
A study on the selective hydrogenation of nitroaromatics to N-arylhydroxylamines using a supported Pt nanoparticle catalyst†
Evert H.
Boymans
a,
P. T.
Witte
b and
D.
Vogt
*a
aSchool of Chemistry, University of Edinburgh, King's Buildings, Joseph Black Building, Room 247 West Mains Road Edinburgh, EH9 3JJ Scotland, UK. E-mail: d.vogt@ed.ac.uk
bBASF Nederland B.V., Strijkviertel 67, 3454 ZG De Meern, the Netherlands
First published on 29th August 2014
Abstract
A supported Pt nanoparticle-based catalyst was used in the chemoselective hydrogenation of nitroarenes to N-arylhydroxylamines (N-AHA). Optimization of NB hydrogenation conditions showed that substantially higher N-PHA yields can be obtained at low temperature. Especially, the influence of an increased hydrogen pressure on selectivity is remarkable. Maximum yields increase from 55% N-PHA at 4 bar H2 to 80% at 23 bar H2 in ethanol. Further optimization led to the use of small amounts of amine additive, TMEDA, with 50 bar H2 raising the maximum yield to 97% N-PHA. The decreased N-PHA hydrogenation rate at high H2 pressure and the presence of TMEDA allow for selective transformation of a range of other nitroarenes containing electron-withdrawing and -donating (reducible) functional groups to their N-AHAs in excellent (more than 90%) yields.
Introduction
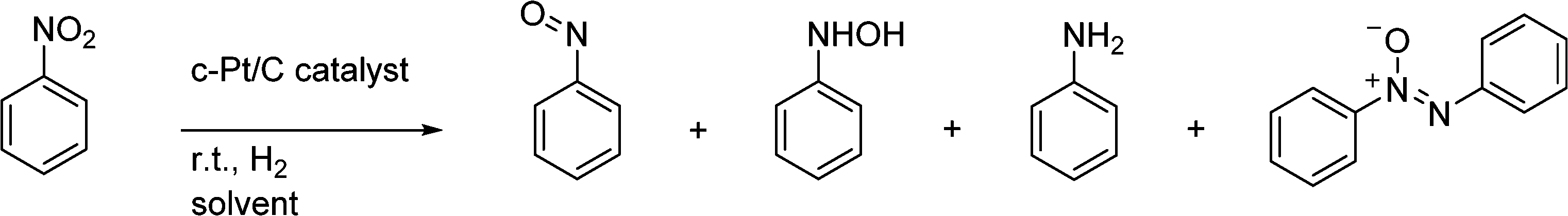
Aniline derivatives are important chemical building blocks which have been produced industrially on a large scale since the 19th century.1 An important route is the hydrogenation of nitrobenzenes (NB) by solid precious metal catalysts (Cu, Pd, Pt) at high temperature. During the past decades, catalyst development has been aimed at the chemoselective hydrogenation of the nitro group in the presence of other reducible groups on the substrate. This is of special interest for the production of pharmaceuticals. A common difficulty in the formation of aniline is the formation and build-up of intermediates. For nitrobenzene, these are N-phenylhydroxylamine (N-PHA), nitrosobenzene (NOB) and their condensation product azoxybenzene (AZOXY), which are summarized in Scheme 1.2,3 This is especially true when working under mild reaction conditions, namely, low temperature (<30 °C) and H2 pressure (<10 bar). Also, electron-withdrawing substituents on the phenyl ring enhance the formation of the N-arylhydroxylamine (N-AHA) intermediates. Suppression of these intermediates is well looked into and has been successful when metal salts that can adopt multiple oxidation states are used, such as vanadium and molybdenum oxides.3–5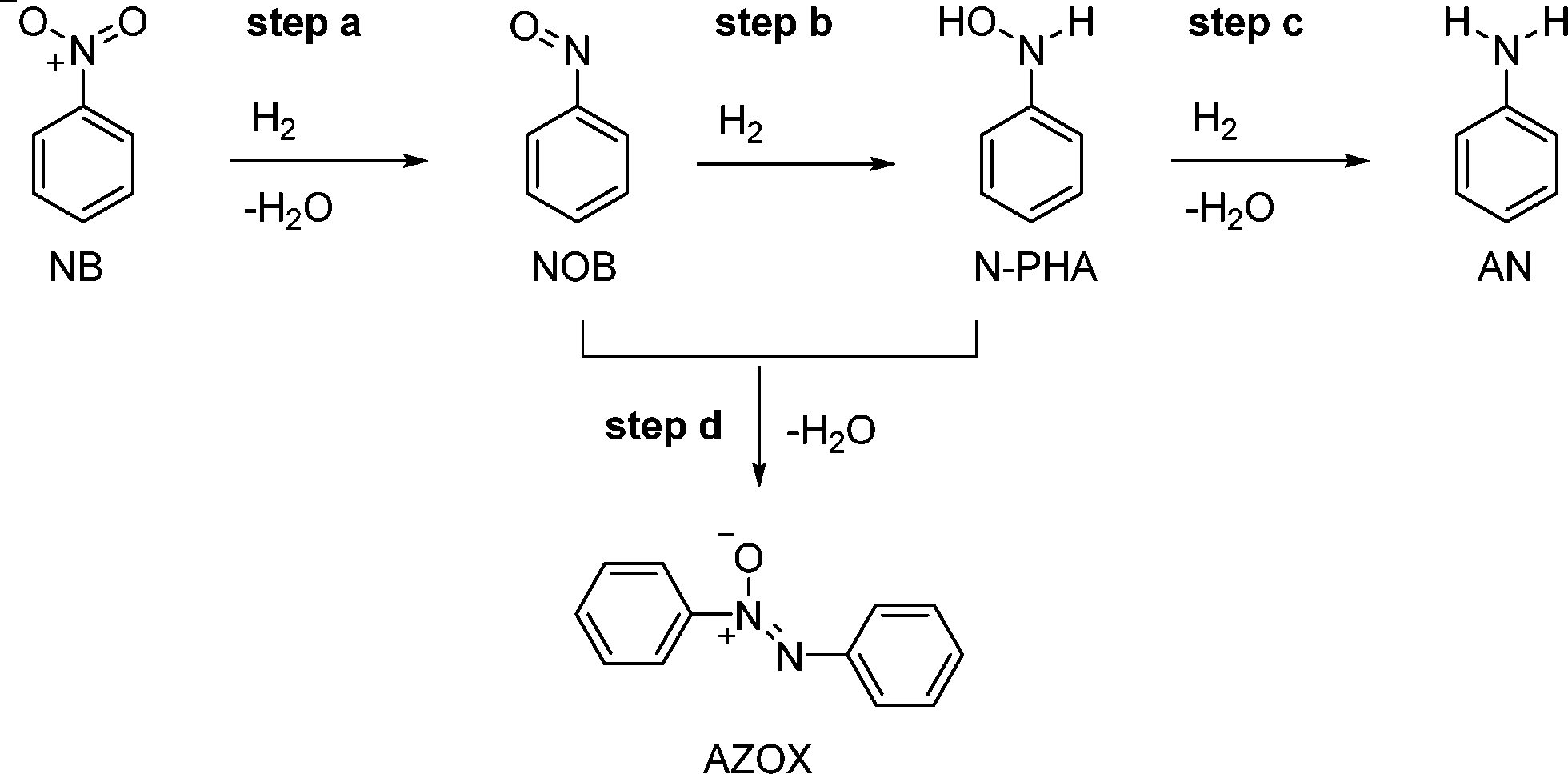 | ||
| Scheme 1 Reaction pathway in the Pt-catalysed hydrogenation of nitrobenzene with intermediates. | ||
Even though considered unwanted in aniline production, arylhydroxylamines are versatile compounds with multiple synthetic applications. For example, in the presence of sulphuric acid, N-PHA can rearrange into aminophenols, called the Bamberger rearrangement.6 The hydroxylamine group can add to double and triple bonds in a gold-catalysed addition reaction.7 Furthermore, N-PHA is suggested as the active intermediate in biologically active substances8,9 and has been reported as a polymerization inhibitor.10
In the literature, a couple of synthetic routes towards N-PHA are reported, but these procedures have limitations related to ease of application, scale-up and fundamental understanding. Firstly, the oldest synthetic procedure is a non-catalytic reduction using Zn in water saturated with ammonium chloride, which results in an isolated yield of 65%.11 In the presence of catalysts, one example shows the selective conversion of nitrobenzene using a novel nitroreductase system giving excellent selectivities but a poor 60% N-PHA isolated yield.12 Alternatively, stoichiometric transfer hydrogenation with hydrazine over a rhodium-on-carbon catalyst gave an 80% N-PHA yield.13 For the catalytic reduction with hydrogen gas, palladium and especially platinum are preferred due to their high activity and N-PHA selectivity compared to similarly carbon-supported metallic Rh, Ir, Ru and Os catalysts.14–16 Moreover, Pt and Pd are far less prone to aromatic ring hydrogenation when compared with, e.g., Ru.17 Platinum supported by silica has the highest reported N-PHA yield of 80% at 5 °C in ethanol in the absence of any additives.15 Metallic Pt as a transition metal (over Ir, Pd and Rh) and the low reaction temperature were found to be crucial for the high N-PHA yield.
It is long known that the addition of dimethylsulfoxide (DMSO) to Pt catalysts can increase selectivity in the three-phase hydrogenation of NB, but unfortunately it also decreased the hydrogenation rates.14 In one example, platinum supported on carbon is used in the hydrogenation of nitrobenzene with a reported selectivity increase from 26% without DMSO additive to 70% with DMSO additive at partial NB conversion.18 This trend was confirmed by Yasuda et al. They found that the addition of amines (such as triethylamine) can increase the N-PHA yield even further to 99% with 1 bar H2 at r.t.19 However, the catalyst poison DMSO is still required in order to obtain high selectivities.
