
Selective sulfoxidation with hydrogen peroxide catalysed by a titanium catalyst†
Lorena
Postigo
a,
Maria
Ventura
b,
Tomás
Cuenca
b,
Gerardo
Jiménez
*b and
Beatriz
Royo
*a
aInstituto de Tecnología Química e Biológica da Universidade Nova de Lisboa. Av. da República, EAN, 2780-157 Oeiras, Portugal. E-mail: broyo@itqb.unl.pt
bDepartamento de Química Orgánica y Química Inorgánica, Universidad de Alcalá, Campus Universitario, 28871 Alcalá de Henares, Spain
First published on 21st August 2014
Abstract
A moisture-tolerant cyclopentadienyl–silsesquioxane titanium complex efficiently catalyses the selective oxidation of various types of sulfides to either sulfoxides (TOFs up to 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 530 h−1) or sulfones with H2O2 (30% in water) under mild conditions.
530 h−1) or sulfones with H2O2 (30% in water) under mild conditions.
Introduction
The development of highly efficient catalytic systems for the oxidation of sulfides is of much current interest because the final products, sulfoxides and sulfones are important intermediates for the synthesis of chemically and biologically significant molecules1 and also in the context of desulfurisation of fuels.2The use of an environmentally benign oxidant such as H2O2 represents an important goal in oxidation reactions since it is non-toxic, cheap and forms water as the only by-product.3 Furthermore, the selective synthesis of sulfoxides over the fully oxidised sulfones represents another important challenge.
In recent years, some homogeneous H2O2-based catalytic systems mediated by transition metals for the oxidation of sulfides have been reported in the literature.4,5 However, most of them present disadvantages, such as the use of excess oxidant, low chemoselectivity, and limited applicability. Notable among these are the efficient catalytic systems based on Ti,6 V,7 and Fe8 using equimolecular amounts of H2O2.
Recently, we disclosed the excellent performance of robust cyclopentadienyl–silsesquioxane titanium complexes in olefin epoxidation using H2O2 as the oxidant.9 For the first time, we demonstrated the applicability of H2O2 as the oxidant in reactions catalysed by titanium silsesquioxanes. Encouraged by these exciting results, we decided to explore further their catalytic activity in oxidation processes. Herein, we report an efficient method for the selective oxidation of sulfides to either sulfoxides or sulfones under green conditions using H2O2 as the oxidant and methanol as solvent with the titanium complex [Ti{η5-C5H4SiMe2OPh7Si7O11-κ2O2}Cl] (1) (Fig. 1). To the best of our knowledge, this is the first report on the oxidation of sulfides by titanium silsesquioxane complexes using aqueous H2O2 as the primary oxidant. So far, the use of titanium silsesquioxanes in oxidation reactions has been limited to catalytic epoxidation of olefins.10–14
