H. S. Matthews, Environ. Sci. Technol., 2007, 41, 6290–6296 CrossRef CAS;
(b) J. P. Longwell, E. S. Rubin and J. Wilson, Prog. Energy Combust. Sci., 1995, 21, 269–360 CrossRef CAS;
(c) M. H. Waldner and F. Vogel, Ind. Eng. Chem. Res., 2005, 44, 4543–4551 CrossRef CAS;
(d) J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Fuel, 2010, 89, 1763–1783 CrossRef CAS PubMed.
-
(a) G. A. Mills and F. W. Steffgen, Catal. Rev.: Sci. Eng., 1974, 8, 159–210 CrossRef;
(b) P. Sabatier and J. B. Senderens, C. R. Seances Acad. Sci., Vie Acad., 1902, 134, 514–516 CAS.
-
(a) M. A. Vannice, J. Catal., 1975, 37, 449–461 CrossRef CAS;
(b) G. A. Somorjai, Catal. Rev.: Sci. Eng., 1981, 23, 189–202 CrossRef CAS.
-
(a) A. Rehmat and S. S. Randhava, Ind. Eng. Chem. Prod. Res. Dev., 1970, 9, 512–515 CrossRef CAS;
(b) M. Krämer, M. Duisberg, K. Stöwe and W. F. Maier, J. Catal., 2007, 251, 410–422 CrossRef PubMed;
(c) H. Habazaki, M. Yamasaki, B. P. Zhang, A. Kawashima, S. Kohno, T. Takai and K. Hashimoto, Appl. Catal., A, 1998, 172, 131–140 CrossRef CAS;
(d) S. Takenaka, T. Shimizu and K. Otsuka, Int. J. Hydrogen Energy, 2004, 29, 1065–1073 CrossRef CAS PubMed.
- A. D. Moeller and C. H. Bartholomew, Ind. Eng. Chem. Prod. Res. Dev., 1982, 21, 390–397 CrossRef CAS.
-
(a) C. H. Bartholomew, Catal. Rev.: Sci. Eng., 1982, 24, 67–112 CrossRef CAS;
(b) D. L. Trimm, Catal. Rev.: Sci. Eng., 1977, 16, 155–189 CrossRef CAS;
(c) J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Chem. Eng. Sci., 2011, 66, 924–934 CrossRef CAS PubMed.
-
M. Sudiro and A. Bertucco, in Natural Gas, ed. P. Pótócnik, Sciyo, 2010, 978-953-307-112-1, Intech Search PubMed.
- A. Duret, C. Friedli and F. Maréchal, J. Cleaner Prod., 2005, 13, 1434–1446 CrossRef PubMed.
-
(a) I. Aigner, C. Pfeifer and H. Hofbauer, Fuel, 2011, 90, 2404–2412 CrossRef CAS PubMed;
(b) T. Gröbl, H. Walter and M. Haider, Appl. Energy, 2012, 97, 451–461 CrossRef PubMed;
(c) M. C. Seemann, T. J. Schildhauer and S. M. A. Biollaz, Ind. Eng. Chem. Res., 2010, 49, 7034–7038 CrossRef CAS.
-
(a) B. Kumar, J. M. Smieja, A. F. Sasayama and C. P. Kubiak, Chem. Commun., 2012, 48, 272–274 RSC;
(b) A. Stamatiou, P. G. Loutzenhiser and A. Steinfeld, Chem. Mater., 2009, 22, 851–859 CrossRef;
(c) A. Stamatiou, P. G. Loutzenhiser and A. Steinfeld, Energy Fuels, 2010, 24, 2716–2722 CrossRef CAS;
(d)
K. D. Dubois and G. Li, in New and Future Developments in Catalysis, ed. S. L. Suib, Elsevier, Amsterdam, 2013, pp. 219–241, 978-0-444-53872-7 Search PubMed;
(e) P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Energy Environ. Sci., 2012, 5, 5902–5918 RSC.
- V. Jiménez, P. Sánchez, P. Panagiotopoulou, J. L. Valverde and A. Romero, Appl. Catal., A, 2010, 390, 35–44 CrossRef PubMed.
- W. Zhang, Q. Ge and H. Xu, Sci. Adv. Mater., 2011, 3, 1046–1051 CrossRef CAS PubMed.
- D. C. Hu, J. J. Gao, Y. Ping, L. H. Jia, P. Gunawan, Z. Y. Zhong, G. W. Xu, F. N. Gu and F. B. Su, Ind. Eng. Chem. Res., 2012, 51, 4875–4886 CrossRef CAS.
-
(a) M. Marafi and A. Stanislaus, Ind. Eng. Chem. Res., 2011, 50, 9495–9501 CrossRef CAS;
(b) M. Marafi and A. Stanislaus, Resour., Conserv. Recycl., 2008, 52, 859–873 CrossRef PubMed.