In our catalyst development studies of supported Pt nanoparticles in the catalytic hydrogenation of nitrobenzene, we found that N-PHA was formed in unusually high amounts (more than 50%) as an intermediate in the preparation of anilines. This occurred when no molybdenum oxide promotor or any other modifier was added.5 For this reason, instead of suppression of N-PHA, we were interested in its selective formation under catalytic hydrogenation conditions. Our goal was to device a methodology for lab-scale preparative production of N-arylhydroxylamines (N-AHA) as easily accessible intermediates for organic syntheses.
In this paper, we report on the selective formation of N-AHAs in the activated carbon-supported platinum nanoparticulate-catalysed hydrogenation of nitrobenzene. Moreover, we will show how reaction conditions, such as hydrogen pressure, temperature and the choice of solvent have an influence on the formation of N-PHA and aniline.
Experimental
Nitrobenzene ACS reagent (>99%), 2-chloronitrobenzene (99%), 4-nitroanisole (97%), 4-nitrobenzonitrile (98%), 4-bromonitrobenzene (99%), 1,3,5-trimethoxybenzene (≥99.0%) and absolute ethanol were purchased from Sigma-Aldrich and used as received. Industrial-grade hydrogen (=99.995%) was supplied by BOC gases. All products were identified by comparison with authentic samples. NanoSelect™ nanoparticulate platinum 0.7% c-Pt/C (surface area BET: 1346 ± 8.5 m2 g−1) and palladium catalyst 0.6% c-Pd/C (BET: 485 ± 9.4 m2 g−1) were dried in an oven at 120 °C for 3 hours before use. A complete catalyst characterisation can be found in the ESI† for both catalysts. 5% Pt/C and 5% Pt/SiO2 were purchased from STREM Chemicals and used as received.Strict reaction conditions have to be applied since N-phenylhydroxylamine thermally disproportionates into nitrosobenzene and aniline. Moreover, N-PHA self-condensates in the presence of oxygen forming the strongly coloured azoxybenzene.
All measurements were performed in the kinetic regime by vigorous stirring at 1500 rpm with an overhead stirrer. Varying the stirring speed between 500 and 3000 rpm did not affect the reaction rate. Catalysts of the NanoSelect™ type prepared via reduction–deposition (0.7% c-Pt/C and 0.6% c-Pd/C) have the Pt and Pd crystallites deposited in an eggshell fashion20,21 on the activated carbon support, so that internal diffusion limitations and heat transfer constraints were not considered.
Autoclave catalytic hydrogenation of nitrobenzene
62 mg of 0.7% c-Pt/C, 2.75 g (22.3 mmol) of nitrobenzene (S/Pt = 10 × 103) and 80 mL of absolute ethanol were added to a 100 mL stainless steel autoclave equipped with a heating jacket, a hydrogen supply system, a sampling unit, and a mechanical stirrer. This mixture was heated to 30 °C after which the reactor was flushed with hydrogen and pressurized to the desired absolute pressure. The reaction was initiated by switching on the mechanical stirrer at 1500 rpm. The pressure was kept constant with a mass-flow controller (which also logs the total gas consumption), and the temperature was kept at 30 °C during the reaction. The reaction was stopped at the required hydrogen uptake (600 mL), after which the hydrogen pressure was released. The reactor was opened and 10% 1,3,5-trimethoxybenzene was added as an internal standard. A 1H NMR spectrum of the reaction mixture was obtained within 1 hour by placing three drops of the reaction mixture into an NMR tube with 0.6 mL of methanol-d4. Reaction profiles were measured from samples which were withdrawn from the autoclave during the reaction and analysed by 1H NMR spectroscopy and/or by GLC measurements, without any further sample treatment.Schlenk tube catalytic hydrogenation of nitrobenzene
62 mg of 0.7% c-Pt/C was added to a 100 mL Schlenk flask, and an inert atmosphere was created by repetitive evacuation and argon introduction; 40 mL of solvent was gently added under a stream of argon. This solution was stirred at 600 rpm with a magnetic PTFE stirring bar for 5 minutes, after which 22.3 mmol of nitrobenzene was slowly added to the reaction mixture. The flask cap was replaced by a septum, and the mixture was degassed three times, replacing argon for hydrogen via introduction of a hydrogen-filled balloon (connected to a needle). This resulted in a total hydrogen pressure of about 1 bar at the start. The mixture was stirred at room temperature, and the balloon was left connected to the flask overnight. Full reduction of nitrobenzene is equal to 1 L of hydrogen consumption under standard conditions. After 16 hours (overnight), the reaction was stopped and the catalyst was filtered off over a funnel with filter paper. Samples were withdrawn from the crude reaction mixture and analysed by GLC and 1H NMR after addition of 10% 1,3,5-trimethoxybenzene as an internal standard.Important: all products were kept at room temperature or lower at all times to prevent thermal decomposition forming by-products such as nitrosobenzene, aniline and azoxybenzene. Furthermore, N-arylhydroxylamines are (potentially) carcinogenic and should be handled with care in a well-ventilated hood at all times!
