
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights into the Nb2O5 and niobium phosphate catalyzed in situ condensation of a fluorescent halochromic assembly†
Stefania
Impellizzeri
a,
Sabrina
Simoncelli
ab,
Chiara
Fasciani
a,
M.
Luisa Marin
ac,
Geniece L.
Hallett-Tapley
a,
Gregory K.
Hodgson
a and
Juan C.
Scaiano
*a
aDepartment of Chemistry, Centre for Catalysis Research and Innovation, University of Ottawa, 10 Marie Curie, Ottawa, Ontario K1N 6N5, Canada. E-mail: Scaiano@photo.chem.uottawa.ca
bINQUIMAE and Departamento de Química Inorgánica, Analítica, y Química Física, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, 1428 Buenos Aires, Argentina
cInstituto Universitario Mixto de Tecnología Química (UPV-CSIC), Universitat Politècnica de València, Avenida de los Naranjos s/n, 46022 Valencia, Spain
First published on 13th August 2014
Abstract
Solid niobium oxides (Nb2O5·nH2O) and niobium phosphate were used as heterogeneous acid catalysts to promote the condensation between a switchable oxazine and a fluorescent coumarin in an aprotic solvent. The catalysts were found to promote the generation of an active methylene from the enamine-based portion of the oxazine, which was followed by a nucleophilic attack on the aldehyde functionality of the coumarin reagent. In the resulting system, the emission of the conjugated fluorophore can be observed at 670 nm and, thus, the processes occurring at the catalyst surface can be monitored in real time by total internal reflection fluorescence microscopy (TIRFM).
A. Introduction
Halochromic compounds switch between different colored states in response to a change in the pH of their surroundings. Often, halochromic behavior is accompanied by the possibility of tuning the structural and the electronic properties of molecules using light (photochromism) or heat (thermochromism).1 Spiropyrans and oxazines, for instance, can undergo isomerization between two states with distinctive characteristics either by light or by chemical stimulation.2 The interplay between the means for interconversion and modulation of these systems offers the possibility of use for advanced applications, such as in optical storage devices,3–6 logic gates7–13 and, if in combination with organic fluorophores, in fluorescence imaging of biological systems with enhanced resolution.14–17In a recent publication we investigated18 the Brønsted acidic properties of several types of Nb2O5·nH2O, including both commercially available and synthesized materials.19–24 Furthermore, we exploited these solids to activate the fluorescence of the halochromic assembly 1 (Fig. 1). The chemical conjugation of a switchable oxazine and a fluorescent coumarin in compound 1 generates a bifunctional system, in which the fluorescence of the latter at 670 nm can be turned on via the acid-induced ring opening reaction of the halochromic unit (compound 2 in Fig. 1).25 Single-molecule fluorescence techniques were successfully used to determine the rate of protonation of 1 → 2 and to obtain a detailed map of the Brønsted acid sites in the catalysts. The switchable assembly can be synthesized in a single step from precursors 3 and 4 (Fig. 2). The conventional protocol consists of refluxing the reagents in a polar protic solvent, such as methanol or ethanol, upon the addition of several equivalents of an acid catalyst (trifluoroacetic acid, TFA) for over 24 hours; with this method, the product can be obtained in moderate yields (ca. 40% after purification by column chromatography).25
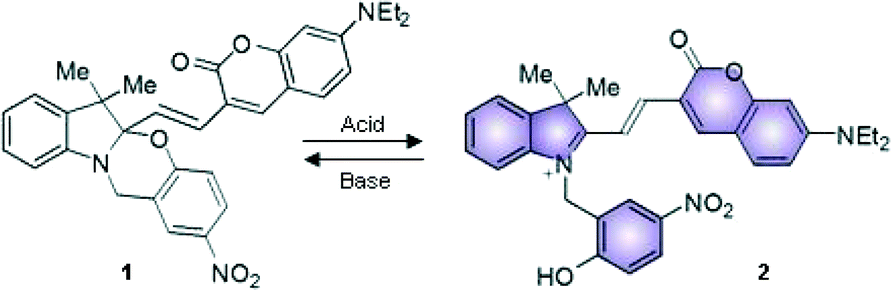 | ||
| Fig. 1 Halochromic interconversion between 1 and 2. | ||
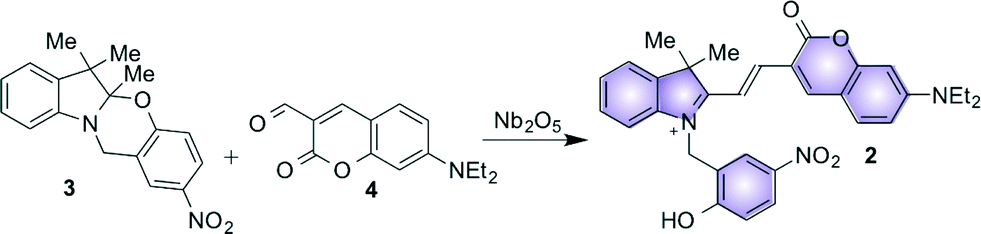 | ||
| Fig. 2 Proposed Nb2O5-catalyzed condensation between oxazine (3) and coumarin (4) to generate the fluorescent halochromic assembly 2. | ||
We envisaged the possibility of using solid niobium oxides and niobium phosphate as potential heterogeneous acid catalysts with activity in polar aprotic solvents, such as acetonitrile (MeCN), to promote the C–C bond formation between 3 and 4 and to directly generate the fluorescent conjugated assembly 2 (Fig. 2). Specifically, we used two commercially available catalysts, hydrated Nb2O5·nH2O (T-I) and niobium phosphate (T-II), and the mesoporous Nb2O5·nH2O (T-III), which was prepared according to our recently published protocol.18 In addition to their known acid properties, these catalysts offer the added advantage of facilitating their removal from the reaction mixture, as compared to the conventional homogeneous catalysts, as well as potential reusability. Moreover, the use of aprotic solvents, such as MeCN, allows for the investigation of the potential solvent-like role of the niobium oxide and niobium phosphate acidic sites in reactions that traditionally require polar protic solvents. The condensation reaction can be carried out at moderate temperatures (45 °C) for approximately 15 hours to obtain 2 in a range of yields that strongly depend on the type of catalyst used in the preparation. The fluorescence of the generated assembly allows the catalyzed condensation between 3 and 4 to be monitored in situ and in real time by total internal reflection fluorescence microscopy (TIRFM) on the catalyst surface.