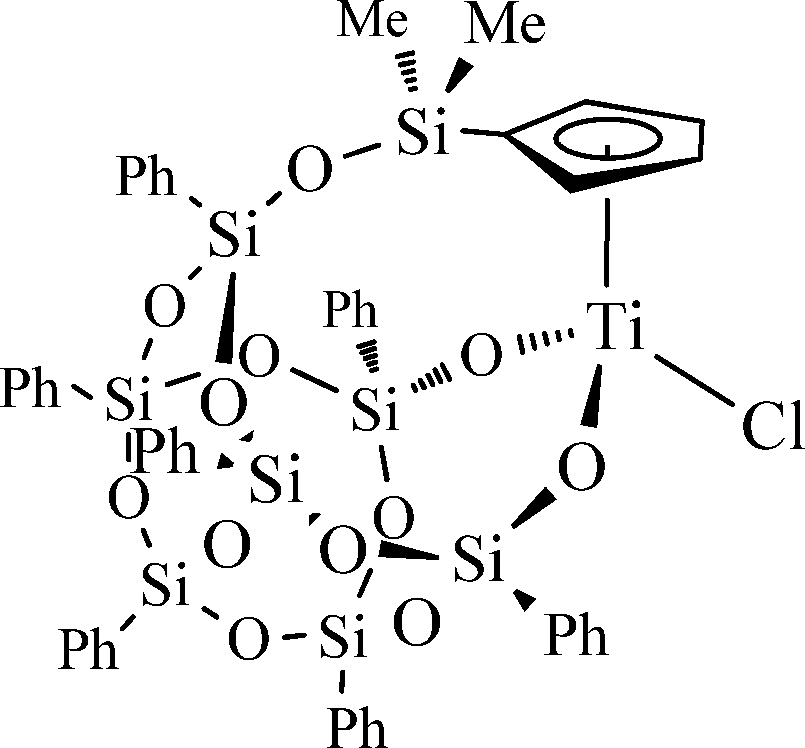 | ||
| Fig. 1 Titanium cyclopentadienyl–silsesquioxane complex (1). | ||
Results and discussion
We first investigated the oxidation of methylphenylsulfide (thioanisole) as a model substrate to explore the potential of 1 as a sulfoxidation catalyst and optimise the reaction conditions. Such oxidation was initially performed in methanol using a 0.5 mol% loading of 1 and 1 equiv. of H2O2 (30% in water) at ambient temperature. We were pleased to find that thioanisole was quantitatively and chemoselectively oxidised to methylphenylsulfoxide in 5 min (Table 1, entry 1). The reaction mixture was examined after 24 h, confirming that no overoxidation process to its corresponding sulfone occurred. In addition, blank experiments confirmed that no reaction took place in the absence of the catalyst.|
|
|||
|---|---|---|---|
| Entry | Cat. (mol%) | % Yieldb | TOFc |
| a All reactions were carried out with 0.54 mmol of thioanisole and 1 equiv. of H2O2 (30% aqueous solution) in MeOH at ambient temperature in 5 min. b Yield determined by 1H NMR spectroscopy using mesitylene (0.26 mmol) as the internal standard. c TOFs (turnover frequencies) calculated at 5 min of reaction. | |||
| 1 | 0.5 | >99 | 2385 |
| 2 | 0.25 | >99 | 4771 |
| 3 | 0.1 | 58 | 6987 |
| 4 | 0.01 | 27 | 32530 |
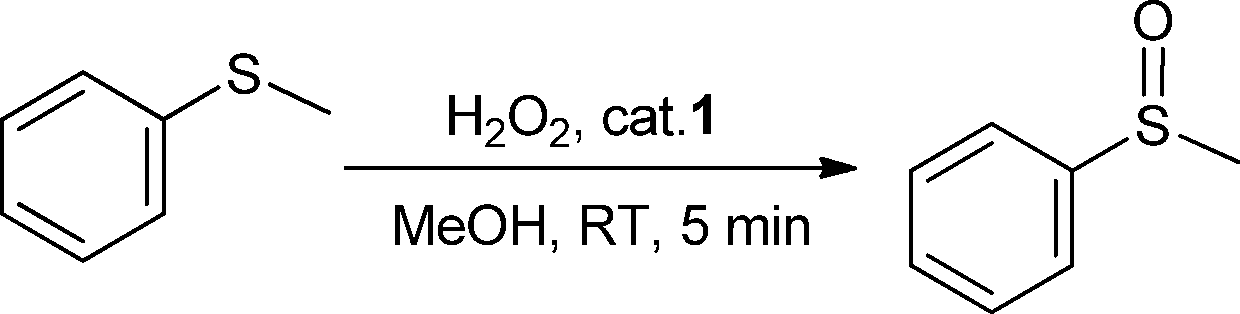
The catalyst loading could be lowered to 0.01 mol% (Table 1, entry 4) without appreciable deterioration of the effectiveness of the catalyst, achieving turnover frequency (TOF) values up to 32530 h−1. These TOF numbers are among the highest reported for titanium-catalysed reactions.6
We investigated the reuse and the selectivity of the catalyst by adding to the reaction mixture new charges of substrate and oxidant in a ratio of 1:1. Interestingly, the catalyst was reused for up to 14 cycles without any loss in its activity and selectivity. The total turnover attained was 2800. No sulfone was present in the reaction mixture after being stirred for ca. 6 h, indicating the remarkable chemoselectivity and stability of our catalyst under these reaction conditions.