GLC analysis was performed on a Shimadzu-2010 gas chromatograph equipped with an FID detector and a 50 m capillary column, the Agilent's HP-PONA with a dimethylpolysiloxane stationary phase. The injector inlet temperature (split) of the GC was set to 70 °C at which most (thermal) decomposition of N-PHA can be avoided.
NMR spectroscopy was performed on a 500 MHz Bruker machine. All 1H NMR spectra were obtained within 1 hour by addition of three drops of the reaction mixture into an NMR tube with 0.6 mL of methanol-d4. All conversion and selectivity data reported in percentages related to substrate and products in this work are molar percentages.
Results and discussion
The catalyst used in these examples was the NanoSelect™ nanoparticulate platinum solid catalyst supported on activated carbon.22 Nanoparticles were determined to be 2.3 ± 0.3 nm in size by transmission electron microscopy (TEM). The TEM images can be found in the ESI.†Scheme 2 represents the simplified reaction profile of the Pt-catalysed hydrogenation based on compounds observed in solution, thus leaving out elementary reaction step a to NOB.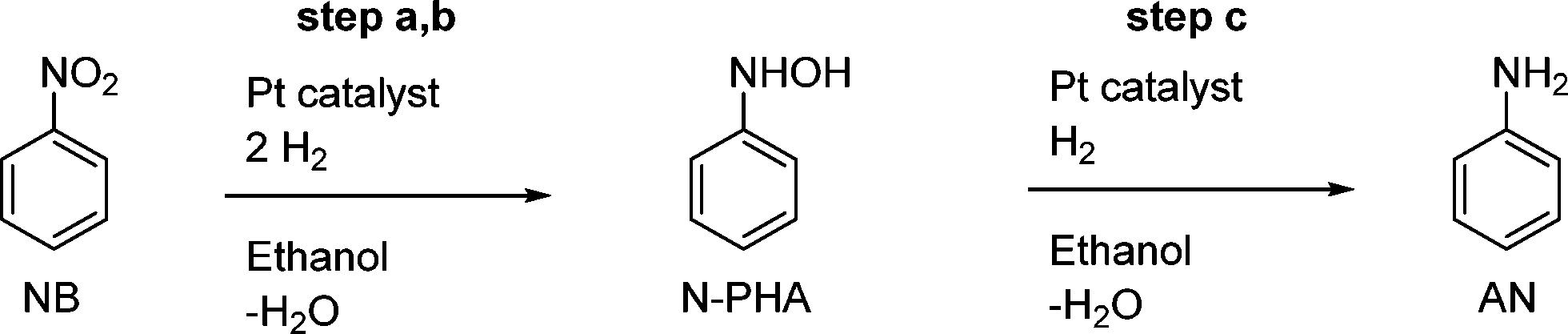 | ||
| Scheme 2 Pt-catalysed hydrogenation of nitrobenzene in ethanol forming N-phenylhydroxylamine at an intermediate stage and aniline. Full conversion will leave aniline as the sole product. | ||
Fig. 1 shows the reaction profile for this reaction as analysed from solution compared with a Pd-catalysed analogous reaction at 4 bar H2 and 30 °C.
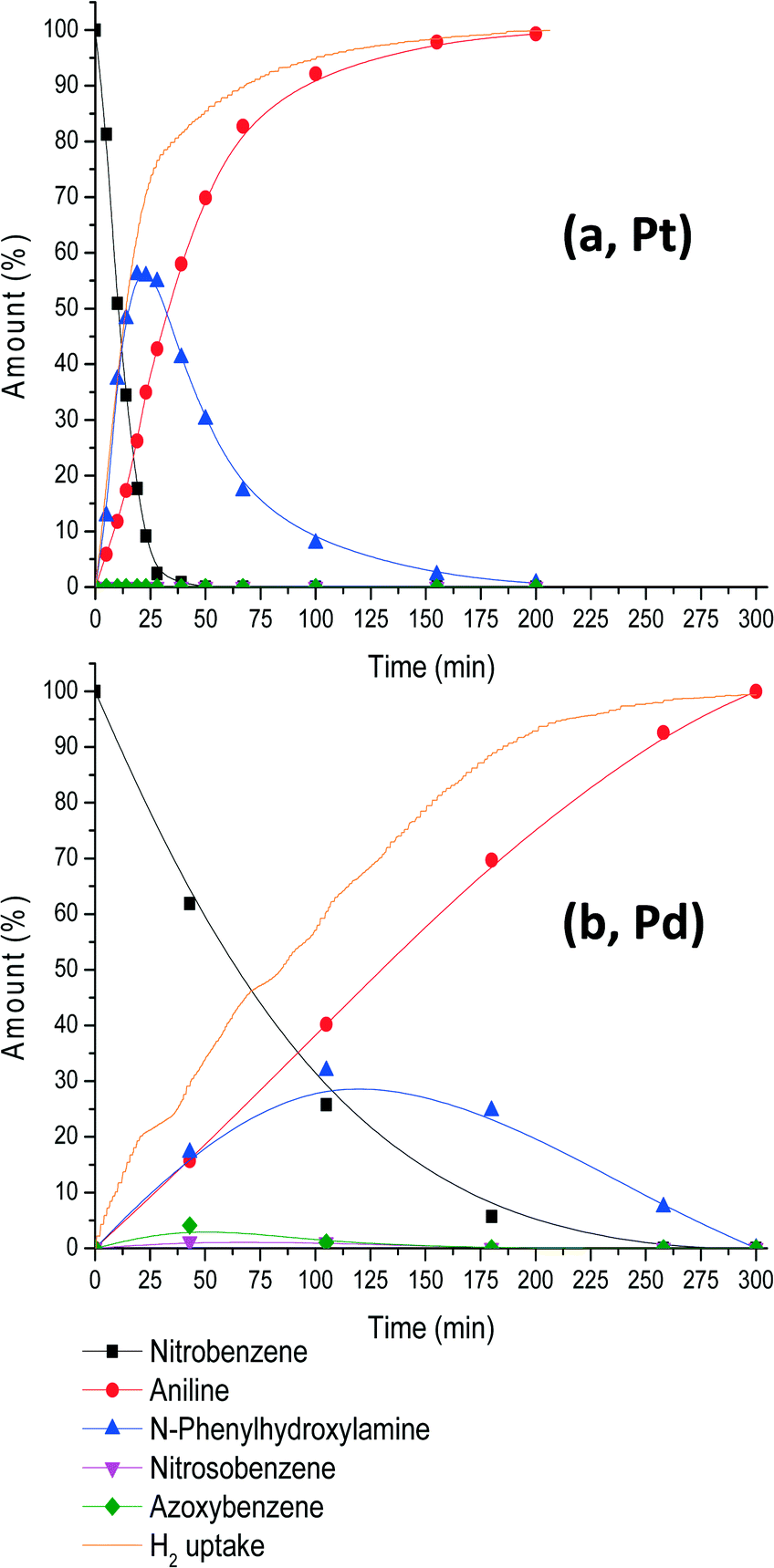 | ||
| Fig. 1 Reaction profile for nitrobenzene hydrogenation in the presence of platinum (a) and palladium (b). Conditions: 22.3 mmol of nitrobenzene (S/C = 10 × 103), 80 mL of EtOH and 4 bar H2 (total pressure) at 30 °C with (a) 62 mg of 0.7% c-Pt/C and (b) 37.1 mg of 0.6% c-Pd/C. Samples were withdrawn from the reactor for GLC and 1H NMR analyses. | ||
The initial reaction rate of the Pt system is fast in terms of nitrobenzene conversion and its corresponding H2 uptake. After complete consumption of nitrobenzene, the reaction slows down significantly and N-PHA, formed up to 55%, is left for hydrogenation. If aniline is the desired product, it is evident that reactions have to be run longer until full hydrogen uptake is reached at 200 minutes. Nitrosobenzene (NOB) is formed on the catalyst's surface but is not detected in solution under these reaction conditions.23 Azoxybenzene (AZOX) formation is largely suppressed because it is formed by the condensation of NOB and N-PHA. Trace amounts of AZOX that are observed by NMR and GLC analyses are probably formed after the samples have been withdrawn from the reactor exposing them to light and oxygen. A measurement of the samples a few days later revealed that all N-PHA had disappeared and AZOX became the main constituent. Therefore, to prevent measuring decomposition products, all measurements were performed within 1 h after sampling.