B. Experimental
Materials
Compounds 1, 3 and 4 were prepared according to literature procedures.25–271. ESI-MS: m/z = 537.9 [M]+. 1H NMR (300 MHz, CD3CN): δ 8.1 (1H, s), 8 (1H, d, 9 Hz), 7.8 (1H, s), 7.3 (1H, d, 9 Hz), 7.08–7.2 (2H, m), 6.68–6.96 (6H, m), 6.5 (1H, s), 4.7 (2H, s), 3.42–3.5 (4H, m), 1.37 (6H, s), 1.16–1.2 (6H, t, 7 Hz). 13C NMR (300 MHz, CD3CN): δ 160.7, 159.6, 150.9, 145.8, 141.2, 140.9, 138.5, 130.8, 129.5, 127.9, 126, 124.2, 123.6, 122.4, 120, 120, 119.8, 119.1, 118.5, 115.7, 109.8, 108.6, 104.2, 96.5, 50.9, 45.1, 41.3, 27.5, 19, 12.5. 3. EI-MS: m/z = 310.1 [M]+. 1H NMR (300 MHz, CD3CN): δ 8.1 (1H, s), 8 (1H, d, 9 Hz), 7.05–7.11 (2H, m), 6.8–6.83 (1H, m), 6.7 (1H, d, 7 Hz), 6.5 (1H, d, 6 Hz), 4.6 (2H, s), 1.6 (3H, s), 1.2 (3H, s). 13C NMR (300 MHz, CD3CN): δ 159.1, 146.5, 140.3, 137.1, 127.6, 123.9, 123.3, 122.3, 120.5, 118.8, 118.1, 108.4, 102.8, 48, 40, 26.1, 18.9, 16.6. 4. EI-MS: m/z = 245.1 [M]+. 1H NMR (300 MHz, CD3CN): δ 10.1 (1H, s), 8.2 (1H, s), 7.4 (1H, d, 7 Hz), 6.6 (1H, d, 8 Hz), 6.5 (1H, s), 3.43–3.48 (4H, q, 5 Hz), 1.21–1.25 (6H, t, 5 Hz). 13C NMR (300 MHz, CD3CN): δ 187.9, 161.8, 158.9, 145.3, 132.5, 114.5, 110.4, 108.4, 97.4, 45.4, 12.4. Commercial niobium oxides types I (T-I, hydrated Nb2O5·nH2O) and II (T-II, niobium phosphate) were kindly provided by CBMM Brazil. Mesoporous Nb2O5·nH2O type III (T-III) was prepared based on our recently published protocol (T-IIIb in ref. 18). The acidity of the niobium oxide materials was estimated using conventional Hammett acidity indicator tests;28,29 the procedure and the indicators have been described elsewhere.18 The pKa ranges of the catalysts are reported in Table S1 of the ESI.†Methods
Acetonitrile was purified using a benchtop solvent purification system (LC Technology Solutions Inc., SPBT-1). All chemicals were purchased from Sigma-Aldrich. All of the reactions were monitored by thin-layer chromatography using aluminum sheets coated with silica (60, F254). NMR spectra were recorded at room temperature with a Bruker Avance 300 spectrometer. Mass spectral analysis was performed with a 6890N Network GC system equipped with a 5973 mass selective detector from Agilent Technologies. ESI mass spectra in positive mode were acquired with a Micromass Q-TOF I. High-resolution EI mass spectra were acquired with a HRes, Concept S1, magnetic sector mass spectrometer at the John L. Holmes Mass Spectrometry Facility at the Department of Chemistry, University of Ottawa. XPS analysis was performed with a Kratos Axis Ultra XPS using monochromated Al X-rays at 140 W at the Centre for Catalysis Research and Innovation, University of Ottawa. Absorbance spectra were recorded using a Cary 50 UV-visible spectrophotometer. HPLC analysis was performed with a Waters Integrity HPLC system coupled to a Waters 996 photodiode array detector. Reverse phase chromatography was performed using a reverse phase Zorbax C18 column and an eluent mixture of MeOH and MeCN (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6) with a flow rate of 0.25 mL min−1 in the presence of a standard (Coumarin 6, C6). For HPLC analysis, 10 equivalents of Et3N were added to each sample (the number of equivalents was calculated with respect to the theoretical yield of the product). Retention times: 11.8 min (4); 13.5 min (3); 17 min (1); 19.7 min (C6,), λobs = 413 nm.
