
Mesoporous carbon nitride grafted with n-bromobutane: a high-performance heterogeneous catalyst for the solvent-free cycloaddition of CO2 to propylene carbonate
Jie
Xu
*,
Fei
Wu
,
Quan
Jiang
and
Yong-Xin
Li
*
Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology, College of Chemistry and Chemical Engineering, Changzhou University, Gehu Road 1, Changzhou, Jiangsu 213164, PR China. E-mail: shine6832@163.com; liyxluck@163.com; Fax: +86 519 86330135; Tel: +86 519 86330135
First published on 29th August 2014
Abstract
A new type of mesoporous-C4N4-based catalyst (n-butBr/mp-C3N4) was prepared by simple grafting of n-bromobutane. The N2 adsorption–desorption and X-ray diffraction characterizations indicate that, in comparison with the parent mp-C3N4, the textual and structural properties have been well retained by n-butBr/mp-C3N4. In the cycloaddition of CO2 with propylene epoxide, the n-butBr/mp-C3N4 exhibits high catalytic conversion as well as a high selectivity to propylene carbonate. The maximum TOF value obtained over n-butBr/mp-C3N4 is 10.7 h−1 at 6 h under 140 °C, which compares favorably to other reported C3N4-based catalysts. In addition to n-bromobutane, mp-C3N4 materials grafted with other alkyl halides also provide high catalytic activities. According to the Fourier transform infrared and X-ray photoelectron spectroscopy measurements, it is speculated that the catalytic active sites of n-butBr/mp-C3N4 are uncondensed amines and Br anions which originate from the reaction between n-bromobutane and the N-containing heterocycles of mp-C3N4.
1. Introduction
Despite notoriety for global warming, carbon dioxide (CO2) is an abundant, economic, and highly functional carbon resource, and the transformation of CO2 as an environmentally benign and economical C1 building block to valuable compounds has attracted increasing attention in the past several decades.1–5 Of particular significance is the cycloaddition of CO2 with epoxides (e.g. propylene oxide, PO) to five-membered cyclic carbonates (e.g. propylene carbonate, PC).6 These products are among the most important chemicals, which are widely used as aprotic polar solvents, electrolyte components in lithium batteries, and chemical intermediates and monomers in the manufacture of fine chemicals.7–9A wide range of catalyst systems have been explored for the cycloaddition processes of CO2 to cyclic carbonates. Up to now, the most efficient catalysts that have been suggested are ionic liquids (ILs), organometallic complexes, phosphines, organic bases,1,3etc. Notwithstanding their high catalytic performances, the crucial issue associated with such homogeneous materials lies in the difficulty in catalyst recovery as well as product purification.10–12 On the other hand, heterogeneous catalysts, including mixed metal oxides, zeolites, smectites, polyoxonetakate, etc., proposed for the catalytic reactions, suffer from low catalytic activity and/or selectivity,9 thus requiring high pressures, temperatures, and an organic solvent (DMF, toluene, or CH2Cl2).13,14 In this context, it is highly desirable to develop a new heterogeneous catalyst that can combine high catalytic performance along with facile catalyst recovery.
As one of the most appealing and versatile metal-free materials, carbon nitride (CN) has recently received tremendous attention in a variety of fields, including fuel cells,15,16 photocatalysis,17,18 gas storage,19,20 and catalysis,21–23 and has been regarded as a potential candidate to complement conventional carbon materials. The success of CN is mainly attributed to its unique combination of various physicochemical properties.24,25 Especially, the incorporation of nitrogen atoms into the carbon nanostructures endows CN with plenty of basic sites in the form of aliphatic and/or aromatic N species, thereby dictating an intrinsically basic function.26,27 Park et al.28,29 prepared a series of mesoporous CN materials via nanocasting approaches, and revealed that mesoporous CN could activate CO2 molecules in various organocatalysis processes, such as the oxidation of cyclic olefins and aromatic alcohols.30 Likewise, Li et al.31 reported that g-C3N4 loaded on SBA-15 demonstrated high catalytic activity in the cycloaddition of CO2 with epoxides to cyclic carbonates. Very recently, adopting urea as a precursor and adjusting its calcination temperatures, Zhang et al.32 have synthesized various CN samples and investigated the influence of the defect sites of synthesized CN materials on their catalytic behavior in the reaction of CO2 and PO.
The above effort evidences the potential catalytic application of CN materials in the cycloaddition of CO2 with epoxides. However, it is worth noting that, in terms of the weight of catalysts and substrates and catalytic conversions, the specific catalytic activities obtained over the reported CN-based catalysts are relatively low. Meanwhile, to achieve a high catalytic conversion, harsh reaction conditions, such as long reaction time and addition of solvent, are frequently demanded. Herein, we report a new catalyst, mesoporous carbon nitride grafted with n-bromobutane (n-butBr/mp-C3N4), for the solvent-free cycloaddition of CO2 with PO to PC. The catalyst is easily prepared and exhibits high and stable catalytic performance, affording a maximum turnover frequency (TOF) as much as 10.7 h−1. The high catalytic activity of n-butBr/mp-C3N4 is mainly attributed to its high surface area, abundant basic sites, and incorporation of Br species.
2. Experimental
2.1. Preparation of mp-C3N4
The mesoporous C3N4 was prepared according to an established nanocasting method reported by Antonietti et al.22 4 g of cyanamide was dissolved in 16 g of aqueous suspensions (40 wt%) of 12 nm silica spheres (Ludox HS40, Aldrich) under vigorous stirring. The mixture was heated in an oil bath at 50 °C under stirring overnight to remove water. Next, the resultant white solid was ground in a mortar, transferred into a covered crucible, and heated at 3 °C min−1 up to 550 °C and then treated for a further 4 h. Afterwards, the as-synthesized yellow powder was ground and immersed into 200 mL of NH4HF2 aqueous solution (4 mol L−1) for 2 d to remove the template. Then, the dispersion was centrifuged and the yellow precipitate was washed using distilled water and ethanol several times. Finally, the yellow sample was dried at 50 °C under vacuum overnight and the mass of the obtained C3N4 was ca. 1.8 g. The resulting C3N4 sample was designated as mp-C3N4.2.2. Preparation of n-butBr/mp-C3N4
1 g of mp-C3N4 and 10 mL of n-bromobutane were added to a stainless steel autoclave. Then, the reactor was pressurized with CO2 to 1.0 MPa and heated to 100 °C under stirring for 10 h. After that, the mixture was centrifuged and the obtained yellow solid was washed with toluene several times. Finally, the sample was dried at 70 °C under vacuum for 2 h and the resultant solid was labeled as n-butBr/mp-C3N4.2.3. Sample characterization
X-ray diffraction (XRD) patterns were recorded with a Rigaku D/max 2500 PC X-ray diffractometer equipped with a graphite monochromator (40 kV, 40 mA) using Ni-filtered Cu-Kα radiation (λ = 1.5418 Å).Nitrogen adsorption–desorption isotherms were measured at −196 °C using a Micromeritics ASAP 2020 analyzer. Prior to the analysis, the samples were degassed (1.33 Pa) at 150 °C for at least 4 h. The specific surface area was calculated according to the Brunauer–Emmet–Teller (BET) method, and pore size distribution was determined by the Barret–Joyner–Halenda method.