To explore the scope of 1 as a sulfoxidation catalyst, we studied the oxidation of a wide variety of substrates with aqueous hydrogen peroxide under the optimised reaction conditions found for thioanisole (0.25 mol% of 1, 1 equiv. of H2O2, in methanol, and at room temperature). As summarised in Table 2, various aryl-alkyl (Table 2, entries 1–6) and dialkyl sulfides (Table 2, entries 7–9) were selectively oxidised in excellent yields (85–98%) within short reaction times (5–60 min). It is worth emphasising the high tolerance displayed by 1 toward sensitive functional groups such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, OH, OMe, N2O, COOEt and COOH. However, when using this protocol for oxidation of thiophene derivatives (benzothiophene and dibenzothiophene), the corresponding sulfones were formed along with the sulfoxides almost simultaneously. The low selectivity observed with diaryl sulfides is a general behaviour in electrophilic oxidation.15
C, OH, OMe, N2O, COOEt and COOH. However, when using this protocol for oxidation of thiophene derivatives (benzothiophene and dibenzothiophene), the corresponding sulfones were formed along with the sulfoxides almost simultaneously. The low selectivity observed with diaryl sulfides is a general behaviour in electrophilic oxidation.15
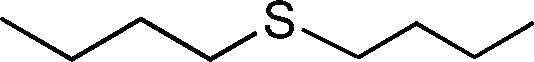

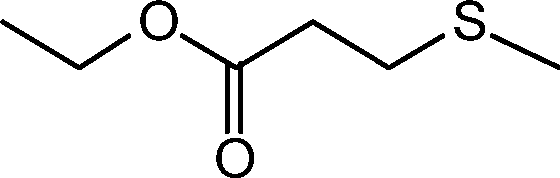
Based on these results, we decided to pursue the oxidation of sulfides to sulfones by increasing the substrate:oxidant molar ratio. Thus, when the oxidation of thioanisole was performed in methanol using 0.5 mol% loading of 1 and 2 equiv. of H2O2 (30% in water) at room temperature, a mixture of the sulfoxide and sulfone was observed. Nevertheless, we were delighted to find that a slight increment of the oxidant amount up to 2.3 equiv. and a reaction temperature of 50 °C led to quantitative oxidation of thioanisole to methylphenylsulfone in a reasonable short reaction time (>99% yield in 3 h, Table 3, entry 1). Remarkably, a quasi-equivalent amount of H2O2 was sufficient to achieve the 99% yield of sulfone, indicating the high efficiency of this oxidation. The effect of the catalyst amount on the reaction rate was evaluated in methanol; decreasing the amount of the catalyst to 0.25 and 0.1 mol% significantly slowed down the oxidation process (Table 3, entries 2 and 3, respectively). The oxidation in different solvents under the optimum reaction conditions was also investigated. As shown in Table 3, methanol and acetonitrile yielded quantitative methylphenylsulfone in 3 h, while lower yields were obtained in dichloromethane and acetone (Table 3, entries 1, 4, 5, and 6, respectively).