:6) with a flow rate of 0.25 mL min−1 in the presence of a standard (Coumarin 6, C6). For HPLC analysis, 10 equivalents of Et3N were added to each sample (the number of equivalents was calculated with respect to the theoretical yield of the product). Retention times: 11.8 min (4); 13.5 min (3); 17 min (1); 19.7 min (C6,), λobs = 413 nm.
Single-molecule fluorescence microscopy
The acid-catalyzed reaction measured at the single-molecule level was performed in a reaction chamber with an area of 4.2 cm2 per well (Thermo Scientific). T-II or T-III was dispersed in methanol and deposited on a clean coverslip (18 mm × 18 mm; Fisher Scientific) through spin-coating (1000 rpm, 40 s). The catalyst-loaded cover glass was then submerged in 1.0 mL of a reaction mixture generated by mixing 255 μL of an 8.2 mM solution of 3 and 800 μL of a 2.6 mM solution of 4 in EtOH. Single-molecule fluorescence imaging was carried out with a TIRF Olympus FV1000 (Olympus, Japan) microscope. A Chroma ZT640 dichroic mirror (Chroma Technology Corporation, Bellows Falls, USA) was used to reflect the 633 nm He–Ne (CW) excitation light into an Olympus Plan Apo oil immersion objective (100×, 1.45 NA, Olympus). The power density of the excitation laser at the sample was estimated to be 40 W cm−2. The Chroma ET600 excitation and Chroma ET655 emission filters (675/50 nm bandpass) allowed for optimum signal detection. The fluorescence signal was collected using the same objective and focused onto a Rolera EM-C2 EMCCD camera (Q-Imaging, Surrey, Canada) with 1004 × 1002 pixels of 8 × 8 μm size operated at 50 ms per frame. The pixel size of the image is 156 nm. Fluorescence spectra were recorded with a fluorescence lifetime imaging system (PicoQuant, Berlin, Germany). The instrument is equipped with a frequency-doubled, picosecond pulse diode laser (637 nm, 93 ps, 40 MHz, LDH-P-FA-640L, PicoQuant). The laser beam was collimated and focused through a fiber-coupling unit. A beam splitter (Z638rdc, Chroma) was used to separate excitation and emission light. The emission signal was collected using a Shamrock SR-163 spectrograph (Andor Technology, South Windsor, USA) with a 690/70 nm bandpass emission filter.C. Results and discussion
For all reactions, 6 mg (0.02 mmol) of 3 and 4.8 mg (0.02 mmol) of 4 were dissolved in 1 mL of EtOH or MeCN. Upon the addition of the catalyst, the mixture was stirred vigorously for 15 h at 45 °C and centrifuged; the supernatant was analyzed by HPLC after the addition of Et3N. Due to its charged nature, the direct product of the condensation (2) possesses a high affinity for the HPLC stationary phase and cannot be eluted into the detection system under reverse phase conditions. Nonetheless, due to its intrinsic halochromic nature, the product (2) can be switched to its closed form (1) by the addition of a base (see Fig. 1), in which the absence of charged centers allows easy quantification of the yields by liquid chromatography. The results are reported in Table 1. Each T-III residue was stirred at room temperature in MeCN in the presence of Et3N for 48 h to encourage desorption of any reagents or products that might have been incorporated into the mesoporous structures. The mixture was again centrifuged (complete detachment was confirmed by lack of coloration of the catalysts), and the supernatant was dried, redissolved in a known volume of MeCN and analyzed by HPLC.| Catalyst | Amount of catalyst | Solvent | Temperature (°C) | % of 1a |
|---|---|---|---|---|
| a The condensation product 2 was quantitatively converted into its neutral closed form (1) in order to perform HPLC analysis. For all reactions, peaks belonging to starting materials 3 and 4 are observed. | ||||
| T-I | 5 mg | MeCN | 45 | 7.5 |
| T-II | 5 mg | MeCN | 45 | 34 |
| T-II | 25 mg | MeCN | 45 | 30 |
| T-III | 5 mg | EtOH | 45 | 7 |
| T-III | 5 mg | MeCN | 45 | 19 |
| T-III | 25 mg | MeCN | 45 | 16 |
| T-III | 50 mg | MeCN | 45 | 19 |
| TFA | 10 eq. | EtOH | 80 | 38 |
| TFA | 10 eq. | MeCN | 45 | No product observed |
| No acid catalyst | — | EtOH or MeCN | 45 | No product observed |
The condensation between 3 and 4 requires several steps based on the mechanism described in Fig. 3. The acid-induced cleavage of the C–O bond at the spirocenter of 3 generates the indolium cation 3a, which forms the enamine 3b upon elimination of a proton. Enamines can be considered as nitrogen analogues of enolates; their reactivity as nucleophiles is due to the dipolar character of one of the resonance structures (3b′), where the carbon at the β position with respect to the nitrogen has a partial negative charge. The aldehyde appendage in 4 then reacts with the active methylene group of the heterocyclic cation of 3b′, followed by proton transfer from the solvent and elimination of H2O, with subsequent formation of a double bond between the two units. The proposed mechanism is in agreement with the conventional synthesis procedure which consists of refluxing the reagents in MeOH or EtOH in the presence of trifluoroacetic acid (TFA).
