
Heterogenization of Pd–NHC complexes onto a silica support and their application in Suzuki–Miyaura coupling under batch and continuous flow conditions†
Alberto
Martínez
a,
Jamin L.
Krinsky
a,
Itziar
Peñafiel
a,
Sergio
Castillón
b,
Konstantin
Loponov
c,
Alexei
Lapkin
c,
Cyril
Godard
*a and
Carmen
Claver
*a
aDepartment of Physical and Inorganic Chemistry, Universitat Rovira i Virgili, C/ Marcel li Domingo s/n, Campus Sescelades, 43007, Tarragona, Spain. E-mail: cyril.godard@urv.cat; Fax: (+34) 977 559563
bDepartment of Analytical and Organic Chemistry, Universitat Rovira i Virgili, C/ Marcel li Domingo s/n, Campus Sescelades, 43007, Tarragona, Spain
cDepartment of Chemical Engineering and Biotechnology, University of Cambridge, New Museum Site, Cambridge CB2 3RA, UK
First published on 11th August 2014
Abstract
The heterogenisation of a new family of Pd–NHC complexes is reported via a straightforward and efficient synthetic procedure. These silica-immobilised materials were successfully applied as catalysts in the Suzuki–Miyaura coupling of aryl chlorides and bromides under mild conditions. The materials exhibited improved stability when the catalytic reaction was run under anhydrous conditions and could be recycled up to five times without significant loss of activity. When the reaction was run within a continuous flow microreactor, these catalysts showed good activity after at least two hours on stream.
Introduction
In the last decade, continuous flow operation has emerged as a powerful tool for chemical synthesis, enabling more efficient transformations in the fine chemicals industrial sector.1 Indeed, continuous flow systems were shown to provide efficient mass transport and mixing2 and to improve heat transfer compared to batch systems, resulting in faster and safer reactions with enhanced product yields.3,4 Furthermore, the combination of improved dynamics and kinetic characteristics of flow chemistry with the use of recyclable catalytic materials represents an innovative synthetic methodology that perfectly fits the current need for more environmentally friendly procedures for transformations catalysed by precious metals such as the Pd-catalysed Suzuki–Miyaura coupling.A desirable strategy would be to fully exploit the features of a specifically designed catalyst in conjunction with modern flow techniques to define useful and practical procedures for the recovery and reuse of the catalysts while obtaining the desired product with minimal cost in terms of time and waste.5 Among the reported catalysts designed for this purpose, many supported molecular6 or particulate7 catalysts have shown high activity and long-term reusability. In these systems, the use of silica supports has proven effective due to their thermal and chemical stability as well as their rigid yet porous structure devoid of swelling properties, a characteristic that makes them compatible with a wide range of solvents.
Pd-catalysed cross-coupling reactions are currently regarded as one of the most important methods for the construction of carbon–carbon bonds.8–10 The application of flow chemistry to these transformations is particularly challenging due to the formation of salts during the reactions, which often leads to rapid clogging of tubular reactors. However, recently several examples of successful metal-catalysed C–C cross coupling reactions under flow conditions have been reported11 employing a broad range of solid supports such as monolithic systems,7,12–18 thin films of palladium nanoparticles,19 Pd/C20 and ionic liquids.21
In the Suzuki–Miyaura process, several methodologies have been reported for the preparation of catalysts under flow conditions. These include immobilisation of a catalyst on a membrane at the centre of a microchannel,22–24 the use of a fluorous-tagged palladium complex25 or the immobilisation of palladium particles on the surface of unfunctionalised and functionalised silica-coated magnetic nanoparticles.26,27 Alternatively, microwave-assisted continuous-flow processes have been described for this reaction.28,29 Organ used capillaries coated with a thin film of palladium,19,30 and Ley and co-workers filled a U-shaped glass tube with the encapsulated catalyst PdEnCat™.31,32 Kirschning and co-workers also reported the use of a homogenous Pd catalyst attached directly through coordination of the metal centre to a polymeric support, namely polyvinylpyridine.12 This system was found to operate via a so-called “release and catch” pathway where the active Pd species is initially released from the support and re-deposited onto the support after the reaction. Excellent results were obtained with this system using a 0.2 mol% catalyst, employing isopropyl alcohol as the solvent and potassium tert-pentoxide as the base. Under flow conditions up to 94% yield could be achieved after 20 hours on stream using aryl chlorides as substrates.
Under batch conditions, soluble Pd catalysts bearing bulky N-heterocyclic carbene (NHC) ligands have proven to be extremely effective for the Suzuki–Miyaura reaction.33,34 Sterically congested NHC ligands are electronically and sterically stabilised and their electron-rich character makes them attractive ligands for a wide range of catalytic transformations. Furthermore, they are resistant to air oxidation and bind very strongly with soft metal centres, which is essential if one is to avoid metal leaching during the catalytic process. Over the last decade, the immobilisation of NHC-based systems has been reported for various types of supports such as polymers, dendrimers, clays and silica.35–40 Various strategies were reported for the anchoring of N-heterocyclic carbenes. In most cases, NHC moieties are covalently attached to the support using alkylic linkers bonded to one or both nitrogen atoms of the imidazolylidene ring (see Scheme 1, complexes 1 and 2, respectively).41–47 These methods usually exhibit advantages in terms of synthetic procedures but they reduce the steric profile of the ligands and hence the performance of the corresponding catalysts. Alternatively, linkers have been attached to the carbon backbone of the NHC ligands, thus avoiding such an alteration but resulting in the need for complex synthetic methods (see Scheme 1, complexes 3 and 4).48–51
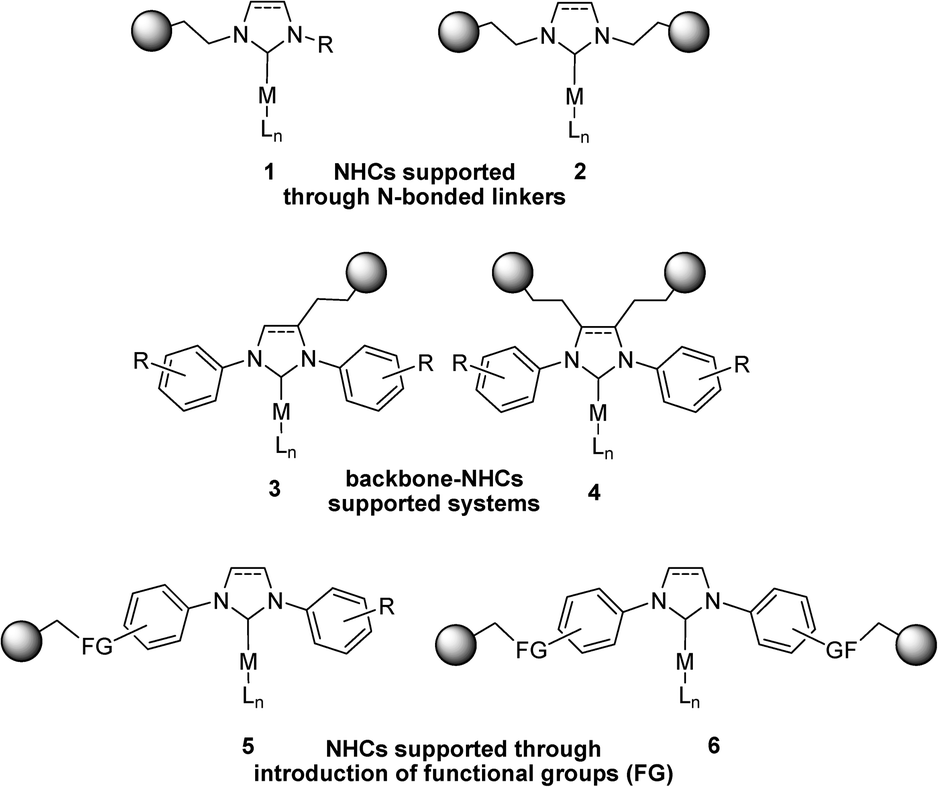 | ||
| Scheme 1 Reported strategies for anchoring NHC-based catalysts onto solid supports. | ||
An interesting alternative for the heterogenisation of such systems involves the introduction of functionality at the N-aryl moieties, as shown in Scheme 1 (complexes 5 and 6). Following this approach, the anchoring can be performed via substitution at the para position of one or both aryl rings of the ligand. Lu and co-workers reported the introduction of an allyl moiety at this position which was subsequently functionalised via thiol–ene methodology and anchored onto an MCM-41 support.52 This supported carbene system was successfully applied in the chemical fixation of carbon dioxide to form cyclic carbonates.