Fourier transform infrared (FT-IR) spectra of the samples were collected in transmission mode from KBr pellets at room temperature on a Bruker Tensor 27 spectrometer with a resolution of 4 cm−1, using 32 scans per spectrum in the region of 400–4000 cm−1. The mass ratio of every sample to KBr was constant at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100.
:100.
X-ray photoelectron spectroscopy (XPS) measurements were performed using a Perkin-Elmer PHI 5000C spectrometer working in the constant analyzer energy mode with Mg Kα radiation as the excitation source. The carbonaceous C 1s line (284.6 eV) was used as the reference to calibrate the binding energies.
CO2 temperature-programmed desorption (CO2-TPD) experiments were performed on a Quantachrome ChemBET-3000 analyzer. 200 mg of the sample was pretreated at 300 °C for 1 h in dry He flow (30 mL min−1), cooled to 50 °C, and then exposed to CO2 for 0.5 h. After purging the sample with He for 0.5 h, the TPD data was recorded from 50 to 550 °C with a ramping rate of 10 °C min−1.
2.4. Catalytic test
The cycloaddition of CO2 with PO to PC was carried out in an 80 mL stainless steel autoclave equipped with a magnetic stirrer. In a typical reaction process, 10 mL of PO, and 0.2 g of the catalyst were added into the reactor. Then, the reactor was pressurized with CO2 to a desired pressure and heated to 140 °C under stirring for 6 h. After the reaction, the autoclave was cooled in ice water and the excess CO2 was vented. The liquid product was separated by centrifugation and analyzed using a GC equipped with a SE-54 capillary column and FID. The filtered catalyst was washed with methanol (50 mL) twice, dried for 2 h under vacuum, and then investigated for its next running. The TOF (mass of synthesized PC per gram catalyst per hour) for each catalyst was calculated as follows:where nPO, MPC, t, and Wcatal. are the molar amount (mmol) of PO, formula weight (g mol−1) of PC, reaction time (h), and the mass of overall catalyst (mg), respectively.
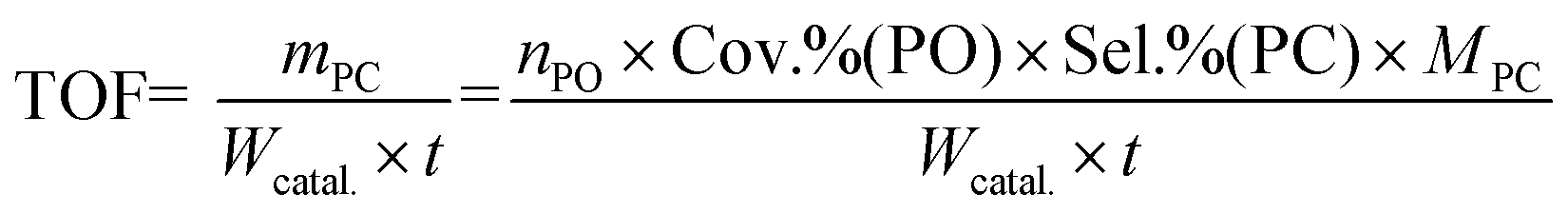
3. Results and discussions
3.1. Structure characterization
The XRD patterns of the bulk C3N4, mp-C3N4, and n-butBr/mp-C3N4 samples are depicted in Fig. 1. All the samples exhibit intensive diffraction peaks at 2θ = 27.4° (d = 0.325 nm), corresponding to the (002) planes, which are characteristic interplanar stacking structures of graphitic-like materials.22,33 Furthermore, a broad peak can be detected near 2θ = 13° (d = 0.680 nm) over C3N4-bulk, which is indexed as (100) planes, i.e. the in-plane structural packing motif.22 For the mesoporous C3N4 samples, the overall intensity of the diffraction peaks decrease, meanwhile the peak at the low angle almost disappears. This means that the intralayer units of C3N4 undergo partial collapse in the mesoporous materials. Nevertheless, the graphitic structures of the parent mp-C3N4 have been well retained in mp-C3N4 after the grafting of n-bromobutane.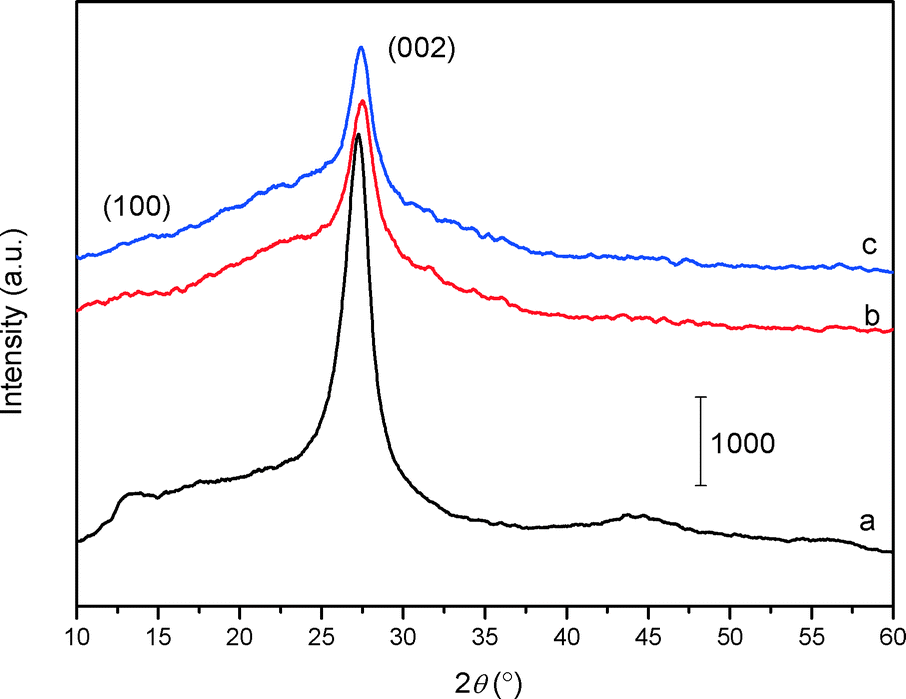 | ||
| Fig. 1 XRD patterns of C3N4-bulk (a), mp-C3N4 (b), and n-butBr/mp-C3N4 (c). | ||
The porous information of mp-C3N4 and n-butBr/mp-C3N4 was analyzed by N2 adsorption–desorption. As shown in Fig. 2A, the pristine mp-C3N4 displays type IV curves with a steep hysteresis loop at p/p0 = 0.45–0.95, indicating that the material has typical mesostructures with a narrow pore size distribution. Moreover, it has also been observed that mp-C3N4 proceeds with further N2 adsorption at p/p0 > 0.95, reflecting that the sample contains a small proportion of macropores. The corresponding pore size distribution of mp-C3N4 (Fig. 2B) shows that the sample has two porous systems. Wherein, the mesopores (~15 nm) could be assigned to the negative templating of the original silica nanoparticles (12–14 nm), while the macropores may originate from the interparticle void. For n-butBr/mp-C3N4, its textual properties including the N2 adsorption–desorption isotherms and pore size distribution have not undergone significant change, except for a minor decline of specific surface area compared with that obtained over mp-C3N4 (Table 1). This confirms that the mesoporous structures of mp-C3N4 have remained after the loading of n-bromobutane.