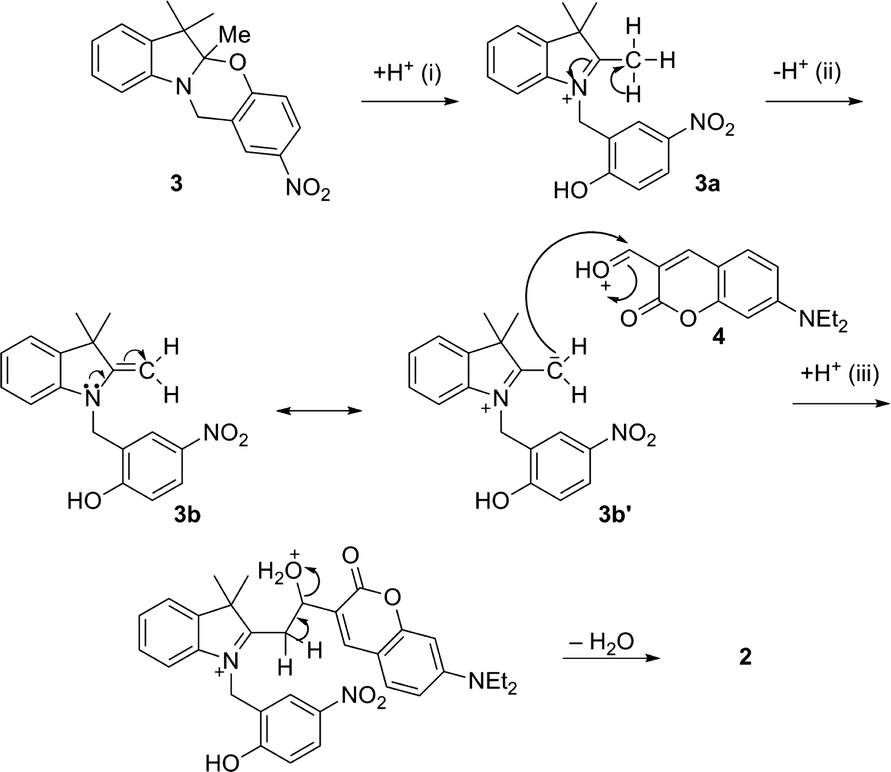 | ||
| Fig. 3 Proposed mechanism for the acid-induced condensation of 3 and 4 to yield 2: acid-induced ring opening of 3 (i), removal of a proton (ii), nucleophilic attack of the aldehyde functionality in 4 from the active methylene in 3b′ and subsequent proton transfer from the solvent (iii), and elimination of H2O. | ||
The purpose of the acid in the homogeneous system is primarily to generate the active protonated open-ring structure (3a) as well as to protonate the reactive carbonyl in 4, while the participation of protic solvents is required for the subsequent series of intermolecular proton transfers (ii and iii). High temperature favors the elimination of water.
Based on these considerations and the results reported in Table 1, we can provide insights into the mechanistic participation of the different catalysts that were tested. Niobium phosphate (T-II) shows the highest activity, followed by the mesoporous Nb2O5 (T-III). Both catalysts possess strong Brønsted-type acidity and thus are able to promote the first step of the reaction and open the spirocenter of 3 and, as previously observed,18 of 1. As listed in Table S1,† the acidity of these catalysts can easily be correlated to the efficacy of the overall reaction.
The use of a protic solvent in the conventional preparation allows for a continuous transfer of protons to/from the solvent itself and therefore plays an active role in the following steps of the reaction (Fig. 3). No product was formed when MeCN was used in combination with a homogeneous acid catalyst (TFA). 1H NMR spectra of 3 in CD3CN and CD3OD (Fig. S1 and S2, ESI†) confirm that upon the addition of TFA the ring-opened species (3a) is generated in both solvents. Nonetheless, the chemical shift of the active methyl Mea in 3a (Fig. 4) is located, as expected, significantly downfield in CD3CN (2.84 ppm, Fig. S1†) with respect to that in CD3OD (1.25 ppm, Fig. S2†). These results seem to indicate that the protons of Mea remain significantly shielded when a protic solvent is used and that, in turn, hydrogen bonding is likely responsible for countering the electron-withdrawing effect of the indolium cation. Addition of CD3OD to 3a in CD3CN promotes hydrogen bonding, and thus the chemical shift of Mea moves upfield and resembles its signal in pure CD3OD (Fig. S3, ESI†).
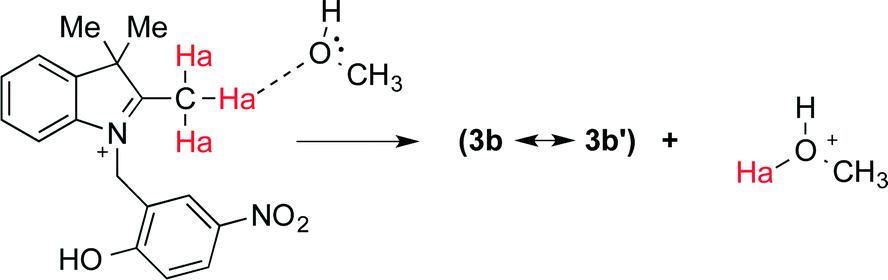 | ||
| Fig. 4 Proposed intermolecular hydrogen bonding between Mea in 3a and MeOH leading to formation of the active methylene in 3b′. | ||
The formation of intermolecular hydrogen bonding with a protic solvent allows for the eventual continuous exchange of protons required to bring the overall reaction to completion. Yet, the presence of niobium oxides or niobium phosphate affords relatively good yields in MeCN. Thus, the efficiency of these catalysts with respect to this particular reaction must be related not only to their intrinsic Brønsted acidity, but also to their ability to participate in intermolecular hydrogen bonding and proton transfer in a similar fashion as in Fig. 4. This ability will, in turn, depend on the hydration of the material18 and/or on the accessibility of such proton-active sites. Hydration would be considerably larger in both T-II and T-III due to the enhanced acidity of these materials as Brønsted acidity is known to be directly correlated to water content. These properties can directly translate into a higher probability of active proton transfer and thus higher reaction yields when the condensation reaction to form 2 is carried out in MeCN.