Pd nanoparticles (15.2 ± 1.6 nm) supported on activated carbon were also used in a similar experiment under the same conditions and substrate-to-catalyst ratio (Fig. 1b). Notably, full conversion to aniline takes place within 300 minutes compared to 200 minutes for the Pt-catalysed hydrogenation. NB is consumed within 260 minutes, whereas for Pt this is only 30 minutes. Consequently, the intermediate build-up of N-PHA is much lower for Pd (30%) compared to Pt (55%). Therefore, further investigation into the selective formation of N-PHA was focused only on Pt-nanoparticle catalysis.
One reason for the high selectivity compared to the literature values given in the introduction is the high substrate-to-catalyst ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The fact that less catalyst and a low metal on support loading are beneficial for the N-PHA yield was already reported by Rylander et al. in 1970.14 In this work, the S/C ratio was set at 10000 to have reasonable conversion times but still high N-PHA selectivity.
000. The fact that less catalyst and a low metal on support loading are beneficial for the N-PHA yield was already reported by Rylander et al. in 1970.14 In this work, the S/C ratio was set at 10000 to have reasonable conversion times but still high N-PHA selectivity.
Reaction temperature and hydrogen pressure were varied to investigate the influence on N-PHA formation with the Pt nanoparticulate catalyst. The results are reported in Tables 1 and 2, respectively. In Table 2, the NB hydrogenation rates R0 have also been included. Intermediates NOB and AZOX are not detected in ethanol under given reaction conditions, so aniline is the only by-product. NB conversion and N-PHA selectivity were determined at partial NB conversion (around 40% of total expected H2 uptake). At this point, the reaction was completely stopped and samples were withdrawn for analysis. The time at which the sample is withdrawn is also reported in the tables and is related to the reaction rate of mainly NB/NOB hydrogenation (steps a and b). The results in Table 1 clearly demonstrate that a decrease in reaction temperature increases the formation of N-PHA. Formation of N-PHA over AN in the early stage of the reaction is favoured at 7, 21 and 30 °C, whereas an increase in the reaction temperature results in a steep selectivity decrease. At 60 °C, N-PHA is formed at almost the same rate as AN, and at 100 °C, its formation becomes drastically suppressed with a selectivity of only 18.4%. These results seem to give a hint towards a higher activation barrier for the final hydrogenation step c to aniline. Lowering the temperature can therefore promote the build-up of the N-PHA intermediate.