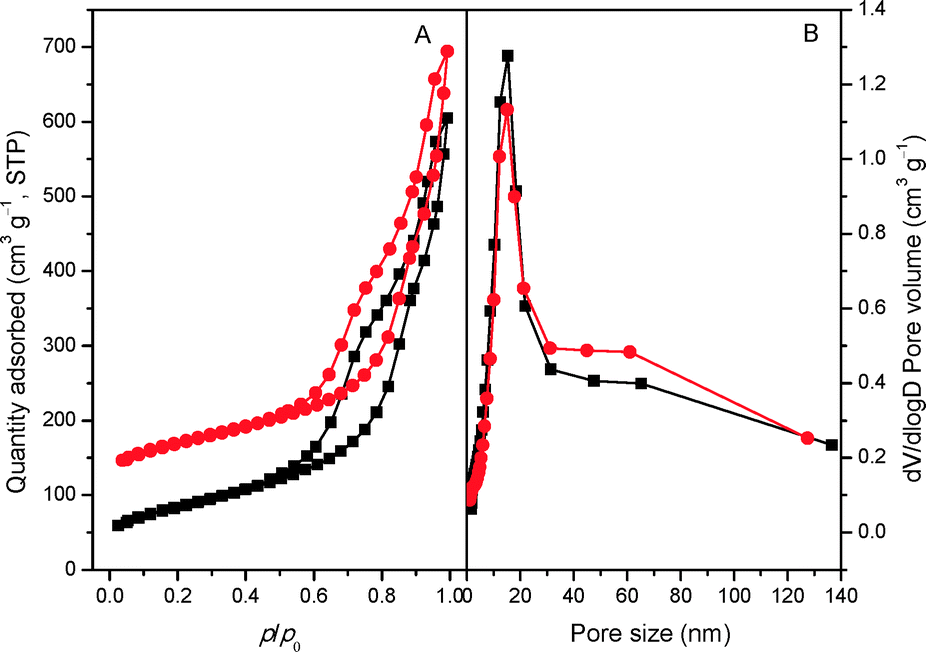 | ||
| Fig. 2 N2 adsorption–desorption isotherms (A) of mp-C3N4 (■) and n-butBr/mp-C3N4 (●) and the corresponding PSD curves (B). The quantity values for n-butBr/mp-C3N4 are shifted by 100 cm3 g−1. | ||
FT-IR and XPS techniques were further employed to investigate the bulk and surface chemical information of the two mp-C3N4 materials, respectively. As described in Fig. 3, the two samples show similar FT-IR spectra. The remarkable multiple bands located in 1200–1600 cm−1 are assigned to the stretching modes of aromatic CN heterocycles.33 The sharp bands at 800 cm−1 are strong evidence of the breathing mode of the triazine units of general C3N4 materials.33,34 Additionally, the broad bands at 3160 cm−1 are associated with the amine species (e.g. terminal –NH2 groups) and/or O–H stretching vibrations of adsorbed water molecules.34,35 Despite these analogies, the FT-IR spectrum of n-butBr/mp-C3N4 gives unique bands at 2220–2260 cm−1, which are ascribed to the typical C–H stretching vibrations, implying that the n-bromobutane has been loaded on the parent mp-C3N4.
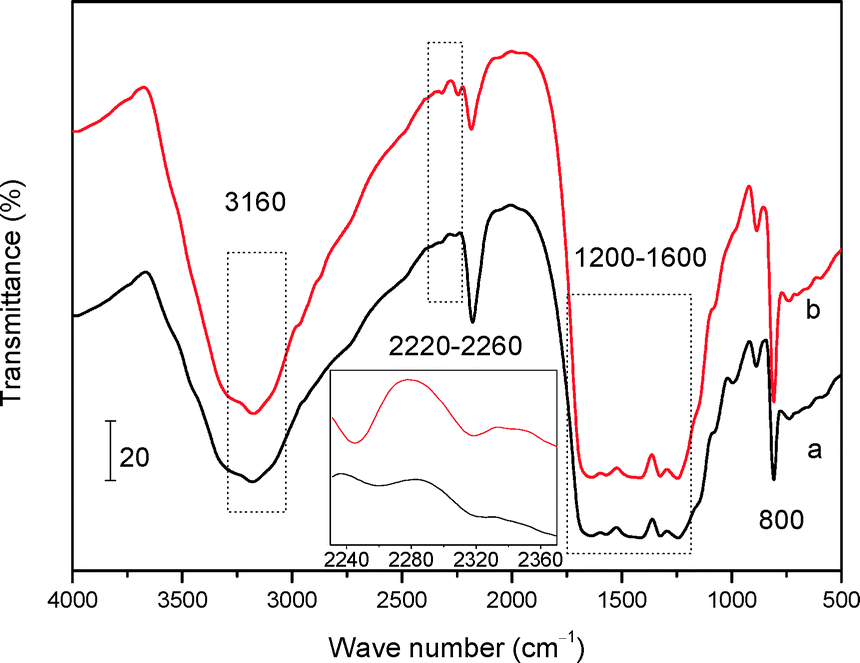 | ||
| Fig. 3 FT-IR spectra of mp-C3N4 (a) and n-butBr/mp-C3N4 (b). The inset is the magnification of the range of 2240–2360 cm−1. | ||
Another interesting finding is the band located at ca. 2320 cm−1, which can be also detected in the spectra of mp-C3N4 and n-butBr/mp-C3N4. Although its intensity is weak, after magnification of the spectra (the inset of Fig. 3), it can be observed that the intensity of the band of 2320 cm−1 acquired on n-butBr/mp-C3N4 is stronger relative to that of the pristine mp-C3N4 sample. The signal is indexed to the trapped CO2 molecules, probably originating from the adsorption and/or absorption of CO2 on the basic surface of mp-C3N4 during the grafting procedure under the high pressure of acidic CO2. This phenomenon agrees well with the result revealed by Park et al.30
The XPS surveys (Fig. 4) show that the main chemical components of mp-C3N4 and n-butBr/mp-C3N4 are carbon and nitrogen, accompanied by a small amount of oxygen that should come from the adsorbed water. Based on the peak areas calculated, the surface molar ratio of C/N in mp-C3N4 is ca. 1.08, slightly higher than that of the bulk sample (0.94, Table 1). The variation probably derives from the addition of silica nanoparticles (as endotemplates), which induce the excessive elimination of N species occurring at the exteria of the C3N4/silica composite during the calcination procedure. A similar phenomenon has also been observed in mesoporous CN materials prepared by other precursors, for instance, guanidinium chloride.34 Besides C and N, the Br element has also been detected in the XPS survey of n-butBr/mp-C3N4. Combining the information and FT-IR result illustrated above, it is apparent that the n-bromobutane compound has been successfully introduced into mp-C3N4.