Although the activities of T-II and T-III are comparable by Hammett acidity testing, the yields obtained when T-III was used as a catalyst are consistently lower than those when T-II was employed. The decreased yield observed when Nb2O5·nH2O T-III was used is strongly dependent on the accessibility of the catalytic protonating sites. Given the mesoporous nature of catalyst T-III, it is likely that a considerable portion of the active proton sites is embedded within the cavities of the solid-state matrix, similar to the known properties of acidic faujasite Y zeolites.30–32 In all cases, the susceptibility of any given substrate to participate in acid catalysis within the zeolite matrix is determined by the ability of the molecule to efficiently diffuse within the pores to the active site of the material. A similar situation can be envisioned within the mesoporous system of Nb2O5·nH2O T-III. In such a case, it is possible that several molecules of substrate 3 can diffuse within the pores and react with the active acid sites of the matrix. Indeed, one major disadvantage of employing porous heterogeneous catalysts involves the ability of substrate molecules to react with the acidic sites at the openings or entry points of the matrix pores. As a result, the three-dimensional pores may become blocked, rendering the remaining catalytic acid sites embedded within the mesoporous structure and thus inaccessible for catalytic conversion, resulting in lower yields of the product, as was observed for the mesoporous catalyst T-III presented in Table 1. Alternatively, the lower yields observed for T-III may also be due to the spatial constraints within the mesoporous system and the inability of the material to accommodate the anticipated large transition state of the oxazine/coumarin adduct. In other words, beyond the acidity of a given site, the traffic that enables molecules to reach the site and ultimately depart from it is the key parameter in the case of mesoporous catalysts.
Interestingly, in the presence of the niobium oxide T-III, the reaction in MeCN is much more efficient than that in EtOH. This is possibly due to the partial solvation effect of EtOH on the active sites of the catalyst; indeed, if EtOH molecules are arranged through intermolecular hydrogen bonding interactions with the proton-like moieties on the surface of the niobium oxide, the activity of the solids may be dramatically reduced.
Catalysis at the single-molecule level
The dynamics of the acid-catalyzed condensation between 3 and 4 was monitored in situ using TIRF microscopy. Previously, we observed that upon protonation, 1 yields a sufficiently fluorescent product (2) that can be easily detected at the single-molecule level over the Nb2O5·nH2O catalyst.18 With an excitation wavelength of 633 nm and an emission bandpass filter of 675/50 nm, only the product of the condensation can be selectively excited and observed (full spectroscopic characterization of 1 and 2 and the starting materials 3 and 4 can be found in the ESI,† Fig. S4 and S5). A 3D image of the accumulated fluorescence intensity (obtained from a 25 s movie) shows spatially scattered distribution of the collected data for the catalytic formation of 2 (Fig. 5). | ||
| Fig. 5 Transmission (left), TIRF microscopy (center) and 3D (right) images of a T-III particle immobilized on a cover glass and immersed in a 2 mM solution of 3 and 4 at 633 nm laser excitation with a 675/50 nm bandpass emission filter (see the ESI† Video S1). The fluorescence image and the 3D representation correspond to the accumulated spot intensity reconstructed from a movie of 500 frames in length (50 ms per frame). | ||
Typical time-dependent fluorescence intensity traces of 2 (Fig. 6) prove that the individual product molecules can be easily distinguished from the background signals caused by scattering or fluorescence of the catalyst surface and from the high concentrated reactant solution. Characterization of the bright spots was performed by measuring their emission spectra in situ (Fig. 6), which is ascribed to product 2.