|
|
|||
|---|---|---|---|
| Entry | Cat. (mol%) | % Yieldb | Solvent |
| a All reactions were carried out with 0.54 mmol of thioanisole and 2.3 equiv. of H2O2 (30% aqueous solution) in MeOH in 3 h at 50 °C. b Yield determined by 1H NMR spectroscopy using mesitylene (0.26 mmol) as the internal standard. c Reaction time: 5 h. | |||
| 1 | 0.5 | >99 | MeOH |
| 2 | 0.25 | 76 | MeOH |
| 3 | 0.1 | 53 | MeOH |
| 4 | 0.5 | >99 | NCMe |
| 5c | 0.5 | 56 | CH2Cl2 |
| 6c | 0.5 | 60 | Acetone |
Further, we explored the scope of the oxidation of sulfides to sulfones with a wide variety of substrates under the optimised reaction conditions using 0.5 mol% of catalyst 1 and 2.3 equiv. of H2O2 in methanol at 50 °C. Various alkyl-aryl (Table 4, entries 1–7), dialkyl (Table 4, entries 8–10), and diaryl (Table 4, entries 11–13) sulfides were oxidised quantitatively to their corresponding sulfones with high efficiency within relatively short periods of time. Results are summarised in Table 4. Significantly, challenging compounds such as benzothiophene (BTP) and dibenzothiophene (DBT) were also oxidised to their corresponding sulfones in quantitative yields (Table 4, entries 12 and 13), although a higher amount of catalyst was required for these substrates (2 mol% of 1).
| Entry | Substrate | t (h) | % Yieldb |
|---|---|---|---|
| a All reactions were carried out with 0.54 mmol of the substrate and 2.3 equiv. of H2O2 (30% aqueous solution) with 0.5 mol% of catalyst 1 in MeOH at 50 °C. b Yield determined by 1H NMR spectroscopy using mesitylene (0.26 mmol) as the internal standard. The numbers in parentheses are isolated yields. c Yield determined by GC analysis, reactions done with 2 mol% of catalyst 1. The numbers in parentheses are isolated yields. | |||
| 1 |
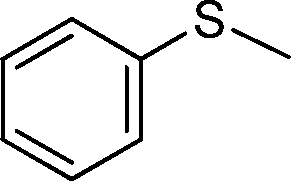
|
3 | >99 (98) |
| 2 |
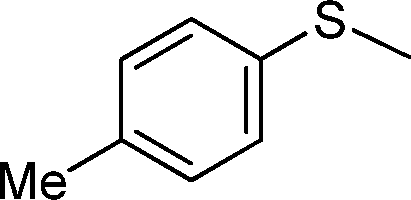
|
3 | >99 (70) |
| 3 |
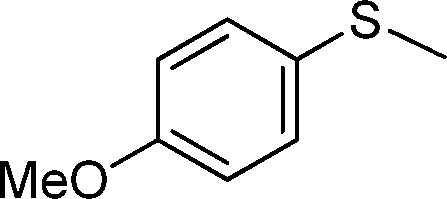
|
3 | >99 (85) |
| 4 |
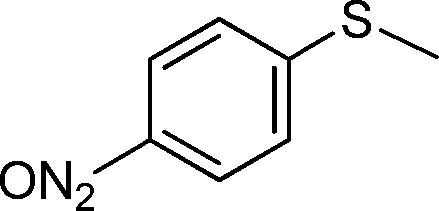
|
5 | >99 (84) |
| 5 |
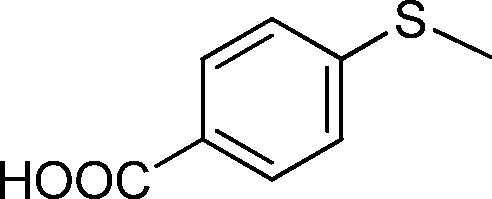
|
3 | >99 (85) |
| 6 |
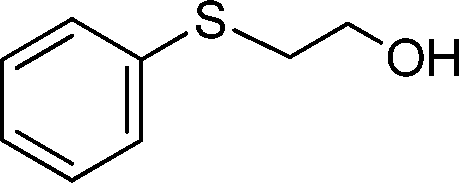
|
0.5 | >99 (95) |
| 7 |
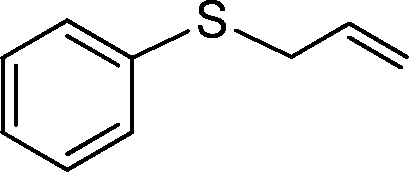
|
3 | >99 (72) |
| 8 |
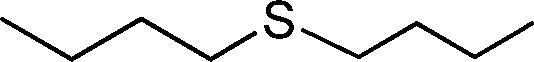
|
3 | >99 (98) |
| 9 |
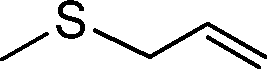
|
3 | >99 (97) |
| 10 |
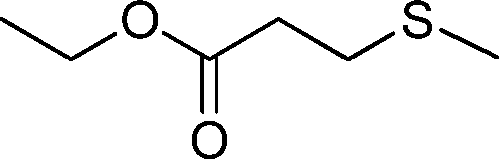
|
3 | >99 (91) |
| 11 |