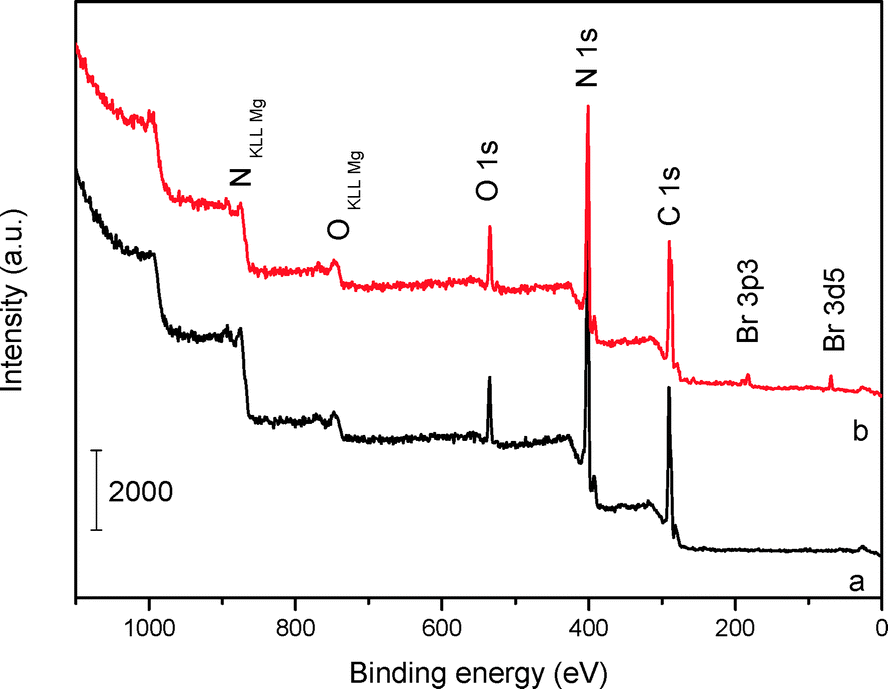 | ||
| Fig. 4 XPS surveys of mp-C3N4 (a) and n-butBr/mp-C3N4 (b). | ||
To further elucidate the detailed chemical states of the two samples, deconvolution of N 1s spectra has been conducted and the result is depicted in Fig. 5. The N spectra can be separated into several peaks, that is, there exist various types of N bonding functions in both mp-C3N4 (Fig. 5A) and n-butBr/mp-C3N4 (Fig. 5B). The main and sharp signal with a binding energy of 400.8 eV is assigned to the aromatic N atoms bonded to two C atoms (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C) in the triazine or heptazine rings.32 The other two peaks at ca. 401.7 and 403.1 eV are attributed to the sp2 N bonded to three atoms (namely tertiary N species, e.g. C–N(–C)–C or C–N(–H)–C), and sp3 terminal N (e.g. uncondensed –NH2) at the edges of graphitic sheets of CN materials, respectively.25,34 In addition, a weak peak with considerably low intensity can also be found at ca. 406.8 eV. The signal with the highest binding energy is associated with the quaternary N species.25 Interestingly, the intensity of quaternary N species becomes much higher from mp-C3N4 to n-butBr/mp-C3N4.
N–C) in the triazine or heptazine rings.32 The other two peaks at ca. 401.7 and 403.1 eV are attributed to the sp2 N bonded to three atoms (namely tertiary N species, e.g. C–N(–C)–C or C–N(–H)–C), and sp3 terminal N (e.g. uncondensed –NH2) at the edges of graphitic sheets of CN materials, respectively.25,34 In addition, a weak peak with considerably low intensity can also be found at ca. 406.8 eV. The signal with the highest binding energy is associated with the quaternary N species.25 Interestingly, the intensity of quaternary N species becomes much higher from mp-C3N4 to n-butBr/mp-C3N4.
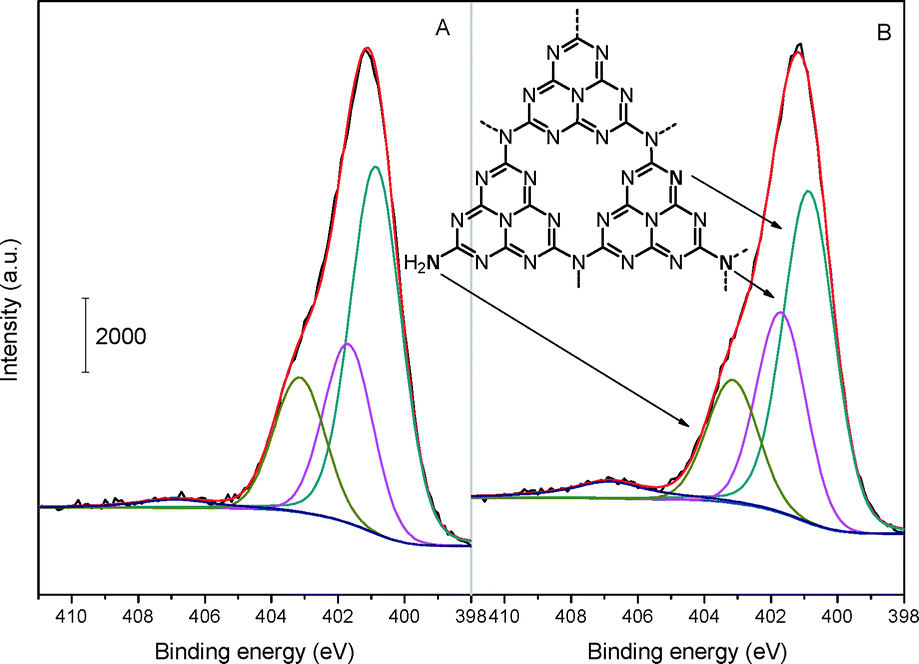 | ||
| Fig. 5 N 1s XP spectra of mp-C3N4 (A) and n-butBr/mp-C3N4 (B). | ||
It is widely recognized that the synthesis of CN materials starting from cyanamide suffers from several consecutive procedures of self-condensation reactions.25,36 During the tandem synthetic routes, there inevitably exist plenty of incompletely uncondensed fragments located at the edges of the graphitic sheets of C3N4 (as confirmed in FT-IR and XPS characterization), or even independent species in the presence of aromatic N-containing compounds, such as pyridine and pyrrole. On the other hand, most ILs are prepared by the reactions between N-containing heterocycles and alkyl halides, triggered by the static force between the active N and halide atoms thereof.37 At this juncture, especially envisioned by the finding from the N 1s XPS, we infer that the n-bromobutane has reacted with the active N-containing heterocycles in mp-C3N4, therein yielding quaternary N compounds and corresponding Br anions.
Concerning the intrinsic presence of N-containing species, CO2-TPD measurement was employed to probe the basicity of mp-C3N4 and n-butBr/mp-C3N4. The thermal conductivity detector (TCD) signal of desorbed CO2 acquired over C3N4-bulk is extremely weak (not shown here), denoting that the basicity in the bulk sample is rather poor, which should be ascribed to its low surface area along with poor porosity (Table 1). By contrast, both mesoporous samples present broad desorption peaks in the range of 160–180 °C (Fig. 6), indicating that the two materials are typical weak bases. These are due to weak chemically and/or physically adsorbed acidic CO2 molecules. Undoubtedly, the mesoporous structures provide mp-C3N4 with numerous N-containing species exposed on its surface, thus facilitating the quantity of accessible basic sites. Moreover, according to peak areas integrated, the density of the basic sites of the pristine mp-C3N4 is 92 μmol CO2 gcatal.−1. For n-butBr/mp-C3N4, the desorption peak shifts to a higher temperature, and meanwhile the basic density improves remarkably (138 μmol CO2 gcatal.−1).