Inspired by the work of Nolan and co-workers, who recently described that para-alkoxy functionalized NHCs provide more active Pd catalysts than their unfunctionalised counterparts in the Buchwald–Hartwig C–N coupling,53 we reported a synthetic strategy for the anchoring of Pd–NHC catalysts via an alkoxy-functionalized ligand.54 Here, we report the synthesis and characterisation of heterogenized Pd–NHC complexes onto amorphous silica via covalent attachment and their application in the Suzuki–Miyaura coupling reaction of aryl chlorides and bromides under batch and continuous flow conditions.
Results and discussion
The synthetic procedure for the preparation of the imidazolium pre-ligands 2a/2b was recently reported by our group and is summarised in Scheme 2.54 This methodology includes the introduction of alkenyl groups through etherification and subsequent photoinitiated thiol–ene addition of mercaptopropyl(triethoxy)silane for the incorporation of triethoxysilyl groups. Using this synthetic route, the two-tethered imidazolium salts 2a/2b were obtained selectively using two equivalents of the thiol. A mixture of one- and two-tethered imidazolium salts 3a/3b and 2a/2b in a ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was also obtained when one equivalent of the thiol was used (Scheme 2). While such a mixture of products is not ideal, the preparation of this material afforded a facile method for testing the effect of the ligand's anchoring mode on the resulting catalyst's activity.
:1 ratio was also obtained when one equivalent of the thiol was used (Scheme 2). While such a mixture of products is not ideal, the preparation of this material afforded a facile method for testing the effect of the ligand's anchoring mode on the resulting catalyst's activity.
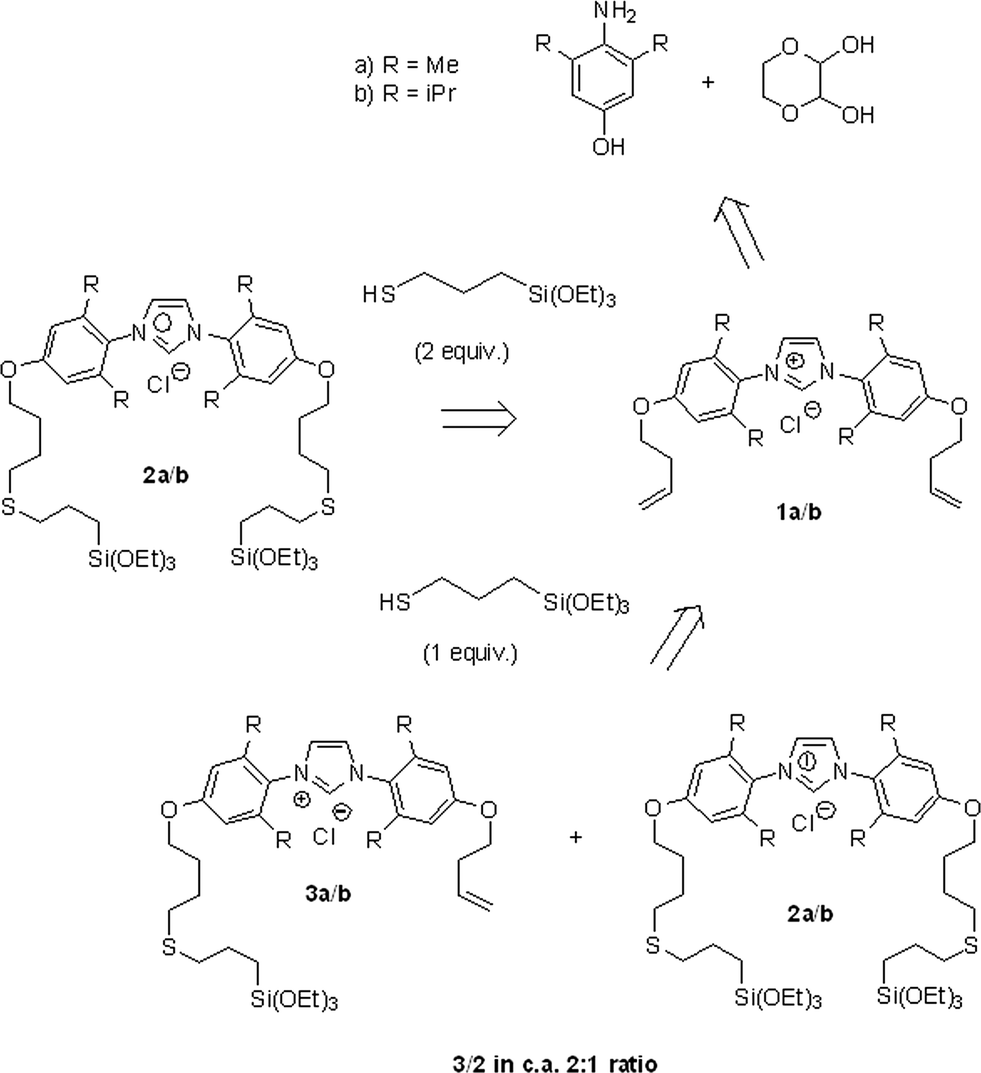 | ||
| Scheme 2 Retrosynthetic scheme for the preparation of the imidazolium pre-ligands 2a, 2b and 3a, 3b.54 | ||
These ligand precursors were then used in the synthesis of the corresponding heterogenised catalysts. The species 2a/2b bearing triethoxysilyl groups were first coordinated to Pd to form complexes 4a/4b by reaction with [Pd(acac)2] in 1,2-DCE at 75 °C for 48 h prior to their immobilization onto silica (see Scheme 3).