| Temperature (°C) | 7 | 21 | 30 | 41 | 60 | 100 |
| Time (min) | 32.5 | 13.5 | 15.9 | 5.0 | 3.5 | 3.0 |
| NB conv. (%) | 59.3 | 62.6 | 72.0 | 57.5 | 52.7 | 50.7 |
| N-PHA select. (%) | 92.6 | 86.9 | 79.1 | 75.3 | 57.7 | 18.4 |
| H2 pressure (bar) | 1.0 | 4.0 | 12.0 | 23.0 | 35 | 50.0 |
| Time (min) | 46.5 | 15.9 | 10.2 | 8.4 | 4.0 | 3.0 |
| NB conv. (%) | 53.0 | 72.0 | 65.1 | 66.2 | 51.2 | 51.7 |
| N-PHA select. (%) | 53.4 | 79.1 | 89.1 | 91.1 | 94.3 | 91.1 |
| R 0 (M s−1 gPt−1) | 0.12 | 0.47 | 0.66 | 0.82 | 1.3 | 1.8 |
As reported in Table 2, an increase in H2 pressure at 30 °C results in a steep increase in N-PHA formation. Selectivity increases from 53.4% at 1 bar to 91.1% at 50 bar H2. Also the rate of N-PHA formation, Ra,b, increases, since the time to reach the specified conversion drops significantly and a first-order rate dependence on H2 is observed from 4 to 50 bar H2 (see Fig. 2). The remarkable lower activity at 1 bar H2 is in line with previously reported work with supported Pt and Pd catalysts.24,25 When the pressure was increased from 23 to 50 bar, the N-PHA selectivity remains at 91.1% with a 94.3% selectivity at 35 bar. An increased individual reaction rate Ra,b (and the decreased rate Rc) results in the dominant N-PHA presence at an intermediate stage. Naturally, aniline will be the sole product at longer reaction times. This relationship between the pressure of molecular hydrogen and N-PHA selectivity has not been reported thus far in the literature. Previously, reported work shows that the intermediate N-PHA concentration does not change over supported Pt catalysts from 10 to 50 bar H2.15,18 Moreover, a reported H2 pressure screening from 0 to 1 bar showed a decrease in selectivity.26
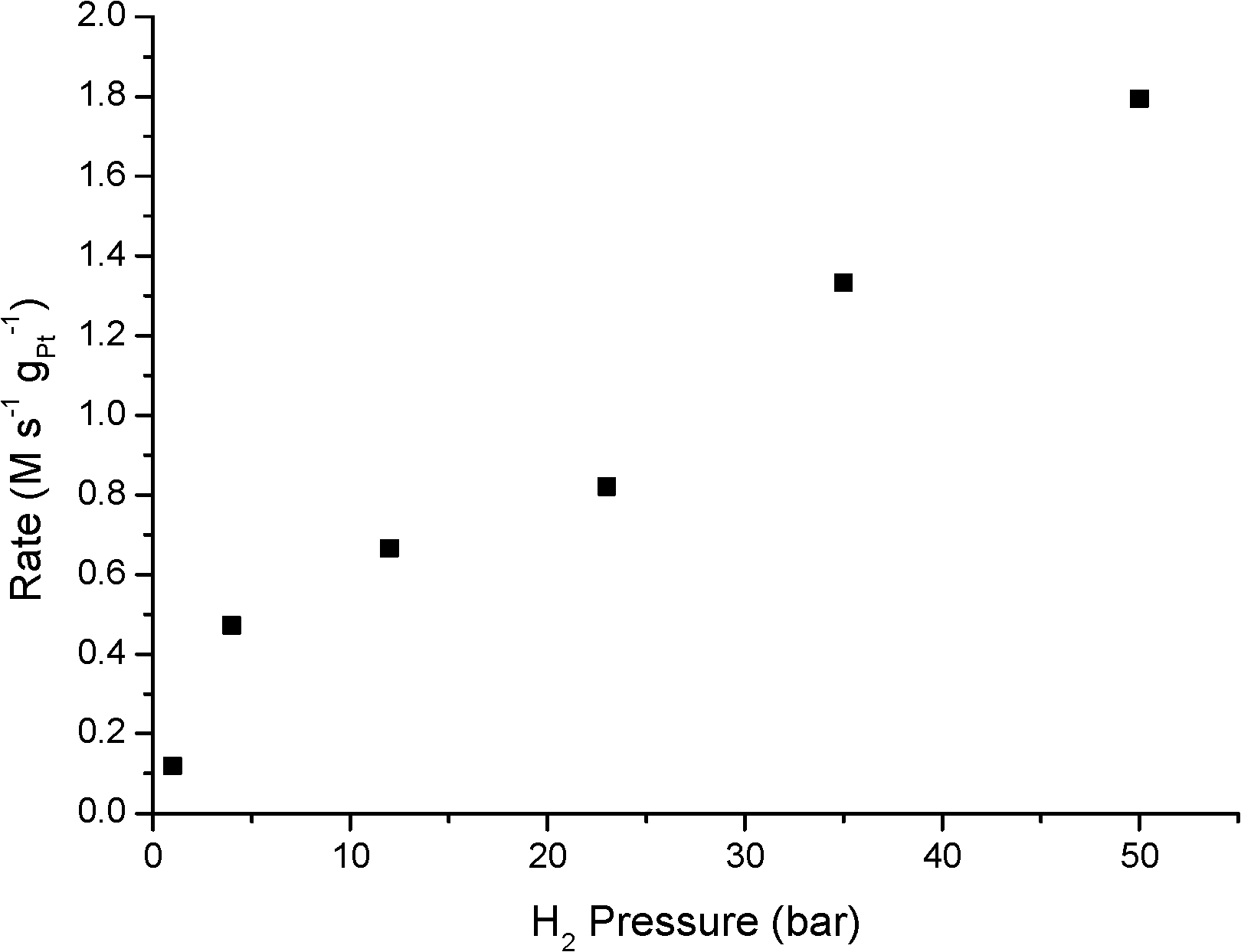 | ||
| Fig. 2 Pt-catalysed hydrogenation of nitrobenzene at 30 °C as a function of H2 pressure. Rate was determined at the data points reported in Table 2, expressed in M s−1 gPt−1. | ||
In order to elucidate why an increased H2 pressure results in higher N-PHA yields, the full reaction profile was monitored for the reaction at 23 bar H2 (Fig. 3). In comparison, the reaction rate, Ra,b, was somewhat higher compared to the reaction rate at 4 bar (Fig. 1). The relative rate of N-PHA formation (Ra,b) and N-PHA hydrogenation (Rc) is Ra,b/Rc = 8.0/0.62 = 12.9 at 23 bar and 30 °C, where at 4 bar H2 this is 3.62/0.82 = 4.5. At 23 bar H2, a maximum N-PHA concentration was reached at 15 min and all NB was consumed within 20 min. At this H2 pressure, full conversion to aniline took 350 min. Surprisingly, the time to full conversion was only 200 minutes at 4 bar H2. The N-PHA hydrogenation rate Rc is actually lower under 23 bar H2 total pressure. This implies negative order dependence in hydrogen on Rc, which is the reason for the increased selectivity to N-PHA. Mechanistically, when N-PHA is adsorbed on a metal surface via its oxygen atom, N–O hydrogenolysis would yield aniline and Pt–O bond hydrogenolysis yields N-PHA. At hydrogen concentrations reaching Pt surface saturation, Pt–O bond dissociation becomes more dominant.15
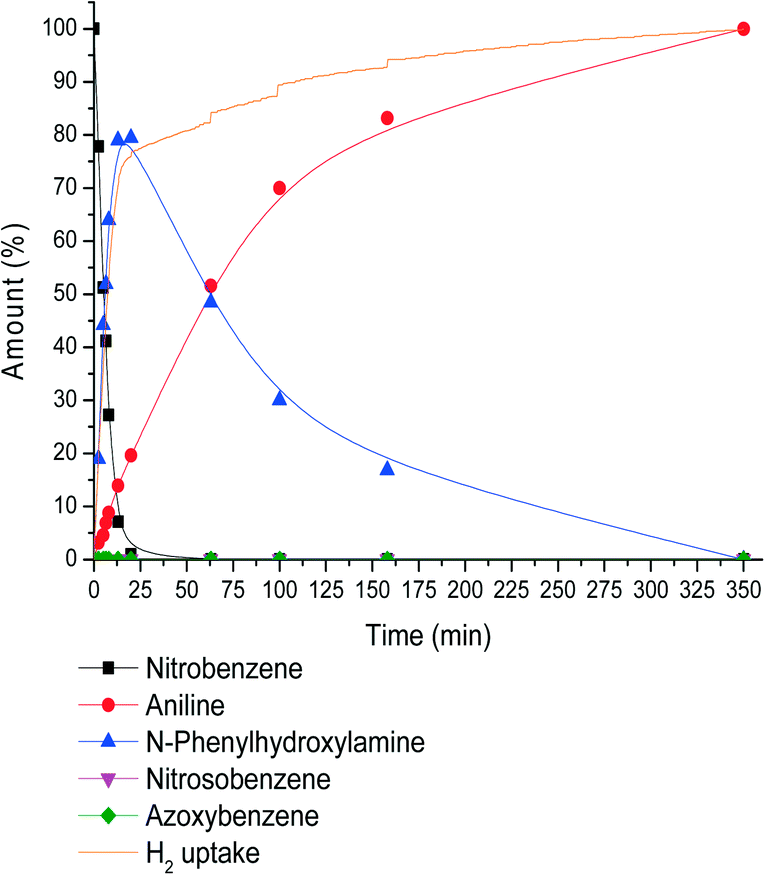 | ||
| Fig. 3 Reaction profile for the Pt-catalysed nitrobenzene hydrogenation at 23 bar H2. Conditions: 62 mg of 0.7% c-Pt/C, 2.75 g (22.3 mmol) of nitrobenzene (S/Pt = 10 × 103) in 80 mL of ethanol at 30 °C. | ||