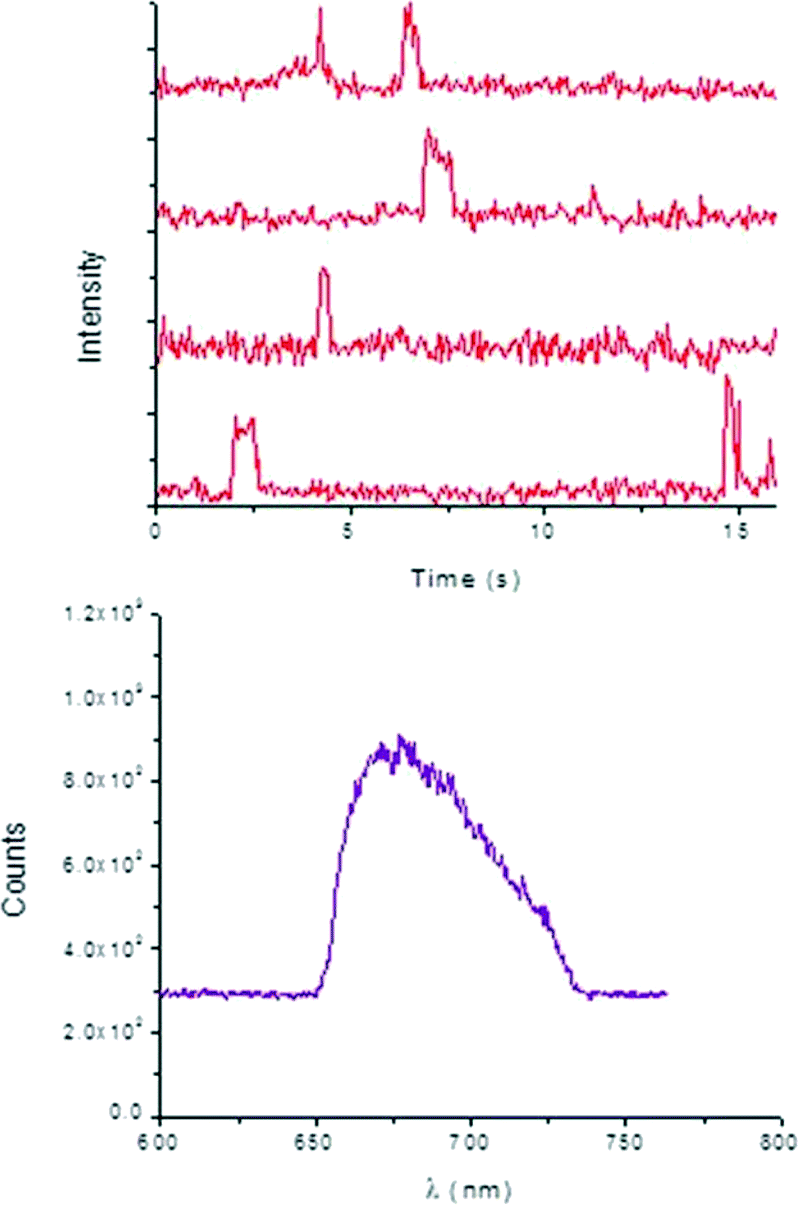 | ||
| Fig. 6 Top: typical fluorescence intensity trajectories observed for the “turn ON” of molecules of 2 over a T-III particle. Bottom: spectral information of the detected bright spots during “turn ON” events measured by passing the epi fluorescence signal through a spectrograph (λEx = 637 nm) and using a 690/70 nm bandpass emission filter installed into the fluorescence lifetime imaging system. | ||
Through the analysis of the number of single “turn ON” events of fluorescent molecules over the catalyst (for a total of six different T-III particles), a rate constant of 3 × 10−15 mol m−2 s−1 was estimated for the reaction between equimolar amounts of 3 and 4. This rate is representative of the overall mechanism described in Fig. 3 which includes the initial adsorption and protonation of 3 and the subsequent condensation between the ring-open active 3b′ with 4 to yield 2. 3D surface maps of the TIRFM sequence (Fig. 7) depict the catalytic “hot spots” of T-III characterized by the highest acidity, and considering the mesoporous nature of the catalyst, easiest accessibility. In Fig. 7D, the positive peaks show sites that lose catalytic activity between the first and the second 12.5 s window (as in Fig. 7A and B). It may appear surprising to observe negative peaks in Fig. 7D as this reflects gain of catalytic activity with time. While the creation of new catalytic sites would be surprising, we believe that as some sites are lost (positive peaks) other sites in their vicinity become active and appear as negative peaks. These are probably less active or less accessible sites that could not compete favorably with the more active sites and thus become active as some of the better sites are lost during reaction.
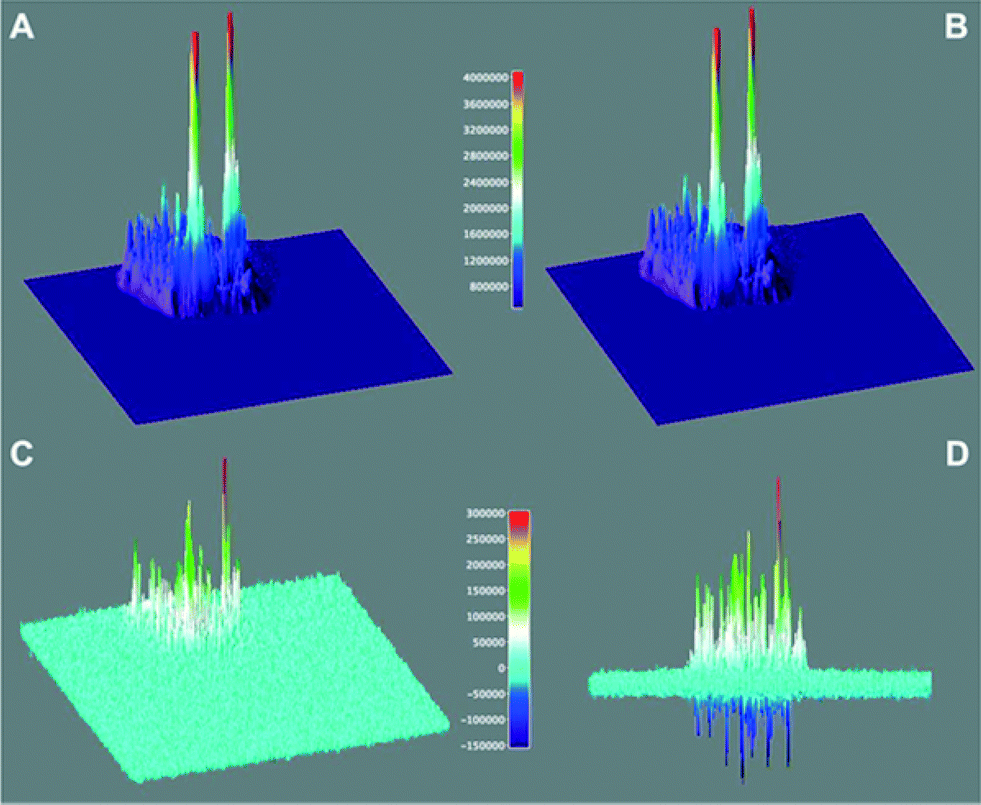 | ||
| Fig. 7 Catalytic activity of a T-III particle for the overall condensation of 3 and 4 to generate 2 (see Fig. 2) monitored by detecting the fluorescence of 2 using TIRFM. Surface maps A and B show the accumulated signal for 12.5 seconds and the subsequent 12.5 seconds, while C and D show the rescaled difference of A minus B (see color-coded legend) from different perspectives. | ||
TIRFM experiments conducted over niobium phosphate (T-II) particles revealed an overall rate of 6 × 10−15 mol m−2 s−1 for the reaction between 3 and 4. In a similar fashion to what is observed for T-III, the 3D surface maps of the TIRFM sequence (Fig. S6 and S7, ESI†) reveal that as the most active or accessible sites are being used, other sites become available for catalysis over time.