- A. Cao, X. Zhang, C. Xu, J. Liang, D. Wu and B. Wei, J. Mater. Res., 2001, 16, 3107–3110 CrossRef CAS.
-
(a) J. Liu and A. T. Harris, AIChE J., 2010, 56, 102–113 CAS;
(b) C. H. See, K. J. MacKenzie, O. M. Dunens and A. T. Harris, Chem. Eng. Sci., 2009, 64, 3614–3621 CrossRef CAS PubMed.
- J. Liu, O. M. Dunens, K. J. Mackenzie, C. H. See and A. T. Harris, AIChE J., 2008, 54, 3303–3307 CrossRef CAS.
-
(a) J. R. A. Sietsma, J. D. Meeldijk, J. P. den Breejen, M. Versluijs-Helder, A. J. van Dillen, P. E. de Jongh and K. P. de Jong, Angew. Chem., Int. Ed., 2007, 46, 4547–4549 CrossRef CAS PubMed;
(b) J. R. A. Sietsma, H. Friedrich, A. Broersma, M. Versluijs-Helder, A. Jos van Dillen, P. E. de Jongh and K. P. de Jong, J. Catal., 2008, 260, 227–235 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
-
(a) B. R. Stanmore, V. Tschamber and J. F. Brilhac, Fuel, 2008, 87, 131–146 CrossRef CAS PubMed;
(b) I. Aarna and E. M. Suuberg, Energy Fuels, 1999, 13, 1145–1153 CrossRef CAS.
- I. Aarna and E. M. Suuberg, Fuel, 1997, 76, 475–491 CrossRef CAS.
-
(a) L. Stobinski, B. Lesiak, L. Kövér, J. Tóth, S. Biniak, G. Trykowski and J. Judek, J. Alloys Compd., 2010, 501, 77–84 CrossRef CAS PubMed;
(b) H. Ago, T. Kugler, F. Cacialli, W. R. Salaneck, M. S. P. Shaffer, A. H. Windle and R. H. Friend, J. Phys. Chem. B, 1999, 103, 8116–8121 CrossRef CAS;
(c) P. Hojati-Talemi, R. Cervini and G. P. Simon, J. Nanopart. Res., 2010, 12, 393–403 CrossRef CAS.
- T. W. Ebbesen, H. Hiura, M. E. Bisher, M. M. J. Treacy, J. L. Shreeve-Keyer and R. C. Haushalter, Adv. Mater., 1996, 8, 155–157 CrossRef CAS.
-
(a) X. Peng, J. Chen, J. A. Misewich and S. S. Wong, Chem. Soc. Rev., 2009, 38, 1076–1098 RSC;
(b) K. Lee, J. J. Zhang, H. J. Wang and D. P. Wilkinson, J. Appl. Electrochem., 2006, 36, 507–522 CrossRef CAS PubMed.
- K. J. MacKenzie, O. M. Dunens, M. J. Hanus and A. T. Harris, Carbon, 2011, 49, 4179–4190 CrossRef CAS PubMed.
- P. Azadi, R. Farnood and E. Meier, J. Phys. Chem. A, 2010, 114, 3962–3968 CrossRef CAS PubMed.
- T. Hou, L. Yuan, T. Ye, L. Gong, J. Tu, M. Yamamoto, Y. Torimoto and Q. Li, Int. J. Hydrogen Energy, 2009, 34, 9095–9107 CrossRef CAS PubMed.
- M. V. Landau, S. V. Savilov, M. N. Kirikova, N. B. Cherkasov, A. S. Ivanov, V. V. Lunin, Y. Koltypin and A. Gedanken, Mendeleev Commun., 2011, 21, 125–128 CrossRef CAS PubMed.
-
(a) A. M. Zhao, W. Y. Ying, H. T. Zhang, H. F. Ma and D. Y. Fang, J. Nat. Gas Chem., 2012, 21, 170–177 CrossRef CAS;
(b) J. Gao, C. Jia, J. Li, F. Gu, G. Xu, Z. Zhong and F. Su, Ind. Eng. Chem. Res., 2012, 51, 10345–10353 CrossRef CAS;
(c) J. Gao, Y. Wang, Y. Ping, D. Hu, G. Xu, F. Gu and F. Su, RSC Adv., 2012, 2, 2358–2368 RSC;
(d) J. Kopyscinski, T. J. Schildhauer, F. Vogel, S. M. A. Biollaz and A. Wokaun, J. Catal., 2010, 271, 262–279 CrossRef CAS PubMed;
(e) J. R. Rostrup-Nielsen, K. Pedersen and J. Sehested, Appl. Catal., A, 2007, 330, 134–138 CrossRef CAS PubMed.
- J. Engbæk, O. Lytken, J. H. Nielsen and I. Chorkendorff, Surf. Sci., 2008, 602, 733–743 CrossRef PubMed.