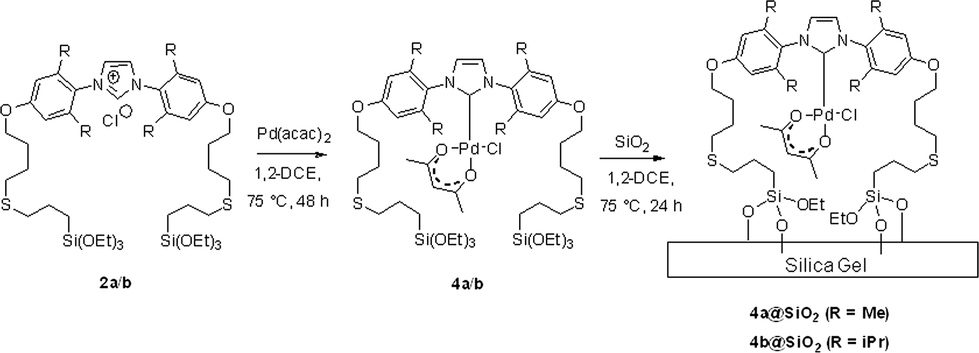 | ||
| Scheme 3 Two-tethered silica gel-bound Pd–NHC complexes 4a@SiO2/4b@SiO2. | ||
During the synthesis, the complexes 4 were not isolated and the crude reaction mixtures containing 4a/4b were used directly for immobilisation onto silica gel, yielding 4a@SiO2/4b@SiO2 after heating the Pd–NHC/silica mixtures at 75 °C for 24 h. To confirm that the formation of complexes 4 was completed the reaction was monitored by 1H NMR spectroscopy which proved the clean, near-quantitative formation of the desired Pd complex under these conditions (see details in ESI).†
Further optimisation showed that the entire synthetic sequence, from thiol addition to alkenyloxy-imidazolium chlorides 1 and then to immobilization of Pd–NHC complexes 4 and 5 onto silica (3 steps), could be performed as a sequential one-pot reaction as shown in Scheme 4.
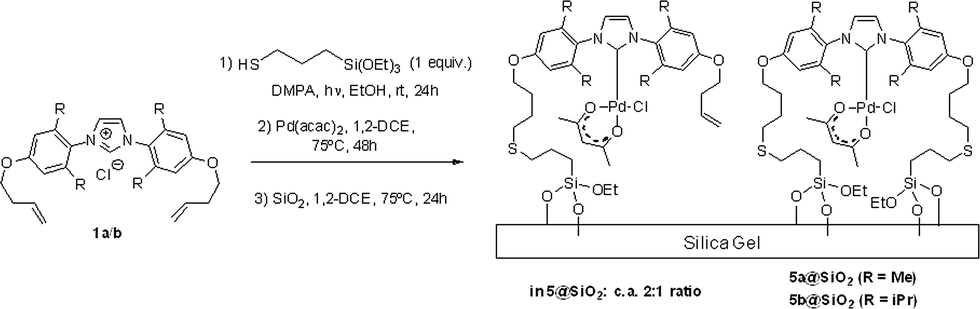 | ||
| Scheme 4 Three-step one-pot sequence used for the synthesis of the one-tethered enriched mixture 5a@SiO2/5b@SiO2. | ||
This methodology was also used for the preparation of the 5a@SiO2/5b@SiO2 materials by reacting 1a/1b with a single equivalent of the thiol reagent. This procedure afforded a mixture of one- and two-tethered imidazolium pre-ligands in a ca. 2:1 ratio, estimated by quantifying the amount of unreacted [Pd(1)(acac)Cl] remaining in solution after the immobilisation procedure.
Finally, the samples of the immobilised catalysts 4@SiO2 and 5@SiO2 were treated with excess MeOSiMe3 in order to cap the remaining accessible hydroxyl groups of the silica surface. The materials obtained are herein labeled 4@SiO2+TMS and 5@SiO2+TMS.
The Pd content of these materials was found to range between 1.7 and 2.6 wt% according to ICP analysis and the results of elemental analysis of 4a@SiO2/4b@SiO2 indicated that the immobilised species are 1:1 NHC–Pd complexes.
To further probe the composition of these materials, a sample of 1b was prepared, incorporating a 13C label at the C2 position by using 13C-enriched paraformaldehyde during the imidazolium-formation step. This modification enabled the electronic environment at the ligand C2 (carbene donor) position to be easily tracked by 13C NMR spectroscopy during the immobilisation and post-immobilisation modification steps. Fig. 1 depicts the 13C{1H} NMR spectra of 1b, 4b, and 5b as well as the CP-MAS NMR spectra of the silica-immobilised 5b@SiO2 and the post-immobilisation modified 5b@SiO2+TMS.
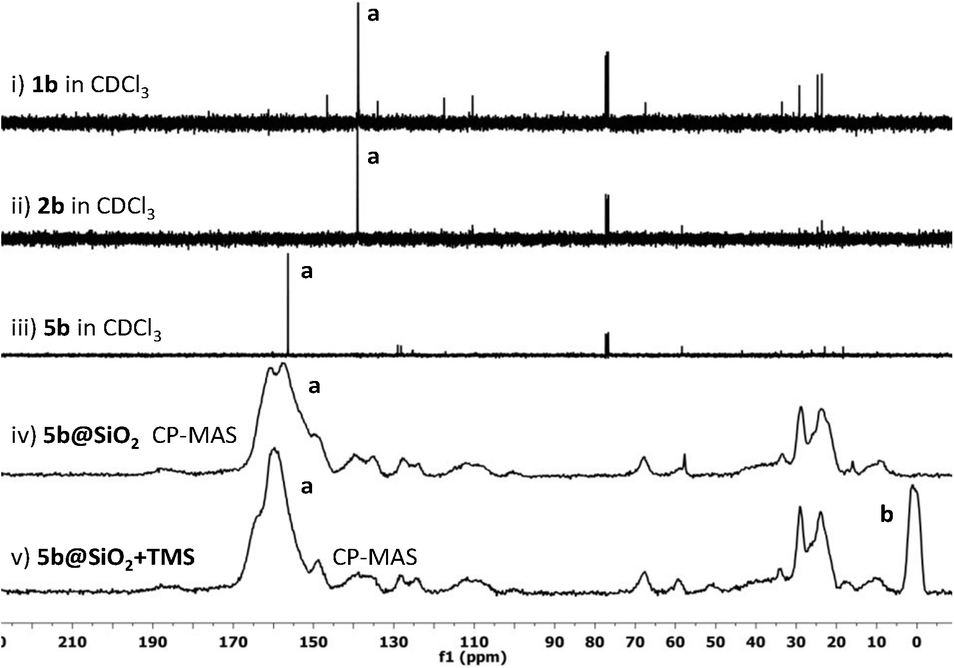 | ||
| Fig. 1 13C{1H} NMR spectra of 13C2-labeled 5b@SiO2+TMS and synthetic precursors: i) imidazolium pre-ligand 1b; ii) triethoxysilyl-functionalised imidazolium pre-ligand 2b; iii) Pd-coordinated imidazolylidene 5b; iv) silica-immobilised Pd-imidazolylidene 5b@SiO2; v) post-immobilisation modified Pd-imidazolylidene (5b@SiO2+TMS). a. The imidazolium/ylidene C2 carbon. b. The peak corresponding to surface SiMe3 groups. | ||
No significant difference in the chemical shift of C2 was observed between the spectrum of the precursor 1b (Fig. 1, i) and that of the product of the thiol–ene reaction 2b (Fig. 1, ii), indicating that the imidazolium C2 carbon's electronic environment is not affected by the introduction of the silyl groups. Upon treatment with Pd, the carbon signal shifted downfield to 160 ppm with the formation of the Pd-coordinated imidazol-ylidene species 5b (Fig. 1, iii). The CP-MAS NMR spectrum of the silica-immobilised 5b@SiO2 (Fig. 1, iv) showed a strong, broad signal at a similar chemical shift, confirming that most of the immobilised ligand is coordinated to Pd. Finally, in the spectrum of the 5b@SiO2+TMS species (Fig. 1, v) the signals arising from the incorporation of SiMe3 groups in the final material were detected at ca. 0 ppm (Fig. 1, v, labeled b). These results clearly show that at least the majority of the Pd–NHC complexes had been successfully anchored to the silica support without catalyst degradation.