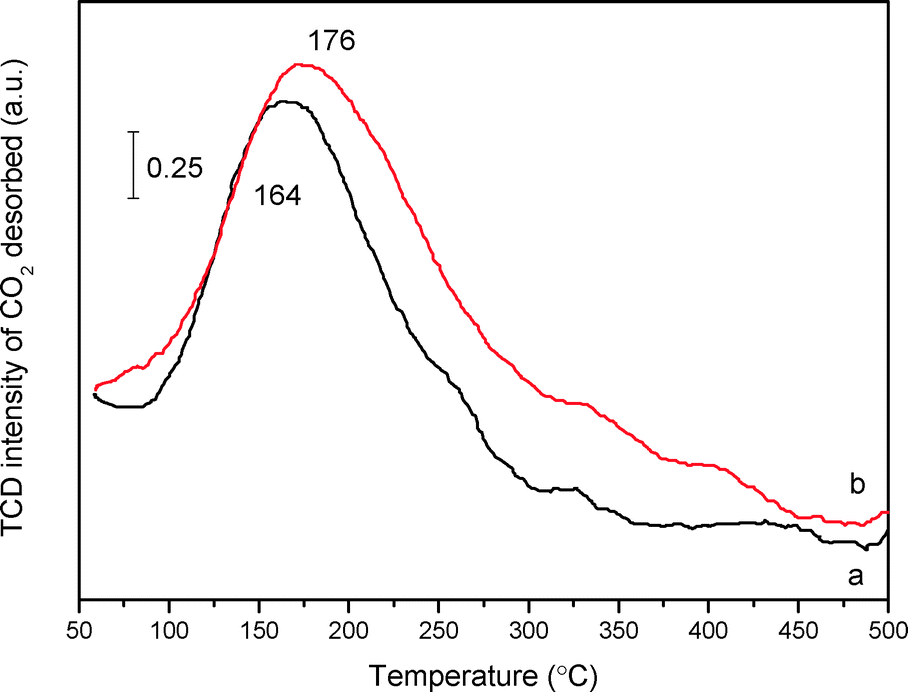 | ||
| Fig. 6 CO2-TPD profiles of mp-C3N4 (a) and n-butBr/mp-C3N4 (b). | ||
As revealed in the above XPS profiles, after the grafting with n-bromobutane, the mp-C3N4 sample has generated quaternary ammonium compounds. In general, quaternary ammonium compounds, e.g. tetrabutylammonium bromide, are typical Lewis base species.38 Regarding this fact, after the introduction of a Br species, the n-butBr/mp-C3N4 possesses basic quaternary ammonium compounds, which thereafter enhance the intensity and quantity of the original basic sites and then stimulate the adsorption of acidic CO2 molecules. Therefore, although the loading of n-bromobutane leads to a minor decrease on the textual parameters of mp-C3N4, the introduction of Br anions enhances the adsorption capacity on CO2 and more importantly, improves its basic intensity simultaneously.
3.2. Catalyst activity
Initially, a blank experiment in the absence of the catalyst was performed and the result revealed that almost no conversion of PO had been achieved (Table 2). After the introduction of mp-C3N4, a minor conversion of 12.5% is obtained, suggesting mp-C3N4 can promote the reaction of CO2 and PO. According to the previous reports,28,32 the catalytic sites are attributed to the uncondensed amino groups (e.g. –NH2 and C–NH–C) which can adsorb and activate CO2 molecules by acid–base coupling. Unfortunately, the catalytic capability of the pure CN material is limited for the cycloaddition reaction. After the grafting of n-bromobutane, the mp-C3N4 catalyst shows an outstanding conversion, affording a maximum yield of PC of 87.7% at 140 °C. Apparently, the incorporation of Br species substantially improves the catalytic activity of the pristine mp-C3N4.| Catalyst | Conv. (%) | Sel. (%) | Yield. (%) |
|---|---|---|---|
| a Reaction conditions: VPO = 10 mL, pCO2 = 2.5 MPa, T = 140 °C, and t = 6 h. b W catal. = 0.2 g. c The molar values of Br are all 0.15 mmol. | |||
| — | <1 | 96.4 | <1 |
| mp-C3N4b | 12.5 | 99.4 | 12.4 |
| n-butBr/mp-C3N4b,c | 88.0 | 99.7 | 87.7 |
| Bu4NBrc | 64.7 | 99.6 | 64.5 |
| mp-C3N4 + Bu4NBrb,c | 82.6 | 99.8 | 82.5 |
Additionally, we have employed the typical quaternary ammonium bromide, i.e. tetrabutylammonium bromide (Bu4NBr) as a catalyst for the catalytic cycloaddition of CO2. For a fair comparison, the mass of Bu4NBr is based on the same molar amount of Br (0.15 mmol) in the n-butBr/mp-C3N4 catalyst. As listed in Table 2, the pure Bu4NBr gives a moderate PC yield of 64.5%, while for the binary catalyst system (mp-C3N4 plus Bu4NBr), the PC yield is up to 82.5%. The above experiments further verify the catalytic promotion of the Br species for the cycloaddition of CO2 with PO. However, it should be noted that, although the acquired catalytic activity with adding quaternary ammonium bromide is superior to the value obtained in n-butBr/mp-C3N4, the catalytic system using bromide is homogeneous, which suffers from intrinsic difficulty in catalyst–product separation and recycling.
In order to acquire an optimal reaction condition, the effects of reaction temperature and CO2 pressures on the catalytic performances have been further evaluated over n-butBr/mp-C3N4. As described in Fig. 7A, under the mild temperature of 110 °C, the catalytic reaction only offers a moderate PC yield of ca. 32%. However, as the temperature is further evaluated, the catalytic activity increases progressively but levels off above 140 °C. In general, the cycloaddition reactions of CO2 with epoxides are typically exothermic processes, and thus an excessive temperate would inhibit the formation of cyclic carbonates. Furthermore, higher temperatures may induce the polymerization of the products and deteriorate the final yields.10,13 Also, it can be observed that the catalytic performance largely depends on the CO2 pressures (Fig. 7B). Improving the temperature leads to a robust increase of PC yield at 0.5–1.5 MPa. Whereas, as the pressure is raised above 2.5 MPa, a significant decrease in PC yield has been found. Such a phenomenon has been reported in other catalytic systems for the cycloaddition of CO2.13,14 A possible explanation is that, under higher pressures, the acidic CO2 dissolves in the basic epoxides and then liquefies resulting from the strong CO2-epoxide complexing. Given the results above, a temperature of 140 °C and CO2 pressure of 2.5 MPa are suitable reaction conditions for achieving the highest PC yield.