-
(a) M. Trepanier, A. Tavasoli, A. K. Dalai and N. Abatzoglou, Appl. Catal., A, 2009, 353, 193–202 CrossRef CAS PubMed;
(b) P. J. Goethel and R. T. Yang, J. Catal., 1987, 108, 356–363 CrossRef CAS.
-
(a) D. A. Saletore and W. J. Thomson, Ind. Eng. Chem. Process Des. Dev., 1977, 16, 70–75 CrossRef CAS;
(b) J. Sehested, S. Dahl, J. Jacobsen and J. R. Rostrup-Nielsen, J. Phys. Chem. B, 2005, 109, 2432–2438 CrossRef CAS PubMed.
- K. B. Kester, E. Zagli and J. L. Falconer, Appl. Catal., 1986, 22, 311–319 CrossRef CAS.
- Z. Liu, B. Chu, X. Zhai, Y. Jin and Y. Cheng, Fuel, 2012, 95, 599–605 CrossRef CAS PubMed.
-
(a) I. Czekaj, F. Loviat, F. Raimondi, J. Wambach, S. Biollaz and A. Wokaun, Appl. Catal., A, 2007, 329, 68–78 CrossRef CAS PubMed;
(b) C. Mirodatos, H. Praliaud and M. Primet, J. Catal., 1987, 107, 275–287 CrossRef CAS.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
-
(a) E. J. Erekson, E. L. Sughrue and C. H. Bartholomew, Fuel Process. Technol., 1981, 5, 91–101 CrossRef CAS;
(b) D. L. Trimm, Appl. Catal., 1983, 5, 263–290 CrossRef CAS.
- J. G. McCarty and H. Wise, J. Catal., 1979, 57, 406–416 CrossRef CAS.
- Note that a separate CO2-based regeneration is preferred over the addition of CO2 during the reaction, as CO2 can be methanated (eqn (2)) in the presence of a Ni-based catalyst and high concentrations of H2 at the temperatures employed.41 That reaction consumes large quantities of H2, and contributes to an overall decrease in CO methanation activity because of the adsorption/dissociation mechanism42 of CO2 methanation over Ni-based catalysts.
- J. L. Figueiredo and D. L. Trimm, J. Catal., 1975, 40, 154–159 CrossRef CAS.
-
(a) F. W. Chang, T. J. Hsiao, S. W. Chung and J. J. Lo, Appl. Catal., A, 1997, 164, 225–236 CrossRef CAS;
(b) D. E. Peebles, D. W. Goodman and J. M. White, J. Phys. Chem., 1983, 87, 4378–4387 CrossRef CAS;
(c) W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- S.-I. Fujita, M. Nakamura, T. Doi and N. Takezawa, Appl. Catal., A, 1993, 104, 87–100 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Description of apparatus and MS technique, additional characterization and reaction data. See DOI: 10.1039/c4cy00696h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
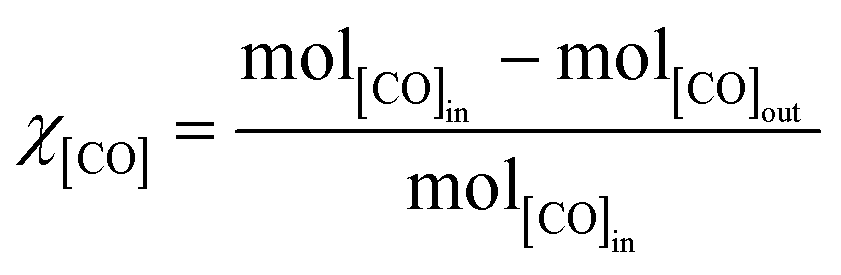
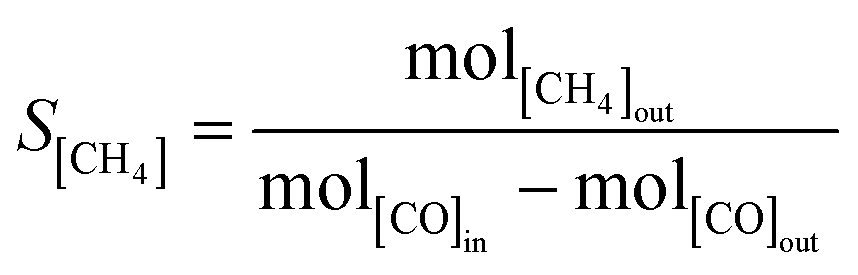
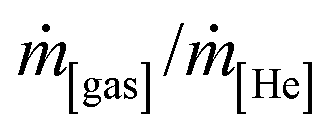 ), where He was used as an internal standard (see Section S3 and Fig. S5 and S6†).
), where He was used as an internal standard (see Section S3 and Fig. S5 and S6†).