Performance of the new silica-supported NHC–Pd complexes in the Suzuki–Miyaura reaction
|
|
||||
|---|---|---|---|---|
| Entrya | Catalyst | Mol% Pd | Time (h) | Yieldb (%) |
| a Conditions: 0.5 mmol of ArBr, 0.6 mmol of ArB(OH)2, 1.0 mmol of Cs2CO3, 1.5 mL of MePh, 0.75 mL of H2O, 60 °C. b By GC-FID, average of two runs. | ||||
| 1 | 4a@SiO2 | 1.5 | 20 | 40 |
| 2 | 5a@SiO2 | 1.5 | 20 | 35 |
| 3 | 4b@SiO2 | 1.0 | 5, 20 | 34, 58 |
| 4 | 4b@SiO2 | 2.0 | 20 | 85 |
| 5 | 5b@SiO2 | 1.5 | 20 | 85 |
| 6 | 4b@SiO2+TMS | 2.0 | 5 | 87 |
| 7 | 5b@SiO2+TMS | 1.0 | 5 | 88 |
| 8 | 5b@SiO2+TMS | 2.0 | 5 | >95 |
The most active catalyst formulation (5b@SiO2+TMS) was used to briefly explore the substrate scope achievable with this system; for full details, see the ESI.†ortho-Substituted coupling partners, activated bromides and chlorides and also heteroaromatic bromides and chlorides furnished the desired biaryl products in over 80% yield, except for the more challenging heteroaromatic halides for which only 50% and 45% yields were obtained in the case of 2-bromo- and 2-chloropyridine, respectively.
However, darkening of the reaction mixtures was observed in all cases, indicating the formation of Pd black and hence catalyst decomposition under these conditions. Indeed, when a series of recyclability tests were performed under these conditions, a rapid decrease in activity was observed after the first cycle and no conversion was achieved after the third cycle (see the ESI).†
To investigate the origin of such unexpected decomposition/deactivation, the soluble complex [Pd(1b)(acac)Cl)] (6b) was used as the precatalyst in the Suzuki–Miyaura coupling under the same reaction conditions used for their supported analogues. The formation of a black precipitate that was observed shortly after the reaction was brought to temperature indicated that catalyst decomposition also occurred in this case, ruling out a deleterious interaction between the metallic centre and the support. It was therefore thought that the use of water as co-solvent in the reaction medium might be detrimental to the stability of these catalysts and new experiments were carried out to investigate their activity and stability under anhydrous reaction conditions.
Several NHC–Pd based heterogenised catalysts have been reported that can operate in nonaqueous solvents. A variety of systems using anhydrous solvent/base such as iPrOH/t-BuOK,55,56 DMF/Cs2CO357 or xylene/K2CO358 have been used in the coupling of arylboronic acids with aryl and benzyl bromides and chlorides. In our case, further optimisation indicated that under anhydrous conditions these catalysts worked best in toluene using cesium carbonate as a base at a reaction temperature of 80 °C.
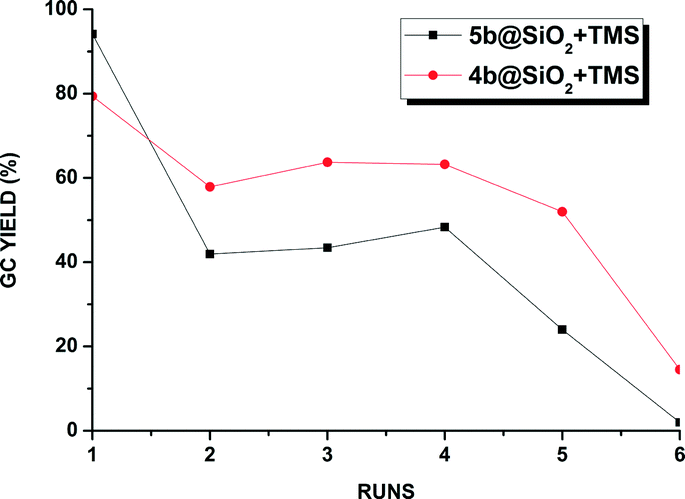
The recyclability of the one-tethered 5b@SiO2+TMS and the two-tethered 4b@SiO2+TMS catalysts was examined in the coupling of 1-bromo-2,4-dimethylbenzene with 4-methoxyphenylboronic acid under these alternative conditions. The results are shown in Fig. 2.
 | ||
| Fig. 2 Performance of the supported catalysts 4b@SiO2+TMS and 5b@SiO2+TMS under anhydrous conditions. | ||
As previously observed under aqueous biphasic conditions, higher activity was achieved using the single tethered 5b@SiO2+TMS in the first run, yielding 95% biaryl product after 12 h. In the next run a large drop in catalyst activity occurred and the coupling product was only obtained in ca. 40% yield. These moderate yields were maintained in the following two cycles before the conversion dropped gradually over the next two runs, resulting in only 3% conversion in the sixth cycle. On the other hand, double tethered 4b@SiO2+TMS, although outperformed in the first cycle by the one-side attached analogue, proved more robust and achieved yields in the 50–60% yield range over the next four cycles before eventually showing signs of degradation after the sixth reuse. No visible formation of precipitated palladium was observed in any case under these conditions and the potential leaching of Pd during each run was measured by ICP measurement (Fig. S2 in the ESI.†). However, no relevant amount of Pd could be detected.