From the literature, it is well known that activity increases when more polar protic solvents are used; therefore, methanol, ethanol, or isopropanol is typically the solvent of choice.27 This higher activity could be explained by their ability to donate a proton besides activated hydrogen on the surface of the catalyst. Ethanol was chosen as the solvent in the results shown above. However, since the results are slightly different compared to the previously reported work, the influence of solvent on the selectivity was also investigated. After an initial screening, we found that in polar amine solvents, the reaction almost completely stops at 2/3 of total H2 uptake. Fig. 4 compares the H2 uptake of two separate hydrogenation experiments, one in ethanol and one in triethylamine. It appeared that the N-PHA hydrogenation is blocked, most probably by competitive adsorption of the strongly coordinating amine on the catalyst surface. In triethylamine, at the plateau, the N-PHA yield is 80% with 10% AN and 10% AZOX. In ethanol, the N-PHA yield was 55% at the transition point (20 min) with 10% NB and 35% AN.
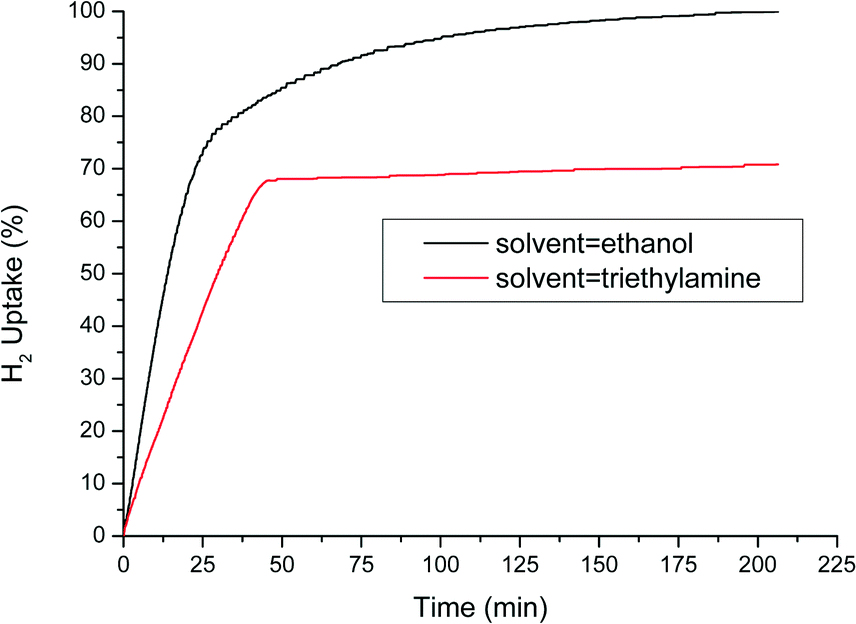 | ||
| Fig. 4 H2-uptake plot for the Pt-catalysed nitrobenzene hydrogenation in 80 mL of (a) ethanol and (b) triethylamine with 62 mg of 0.7% c-Pt/C, 2.75 g (22.3 mmol) of nitrobenzene (S/Pt = 10 × 103) and 4 bar H2 at 30 °C. | ||
To test the influence of solvent, a range of Pt-catalyzed NB hydrogenation experiments was performed at room temperature in combination with H2 supply from a balloon. Table 3 contains the results obtained after a reaction time of 16 h. The formation of NOB is important when looking at high N-PHA selectivity. NOB formation seems to go along with more AZOX, since AZOX is formed from the reaction of NOB with N-PHA (Scheme 1). In comparison, self-condensation of N-PHA is much slower and appears to require oxygen, which is why entries 1–4 in Table 3 show no AZOX formation. That is, they do not release NOB into solution; thus, N-PHA can be formed in significant amounts without AZOX formation. Note that azobenzene was not detected in any of the reactions reported in this work. When NB was hydrogenated in triethylamine at 20 bar H2, 16.5% AZOX was still formed, which means that NOB is still formed at elevated pressures.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | NB conv. (%) | NOB (%) | N-PHA (%) | AN (%) | AZOX (%) | N-PHA select. (%) |
| 62 mg of 0.7% c-Pt/C, 22.3 mmol of nitrobenzene (mol ratio, S/Pt = 10 × 103), 40 mL of solvent, 1 bar H2 total pressure (balloon) at r.t. 1H NMR yields, 10% 1,3,5-trimethoxybenzene used as an internal standard.a S/Pt = 1.0 × 103. | |||||||
| 1 | Ethanol | 51.6 | 0 | 31.4 | 20.2 | 0 | 60.9 |
| 2 | Isopropanol | 58.1 | 0.9 | 34.7 | 22.6 | 0 | 59.6 |
| 3 | Toluene | 34.1 | 0 | 15.8 | 18.4 | 0 | 46.3 |
| 4 | n-Hexane | 31.8 | 0 | 1.3 | 30.6 | 0 | 4.1 |
| 5 | Triethylamine | 80.8 | 1.2 | 69.3 | 3.6 | 6.8 | 85.7 |
| 6a | 5% Pt/C | 65.2 | 0.7 | 52.4 | 4.7 | 7.5 | 80.2 |
| 7a | 5% Pt/SiO2 | 91.9 | 4.1 | 30.3 | 50.6 | 6.9 | 33.0 |
| 8 | Pyridine | 30.3 | 0 | 29.9 | 0 | 0.4 | 98.7 |
| 9 | Piperidine | 19.1 | 3.3 | 12.9 | 0 | 2.9 | 67.5 |
| 10 | n-Butylamine | 56.1 | 2.5 | 28.7 | 0 | 24.9 | 51.2 |
| 11 | Aniline | 39.2 | 0 | 16.1 | 23.1 | 0 | 41.1 |
As reported in Table 3, ethanol and isopropanol show, respectively, 51.6% and 58.1% conversion in 16 hours. In comparison, n-hexane, as a non-polar solvent, showed a somewhat lower activity with a conversion of only 31.8%, and the formation of N-PHA is largely suppressed. Among the amine solvents, triethylamine is most activating (high Ra,b) with a relatively high selectivity of 85.7% but with still considerable NB conversion in 16 h. Variation of the H2 pressure in experiments performed at 1 and 23 bar H2 in triethylamine showed that the overall yield was limited to 80% N-PHA due to NOB and AZOX formation. These yields of up to 80% N-PHA are the same as the results obtained in ethanol at high H2 pressure at 30 °C, but in ethanol, aniline is the only by-product.