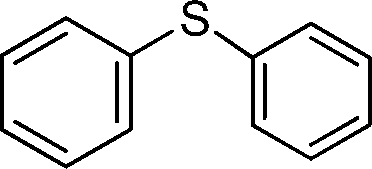
|
5 | >99 (60)c |
| 12 |
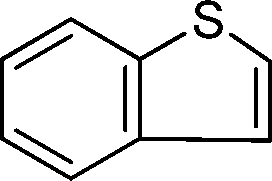
|
5 | >99 (56)c |
| 13 |
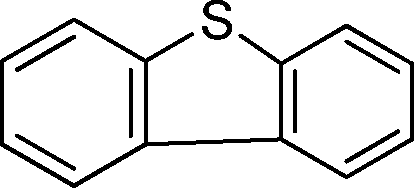
|
5 | >99 (70)c |
The generally accepted mechanism for titanium-based oxidation catalysts involves the formation of a peroxo-titanium active species, in which the peroxo group is η2 coordinated to the metal centre.6 In order to gain insight into the species formed during the course of the reaction, catalyst 1 was treated with a 10-fold excess of H2O2 (30% in water) in acetonitrile at ambient temperature, and the reaction was followed by MS-ESI. The MS-ESI spectrum of 1 displays an intense peak at m/z 1096.8 corresponding to the titanium ion [C49H45O12Si8Ti]+, [M-Cl]+. Immediately after addition of H2O2, a new intense peak at m/z 1146.9 appeared in the mass spectrum while the peak at 1096.8 completely disappeared from the spectrum. The peak at m/z 1146.9 displayed the characteristic pattern arising from the natural abundance of titanium isotopes and it was assigned to the aquo peroxo-titanium species [M(O–O)(H2O)]+, which might be the catalytically active species.† The MS-ESI experiments were performed in acetonitrile instead of methanol (the solvent used in the catalytic assays) due to the poor solubility of the titanium species in methanol. Recently, Licini and Zonta have reported an elegant mechanistic study of the oxidation of sulfides with H2O2 in methanol using a titanium(IV) aminotriphenolate complex.16 Based on kinetic experiments and theoretical calculations, they proved the fundamental role of methanol in the activation of H2O2 in sulfoxidation. In order to clarify if MeOH could coordinate to the titanium centre of 1, we decided to study the stability of 1 in methanol by preparing a solution of 1 in deuterated chloroform, adding some drops of methanol, and monitoring the sample by NMR spectroscopy. We observed that complex 1 was maintained intact after 24 h in methanol at room temperature. Complex 1 also exhibited remarkable stability towards water; wet methanol solutions of 1 could be kept for 48 h without exhibiting any decomposition. These observations corroborate that methanol does not coordinate to complex 1. The use of methanol does not seem to accelerate the oxidation reaction, since similar activity of catalyst 1 was found when the reactions were performed in acetonitrile (Table 3, entries 1 and 4, respectively).