Reusability experiments
Heterogeneous catalysts have the advantage of allowing their easy and inexpensive removal from reaction mixtures by filtration or centrifugation. Due to these reasons, a catalyst can often be recovered and used for multiple-reaction cycles, provided that its efficiency has not been compromised by previous uses. Thus, the reusability of T-II was tested for the condensation between 3 and 4. Indeed, T-II resulted to be the most efficient catalyst for generating 2, and the lack of surface adsorption of the latter on the heterogeneous surface allowed simpler and faster separation than the mesoporous T-III. In all instances, after centrifugation, separation and analysis of the supernatant phase, T-II was washed three times with MeCN, centrifuged, dried overnight in a fumehood and used “as is”. The recyclability results are depicted in Fig. 8.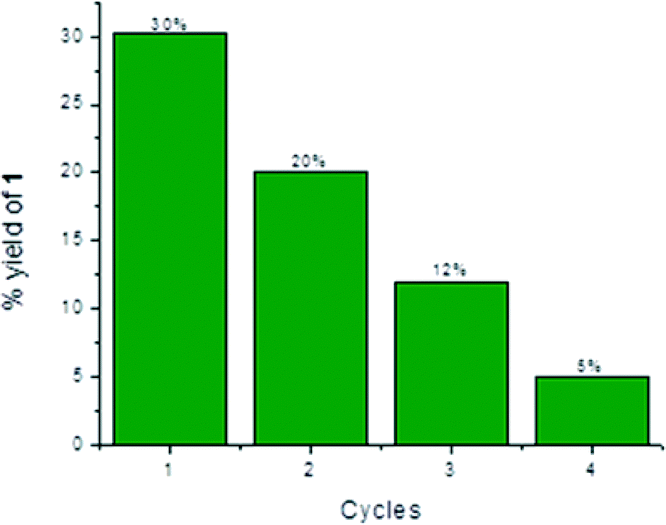 | ||
| Fig. 8 Recyclability of T-II for the acid-catalyzed condensation of 3 and 4. | ||
Conclusions
We have successfully demonstrated that solid niobium phosphate and mesoporous niobium oxide (T-II and T-III, respectively) have the ability of promoting complex reactions, such as the condensation between enamine-like moieties and aldehydes. In particular, the role of these catalysts in several reaction steps was investigated. Based on our results, we conclude that in addition to the known and previously reported Brønsted acidity (required in the first step of the reaction), niobium oxides and niobium phosphate can adopt solvent-like behavior, specifically in the formation of hydrogen bonding interactions and through participation in intermolecular proton transfers. The reaction can also be monitored using single-molecule techniques by selecting reagents that once assembled can generate a species with a suitable emission wavelength and quantum yield for detection. Using TIRFM to monitor the fluorescence events on the catalyst surface, the rate constant for the reaction between 3 and 4 to yield 2 was found to be 3 × 10−15 mol m−2 s−1. In addition, 3D surface mapping techniques were used to illustrate catalytic hot spots of the niobium oxide T-III and their evolution over time.Acknowledgements
The authors wish to thank the Natural Sciences and Engineering Research Council (NSERC) and the Canada Research Chairs program. The University of Ottawa International Office provided grants to study niobium. Thanks are due to the Government of Canada and NSERC for a Banting Postdoctoral Fellowship to S. Impellizzeri and a Vanier Scholarship to C. Fasciani. S. Simoncelli acknowledges a DFAIT fellowship from ELAP (Emerging Leaders in the Americas Program) to support her visit to Canada. M. L. Marin thanks the financial support of the Generalitat Valenciana (BEST/2012/233) and the Distinguished Visiting Professor program of the University of Ottawa. We thank Dr. José-Carlos Netto-Ferreira of INMETRO (Rio de Janeiro, Brazil) for providing chemicals from CBMM.Notes and references
- B. L. Feringa, Molecular Switches, Wiley-VCH, Weinheim, 2001 Search PubMed.
- J. C. Crano and R. J. Guglielmetti, Organic Photochromic and Thermochromic Compounds, Plenum Press, New York, 1999 Search PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1683–1890 CrossRef CAS PubMed.
- D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 245, 843–845 CAS.
- A. S. Dvornikov, J. Malkin and P. M. Rentzepis, J. Phys. Chem., 1994, 98, 6746–6752 CrossRef CAS.