Little is reported about the role of solvents and their individual performance in the three-phase hydrogenation of nitrobenzene. Quantum mechanical PM3 calculations of solvent–substrate adducts have been performed in the literature to correlate empirical reaction rates with effective charges on NB/NOB/N-PHA atoms when dissolved in ethanol.28 Naturally, ethanol can form hydrogen bonds with the atoms of the nitrogen group of all the NB hydrogenation intermediates presented in Scheme 1. Interestingly, the N-PHA–ethanol adduct showed a lowered effective positive charge of the nitrogen atom (qef = 0.119) compared to a solvent-free environment (qef = 0.127). This was not the calculated result for NB and NOB ethanol adducts, where the effective charge on nitrogen increased. This could indicate a stabilizing effect of N-PHA in alcoholic solvents.28 Since hexane and toluene are not capable of forming hydrogen bonds with N-PHA, N-PHA is detected in much lower quantities during the course of the reaction.
Besides nitrobenzene solubility, naturally also the hydrogen solubility is of importance, especially since an increase in the pressure of hydrogen resulted in an increase in N-PHA selectivity. However, the H2 solubility in hexane is much higher than in ethanol, isopropanol and toluene, but the selectivity is substantially lower.27 The different levels of hydrogen dissolved in the liquid seem to be of minor importance when comparing the solvents.
To investigate how the NanoSelect™ catalyst compares with other commercially available Pt catalysts, 5% Pt/C and 5% Pt/SiO2 were used as reported in Table 3, entries 6 and 7. More Pt (S/Pt = 1000) was introduced, as much lower activities were observed. Besides being intrinsically less active, reasonable selectivities were obtained with 5% Pt/C and 5% Pt/SiO2, yielding 52.4% and 30.3% N-PHA, respectively. Comparison of hydrogenation activity and N-PHA selectivity between 0.6% c-Pd, 0.7% c-Pt/C and 5% Pt/C at varied H2 pressure is reported in the ESI† in a separate table. It shows that the response when increasing H2 pressure from 4 to 23 bar at 30 °C is similar for the 5% Pt/C catalyst. However, it is still at a much lower initial rate of 0.036 M s−1 gPt−1 compared with 0.82 M s−1 gPt−1 for the colloidal 0.7% c-Pt/C at 23 bar and 30 °C.
When other nitroarenes were tested, we combined both strategies so as to obtain maximum yields of the corresponding N-AHAs using the NanoSelect™ Pt catalyst. This meant adding small amounts of amine to the reactions in EtOH at high pressure (50 bar H2). As an amine derivative, N,N,N′,N′-tetramethylethylenediamine (TMEDA) was found very efficient when added in small quantities to the reaction mixture. 1% TMEDA was sufficient; addition of more did not result in a further increased N-PHA yield. A solvent mixture of EtOH and THF (1/1, v/v) was chosen because of the poor solubility of some of the substituted nitrobenzene substrates tested, e.g. 4-nitroanisole in absolute ethanol. The experimental procedure is fully described in the ESI.† When nitrobenzene was hydrogenated under these conditions, the N-PHA yield increased to 97.1% as reported in Table 4, entry 1.
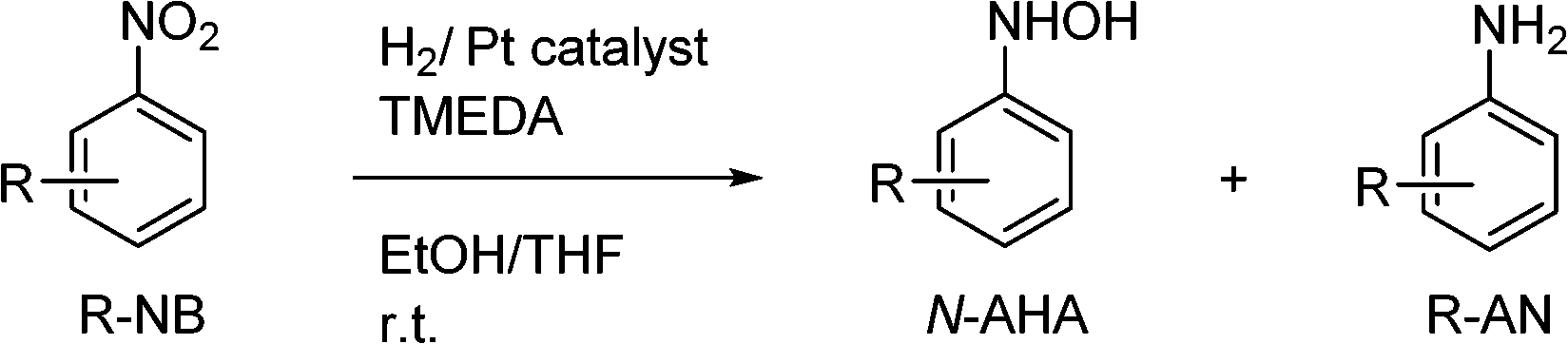
2-Chloronitrobenzene was chosen as a substrate with an electron-withdrawing functional group. As expected, the selectivity for the hydrogenation of 2-chloronitrobenzene to N-2-chlorophenyl hydroxylamine is high with 97.2% (Table 4, entry 2). No hydrogenolysis of the C–Cl bond was observed, not even after 20 h of reaction. Notably, even the C–Br was retained under set hydrogenation conditions with a 98.6% maximum formation of 4-bromo-N-phenylhydroxylamine.
4-Nitrobenzonitrile was chosen as it contains another electron-withdrawing, but reducible, substituent (Table 4, entry 4). Full conversion was reached in only 13 minutes with a maximum N-AHA yield of 98%.
4-Nitroanisole is more challenging due to the electron-donating methoxy substituent, making the nitro functional group less electrophilic. A lowered electrophilicity means lower activity when hydrogenated with the strongly nucleophilic Pt catalyst. For this reason, 96 minutes were required for full conversion of 4-nitroanisole with a corresponding maximum yield of 91.4% 4-methoxy-N-phenylhydroxylamine (Table 4, entry 5).