Conclusions
In summary, the titanium complex 1 acts as a highly efficient catalyst for the chemoselective oxidation of sulfides either to sulfoxides or to sulfones with near-stoichiometric amount of aqueous H2O2. This method represents the first example of a titanium silsesquioxane complex catalysing the oxidation of sulfides under environmentally friendly (H2O2/methanol) and mild conditions. It is worth noting that catalyst 1 showed remarkable stability towards water, being reused for up to 14 cycles without appreciable loss in its activity. We are currently investigating the potential of this type of catalyst in other oxidation processes with hydrogen peroxide.Experimental section
Materials and methods
The compound Ti(η5-C5H4SiMe2Cl)Cl3 was synthesised according to the method described in the literature.17 All other reagents were used as received from commercial suppliers without further purification. All manipulations were carried out under a nitrogen atmosphere using standard Schlenk techniques and solvents were purified from appropriate drying agents. Deuterated solvents were degassed and stored over molecular sieves. 1H, 13C, and 29Si NMR spectra were recorded on a Bruker Avance III 400 MHz. Electrospray mass spectra (MS-ESI) were recorded on a Micromass Quattro LC instrument; nitrogen was employed as drying and nebulising gas.Catalytic oxidation of sulfides to sulfoxides
To a stirred mixture of catalyst, sulfide (0.54 mmol), and mesitylene (0.26 mmol, 37 μl) in MeOH (1.25 mL), 1 equiv. of H2O2 (30% aqueous solution, 60 μl) was added. The course of the reaction was monitored by 1H NMR or analyzed by GC. In the latter case, aliquots of 0.1 mL were taken at regular intervals, treated with MnO2 in 0.4 mL of CH2Cl2, and filtered over Celite previously to be analysed. The products were characterized by 1H and 13C NMR spectroscopy.Catalytic oxidation of sulfides to sulfones
A procedure similar to that described for sulfoxides was adopted by using 2.3 equiv. of H2O2 (30% aqueous solution, 140 μl) and heating to 50 °C.To isolate the final sulfone or sulfoxide, the above-mentioned reactions were performed without mesitylene and kept until completion of the reaction. The final mixture was treated with MnO2 in 2 mL of CH2Cl2, filtered over a Celite pad, and volatile liquids were removed under vacuum to afford the corresponding sulfone or sulfoxide. The data produced were identified by comparison of the NMR spectra with the reported data.18
Acknowledgements
We gratefully acknowledge financial support from FCT of Portugal, POCI 2010 and FEDER through projects PTDC/QEQ-QIN/0565/2012, CRUP and the Ministerio de Educación y Ciencia for Bilateral Action Program. We thank FCT for REDE/1517/RMN/2005. M.V. thanks the University of Alcalá for a fellowship. C. M. Almeida is acknowledged for providing data from Mass Spectrometry Services at ITQB.Notes and references
- (a) N. S. Simpkins, in Sulphones in Organic Synthesis, ed. J. E. Baldwin and P. D. Magnus, Pergamon, Oxford, 1993 Search PubMed; (b) I. Fernández and N. Khiar, Chem. Rev., 2003, 103, 3651–3705 CrossRef; (c) P. Kowalski, K. Mitka, K. Ossowska and Z. Kolarska, Tetrahedron, 2005, 61, 1933–1953 CrossRef CAS; (d) E. Wojaczynska and J. Wojaczynski, Chem. Rev., 2010, 110, 4303–4356 CrossRef CAS.
- (a) V. Babich and J. A. Moulijn, Fuel, 2003, 82, 607–631 CrossRef; (b) J. Xu, S. Zhao, W. Chen, M. Wang and Y.-F. Song, Chem. – Eur. J., 2012, 18, 4775–4781 CrossRef CAS.
- (a) C. W. Jones, in Applications of Hydrogen Peroxide and Derivatives, Royal Society of Chemistry, Cambridge, 1999 Search PubMed; (b) R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC; (c) B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2473 CrossRef CAS; (d) G. Grigoropoulor, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1–7 RSC.