|
|
|||
|---|---|---|---|
| Entry | ArB(OH)2 | Ar'X | Isolated yield (%) |
| a Conditions: 1 mmol of ArX, 1.2 mmol of ArB(OH)2, 2.0 mmol of Cs2CO3, 4 mL of toluene anh., 80 °C, 12 h. | |||
| 1 |
|
|
82 |
| 2 |
|
|
90 |
| 3 |
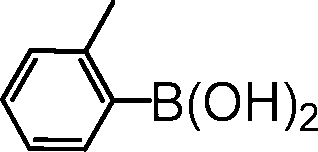
|
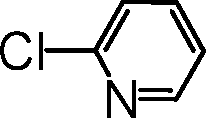
|
43 |
| 4 |
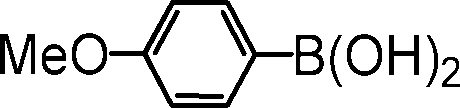
|
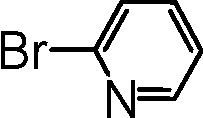
|
55 |
| 5 |
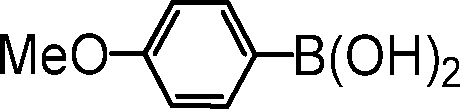
|
|
91 |
| 6 |
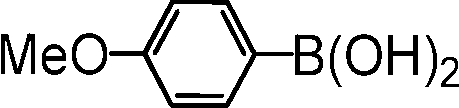
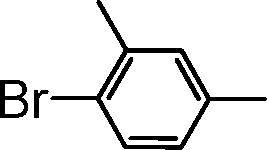
|
|
92 |
| 7 |
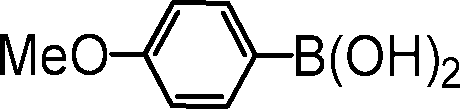
|
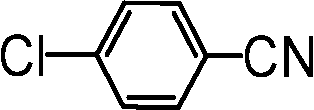
|
83 |
Evaluation of the performance of the new catalysts under continuous flow conditions
Next, performance of the silica-supported 4b@SiO2+TMS and 5b@SiO2+TMS was assessed under continuous flow operation. Dry methanol was used as a solvent because it offered the best compromise between the catalyst activity and the solubility of the reactants and by-product salts (Fig. 3a, experiments 1 and 2); for comparison, wet acetonitrile was also used as a solvent. Two solutions with different chemical compositions were used in the packed bed flow reactor and the catalyst and the corresponding amounts of Pd in the catalytic bed (see the ESI).†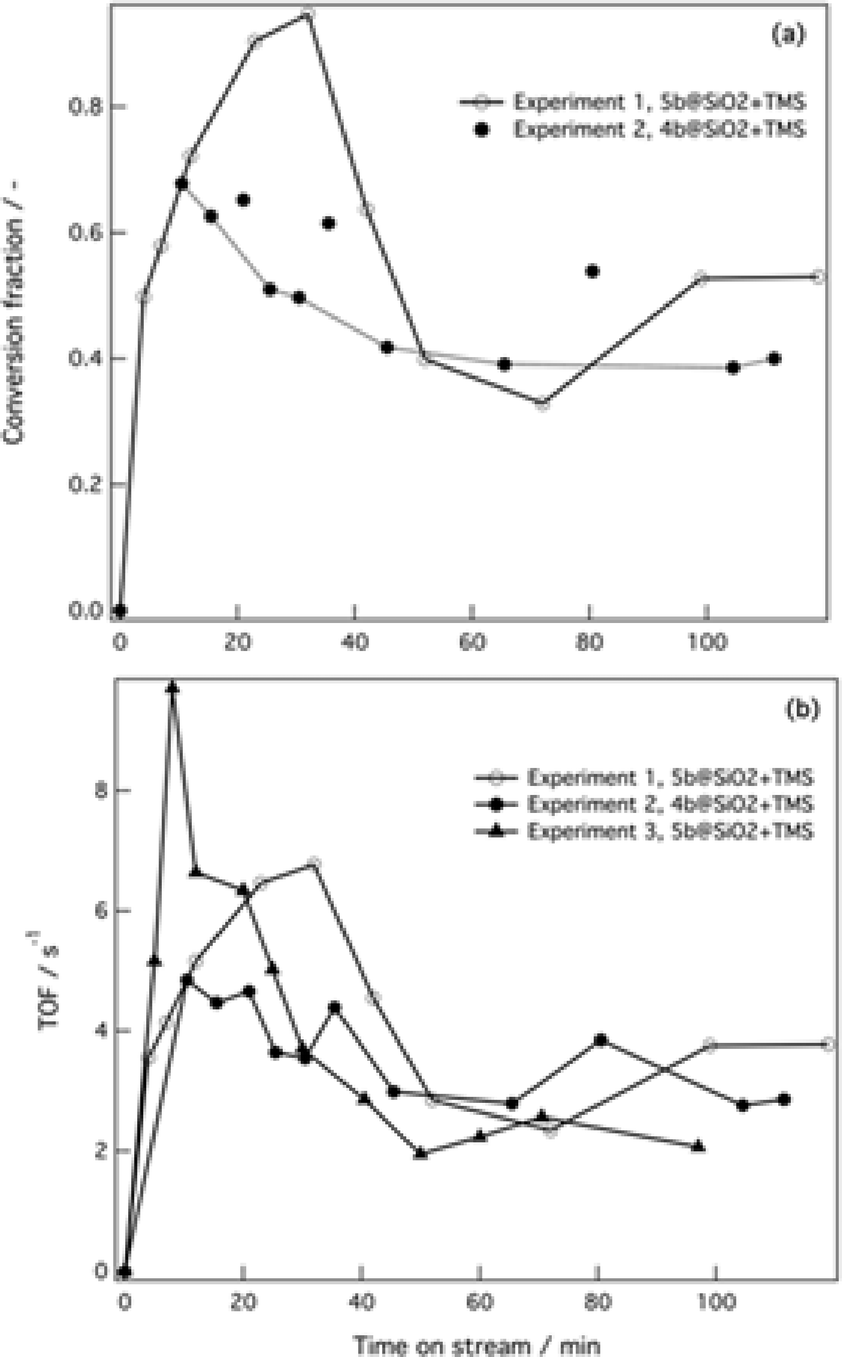 | ||
| Fig. 3 Experiments 1 and 2 took place in dry MeOH under anhydrous conditions. For both experiments solution I was 0.2 M in the corresponding ArBr and solution II was 0.4 M in base ( Cs2CO3) and 0.2 M in boronic acid. Experiment 3 was carried out in wet acetonitrile, solution I was 1 M in the ArBr and solution II was 2 M in base (K3PO4) and 1 M in boronic acid. (a) Conversion of the ArBr to the Suzuki coupling product vs. time on stream. (b) TOF vs. time on stream. Experiment numbers correspond to Table S3†; lines are given as guides to the eyes. C0 = 0.1 M in the reactor; u1 = u2 = 0.15 mL min−1 in both experiments. GC conversion; mesitylene was used as the internal standard. | ||
Different reaction parameters were used in the flow tests. For all the experiments, the temperature inside the flow reactor was set at 65 °C, the total flow rate was 0.30 mL min−1 and the residence time was 3.5 min for experiments 1 and 2 and 1.9 min for experiment 3.
The flow experimental conditions clearly show the trend of catalyst activation–deactivation under the reaction conditions. In all three experiments we observed an initial incubation period, which could indicate the possible generation of palladium nanoclusters, although no decomposition was apparent during the experiments.59 Full conversion of the bromide substrate was achieved with the 5b@SiO2+TMS catalyst after 40 min time-on-stream (see Fig. 3a, empty circles). Then, as also observed under batch conditions, conversion dropped to ca. 40–50%, and this level of activity was maintained over the following 90 min.