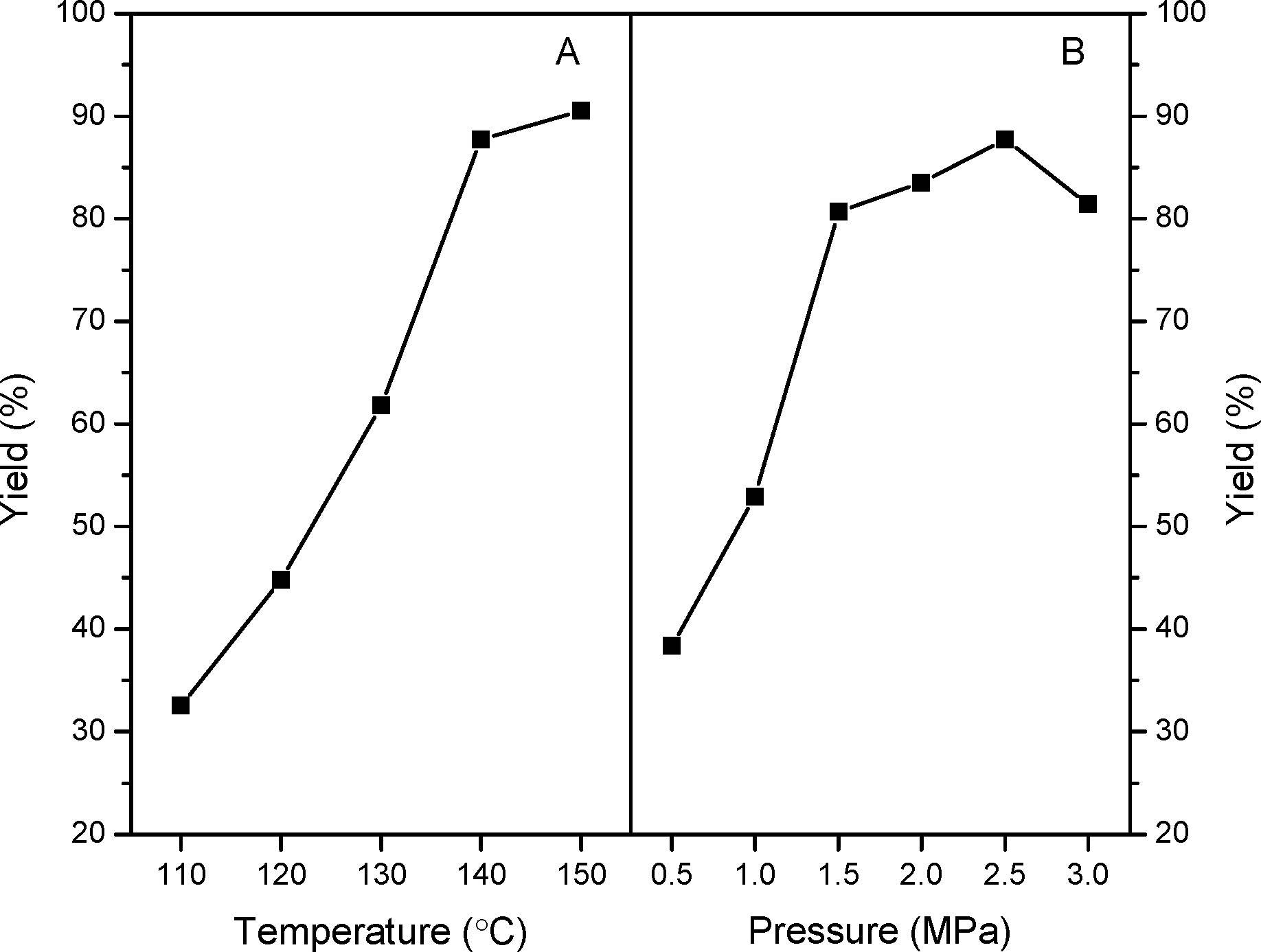 | ||
| Fig. 7 Effects of reaction temperature (A) and pressure (B) on the catalytic performances of n-butBr/mp-C3N4. Reaction conditions: Wcatal. = 0.2 g, VPO = 10 mL, and t = 6 h. | ||
Another important issue associated with the performance of a heterogeneous catalyst is its recovery and reusability. Regarding this point, a series of respective experiments have been conducted. As summarized in Fig. 8, the selectivity for the target PC is above 99%. In terms of conversion, after the first run, the catalyst exhibits an apparent decline accounting for ca. 24% of the original value. As discussed above, the synthesized mp-C3N4 and n-butBr/mp-C3N4 contain a certain amount of uncondensed N-containing compounds, which can activate CO2 molecules. However, some N-containing species may be physically adsorbed on the surface of the catalyst, and are liable to leach when subjected to the catalytic reaction and following recycling process (rinse for several times). Furthermore, it is worth noting that, due to its high boiling point, a small amount of n-bromobutane could not be thoroughly eliminated in the drying procedure, and thus was physically adsorbed on the surface of the n-butBr/mp-C3N4. Consequently, the residual bromide partly contributed to the catalytic activity during the first run. Despite the initial loss, the subsequent catalytic reactions still demonstrate remarkably stable activity.
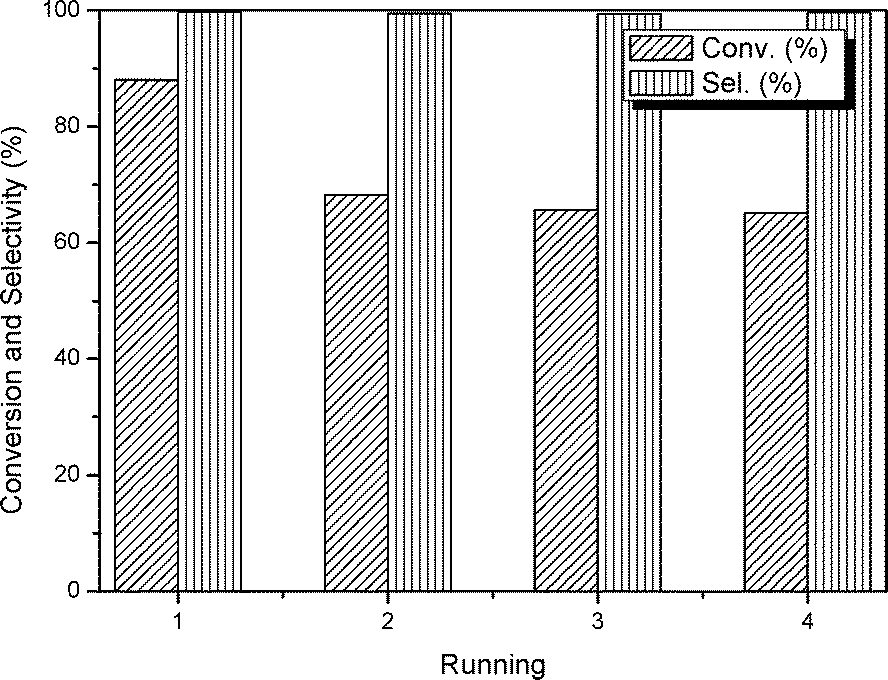 | ||
| Fig. 8 Evolution of catalytic activities during four consecutive tests over n-butBr/mp-C3N4. Reaction conditions: Wcatal. = 0.2 g, VPO = 10 mL, pCO2 = 2.5 MPa, T = 140 °C, and t = 6 h. | ||
As stated above, it is well known that ILs have been regarded as the most promising and robust catalysts for the cycloaddition of CO2 with epoxides, wherein, the ILs with halide anions (Cl−, Br−, and I−) are the common and representative formulations. Moreover, the actual components of such ILs are mainly the halide anions, which have been proven to be able to activate the epoxides via nucleophilic attack. In addition, it has been reported that alkyl ammonium bromides, such as tetraethyl ammonium bromide, can enhance the performance of CO2 activation reactions as promoters or sole catalysts.2,8,39,40 Herein, combining the characterization results above and the reports published previously,8,12,13 a possible mechanism for the cycloaddition of CO2 with PO catalyzed by the present n-butBr/mp-C3N4 has been proposed, as illustrated in Scheme 1. First and foremost, owing to its high surface area and mesoporous architecture, there exist a large number of defect sites in the presence of uncondensed amines and N-containing heterocycles at the edges of graphitic sheets of mp-C3N4. Among them, the N-containing heterocycles react with n-bromobutane, forming quaternary-like N compounds and Br− anions. Next, the CO2 molecule is adsorbed by the amine through an acid–base interaction (step I). After that, the Br− anion attacks a carbon atom of PO and leads to the ring opening, generating a bromoalkoxy anion (step II). The subsequent bromoalkoxy anion nucleophilically attacks the adsorbed CO2 molecule (CO2 inserting reaction) and turns into a linear bromocarbonate (step III). Finally, the bromocarbonate transforms into PC via intramolecular substitution of the bromine (step IV). In this sense, the active amines and Br anions located at the edges of the graphitic sheets of mp-C3N4 essentially promote the adsorption of CO2 and activation of PO reagents, respectively.