- Selected references for sulfide oxidation to sulfoxides: (a) S. Hussain, D. Talukdar, S. K. Bharadwaj and M. K. Chaudhuri, Tetrahedron Lett., 2012, 53, 6512–6515 CrossRef CAS; (b) K. Kamata, T. Hirano, R. Ishimoto and N. Mizuno, Dalton Trans., 2010, 39, 5509–5518 RSC; (c) M. Mba, L. J. Prins and G. Licini, Org. Lett., 2007, 9, 21–24 CrossRef CAS; (d) P. Adão, F. Avecilla, M. Bonchio, M. Carraro, J. C. Pessoa and I. Correia, Eur. J. Inorg. Chem., 2010, 5568–5578 CrossRef; (e) P. Adão, M. L. Kuznetsov, S. Barros, A. M. Martins, F. Avecilla and J. C. Pessoa, Inorg. Chem., 2012, 51, 11430–11449 CrossRef; (f) E. P. Talsi and K. P. Bryliakov, Appl. Organomet. Chem., 2013, 27, 239–244 CrossRef CAS; (g) O. Hamelin, S. Mégane, F. Charnay, M. Chavarot, J.-L. Pierre, J. Pécaut and M. Fontecave, Inorg. Chem., 2008, 47, 6413–6420 CrossRef CAS; (h) C. Yang, Q. Jin, H. Zhang, J. Zhu, B. Yu and J. Deng, Green Chem., 2009, 11, 1401–1405 RSC; (i) C. A. Gamelas, T. Lourenço, A. P. da Costa, A. L. Simplicio, B. Royo and C. C. Romão, Tetrahedron Lett., 2008, 49, 4708–4712 CrossRef CAS; (j) K. Jeyakumar and D. K. Chand, Tetrahedron Lett., 2006, 47, 4573–4576 CrossRef CAS.
- Selected references for sulfide oxidation to sulfones: (a) K. Jeyakumar and D. K. Chand, Tetrahedron Lett., 2006, 47, 4573–4576 CrossRef CAS; (b) K. Bahrami, M. M. Khodaei, V. Shakibaian, D. Khaledian and B. H. Yousefi, J. Sulfur Chem., 2012, 33, 155–163 CrossRef CAS; (c) E. Barker and T. Ren, Tetrahedron Lett., 2004, 45, 4681–4683 CrossRef; (d) L. Xu, J. Cheng and M. L. Trudell, J. Org. Chem., 2003, 68, 5388–5391 CrossRef CAS.
- (a) C. Zonta, E. Cazzola, M. Mba and G. Licini, Adv. Synth. Catal., 2008, 350, 2503–2506 CrossRef CAS; (b) G. Licini, M. Mba and C. Zonta, Dalton Trans., 2009, 5265–5277 RSC; (c) M. Mba, L. J. Prins, C. Zonta, M. Cametti, A. Valkonen, K. Rissanen and G. Licini, Dalton Trans., 2010, 39, 7384–7392 RSC; (d) M. K. Panda, M. M. Shaikh and P. Ghosh, Dalton Trans., 2010, 39, 2428–2440 RSC; (e) W. Al-Maksoud, S. Daniele and A. B. Sorokin, Green Chem., 2008, 10, 447–451 RSC; (f) A. M. Cojocariu, P. H. Mutin, E. Dumitriu, F. Fajula, A. Vioux and V. Hulea, Chem. Commun., 2008, 5357–5359 RSC.
- (a) P. Kowalski, K. Mi, C. Drago, L. Caggiano and R. F. Jackson, Angew. Chem., Int. Ed., 2007, 9, 21–24 Search PubMed; (b) T. S. Smith and V. L. Pecoraro, Inorg. Chem., 2002, 41, 6754–6760 CrossRef CAS; (c) M. Mba, M. Pontini, S. Lovat, C. Zonta, G. Bernardinelli, P. E. Kündig and G. Licini, Inorg. Chem., 2008, 47, 8616–8618 CrossRef CAS.