The 4b@SiO2+TMS catalyst also showed an incubation period after which the mixture that came out of the reactor reached a maximum conversion of 70% after 10 min on stream. In this case a smaller drop in conversion occurred compared with the single tethered catalyst, and conversion remained stable at 40–50% after two hours under continuous flow. When a biphasic aqueous–organic mixture was used (experiment 3, Fig. 3b), higher initial activities (expressed as turnover frequency, TOF) were achieved, followed by a decrease of 80% in catalytic activity after 45 minutes in flow, dropping from 10 to 2 s−1. Traces of the deboronation by-product as well as an unidentified by-product (possibly arising from homo-coupling) were observed for both catalysts. However, the selectivity of the ArBr substrate towards the Suzuki coupling product was found to be 93–96%. The color of the supported catalysts changed from the initial yellow to dark orange-brown over time. However, no visible Pd black was observed in the outlet flow.
The observed concentration vs. time-on-stream trends for several experiments show fluctuations in concentrations. These are correlated with periodic increases in the pressure drop within the packed catalyst bed caused by salt formation and its removal from the packed bed with the flow. The estimated residence time in the single-phase experiments is 3.5 min, resulting in 50–60% conversion. The considerably faster reaction in flow compared to batch conditions is due to the high ratio of catalyst to reactant in the packed-bed microreactor. Despite the observed fluctuations the overall conversion was relatively stable over ca. 90 min on stream.
Conclusions
In the present work, the synthesis and characterisation of a new family of silica-immobilised Pd–NHC precatalysts are reported, following a highly modular synthesis of the functionalised ligands recently reported by our group. The introduction of trialkoxysilyl groups to the imidazolium architecture allowed the covalent attachment of the Pd complexes onto the silica support. These supported catalysts were characterized by ICP, elemental analysis, and solid state 13C and 29Si NMR. These catalysts were active in the Suzuki–Miyaura reaction of aryl chlorides and bromides bearing sterically hindered substituents. The stability of these catalysts improved under anhydrous conditions and the coupling of 1-bromo-2,4-dimethylbenzene with 4-methoxyphenylboronic acid was carried out five successive times by simply filtering and washing the catalyst. Finally, 4b@SiO2+TMS and 5b@SiO2+TMS catalysts were evaluated under continuous flow operation. Moderate conversions were achieved at a relatively short residence time of 3.5 min and conversion was stable for two hours in flow, confirming the increased robustness of these systems under anhydrous reaction conditions.Experimental section
General considerations
Reactions were carried out using standard bench-top techniques unless the use of a Schlenk flask is specified, in which case inert atmosphere techniques were used. Where stirring of the reaction mixture is indicated, magnetic stirring using a Teflon-coated stir bar is employed throughout. Commercially supplied compounds were used without further purification. Dry solvents were prepared by distillation from CaH2/NaH or P2O5 or collected from an MBraun SPS800 solvent purification system. Photochemical reactions were performed using a Philips HPL-N 125 W high-pressure mercury lamp which can be purchased at most commercial lighting stores. Solution NMR spectra were obtained at the Servei de Recursos Científics i Tècnics (SRCT) of the URV using a 400 MHz Varian Mercury VX400 spectrometer and calibrated to the residual solvent peaks. CP-MAS spectra were recorded at the Servei de Ressonància Magnètica Nuclear (SeRMN), Universitat Autònoma de Barcelona on a 400 MHz spectrometer at a 12 kHz rotation speed (and calibrated to an external adamantane standard). Chemical shifts in the 1H and 13C{1H} NMR spectra are reported relative to TMS. ICP analyses were conducted at the SCRT using an ICP-OES Spectro Arcos instrument. Samples were digested in concentrated HNO3 under microwave irradiation before being diluted for analysis. HR-MS (ESI-TOF) analyses were also performed at the SCRT on an Agilent Time-of-Flight 6210 spectrometer. GC-MS and GS-FID analyses were conducted on Shimadzu GC-MS-QP2010 and Agilent 6850 instruments, respectively, fitted with HP-5 capillary columns. Elemental analyses were performed at the Centro de Microanálisis Elemental de la Universidad Complutense de Madrid or at the Unitat d'Anàlisi Química i Estructural-Serveis Tècnics de Recerca, Universitat de Girona. Aside from solvents, reagents obtained from commercial sources were used without further purification. Silica used for catalyst immobilization was 60 mesh chromatography-grade (purchased from SDS), dried for 1 h at 80 °C and 10−3 mbar prior to use. [Pd(acac)2]60 was prepared according to literature procedures. Preparative procedures for the imidazolium salt ligands and precatalysts 1a/1b, 2a/2b and 3a/3b, characterization data for all compounds and computational details can be found in the ESI.†Synthesis of supported catalysts
4-methoxy-2′,4′-dimethylbiphenyl. The reagents 4-bromo-m-xylene (136 μL, 1.0 mmol, 1.0 equiv.), 4-methoxyphenylboronic acid (182 mg, 1.2 mmol, 1.2 equiv.), Cs2CO3 (652 mg, 2.0 mmol, 2.0 equiv.), (5b@SiO2+TMS) (60 mg, 0.01 equiv.) were used as described in the general procedure for catalytic SM reaction runs. Purified by column chromatography using 25:1 n-hexane/EtOAc as the eluant. 1H NMR (400 MHz, CDCl3): δ 7.24 (d, J = 8.8 Hz, 2H), 7.12 (d, J = 7.7 Hz, 1H), 7.09 (s, 1H), 7.05 (d, J = 7.7 Hz, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.85 (s, 3H), 2.36 (s, 3H), 2.25 (s, 3H). NMR data match literature values.61 MS (EI): m/z = 212 (H+), 197, 181.
2,4-dimethyl-2′-methylbiphenyl. The reagents 4-bromo-m-xylene (136 μL, 1.0 mmol, 1.0 equiv.), 2-methylphenylboronic acid (164 mg, 1.2 mmol, 1.2 equiv.), Cs2CO3 (652 mg, 2.0 mmol, 2.0 equiv.), (5b@SiO2+TMS) (60 mg, 0.01 equiv.) were used as described in the general procedure for catalytic SM reaction runs. Purified by column chromatography using n-hexane as the eluant. 1H NMR (400 MHz, CDCl3): δ 7.26–7.19 (m, 3H), 7.11–7.09 (m, 2H), 7.04 (br d, J = 7.6 Hz, 1H), 7.00 (d, J = 7.6 Hz, 1H), 2.37 (s, 3H), 2.06 (s, 3H), 2.03 (s, 3H). MS (EI): m/z = 196 (M+), 181, 165, 152. NMR and MS peaks match literature values.62
2-(2-methylphenyl)pyridine. The reagents 2-bromopyridine (96 μL, 1.0 mmol, 1.0 equiv.), 2-methylphenylboronic acid (164 mg, 1.2 mmol, 1.2 equiv.), Cs2CO3 (652 mg, 1.0 mmol, 2.0 equiv.), (5b@SiO2+TMS) (30 mg, 0.01 equiv.) were used as described in the general procedure for catalytic SM reaction runs. Purified by column chromatography using 10
:1 n-hexane/EtOAc as the eluant. 1H NMR (400 MHz, CDCl3): δ 8.71 (d d d, J1 = 4.9 Hz, J2 = 1.8 Hz, J3 = 0.9 Hz, 1H), 7.78 (d d d, J1 = 7.7 Hz, J2 = 7.7 Hz, J3 = 1.8 Hz, 1H), 7.43 (d d d, J1 = 7.8 Hz, J2 = 1.1 Hz, J3 = 1.0 Hz, 1H), 7.41–7.39 (m, 1H), 7.32–7.26 (m, 4H), 2.03 (s, 3H). HR-MS: m/z = 170.1000, calcd. for C12H12N [M–H+]: 170.0966.