- A. S. Dvornikov, E. P. Walker and P. M. Rentzepis, J. Phys. Chem. A, 2009, 113, 13633–13644 CrossRef CAS PubMed.
- A. P. de Silva, N. D. McClenaghan and C. P. McCoy, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, pp. 156–185 Search PubMed.
- V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, Wiley-VCH, Weinheim, 2008 Search PubMed.
- F. M. Raymo and S. Giordani, Org. Lett., 2001, 3, 3475–3478 CrossRef CAS PubMed.
- F. M. Raymo, Adv. Mater., 2002, 14, 401–414 CrossRef CAS.
- F. M. Raymo, S. Giordani, A. J. P. White and D. J. Williams, J. Org. Chem., 2003, 68, 4158–4169 CrossRef CAS PubMed.
- F. M. Raymo, R. J. Alvarado, S. Giordani and M. A. Cejas, J. Am. Chem. Soc., 2003, 125, 2361–2364 CrossRef CAS PubMed.
- S. Silvi, E. C. Constable, C. E. Housecroft, J. E. Beves, E. L. Dunphy, M. Tomasulo, F. M. Raymo and A. Credi, Chem. – Eur. J., 2009, 15, 178–185 CrossRef CAS PubMed.
- J. Cusido, S. Impellizzeri and F. M. Raymo, Nanoscale, 2011, 3, 59–70 RSC.
- F. M. Raymo, J. Phys. Chem. Lett., 2012, 3, 2379–2385 CrossRef CAS.
- E. Deniz, M. Tomasulo, J. Cusido, I. Yildiz, M. Petriella, M. L. Bossi, S. Sortino and F. M. Raymo, J. Phys. Chem. C, 2012, 116, 6058–6068 CAS.
- M. Petriella, E. Deniz, S. Swaminathan, M. J. Roberti, F. M. Raymo and M. L. Bossi, Photochem. Photobiol., 2013, 89, 1391–1398 CrossRef CAS PubMed.
- M. L. Marin, G. Hallett-Tapley, S. Impellizzeri, C. Fasciani, S. Simoncelli, J. C. Netto-Ferreira and J. C. Scaiano, Catal. Sci. Technol., 2014, 4, 3044–3052 Search PubMed.
- I. Nowak and M. Ziolek, Chem. Rev., 1999, 99, 3603–3624 CrossRef CAS PubMed.
- S. Okazaki and A. Kurosaki, Catal. Today, 1990, 8, 113–122 CrossRef.
- S. Okazaki and N. Wada, Catal. Today, 1993, 16, 349–359 CrossRef CAS.
- T. Ushikubo, I. Iizuka, H. Hattori and K. Tanabe, Catal. Today, 1993, 16, 291–295 CrossRef CAS.
- Q. Sun, A. Auroux and J. Shen, J. Catal., 2006, 244, 1–9 CrossRef CAS PubMed.
- I. A. L. Bassan, D. R. Nascimento, R. A. S. San Gil, M. I. P. da Silva, C. R. Moreira, W. A. Gonzalez, A. C. Faro Jr, T. Onfroy and E. R. Lachter, Fuel Process. Technol., 2013, 106, 619–624 CrossRef CAS PubMed.
- E. Deniz, S. Sortino and F. M. Raymo, J. Phys. Chem. Lett., 2010, 1, 3506–3509 CrossRef CAS.
- M. Tomasulo, S. Sortino, A. J. P. White and F. M. Raymo, J. Org. Chem., 2005, 70, 8180–8189 CrossRef CAS PubMed.
- J.-S. Wu, W.-M. Liu, X.-Q. Zhuang, F. Wang, P.-F. Wang, S.-L. Tao, X.-H. Zhang, S.-K. Wu and S.-T. Lee, Org. Lett., 2006, 9, 33–36 CrossRef PubMed.
- I. Matsuzaki, M. Nitta and K. Tanabe, J. Res. Inst. Catal., Hokkaido Univ., 1969, 17, 46–53 CAS.
- M. Yurdakoc, M. Akcay, Y. Tonbul and K. Yurdakoc, Turk. J. Chem., 1999, 23, 319–327 CAS.
- A. Corma and A. Martinez, Adv. Mater., 1995, 7, 137–144 CrossRef CAS.
- S. Corrent, P. Hahn, G. Pohlers, T. J. Connolly, J. C. Scaiano, V. Fornés and H. García, J. Phys. Chem. B, 1998, 102, 5852–5858 CrossRef CAS.
- J. C. Scaiano and H. García, Acc. Chem. Res., 1999, 32, 783–793 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Hammett acidity (H0) of the niobium catalysts employed for the condensation of 3 and 4; 1H NMR of 3 in CD3CN before and after the addition of TFA; 1H NMR of 3 in CD3OD before and after the addition of TFA; 1H NMR (detail) of 3a in CD3CN after the addition of CD3OD; normalized absorption spectra of compounds 1–4; emission spectra of compounds 1–4; transmission electron microscopy, TIRF microscopy and 3D images of a T-II particle for the reaction between 3 and 4; catalytic activity of a T-II particle for the overall condensation of 3 and 4 to generate 2 monitored by detecting the fluorescence of 2 using TIRFM; XPS data and survey result for 3a; ESI-MS spectrum of 1; EI-MS spectrum of 3; and EI-MS spectrum of 4. See DOI: 10.1039/c4cy00703d |
| This journal is © The Royal Society of Chemistry 2015 |