- (a) J. Legros and C. Bolm, Chem. – Eur. J., 2005, 11, 1086–1092 CrossRef CAS; (b) J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2003, 42, 5487–5489 CrossRef CAS; (c) W. D. Kerber, B. Ramdhanie and D. P. Goldberg, Angew. Chem., Int. Ed., 2007, 46, 3718–3721 CrossRef CAS; (d) E. Baciocchi, M. F. Gerini and A. Lapi, J. Org. Chem., 2004, 69, 3586–3589 CrossRef CAS.
- M. Ventura, M. E. G. Mosquera, T. Cuenca, B. Royo and G. Jiménez, Inorg. Chem., 2012, 51, 6345–6349 CrossRef CAS PubMed.
- H. C. L. Abbenhuis, S. Krijnen and R. A. van Santen, Chem. Commun., 1997, 331–332 RSC.
- M. Crocker, R. H. M. Herold, A. G. Orpen and M. T. A. Overgaag, J. Chem. Soc., Dalton Trans., 1999, 3791–3804 RSC.
- M. D. Skowronska-Ptasinska, M. L. Vorstenbosch, R. A. van Santen and H. C. L. Abbenhuis, Angew. Chem., Int. Ed., 2002, 41, 637–639 CrossRef CAS.
- T. Giovenzana, M. Guidotti, E. Lucenti, A. O. Biroli, L. Sordelli, A. Sironi and R. Ugo, Organometallics, 2010, 29, 6687–6694 CrossRef CAS.
- (a) K. Wada, S. Sakugawa and M. Inoue, Chem. Commun., 2012, 48, 7991–7993 RSC; (b) S. Sakugawa, K. Wada and M. Inoue, J. Catal., 2010, 275, 280–287 CrossRef CAS PubMed.
- M. Bonchio, S. Campestrini, V. Conte, F. D. Furia and S. Moro, Tetrahedron, 1995, 51, 12363–12372 CrossRef CAS.
- C. Zonta and G. Licini, Chem. – Eur. J., 2013, 19, 9438–9441 CrossRef CAS PubMed.
- S. Ciruelos, T. Cuenca, P. Gómez-Sal, A. Manzanero and P. Royo, Organometallics, 1995, 14, 177–185 CrossRef CAS.
- (a) W. E. Truce and R. H. Knope, J. Am. Chem. Soc., 1955, 77, 5063–5067 CAS; (b) R. W. Brown, J. Org. Chem., 1991, 56, 4974–4976 CrossRef CAS; (c) V. Mojr, V. Herzig, M. Budesíncky, R. Cibulka and T. Draus, Chem. Commun., 2010, 46, 7599–7601 RSC; (d) X. Shi, X. Han, W. Ma, J. Wei, J. Li, Q. Zhang and A. Chen, J. Mol. Catal. A: Chem., 2011, 341, 57–62 CrossRef CAS PubMed; (e) S. P. Das, J. J. Boruah, H. Chetry and N. S. Islam, Tetrahedron Lett., 2012, 53, 1163–1168 CrossRef CAS PubMed; (f) S. Wang, L. Wang, M. Dakovic, Z. Popovic, H. Wu and Y. Liu, ACS Catal., 2012, 2, 230–237 CrossRef CAS; (g) X. Xue, W. Zhao, B. Ma and Y. Ding, Catal. Commun., 2012, 29, 73–76 CrossRef CAS PubMed; (h) Y. Imada, M. Takagishi, N. Komiya and T. Noata, Synth. Commun., 2013, 43, 3064–3071 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and MS-ESI spectra of the compounds. See DOI: 10.1039/c4cy00965g |
| This journal is © The Royal Society of Chemistry 2015 |