2-(4-methoxyphenyl)pyridine. The reagents 2-bromopyridine (96 μL, 1.0 mmol, 1.0 equiv.), 4-methoxyphenylboronic acid (182 mg, 1.2 mmol, 1.2 equiv.), Cs2CO3 (652 mg, 1.0 mmol, 2.0 equiv.), (5b@SiO2+TMS) (30 mg, 0.01 equiv.) were used as described in the general procedure for catalytic SM reaction runs. Purified by column chromatography using 10
:1 n-hexane/EtOAc as the eluant. 1H NMR (400 MHz, CDCl3): δ 8.65 (d d d, J1 = 4.9 Hz, J2 = 1.8 Hz, J3 = 1.0 Hz, 1H), 7.95 (d, J = 9.0 Hz, 2H), 7.75–7.66 (m, 2H), 7.18 (d d d, J1 = 7.2 Hz, J2 = 4.9 Hz, J3 = 1.3 Hz, 1H), 7.00 (d, J = 9.0 Hz, 2H), 3.87 (s, 3H). HR-MS: m/z = 186.0889, calcd. for C12H12NO [M–H+]: 186.0915.
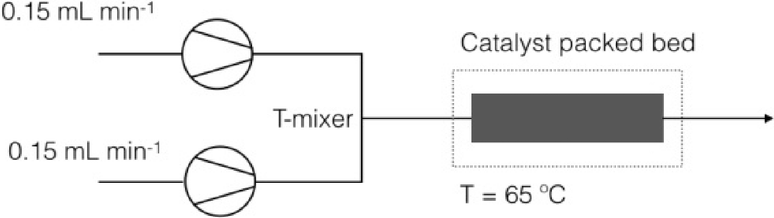 | ||
| Fig. 4 Scheme of a Vapourtec reactor. | ||
Two feed solutions were deoxygenated by bubbling N2 for 1 h prior to reactions and then were pumped by two HPLC pumps into the T-connection (PTFE, 0.5 mm through holes, Upchurch Scientific) via PFA tubing (1.6 mm in OD, 1.2 mm in ID) at equal flow rates (u1 = u2 = 0.15 mL min−1). The merged flow was then introduced to the inlet of the packed-bed column assembly (borosilicate glass with PTFE end pieces, 6.6 mm in ID × 100 mm in length, OmniFit).
The column was packed with a catalyst and has a catalytic bed length of 2.3 cm, which corresponds to a packed bed volume of 0.787 mL. The measured amount of the catalyst was diluted with QuadraSil AP (Johnson Matthey) spherical silica beads. The void volume in the catalytic bed in the approximation of random close packing of ideal spheres is 0.295 mL (the void fraction is about 0.375, neglecting the porosity of the support). The mean residence times in the packed bed were found to be 3.5 and 1.9 minutes in the cases of the single phase and biphasic aqueous–organic flow, respectively (calculated from the residence time distribution curves measured for the QuadraSil AP using a standard tracer technique). For the loadings of the catalysts used in this study this void corresponds to an initial molar ratio of Ar–Br:Pd ≈ 2.1 in the reactor in the case of the biphasic aqueous–organic flow or 0.4 in the case of the single phase conditions. The catalytic loadings in the reactor are still quite high even if we assume the porosity of the support to be about 60% (ArBr:Pd ≈ 4.2 or 0.9 for the biphasic and single phase conditions, respectively).
Acknowledgements
The authors are grateful to the 7th Framework Program (NMP2-LA-2010-246461; SYNFLOW) and the Spanish Ministerio de Economía y Competitividad (CTQ2010-14938/BQU, and a Ramon y Cajal fellowship to C. Godard) for financial support.References
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- V. Kumar, M. Paraschivoiu and K. D. P. Nigam, Chem. Eng. Sci., 2011, 66, 1329–1373 CrossRef CAS PubMed.
- L. Dalla-Vechia, B. Reichart, T. Glasnov, L. S. M. Miranda, K. C. O. Kappe and R. O. M. A. de Souza, Org. Biomol. Chem., 2013, 11, 6806–6813 CAS.
- D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed.
- C. Pavia, E. Ballerini, L. A. Vilona, F. Fiacalone, C. Aprile, L. Vaccaro and M. Gruttadauria, Adv. Synth. Catal., 2013, 355, 2007–2018 CrossRef CAS PubMed.
- J. M. Muñoz, J. Alcázar, A. de la Hoz and A. Díaz-Ortíz, Adv. Synth. Catal., 2012, 354, 3456–3460 CrossRef PubMed.
- K. Mennecke, W. Sodolenko and A. Kirschning, Synthesis, 2008, 10, 1589–1599 Search PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- N. Miyaura, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, New York, 2004 Search PubMed.
- F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 115, 2419–2440 Search PubMed.
- T. Noel and S. L. Buchwald, Chem. Soc. Rev., 2011, 40, 5010–5029 RSC.
- K. Mennecke and A. Kirschning, Synthesis, 2008, 20, 3267–3272 Search PubMed.
- A. Gömann, J. A. Deverell, K. F. Munting, R. C. Jones, T. Rodemann, C. A. J. Canty, J. A. Smith and R. M. Guijt, Tetrahedron, 2009, 65, 1450–1454 CrossRef PubMed.
- R. C. Jones, A. J. Canty, J. A. Smith, R. M. Guijt, T. Rodemann, J. A. Smith and V.-A. Tolhurst, Tetrahedron, 2009, 65, 7474–7481 CrossRef CAS PubMed.
- K. Mennecke and A. Kirschning, Beilstein J. Org. Chem., 2009, 5, 21 Search PubMed.
- U. Kunz, A. Kirschning, H.-L. Wen, W. Sodolenko, R. Cecilia, C. O. Kappe and T. Turek, Catal. Today, 2005, 105, 318–324 CrossRef CAS PubMed.
- N. Karbass, V. Sans, E. García-Verdugo, M. I. Burguete and S. V. Luis, Chem. Commun., 2006, 3095–3097 RSC.
- N. Nikbin, M. Ladlow and S. V. Ley, Org. Process Res. Dev., 2007, 11, 458–462 CrossRef CAS.
- G. Shore, S. Morin and M. G. Organ, Angew. Chem., Int. Ed., 2006, 45, 2761–2766 CrossRef CAS PubMed.
- D. A. Snyder, C. Noti, P. H. Seeberger, F. Schael, T. Bieber, G. Rimmel and W. Ehrfeld, Helv. Chim. Acta, 2005, 88, 1–9 CrossRef CAS PubMed.