Entry 6 in Table 4 shows the high chemoselectivity for the nitro group in the hydrogenation of 3-nitrostyrene. An 89.3% yield of the corresponding N-AHA was obtained without much hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (7.5% of 3-ethylaniline is formed).
C bond (7.5% of 3-ethylaniline is formed).
This selected group of nitroaromatics was chosen, but we believe that a much broader group of substituted N-phenylhydroxylamines could be synthesized with high yields.
Finally, a “hot filtration” experiment was carried out to exclude possible leaching of large amounts of platinum into solution. At high pressure and in the presence of TMEDA, the reactivity in terms of nitrobenzene hydrogenation stopped completely after catalyst filtration from the reaction solution and no sign of any additional reactivity was observed (see the ESI†).
Conclusions
In the Pt-catalysed hydrogenation of nitrobenzenes using Pt nanoparticles of the NanoSelect™ type, the selectivity towards the intermediate N-arylhydroxylamines has been optimized. Hydrogenation of nitrobenzene to aniline in ethanol at 4 bar H2 pressure already showed reasonable 79.1% intermediate selectivity of N-PHA, with a maximum intermediate yield of 55%. Increasing the hydrogen pressure to 23 bar significantly increased the intermediate formation to 80% so that N-PHA can be obtained in good yields at an intermediate stage. This is explained by the negative order in H2 concentration for the (over)hydrogenation rate of N-PHA. Performing the experiments in different solvents showed that polar solvents are more activating and selective towards N-PHA because they are capable of forming hydrogen bonds with the substrate. Moreover, amine solvents such as triethylamine and pyridine were found especially selective, because they suppress further N-PHA hydrogenation to a large extent (step c) by competitive adsorption. However, formation of AZOX side products makes the use of amine solvents undesirable.In summary, to obtain high yields of N-PHA in the Pt-catalysed hydrogenation of nitrobenzene, firstly a low temperature is favourable, secondly, hydrogen bonding of N-PHA with the solvent and finally N-PHA substitution from the surface by amines and a high concentration of molecular hydrogen.
With this knowledge, the conditions were optimized in order to convert a variety of nitroarene substrates to N-AHAs. Experiments performed at high H2 pressure (50 bar) with TMEDA as amine additive resulted in excellent (more than 90%) N-AHA yields.
Acknowledgements
We would like to thank BASF Nederland B.V. for their financial support and scientific contribution to this work.Notes and references
- M. Bohnet, Ullmann's encyclopedia of industrial chemistry, Wiley-VCH, Weinheim, 6th edn, completely rev., 2003 Search PubMed.
- F. Z. Haber, Electrochemistry, 1898, 22, 506 Search PubMed.
- P. Baumeister, H. U. Blaser and M. Studer, Catal. Lett., 1997, 49, 219–222 Search PubMed.
- K. Mobus, D. Wolf, H. Benischke, U. Dittmeier, K. Simon, U. Packruhn, R. Jantke, S. Weidlich, C. Weber and B. S. Chen, Top. Catal., 2010, 53, 1126–1131 Search PubMed.
- E. Boymans, S. Boland, P. T. Witte, C. Muller and D. Vogt, ChemCatChem, 2013, 5, 431–434 Search PubMed.
- E. Bamberger, Ber. Dtsch. Chem. Ges., 1894, 27, 1548–1557 CrossRef CAS.
- Y. Z. Wang, L. W. Ye and L. M. Zhang, Chem. Commun., 2011, 47, 7815–7817 Search PubMed.
- C. K. Svensson, Chem. Res. Toxicol., 2003, 16, 1035–1043 CrossRef CAS PubMed.
- P. A. Vyas, S. Roychowdhury, P. M. Woster and C. K. Svensson, Biochem. Pharmacol., 2005, 70, 275–286 Search PubMed.
- V. V. Perez, J. F. Martin and P. V. Roling, EP0240297, 1987.
- O. Kamm, Org. Synth., 1925, 4, 57–58 Search PubMed.
- H.-H. Nguyen-Tran, G.-W. Zheng, X.-H. Qian and J.-H. Xu, Chem. Commun., 2014, 50, 2861–2864 Search PubMed.
- P. W. Oxley, B. M. Adger, M. J. Sasse and M. A. Forth, Org. Synth., 1989, 67, 187–192 Search PubMed.
- P. N. Rylander, I. M. Karpenko and G. R. Pond, Ann. N. Y. Acad. Sci., 1970, 172, 266–275 Search PubMed.
- L. Pernoud, J. P. Candy, B. Didillon, R. Jacquot and J. M. Basset, in Studies in Surface Science and Catalysis, ed. F. V. M. S. M. Avelino Corma and G. F. José Luis, Elsevier, 2000, vol. 130, pp. 2057–2062 Search PubMed.
- M. Tamura, K. Kon, A. Satsuma and K. Shimizu, ACS Catal., 2012, 2, 1904–1909 Search PubMed.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 191, 187–207 Search PubMed.
- S. L. Karwa and R. A. Rajadhyaksha, Ind. Eng. Chem. Res., 1987, 26, 1746–1750 Search PubMed.
- Y. Takenaka, T. Kiyosu, J. C. Choi, T. Sakakura and H. Yasuda, Green Chem., 2009, 11, 1385–1390 Search PubMed.
- P. T. Witte, S. Boland, F. Kirby, R. van Maanen, B. F. Bleeker, D. A. M. de Winter, J. A. Post, J. W. Geus and P. H. Berben, ChemCatChem, 2013, 5, 582–587 Search PubMed.
- G. Vile, N. Almora-Barrios, S. Mitchell, N. Lopez and J. Perez-Ramirez, Chem. – Eur. J., 2014, 20, 5926–5937 Search PubMed.
- P. T. Witte, P. H. Berben, S. Boland, E. H. Boymans, D. Vogt, J. W. Geus and J. G. Donkervoort, Top. Catal., 2012, 55, 505–511 Search PubMed.
- G. Richner, J. A. van Bokhoven, Y. M. Neuhold, M. Makosch and K. Hungerbuhler, Phys. Chem. Chem. Phys., 2011, 13, 12463–12471 Search PubMed.
- V. Holler, D. Wegricht, I. Yuranov, L. Kiwi-Minsker and A. Renken, Chem. Eng. Technol., 2000, 23, 251–255 Search PubMed.
- S. B. Tong, K. F. Odriscoll and G. L. Rempel, Can. J. Chem. Eng., 1978, 56, 340–345 Search PubMed.
- L. Spiegler, US2765342 A, 1956.
- R. A. Rajadhyaksha and S. L. Karwa, Chem. Eng. Sci., 1986, 41, 1765–1770 Search PubMed.
- L. B. Kochetova and M. V. Klyuev, J. Mol. Liq., 2001, 91, 255–260 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Results of the Pt leaching experiment, catalyst synthesis, detailed experimental procedure including NMR data of all the N-arylhydroxylamines, and TEM images. See DOI: 10.1039/c4cy00790e |
| This journal is © The Royal Society of Chemistry 2015 |