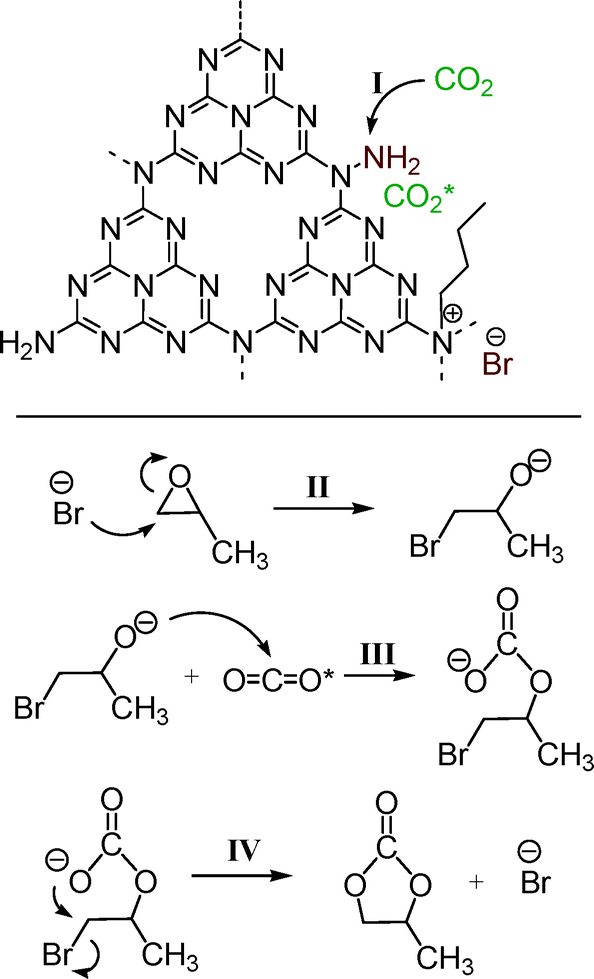 | ||
| Scheme 1 A possible mechanism for the cycloaddition of CO2 with PO catalyzed by n-butBr/mp-C3N4. | ||
Since n-bromobutane can react with the active N-containing species of mp-C3N4 material, we then prepared a series of mp-C3N4 catalysts grafted with various alkyl halides including n-bromopropane, n-chloropropane, n-chlorobutane, and n-bromopentane, and examined their catalytic performances for the cycloaddition of CO2 with PO to PC. As expected, all the synthesized mp-C3N4 catalysts display notable catalytic conversions and selectivities (Table 3). A further comparison reveals that the mp-C3N4 catalysts loaded with a Br-derived alkane (entries 1 and 3) have superior catalytic activities to those with a Cl-derived alkane (entries 2 and 4). This difference is in accord with the nucleophilicity of halide anions; the leaving ability of Br− is higher than Cl−, therein facilitating the intramolecular cyclization step8,13 (step IV of Scheme 1). In this case, it can be deduced that mp-C3N4 grafted by alkyl iodides would provide much higher activity than the present catalysts. Another important factor contributing to the catalytic activity is the length of the alkyl chain. Alkyl halides with long alkyl chains suffer from steric hindrance during the reaction with the N-containing heterocycles of mp-C3N4, which coincides well with the order of activities (entries 1, 3, and 5) listed in Table 3.
| Entry | Alkyl halide | Conv. (%) | Sel. (%) | Yield. (%) |
|---|---|---|---|---|
| a Reaction conditions: Wcatal. = 0.2 g, VPO = 10 mL, pCO2 = 2.5 MPa, T = 140 °C, and t = 6 h. | ||||
| 1 |

|
92.7 | 99.8 | 92.6 |
| 2 |

|
82.0 | 99.5 | 81.6 |
| 3 |

|
88.0 | 99.7 | 87.7 |
| 4 |

|
69.6 | 99.6 | 69.3 |
| 5 |

|
80.2 | 99.5 | 79.8 |
Table 4 lists the catalytic performances of several CN-based materials in the cycloaddition of PO to PC. To the best of our knowledge, the information has covered all recently reported results involving the relevant catalytic systems. Considering that the reaction conditions for the catalyst deviate from each other, the TOF values were calculated for a fair comparison. The CN-based catalysts are synthesized using various precursors, including dicyandiamide, melamine, urea, and aldehyde resins. Every CN material demonstrates catalytic ability to the reaction of CO2 and PO. In sharp contrast with cyanamide, melamine is very difficult to dissolve in most solvents, which results in its poor dispersion with the hydrophilic mesoporous silica template.34,41 Consequently, the surface areas and pore volumes (0.1–0.5 cm3 g−1) achieved over the final MCN (mesoporous carbon nitride) materials are not very high. On the other hand, to expose the abundant accessible active sites, a high surface area and large pore volume are largely demanded. In this sense, the low catalytic activity of MS-MCN and MG-MCN could be due to their underdeveloped porosity. This explanation can also account for the low TOF values obtained in UF-MCN and u-g-C3N4-480. In Table 4, the highest activity is provided by g-C3N4/SBA15, affording a TOF value of 28.1 h−1, which is mainly attributed to its extremely high textual parameters together with abundant active sites. In addition, the contribution of Zn2+ to the catalytic activity should not be neglected. For the catalytic system of n-butBr/mp-C3N4, the final TOF is 10.7 h−1, and is therefore ranked moderate among these CN-based catalysts. Nevertheless, it should be pointed that the amount of PO fed in the present study is the highest among all the entries in Table 4, and the reaction proceeds in the absence of any solvent. Hence, taking into account both TOF value and stable recyclability, it can be anticipated that n-butBr/mp-C3N4 would serve as a potential high-performance catalyst for the cycloaddition of CO2 with epoxides to cyclic carbonates.