- M. T. Rahman, T. Fukuyama, N. Kamata, M. Sato and I. Ryu, Chem. Commun., 2006, 2236–2238 RSC.
- Y. Uozumi, Y. M. A. Yamada, T. Beppur, N. Fukuyama, M. Ueno and T. Kitamor, J. Am. Chem. Soc., 2006, 128, 15994–15995 CrossRef CAS PubMed.
- Y. M. A. Yamada, T. Watanabe, K. Torii and Y. Uozumi, Chem. Commun., 2009, 5594–5596 RSC.
- Y. M. A. Yamada, T. Watanabe, T. Beppu, N. Fukuyama, K. Torii and Y. Uozumi, Chem. – Eur. J., 2010, 16, 11311–11319 CrossRef CAS PubMed.
- A. B. Theberge, G. Whyte, M. Frenzel, L. M. Fidalgo, R. C. R. Wootton and W. T. S. Huck, Chem. Commun., 2009, 6225–6227 RSC.
- S. Ceylan, C. Firese, C. Lammel, K. MAzac and A. Kirschning, Angew. Chem., Int. Ed., 2008, 47, 8950–8953 CrossRef CAS PubMed.
- S. Ceylan, L. Coutable, J. Wegner and A. Kirschning, Chem. – Eur. J., 2011, 17, 1844–1893 CrossRef PubMed.
- P. He, S. J. Haswell and P. D. I. Fletcher, Lab Chip, 2004, 4, 38–41 RSC.
- P. He, S. J. Haswell and P. D. I. Fletcher, Appl. Catal., 2004, 274, 111–114 CrossRef CAS PubMed.
- E. Comer and M. G. Organ, J. Am. Chem. Soc., 2005, 127, 8160–8167 CrossRef CAS PubMed.
- C. Ramarao, S. V. Ley, S. C. Smith, I. M. Shirley and N. DeAlmeida, Chem. Commun., 2002, 1132–1133 RSC.
- I. R. Baxendale, C. M. Griffiths-Jones, L. S. V. Ley and G. K. Tranmer, Chem. – Eur. J., 2006, 12, 4407–4416 CrossRef CAS PubMed.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- C. Valente, S. Calimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed.
- V. Sans, F. Gelat, M. I. Burguete, E. Garcia-Verdugo and S. V. Luis, Catal. Today, 2012, 196, 137–147 CrossRef CAS PubMed.
- Z. S. Quershi, S. A. Revankar, M. V. Khedkar and B. M. Bhanage, Catal. Today, 2012, 198, 148–153 CrossRef PubMed.
- A. Monge-marcet, R. Pleixats, T. Parella, X. Catooën and M. W. C. Man, J. Mol. Catal. A: Chem., 2012, 357, 59–66 CrossRef CAS PubMed.
- G. Borja, A. Monge-marcet, R. Pleixats, T. Parella, X. Catooën and M. W. C. Man, Eur. J. Org. Chem., 2012, 3625–3635 CrossRef CAS PubMed.
- W. Gil and A. M. Treciak, Coord. Chem. Rev., 2011, 255, 473–483 CrossRef CAS PubMed.
- W. J. Sommer and M. Weck, Coord. Chem. Rev., 2007, 251, 860–873 CrossRef CAS PubMed.
- D.-H. Lee, J.-H. Kim, B.-H. Jun, H. Kang, P. J. Park and L. Y.-S. Lee, Org. Lett., 2008, 10, 1609–1612 CrossRef CAS PubMed.
- M. Wang, P. Li and L. Wang, Eur. J. Org. Chem., 2008, 2251–2261 Search PubMed.
- L. Wan and C. Cai, Catal. Lett., 2012, 142, 1134–1140 CrossRef CAS.
- M. K. Samantaray, J. Alauzun, D. Gajan, S. Kavitake, A. Mehdi, L. Veyre, M. Lelli, A. Lesage, L. Emsley, C. Copéret and C. Thieuleux, J. Am. Chem. Soc., 2013, 135, 3193–3199 CrossRef CAS PubMed.
- G. Lázaro, F. J. Fernández-Alvarez, M. Iglesias, C. Horna, E. Vispe, R. Sancho, F. J. Lahoz, M. Iglesias, J. J. Pérez-torrente and L. A. Oro, Catal. Sci. Technol., 2014, 4, 62–70 Search PubMed.
- Y. Hou, X. Ji, G. Liu, J. Tang, J. Zheng, Y. Liu, W. Zhang and M. Jia, Catal. Commun., 2009, 10, 1459–1462 CrossRef CAS PubMed.
- G. Villaverde, A. Corma, M. Iglesias and F. Sánchez, ChemCatChem, 2011, 3, 1320–1328 CrossRef CAS PubMed.
- C. Schürer, S. Gessler, N. Buschmann and S. Blechert, Angew. Chem., Int. Ed., 2000, 39, 3898–3901 CrossRef.
- M. Mayr, R. M. Buchmeiser and K. Wurst, Adv. Synth. Catal., 2002, 344, 712–719 CrossRef CAS.
- J.-M. Collinson, J. D. E. T. Wilton-Ely and S. Díez-González, Chem. Commun., 2013, 49, 11358–11360 RSC.
- P. Li, W. A. Hermmann and F. E. Kühn, ChemCatChem, 2013, 5, 3324–3329 CrossRef CAS PubMed.
- H. Zhou, Y.-M. Wang, W.-Z. Zhang, J.-P. Qu and X.-B. Lu, Green Chem., 2011, 13, 644–650 RSC.
- S. Meiries, K. Speck, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 330–339 CrossRef CAS.
- J. L. Krinsky, A. Martínez, C. Godard, S. Castillón and C. Claver, Adv. Synth. Catal., 2014, 356, 460–474 CrossRef CAS PubMed.
- H. Q. Yang, X. J. Han, G. A. Li, Z. C. Ma and Y. J. Hao, J. Phys. Chem. C, 2010, 114, 22221–22229 CAS.
- H. Q. Yang, G. A. Li, Z. A. Ma, J. B. Chao and Z. Q. Guo, J. Catal., 2010, 276, 123–133 CrossRef CAS PubMed.
- J. Y. Shin, B. S. Lee, Y. Jung, S. J. Kim and S. G. Lee, Chem. Commun., 2007, 5238–5240 RSC.
- T. Sasaki, M. Tada, C. Zhong, T. Kume and Y. Iwasawa, J. Mol. Catal. A: Chem., 2008, 279, 200–209 CrossRef CAS PubMed.
- J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Domínguez, A. Leyva-Pérez and A. Corma, Science, 2012, 338, 1452–1455 CrossRef CAS PubMed.
- G. Kunstle and H. Siegl, US Pat. 3960909, 1976 Search PubMed.
- K. Pati and R. S. Liu, Chem. Commun., 2012, 48, 6049–6051 RSC.
- W. K. Chow, C. M. So, C. P. Lau and F. Y. Kwong, J. Org. Chem., 2010, 75, 5109–5112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00829d |
| This journal is © The Royal Society of Chemistry 2015 |