| Catalyst | T (°C) | t (h) | V PO (mL) | W catal. (mg) | Yield (%) | TOF (h−1) |
|---|---|---|---|---|---|---|
| a Prepared using disk-shaped 2D hexagonal mesoporous silica as a hard template. b Melamine as a precursor. c Urea and formaldehyde resin as precursors. d Melamine and glyoxal as precursors. e The catalyst was prepared using SBA-15 as a catalytic support and dicyandiamide as a precursor through a chemical-vapor-deposition method. Zn2+ was further doped into g-C3N4/SBA15 as an additive. f Prepared using urea as a starting material without addition of any template. | ||||||
| MS-MCNa,b,28 | 140 | 10 | 1.5 | 20 | 30.6 | 3.3 |
| UF-MCNa,c,28 | 140 | 10 | 1.5 | 20 | 25.8 | 2.8 |
| MG-MCNa,d,28 | 140 | 10 | 1.5 | 20 | 24.5 | 2.7 |
| g-C3N4/SBA15e,31 | 150 | 1.5 | 3 | 100 | 96.1 | 28.1 |
| u-g-C3N4-480f,32 | 130 | 4 | 1.5 | 50 | 23.7 | 2.6 |
| n-butBr/mp-C3N4 | 140 | 6 | 10 | 200 | 87.7 | 10.7 |
Conclusions
In summary, n-bromobutane was successfully grafted on the mp-C3N4, and the high surface area (248 m2 g−1) and large pore volume (0.92 cm3 g−1), along with mesoporous structures (ca. 15 nm), have been well retained over n-butBr/mp-C3N4. As a heterogeneous catalyst, the n-butBr/mp-C3N4 shows high catalytic activity in the cycloaddition of CO2 with PO to PC in the absence of any solvent, affording a maximum PC yield of 87.7% at 6 h. Moreover, other alkyl halides can be incorporated with mp-C3N4, with the synthesized grafted samples also demonstrating high catalytic conversions (≥70%). The catalytically active sites are suggested as the amine species located at the edges of graphitic sheets and halide anions, which promote the adsorption of CO2 and activate the PO molecules, respectively. The formation of halide anions might be contributed from the reaction of N-containing heterocycles between alkyl halides. We think that these findings would constitute a new insight for the development of multifunctional mesoporous C3N4 materials as heterogeneous catalysts for the cycloaddition of CO2 and even wider organocatalysis processes.Acknowledgements
This work was supported by National Natural Science Foundation of China (21203014), Open Foundation of Jiangsu Key Laboratory of Fine Petrochemical Engineering (KF1201), Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology (BM2012110), and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- W.-L. Dai, S.-L. Luo, S.-F. Yin and C.-T. Au, Appl. Catal., A, 2009, 366, 2–12 CrossRef CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- C.-X. Miao, J.-Q. Wang, Y. Wu, Y. Du and L.-N. He, ChemSusChem, 2008, 1, 236–241 CrossRef CAS PubMed.
- K. M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tsang, ChemSusChem, 2008, 1, 893–899 CrossRef CAS PubMed.
- W.-L. Dai, B. Jin, S.-L. Luo, S.-F. Yin, X.-B. Luo and C.-T. Au, J. CO2 Util., 2013, 3–4, 7–13 CrossRef CAS.
- S.-F. Yin and S. Shimada, Chem. Commun., 2009, 1136–1138 RSC.
- W.-L. Dai, S.-F. Yin, R. Guo, S.-L. Luo, X. Du and C.-T. Au, Catal. Lett., 2010, 136, 35–44 CrossRef CAS.
- Y. Zhou, S. Hu, X. Ma, S. Liang, T. Jiang and B. Han, J. Mol. Catal. A: Chem., 2008, 284, 52–57 CrossRef CAS.
- S. Udayakumar, M.-K. Lee, H.-L. Shim, S.-W. Park and D.-W. Park, Catal. Commun., 2009, 10, 659–664 CrossRef CAS.
- L. Han, H. Li, S.-J. Choi, M.-S. Park, S.-M. Lee, Y.-J. Kim and D.-W. Park, Appl. Catal., A, 2012, 429, 67–72 CrossRef.
- X. Zhang, D. Wang, N. Zhao, A. S. Al-Arifi, T. Aouak, Z. A. Al-Othman, W. Wei and Y. Sun, Catal. Commun., 2009, 11, 43–46 CrossRef CAS.
- W.-L. Dai, L. Chen, S.-F. Yin, S.-L. Luo and C.-T. Au, Catal. Lett., 2010, 135, 295–304 CrossRef CAS.
- J. Sun, W. Cheng, W. Fan, Y. Wang, Z. Meng and S. Zhang, Catal. Today, 2009, 148, 361–367 CrossRef CAS.
- L.-F. Xiao, F.-W. Li, J.-J. Peng and C.-G. Xia, J. Mol. Catal. A: Chem., 2006, 253, 265–269 CrossRef CAS.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 CAS.
- J. Liang, Y. Zheng, J. Chen, J. Liu, D. Hulicova-Jurcakova, M. Jaroniec and S. Qiao, Angew. Chem., 2012, 124, 3892–3896 CrossRef PubMed.
- F. Su, S. C. Mathew, L. Möhlmann, M. Antonietti, X. Wang and S. Blechert, Angew. Chem., Int. Ed., 2011, 50, 657–660 CrossRef CAS PubMed.
- J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Fu and X. Wang, Energy Environ. Sci., 2011, 4, 675–675 CAS.
- Q. Li, J. Yang, D. Feng, Z. Wu, Q. Wu, S. S. Park, C.-S. Ha and D. Zhao, Nano Res., 2010, 3, 632–642 CrossRef CAS.
- S. S. Park, S.-W. Chu, C. Xue, D. Zhao and C.-S. Ha, J. Mater. Chem., 2011, 21, 10801–10807 RSC.
- J. Xu, K. Shen, B. Xue, Y.-X. Li and Y. Cao, Catal. Lett., 2013, 143, 600–609 CrossRef CAS.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Chem. Commun., 2006, 4530–4532 RSC.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- X. Jin, V. V. Balasubramanian, S. T. Selvan, D. P. Sawant, M. A. Chari, G. Q. Lu and A. Vinu, Angew. Chem., Int. Ed., 2009, 48, 7884–7887 CrossRef CAS PubMed.
- J. Xu, K.-Z. Long, T. Chen, B. Xue, Y.-X. Li and Y. Cao, Catal. Sci. Technol., 2013, 3, 3192–3199 CAS.
- M. B. Ansari, B.-H. Min, Y.-H. Mo and S.-E. Park, Green Chem., 2011, 13, 1416–1421 RSC.
- B.-H. Min, M. B. Ansari, Y.-H. Mo and S.-E. Park, Catal. Today, 2013, 204, 156–163 CrossRef CAS.
- M. B. Ansari, H. Jin and S.-E. Park, Catal. Sci. Technol., 2013, 3, 1261–1266 CAS.
- Z. Huang, F. Li, B. Chen, T. Lu, Y. Yuan and G. Yuan, Appl. Catal., B, 2013, 136, 269–277 CrossRef.
- Q. Su, J. Sun, J. Wang, Z. Yang, W. Cheng and S. Zhang, Catal. Sci. Technol., 2014, 4, 1136–1138 Search PubMed.
- M. J. Bojdys, J.-O. Müller, M. Antonietti and A. Thomas, Chem. – Eur. J., 2008, 14, 8177–8182 CrossRef CAS PubMed.
- J. Xu, H.-T. Wu, X. Wang, B. Xue, Y.-X. Li and Y. Cao, Phys. Chem. Chem. Phys., 2013, 15, 4510–4517 RSC.
- S. N. Talapaneni, S. Anandan, G. P. Mane, C. Anand, D. S. Dhawale, S. Varghese, A. Mano, T. Mori and A. Vinu, J. Mater. Chem., 2012, 22, 9831–9840 RSC.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- D. Zhao, M. Wu, Y. Kou and E. Min, Catal. Today, 2002, 74, 157–189 CrossRef CAS.
- D. Bai, H. Jing, Q. Liu, Q. Zhu and X. Zhao, Catal. Commun., 2009, 11, 155–157 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- Y. Du, Y. Wu, A.-H. Liu and L.-N. He, J. Org. Chem., 2008, 73, 4709–4712 CrossRef CAS PubMed.
- J. Xu, T. Chen, X. Wang, B. Xue and Y.-X. Li, Catal. Sci. Technol., 2014, 4, 2126–2133 CAS.
| This journal is © The Royal Society of Chemistry 2015 |